

Abstract
Lysosomal membrane permeabilization (LMP) contributes to tissue involution, degenerative diseases, and cancer therapy. Its investigation has, however, been hindered by the lack of sensitive methods. Here, we characterize and validate the detection of galectin puncta at leaky lysosomes as a highly sensitive and easily manageable assay for LMP. LGALS1/galectin-1 and LGALS3/galectin-3 are best suited for this purpose due to their widespread expression, rapid translocation to leaky lysosomes and availability of high-affinity antibodies. Galectin staining marks individual leaky lysosomes early during lysosomal cell death and is useful when defining whether LMP is a primary or secondary cause of cell death. This sensitive method also reveals that cells can survive limited LMP and confirms a rapid formation of autophagic structures at the site of galectin puncta. Importantly, galectin staining detects individual leaky lysosomes also in paraffin-embedded tissues allowing us to demonstrate LMP in tumor xenografts in mice treated with cationic amphiphilic drugs and to identify a subpopulation of lysosomes that initiates LMP in involuting mouse mammary gland. The use of ectopic fluorescent galectins renders the galectin puncta assay suitable for automated screening and visualization of LMP in live cells and animals. Thus, the lysosomal galectin puncta assay opens up new possibilities to study LMP in cell death and its role in other cellular processes such as autophagy, senescence, aging, and inflammation.
Keywords: breast involution, C. elegans, cell death, galectin, lysosome
Abbreviations
- AF
Alexa Fluor
- C. elegans
Caenorhabditis elegans
- BAX
BCL2-associated X protein
- CD63
CD63 molecule
- CTSB
cathepsin B
- DPBS
Dulbecco's phosphate-buffered saline
- EEA1
early endosome antigen 1
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GOLGA1
golgin A1
- HRP
horseradish peroxidase
- HSPA9
heat shock 70kDa protein 9 (mortalin)
- L4
stage 4 larvae
- LAMP
lysosomal-associated membrane protein
- LGALS
lectin galactoside-binding soluble (galectin)
- LLOMe
L-leucyl-L-leucine methyl ester
- LMNB1
lamin B1
- LMP
lysosomal membrane permeabilization
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MOMP
mitochondrial outer membrane permeabilization
- NUC-1
abnormal NUClease (C. elegans homolog of DNAse II)
- PDIA2
protein disulfide isomerase family A, member 2
- RAB5
RAB5, member RAS oncogene family
- SCID/FOX
FOX CHASE severe combined immunodeficiency
- TNF
tumor necrosis factor
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- YFP
yellow fluorescent protein
Introduction
Lysosomes serve as cellular recycling centers for cargo received mainly through autophagy and endocytosis. For this purpose, they are filled with hydrolases capable of degrading most cellular macromolecules. Consequently, lysosomal membrane permeabilization (LMP) and the subsequent leakage of lysosomal hydrolases into the cytosol can lead to so-called “lysosomal cell death,” which can present with necrotic, apoptotic or apoptosis-like features depending on the extent of the leakage and the cellular context, and is often misnamed as autophagic cell death due to the accumulation of autophagosomes.1-6 Lysosomal cell death is highly conserved in evolution from yeast, roundworm, and fruit fly to mammals and has important physiological functions, e.g. in mammary gland involution and immune tolerance.7-9 It also contributes to pathologies of various degenerative and bacterial diseases as well as efficacies of old and new cancer therapies.10-20 This mode of cell death remains, however, poorly understood largely due to the lack of appropriate detection methods. Currently, the most sensitive microcopy-based method for the detection of LMP is based on the release of fluorescently labeled dextran molecules from the lysosomes into the cytosol. This method requires the loading of dextran molecules into the lysosomes by endocytosis and carries therefore a risk of interfering with normal lysosomal function. Alternatively, the appearance of endogenous lysosomal enzymes, e.g. cathepsin proteases, in the cytosol can be detected by immunocytochemistry in intact cells and by immunoblotting or enzyme activity assays after cellular fractionation. The immunocytochemistry-based detection offers only a limited sensitivity as it fails to detect small amounts of released lysosomal hydrolases that could be sufficient to trigger cell death. While relatively sensitive, the detection of released enzymes in the cytosolic fraction carries a risk of artifacts caused by sample processing during the extraction of the cytosol. Moreover, none of these methods is suitable for immunohistochemistry or detection of individual damaged lysosomes and their subsequent fate, e.g. recovery or removal by autophagy (lysophagy). It should also be noted that even transmission electron microscopy without preloading of the lysosomes with e.g. gold-albumin fails to detect partial lysosomal leakage, which does not change the ultrastructure of lysosomes or other cellular compartments.21,22 Consequently, a better assay for LMP is urgently needed.
Galectins are soluble carbohydrate-binding lectins defined by their ability to bind β-galactoside sugars with one or 2 conserved carbohydrate-recognition domains.23 To date, 10 human galectins with different expression patterns and sugar binding affinities have been identified, and highly conserved members of this family are present in organisms from roundworms to mammals.24 Galectins are present in the cytosol and nucleus as well as in the extracellular space. The binding of extracellular galectins to cell surface glycans can modulate cellular behavior by regulating transmembrane signaling as well as cell-cell and cell-matrix interactions whereas the physiological role of galectins in the cytosol and nucleus, which are devoid of β-galactoside sugars, has remained largely mysterious,25 except for the recently identified role of cytosolic LGALS8/galectin-8 in autophagy-mediated defense against the invasion of the host cytosol by Salmonella typhimurium.26 An idea to develop a galectin-based method to detect LMP was inspired by reports demonstrating the translocation of ectopic LGALS3/galectin-3 to phagosomal membranes upon bacteria-induced rupture,27-29 and to the early endosomal vesicles upon adenoviral entry.30 The endo-lysosomal glycocalyx being especially rich in β-galactosides, we reasoned that galectins could also be recruited to the sites of endo-lysosomal leakage. Supporting this hypothesis, recent studies have demonstrated that even in the absence of bacteria or viruses, some galectins can translocate to damaged endo-lysosomes before their autophagy-mediated removal.26,31 Prompted by these data, we investigated the sensitivity and specificity of galectin puncta formation as a novel assay for LMP. We show that LGALS1/galectin-1 and LGALS3 puncta assays are superior to existing LMP assays in sensitivity and applicability. Furthermore, they detect LMP even in the absence of cell death, allow the identification of a fragile subpopulation of lysosomes and open a range of new possibilities to study LMP in cells, tissues, and live animals.
Results
LGALS1 is highly expressed in most normal tissues and cancer cell lines
To identify which galectin would be most suitable for a universal LMP assay, we compared their expression using publicly available data. According to the Human Protein Atlas RNA-Seq data for a panel of normal tissue samples and cancer cell lines (www.proteinatlas.org, Version 12, Ensembl version 73.3732) LGALS1 mRNA was most highly expressed in around 70% of the samples, followed by LGALS3 mRNA (Fig. 1A and B). In contrast, the levels of LGALS8 and LGALS9 mRNAs, as well as other human LGALS mRNAs, were very low or undetectable in most samples (Fig. 1A and B and data not shown). Data extracted from the published proteome analysis of the NCI-60 cancer cell panel33 confirmed that LGALS1 and LGALS3 proteins were expressed in most cell lines, LGALS1 levels being higher in around 90% of cases (Fig. 1C). Thus, endogenous LGALS1 and LGALS3 appear promising as general markers of LMP.
Figure 1.
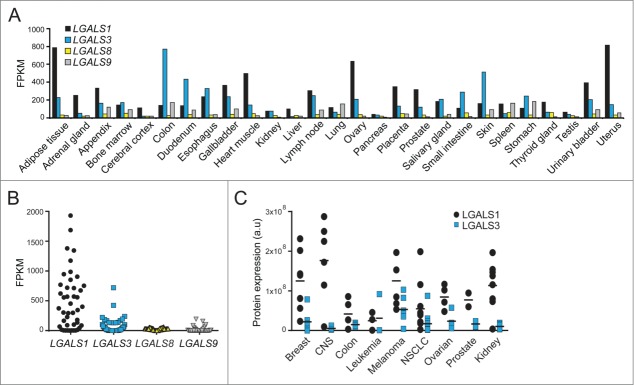
Expression profiles of LGALS mRNAs and protein products. (A and B) Expression levels of LGALS1, LGALS3, LGALS8, and LGALS9 mRNA in indicated normal human tissues (A) and 44 cancer cell lines (B) were extracted from RNA-Seq data published in the Human Protein Atlas (www.proteinatlas.org). FPKM, fragments per kb of transcript per million mapped fragments. (C) Expression levels of LGALS1 and LGALS3 proteins in 51 human cancer cell lines were extracted from published mass spectrometry data.33 CNS, central nervous system; NSCLC, non-small cell lung cancer.
Endogenous LGALS1 and LGALS3 translocate to damaged lysosomes
Next, we investigated whether endogenous LGALS1 or LGALS3 translocated to damaged lysosomes. For this purpose, we stained paraformaldehyde-fixed MCF7 breast cancer cells and U2OS osteosarcoma cells, which have different expression levels of LGALS1 and LGALS3 (Fig. 2A), with commercially available LGALS1 and LGALS3 antibodies. In untreated control cells both antibodies resulted in diffuse mainly cytosolic stainings, whose specificities were confirmed by siRNA-mediated depletion of the respective mRNAs (Fig. 2B). After treatment with L-leucyl-L-leucine methyl ester (LLOMe), which induces lysosome-specific membrane damage,34 stainings for both galectins changed from diffuse to punctate (Fig. 2C). A similar staining pattern was observed with an independent LGALS3 antibody and in cells fixed with methanol (Fig. S1A and B).
Figure 2.

For figure legend, see page 1412.
In MCF7 cells costained for LGALS1 and LGALS3, puncta of the relatively highly expressed LGALS3 appeared as early as 10 min after LLOMe treatment, whereas the relatively low expressed LGALS1 formed readily detectable puncta 75 min after the treatment (Fig. 2C). By 6 h, the vast majority of the cells had both LGALS1 and LGALS3 puncta. LGALS1 puncta appeared with similar kinetics in U2OS cells (Fig. 2C) whereas LGALS3 puncta were barely detectable due to low levels of expression (Fig. 2A and data not shown). It should be noted that cell death was not detected in MCF7 or U2OS cells treated with LLOMe for 6 h (Fig. 2C), and even after 24 and 28 h treatment, respectively, only 2.6% of MCF7 cells and 13.8% of U2OS cells had lost plasma membrane integrity (Fig. 2D).
To ensure that LGALS1 and LGALS3 labeled damaged lysosomes specifically, we examined their colocalization with lysosomes and other organelles. Endogenous LGALS1 colocalized with enhanced green fluorescent protein (EGFP)-tagged CD63/LAMP3, a marker of lysosomal membranes, in LLOMe-treated HeLa-EGFP-CD63 cells (Fig. 2E). Similarly, more than 60% of endogenous LGALS1 and LGALS3 colocalized with endogenous LAMP2 and LAMP1, respectively, in LLOMe-treated MCF7 cells (Fig. 2F). Contrary to lysosomes, galectin puncta did not colocalize with the Golgi apparatus, mitochondria, endoplasmic reticulum (ER), or early endosomes (Fig. 2G). Taken together, this sensitive assay reveals that LMP does not necessarily lead to cell death and that not all lysosomes are equally sensitive to LMP.
LGALS3 colocalizes with other galectins and binds to N-acetyllactosamine in damaged lysosomes
To examine whether different galectins mark the same damaged lysosomes, we next compared their localization by immunocytochemistry and live-cell imaging. LLOMe induced a significant colocalization of endogenous LGALS1 with LGALS3 and vice versa in MCF7 cells (Figs. 2C and 3A). Similar overlap was observed between ectopic yellow fluorescent protein (YFP)-LGALS1 or EGFP-LGALS3 and mCherry-LGALS3 in U2OS cells (Fig. 3B). Due to the low expression levels and lack of suitable antibodies for LGALS8 and LGALS9, we studied their localization only in cells expressing tagged ectopic proteins. YFP-LGALS8 was diffusely distributed in untreated cells and colocalized with mCherry-LGALS3 puncta upon LLOMe treatment, whereas YFP-LGALS9 formed some mCherry-LGALS3-negative puncta even in untreated cells (Fig. 3B). LLOMe treatment induced the formation of additional YFP-LGALS9 puncta, the majority of which colocalized with mCherry-LGALS3 (Fig. 3B). YFP-LGALS8 and EGFP-LGALS3 puncta appeared approximately simultaneously with mCherry-LGALS3 puncta, whereas the appearance of YFP-LGALS1 and YFP-LGALS9 puncta was repeatedly slightly slower and faster than mCherry-LGALS3, respectively (Movies S1–S4). After 120 min, all galectins colocalized suggesting that they mark the same pool of leaky lysosomes (Fig. 3B).
Figure 3.

LGALS3 binds N-acetyllactosamine and colocalizes with LGALS1, LGALS8, and LGALS9 on damaged lysosomes. (A) Representative confocal images of MCF7 cells treated with 2 mM LLOMe for 6 h and costained with rabbit anti-LGALS1 (red) and mouse anti-LGALS3 antibodies (green), and Manders' coefficients for colocalization of LGALS1 with LGALS3 and vice versa. Error bars are SEMs for ≥ 26 cells. (B) Representative confocal images of U2OS-mCherry-LGALS3 cells (red) transiently transfected with plasmids coding for YFP-LGALS1, -8, -9 or EGFP-LGALS3 (green) and left untreated or treated with 2 mM LLOMe for 2 h before fixation. See also Movies S1–S4 for live-cell imaging. (C) Representative confocal images of U2OS cells transiently transfected with wild type EGFP-LGALS3, its R186S mutant (deficient in N-acetyllactosamine binding) or its G182A mutant (deficient in Galβ1-3GlcNAc binding), and treated with 2 mM LLOMe for 30 min before fixation. ***, P< 0.001. Scale bars: 10 µm.
The lumenal tails of many lysosomal membrane proteins, such as LAMP1 and LAMP2, form an N-acetyllactosamine-rich carbohydrate shield to which galectins might bind with their carbohydrate recognition domains upon LMP.35,36 To test this hypothesis, we took advantage of the LGALS3R186S mutant with approximately 40-fold reduced binding to N-acetyllactosamine, and the G182A mutation that mainly reduces the interaction with Galβ1-3GlcNAc-containing sugars with only a small effect on binding to N-acetyllactosamine and major cellular glycans.37,38 Akin to the wild type EGFP-LGALS3, the LGALS3G182A mutant formed puncta in LLOMe-treated U2OS cells, whereas the puncta formation was severely reduced by the R186S mutation (Fig. 3C). This suggests that LGALS3 puncta formation upon LMP is mediated by the binding of its carbohydrate-binding domain to N-acetyllactosamine-containing glycans.
Galectin puncta form regardless of the mode of lysosomal damage
To test whether galectin puncta serve as general markers for lysosomal leakage, we investigated their formation after treatment with known LMP inducers, i.e. cationic amphiphilic drugs terfenadine and siramesine,18 microtubule-disrupting agent vincristine,19 DNA-damaging drug etoposide,19 and hydrogen peroxide.39 Automated microscopy and quantification revealed an increase in mCherry-LGALS3 puncta formation after all treatments (Fig. 4A and B). Cationic amphiphilic drugs, which trigger lysosomal cell death at the concentrations used here,18,40,41 induced mCherry-LGALS3 puncta formation clearly before rounding of the cells (Fig. 4A and B). LLOMe induced extensive mCherry-LGALS3 puncta formation in the majority of cells already after 30 min (Fig. 4A and B). The appearance of the puncta was accompanied by early rounding and death of approximately 30% of the cells, but the large majority of the LGALS3 puncta-positive cells remained adherent and appeared healthy throughout the experiment (Fig. 4A and B).
Figure 4.

The galectin puncta assay is suitable for automated analysis of LMP. (A and B) U2OS-mCherry-LGALS3 cells were treated as indicated for 24 h (A) or for indicated times (B). After staining the nuclei with Hoechst 33342, cells were analyzed in a ScanR automated microscopy system to quantify the number of puncta inside a “cell mask” defined by a fixed radius around each nucleus. Representative images (A), quantification of percentage of cells with >3 mCherry-LGALS3 puncta (B, top), number of mCherry-LGALS3 puncta per cell (B, middle), and percentage of rounded cells (B, bottom) are shown. Error bars are SDs for means of 2 duplicate experiments with an average of > 400 cells/sample. (C and D) Representative confocal images of MCF7 cells treated with 200 nM thapsigargin or vehicle control (H2O) for 24 h (C) or 300 nM vincristine or vehicle control (DMSO) for 48 h (D) and costained with mouse anti-LGALS3 (green) and rabbit anti-PDIA2 (red) or anti-GOLGA1 (red) antibodies. Nuclei are visualized with Hoechst 33342 (blue) in merged images. RGB color intensity for entire panel (D) was equally enhanced with Photoshop. Scale bars: 10 µm.
Galactose-containing complex glycoproteins and glycolipids are formed in the lumen of the ER or Golgi. Thus, we examined whether LGALS3 translocates to these organelles when they are stressed. No LGALS3 puncta were observed in cells treated with thapsigargin (Fig. 4C), which induces ER stress,42 and in cells treated with vincristine, which disperses the Golgi apparatus eventually leading to LMP,19,43 the puncta did not colocalize with the trans-Golgi resident protein GOLGA1 (golgin A1) (Fig. 4D). Taken together, these data indicate that galectin puncta formation can be used as a rapid and specific assay for LMP in various experimental settings, including high-throughput screening.
The galectin puncta assay is superior to existing LMP assays
Next, we compared LGALS1 and LGALS3 puncta formation to microscope-based methods available to detect LMP, i.e. leakage of cathepsins or preloaded fluorescent dextran from lysosomes to the cytosol. LGALS1 and LGALS3 puncta were observed in almost all cells treated with LLOMe or terfenadine, when diffuse cytosolic CTSB (cathepsin B) staining was only barely detectable in a few cells (Fig. 5A and B). LGALS3 puncta also preceded or coincided with detectable release of dextran in LLOMe-treated cells (Fig. 5C and Movie S5). Cytosolic dextran was undetectable in some cells with abundant LGALS3 puncta even 19 h after the addition of LLOMe (Fig. 5D). Galectin puncta formation was also a more sensitive measure of LMP than leakage of fluorescent dextran when analyzed by the ScanR automated microscopy system suitable for larger screens (Fig. 5E). Thus, the galectin puncta assay is more sensitive and user friendly than the current microscopy-based assays for the detection of LMP.
Figure 5.

The galectin puncta assay is more sensitive than existing methods to detect LMP. (A and B) Representative confocal images of U2OS (A) and MCF7 (B) cells that were treated with 2 mM LLOMe or 8 µM terfenadine for indicated times, fixed in ice-cold methanol and costained with mouse anti-CTSB antibodies (green) and rabbit anti-LGALS1 or rat anti-LGALS3 antibodies (red). Note that higher gain settings were used for terfenadine-treated cells. (C) U2OS cells transiently transfected with EGFP-LGALS3 (green) and preloaded for approximately 18 h with AF594-dextran (10 kDa; red) were treated with 2 mM LLOMe for indicated times and analyzed by live-cell confocal microscopy. Examples of cells with EGFP-LGALS3 puncta without clearly visible AF594-dextran in the cytosol are indicated by arrowheads. See also Movie S5. (D) U2OS-mCherry-LGALS3 cells (red) preloaded with fluorescein-dextran (10 kDa; green) for 6 h were treated with 1.5 mM LLOMe for 19 h and examined by live-cell confocal microscopy. Examples of cells with mCherry-LGALS3 puncta but without clearly visible fluorescein-dextran in the cytosol are indicated by arrowheads. (E) U2OS-mCherry-LGALS3 cells loaded with AF488-dextran (10 kDa) for approximately 18 h were treated with 1.5 mM LLOMe and analyzed in the ScanR automated microscopy system. Images were acquired every 60 ec. Scale bars: 10 µm.
Staining for LGALS3 and active BAX defines the sequence of LMP and mitochondrial outer membrane permeabilization (MOMP)
In order to define the primary and secondary cell death pathways activated by different treatments, it is of great interest to determine the sequence of their activation in individual cells.44-46 Therefore, we treated MCF7 cells with TNF and terfenadine, which trigger apoptosis and lysosomal cell death in these cells, respectively,18,47 and stained them for LGALS3 and activated BAX (BCL2-associated X protein), a marker for MOMP and apoptosis.48 As expected due to the crosstalk between the death pathways, both treatments eventually induced BAX activation and LGALS3 puncta (Fig. 6A). However, terfenadine-treated cells became positive for LGALS3 puncta before the appearance of activated BAX, whereas TNF activated BAX in the absence of LGALS3 puncta in the majority of cells (Fig. 6A). Nearly all cells treated with LMP-inducing LLOMe for 2 to 6 h had abundant LGALS3 puncta while only less than 5% were positive for both markers (Fig. 6A). At 24 h, more than 20% of the LLOMe-treated cells had cleared the galectin puncta indicating that cells can, indeed, survive LMP (Fig. 6A). Taken together, costaining for LGALS3 and active BAX is a useful method to define the sequence of LMP and apoptosis induction.
Figure 6.

For figure legend, see page 1417.
LMP induces MAP1LC3B puncta formation at the site of lysosomal damage
Recently, LGALS3-positive leaky lysosomes were reported to be removed by autophagy (lysophagy).49 To confirm this and investigate how rapidly autophagosomes are recruited to galectin puncta, we treated U2OS-mCherry-LGALS3 cells which had been transiently transfected with an autophagosomal marker, EGFP-tagged rat MAP1LC3B (microtubule-associated protein 1 light chain 3 β), with LLOMe. LLOMe treatment induced a very rapid increase in both mCherry-LGALS3 and EGFP-MAP1LC3B puncta. mCherry-LGALS3 puncta were abundant already 2 min after the treatment and a few minutes later many of them became decorated by EGFP-MAP1LC3B as well (Fig. 6B and Movie S6). At the 2 h time point most mCherry-LGALS3 puncta were also positive for EGFP-MAP1LC3B, some of them being surrounded by a ring-like EGFP-MAP1LC3B-positive structure (Fig. 6C). Thus, the combination of galectin and LC3 puncta assays allows the study of the sequence of lysosomal damage and autophagosome formation and supports the notion that leaky lysosomes are rapidly engulfed by autophagosomes.49
Galectin staining detects LMP in paraffin-embedded tissue sections
Next, we examined whether galectin immunostaining was suitable for detecting LMP in tissue samples, which is practically impossible with available assays. First, we showed that the galectin antibodies retained their antigen-binding capacity and specificity in formaldehyde-fixed and paraffin-embedded pellets of MCF7 cells (Fig. S3A). Then, we tested whether the ability of terfenadine and siramesine to inhibit the growth of tumor xenografts in mice18,40 was associated with lysosomal leakage, as so far documented only in cultured cells. Immunostaining of paraffin-embedded tumor samples revealed only sporadic LGALS3 puncta in vehicle-treated tumors, whereas cells with multiple LAMP1-positive LGALS3 puncta were abundant in siramesine- and terfenadine-treated tumors indicating that these drugs induce LMP also in vivo (Fig. 7A).
Figure 7.

The galectin puncta assay detects LMP in paraffin-embedded tissue samples. (A) Sections of MCF7 breast cancer xenografts collected from SCID/FOX mice 48 h after p.o. treatment with 100 mg/kg terfenadine or siramesine or vehicle (200 µl of 0.5% methyl cellulose 15 in 0.9% NaCl solution) were costained with rat anti-LGALS3 (red) and mouse anti-LAMP1 antibodies (green) and Hoechst 33342 (blue) and analyzed by confocal microscopy. Representative maximum projection images of z-stacks are shown. Examples of puncta positive for both LGALS3 and LAMP1 are indicated by arrowheads. Two biological replicas showed similar results. See Figure S3A for images of galectin puncta formation in paraffin-embedded MCF7 cell pellets. (B) Sections of lactating (10 d) and involuting (24 h) mouse mammary gland tissues from C57B6/J wild type mice were stained with rabbit anti-LGALS1 or rat anti-LGALS3 antibodies. Representative images of chromogenic staining with HRP-linked secondary antibodies and diaminobenzidine (brown) and hematoxylin counterstain (blue) are shown. Black arrowheads indicate examples of cells with LGALS1 or LGALS3 puncta. Two biological replicas showed similar results. See Figure S3B for lower magnifications of the LGALS1 samples and control IgG staining. (C) Representative maximum projection images of confocal microscopy z-stacks of mammary gland sections [described in (B)] stained with rat anti-LGALS3 (red) and mouse anti-LAMP1 antibodies (green). Nuclei were labeled with Hoechst 33342 (blue; in merged images). Two biological replicas showed similar results. Scale bars: 10 µm.
Next we investigated galectin puncta formation during mouse mammary gland involution, a process associated with lysosomal cell death.7 LGALS1 and LGALS3 staining in alveolar epithelial cells from lactating glands showed a diffuse cytosolic staining pattern, whereas in involuting glands both galectins formed cytosolic puncta suggestive of leaky lysosomes (Fig. 7B). The interpretation of LGALS1 staining was greatly complicated by abundant LGALS1 staining of intraalveolar material, alveolar epithelial cell surface, and large cytosolic structures (Fig. 7B and Fig. S3B) which resembled large phagosomes/phagolysosomes characteristic of involuting glands.8 Thus, we continued with the confocal analysis of LGALS3 puncta, which colocalized with small LAMP1-positive vesicles, but not with the enlarged LAMP1-positive structures (Fig. 7C). Thus, lysosomal leakage during mammary gland involution appears to start from the small lysosomes rather than the enlarged phagolysosomes as suggested recently.8 These data indicate that the galectin puncta assay is suitable for paraffin-embedded tissue sections.
LGALS3 decorates leaky lysosomes in heat-stressed C. elegans
Heat shock and other stresses induce LMP in the intestinal cells of C. elegans lacking an intracellular serpin, SRP-6, a protease inhibitor that inhibits LMP.12 We first confirmed that heat shock (3 h at 37°C) caused LMP even in L4 larvae of a Pced-1nuc-1-mcherry reporter strain with intact SRP-6, as defined by a significant decrease in lysosomes positive for the lysosomal nuclease NUC-1 (abnormal NUClease) (Fig. 8A). In contrast, the leakage of NUC-1-mCherry was less clear in hypodermal cells of heat-shocked adult worms (Fig. 8B). Expression of Phyp7gfp-lgals3 made it, however, possible to detect LMP in these cells after sublethal heat shock (Fig. 8B and C). Thus, the galectin puncta assay allows the highly sensitive detection of LMP in C. elegans, demonstrating for the first time LMP in live animals without a predisposing genetic defect.
Figure 8.
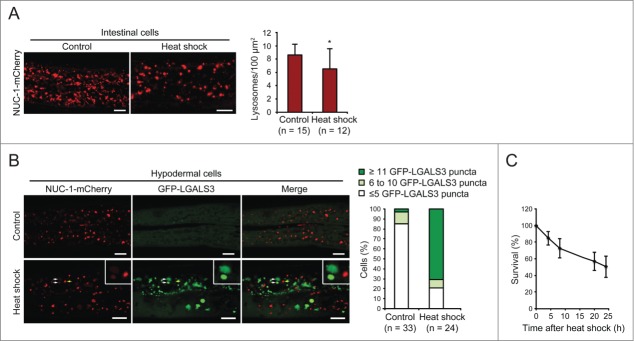
GFP-LGALS3 puncta mark heat-shock-induced leaky lysosomes in C. elegans. (A) L4 stage C. elegans larvae expressing the lysosomal marker NUC-1-mCherry were incubated at 20°C (control) or 37°C (heat shock) for 3 h, allowed to recover at 20°C for 3 h and imaged by confocal microscopy. Three randomly chosen intestinal areas of approximately 800 µm2 in each worm were analyzed for the number of NUC-1-mCherry-positive lysosomes. Representative confocal images and average number of lysosomes per 100 µm2 are shown. Error bars are SDs for means of 3 areas in ≥ 12 worms per condition. *, P< 0.05. (B) Adult C. elegans worms (L4 + 24 h) expressing NUC-1-mCherry and GFP-LGALS3 were treated and imaged as in (A) (recovery time was 4 h). The posterior parts of Hyp7 cells (n ≥ 24) were analyzed for the number of GFP-LGALS3 puncta. Representative images (left) and the percentage of cells with ≤5, 6 to 10, or ≥11 large (>1.5 µm2) GFP-LGALS3 puncta (right) are shown. White arrows indicate examples of GFP-LGALS3-positive leaky lysosomes with reduced NUC-1-mCherry content and the yellow arrow shows an example of a GFP-LGALS3-negative NUC-1-mCherry-positive intact lysosome. (C) The survival of worms treated as in (B) was analyzed by their responsiveness to picker touch at indicated time points after the heat shock. Error bars are SDs for means of 4 independent experiments with approximately 50 worms each. Scale bars: 10 µm.
Discussion
Our understanding of cellular processes such as apoptosis and autophagy has exploded over the last 2 decades largely due to the development of good biomarkers, assays, and standardized guidelines for detection.50,51 Meanwhile, the field of lysosomal cell death has lagged behind due to the lack of appropriate and user-friendly methods to detect limited LMP, the hallmark of lysosomal cell death. Here, we validate the lysosomal galectin puncta assay as a sensitive and specific LMP detection method that overcomes most of the current limitations. Upon lysosomal membrane damage, LGALS1, LGALS3, LGALS8, and LGALS9 translocate rapidly from their diffuse cytosolic distribution to defined puncta,26,31 which colocalize with damaged lysosomes and with each other. This event can be easily visualized with commercially available highly specific antibodies against endogenous LGALS1 and LGALS3, which are expressed in detectable levels in most tissues and cell types, or by employing cells expressing fluorescently tagged LGALS1, LGALS3, LGALS8, or LGALS9. Importantly, galectin puncta are practically absent in healthy cells, and their formation occurs regardless of the mode of lysosomal damage. This suggests that the translocation of galectins to lysosomes is a general consequence of the disruption of the lysosomal membrane. Furthermore, the galectin puncta appear rapidly after the treatment with LMP-inducing agents and persist for several hours regardless of the fate of the cells, i.e. whether they are destined to die or capable of recovery due to limited damage followed by regained lysosomal function or autophagic removal of damaged lysosomes. This ensures that galectin puncta are easy to detect even in larger screens in which the optimal conditions for each treatment cannot be defined beforehand. It should also be noted that the galectin puncta assay based on commercially available antibodies is easily adaptable to most cell biology laboratories because it does not require highly specialized expertise or equipment.
In addition to its superior sensitivity, the galectin puncta assay has several advantages over the current methods to detect LMP, i.e. the release of lysosomal hydrolases or preloaded fluorophore-labeled dextran. The de novo formation of defined puncta by galectins is more suitable for screening applications using automated image analysis than the detection of a slow decline in the intensity of preexisting puncta (lysosomal hydrolases or fluorescent dextran) or leakage of a few molecules into the large volume of the cytosol. Moreover, the ability of galectins to mark individual damaged lysosomes opens up the possibility to characterize the subpopulation of lysosomes with the highest susceptibility to membrane destabilization and follow their fate. And most importantly, the compatibility of the galectin puncta assay with immunohistochemical methods now makes it possible to detect LMP in tissue sections in a reliable manner allowing the investigation of the role of LMP in various physiological and pathological processes in vivo as demonstrated here in tumor xenografts treated with novel anti-cancer drugs and in involuting mouse mammary gland.
Due to their widespread expression in tissues and cell lines and availability of specific antibodies that circumvent the need for cell manipulation, endogenous LGALS1 and LGALS3 are best suited for LMP detection. Endogenous LGALS1 and LGALS3 mark the same population of leaky lysosomes, with a higher percentage of LGALS1 colocalizing with LGALS3 than vice versa in LLOMe-treated MCF7 cells. The significantly stronger signal obtained with LGALS3 antibodies in MCF7 cells may explain this difference. Possibly for the same reason, LGALS3 puncta become detectable clearly before LGALS1 puncta. The faster translocation kinetics of LGALS3 are, however, supported by a slight delay in LGALS1 puncta formation observed also when comparing the puncta formation of fluorescently tagged ectopic LGSLS1, LGALS3, LGALS8, and LGALS9 after LLOMe treatment. The slower kinetics of YFP-LGALS1 could, however, be due to a sterical hindrance caused by the YFP-tag, which with its molecular mass of 27 kDa is almost twice the size of LGALS1, by far the smallest of the 4 examined galectins. This could also explain why YFP-LGALS1 has previously been reported not to localize to phagosomes damaged by Salmonella typhimurium26 whereas we observed clear puncta formation by endogenous LGALS1 after lysosomal damage.
In spite of its putatively slower translocation to leaky lysosomes, we recommend endogenous LGALS1 as the first choice for LMP detection because of its almost ubiquitous expression in tissues and cell lines. Its very high expression in e.g. adipose and uterine tissues, may, however, cause high background staining that can complicate the detection of the specific puncta. It should also be noted that in involuting mammary gland, where LGALS1 is abundant inside the alveolar lumen and on the surface of alveolar epithelium, as well as in other tissues with abundant extracellular LGALS1, endocytosed LGALS1 may cause punctate staining without the permeabilization of endo-lysosomal membranes. In such cases, staining for LGALS3, LGALS8, or LGALS9 may be more appropriate, naturally with the same precautions. We also show that fluorescent ectopic galectins are useful in experimental situations where the detection of endogenous galectins is not possible (e.g. imaging of live cells or animals) or is too costly and labor-demanding (e.g. high throughput screen). Immunostaining should, however, be the method of choice whenever possible because galectin overexpression may have as yet unknown consequences. This is supported by recent reports of a role for LGALS8 and possibly LGALS3 as recognition molecules for the autophagy-mediated removal of ruptured phagosomes and late endosomes, respectively.26,52 Thus, galectins recruited to leaky lysosomes may also enhance their autophagy-mediated removal (so called lysophagy) thereby limiting LMP and cell death.31,41,49 To ensure that galectin puncta, indeed, mark leaky lysosomes, it is important to verify their colocalization with lysosomal markers, and when feasible, LMP should be confirmed with other methods. Furthermore, the sensitive galectin puncta assay demonstrates that limited LMP does not necessarily lead to cell death. Thus, additional assays should be used to prove a connection between LMP and cell death. On the other hand, the ability of cells to limit and survive LMP suggests that minor leakage of lysosomal enzymes may play an as yet unrecognized role in various cellular processes not associated with cell death.
The mechanism by which galectins translocate to leaky lysosomes is not fully understood. Taking into account their high-affinity binding to N-acetyllactosamine and relatively small size (< 40 kDa) that is comparable to molecules that escape out of lysosomes upon LMP, 53,54 it is tempting to speculate that they enter leaky lysosomes and get trapped there upon binding to N-acetyllactosamine-rich luminal tails of lysosomal membrane proteins such as LAMP1 and LAMP2.35,36 Supporting this notion, we found that the R186S mutation in the carbohydrate-binding domain of LGALS3, which abolishes LGALS3 binding to N-acetyllactosamine,37,38 strongly reduced LGALS3 puncta formation after lysosomal damage. This mutation as well as a mutation in a glycosyltransferase gene required for the formation of complex and hybrid N-glycans have also been shown to abolish LGALS3 localization to phagosomes lysed by Shigella flexneri in Chinese hamster ovary cells.28
In summary, the galectin puncta assay presented here is a straightforward, inexpensive, sensitive, and robust method for the detection of LMP. The assay is extremely versatile and can be used for fixed cells, live-cell imaging and automated screens, and combined with apoptosis markers to discern whether LMP is a primary or secondary event during cell death or with autophagosome markers to assess whether the damaged lysosomes are removed by autophagy. Importantly, it also works in paraffin-embedded tissue samples and allows for the first time the detection of individual damaged lysosomes. Thereby, it can pave the way to the answers for many remaining questions regarding the regulation of lysosomal cell death and its connection to so-called autophagic cell death. Finally, this assay may open up an entirely new research area focused on the role of nonlethal LMP for example as a cause of inflammation and senescence in lysosomal storage disorders and other degenerative diseases. Based on the above, the galectin puncta assay can be recommended as the new gold standard for LMP detection.
Materials and Methods
Cell culture and treatments
HeLa human cervix carcinoma cells and U2OS human female osteosarcoma cells were obtained from the ATCC. The MCF7 cell line used here is the S1 subclone of MCF7 human female breast adenocarcinoma cells selected for high TNF sensitivity.47 U2OS-mCherry-LGALS3 cells30,55 were kindly provided by Dr. Harald Wodrich (Laboratoire de Microbiologie Fondamentale et Pathogénicité, Bordeaux, France). HeLa-EGFP-CD63 cells56 were kindly provided by Dr. J. Paul Luzio (University of Cambridge, UK).
All cell lines were cultured in RPMI 1640 with 6% fetal calf serum, 23 U/ml penicillin, and 23 U/ml streptomycin (Life Technologies, 61870-010, 10270-106, 15140-122) at 37°C in a humidified atmosphere with 5% CO2. All cell lines were regularly tested and found negative for mycoplasma as analyzed by PCR and Hoechst staining.
Human TNF was kindly provided by Dr. Anthony Cerami (Kenneth Warren Laboratories, Tarrytown, NY, USA) and siramesine by Dr. Christiane Volbracht (H. Lundbeck A/S, Valby, Denmark). DMSO was obtained from Amresco (N182), LLOMe from Santa Cruz Biotechnology (sc-285992), and etoposide (E1383), hydrogen peroxide (349887), terfenadine (T9652), thapsigargin (T9033) and vincristine (V8879) from Sigma-Aldrich.
Transfections
pETM eukaryotic expression plasmids encoding YFP-LGALS1, -LGALS3 (isoform 1), -LGALS8 and -LGALS926 were kindly provided by Dr. Felix Randow (MRC Laboratory of Molecular Biology, Cambridge, UK). pCDH-EF1-EGFP-LGALS3 and pCDH-EF1-EGFP-MAP1LC3B were created by cloning human LGALS3 (isoform 1) and rat map1lc3b DNA (without start codon) sequences, respectively, with N-terminal EGFP tags into the lentiviral pCDH-EF1-MCS-IRES-Neo vector (Systems Biosciences, CD533A-2). The R186S and G182A mutants of LGALS3 were generated by site-directed mutagenesis using PfuUltra II fusion HS polymerase (Agilent Technologies, 600672). The inserts were verified by sequencing (LGC Genomics). Transient cDNA transfections were performed using Lipofectamine LTX with PLUS Reagent (Life Technologies, 15338-100) or Fugene HD (Roche, 04709713001) 48 h before the experiments.
For RNA interference, cells were reverse-transfected with 20 nM LGALS1 siRNA (sense, CCAACACCAUCGUGUGCAA[dT][dT]; antisense, UUGCACACGAUGGUGUUGG[dT][dT]), LGALS3 siRNA (sense, CAGAAUUGCUUUAGAUUUC[dT][dT]; antisense, GAAAUCUAAAGCAAUUCUG[dT][dT]) or Allstar Negative Control siRNA (Qiagen, 1027281) using Lipofectamine RNAiMAX (Life Technologies, 13778-075) for U2OS or Oligofectamine (Life Technologies, 12252-011) for MCF7.
Analysis of cell survival
Cells were plated in 96-well plates (5000 cells/well) and allowed to attach for 18 h before the indicated treatments, after which they were stained with 0.333 µg/ml propidium iodide (Sigma-Aldrich, P4864) and 0.1 µg/ml Hoechst 33342 (Life Technologies, H1399) for 15 min at 37°C to label dead and all cells, respectively. Samples were analyzed with the Celigo Cell Imaging Cytometer (Brooks Automation, Chelmsford, MA, USA) to image the entire well surface and quantified with the Celigo software (version 2.1, Brooks Automation).
Immunoblotting
Cells were lysed in Laemmli sample buffer (125 mM Tris, pH 6.7, 20% glycerol, 140 mM SDS (Sigma-Aldrich, L3771)) supplemented with 3x cOmplete protease inhibitor cocktail (Roche, 11697498001) and 40 µg/ml pepstatin A (Roche, 10359053001) at 10,000 cells/µl. After addition of 0.1 M dithiothreitol and bromophenol blue (Sigma-Aldrich, D9779 and B0126), boiling and separation by 4-20% gradient SDS-PAGE, proteins were transferred onto nitrocellulose membranes using the Bio-Rad Trans-Blot Turbo system. Membranes were blocked with Dulbecco's phosphate-buffered saline (DPBS; Life Technologies, 14190-094) containing 5% milk and 0.1% Tween-20 (Sigma-Aldrich, 274348) and incubated with rabbit anti-LGALS1 (Abcam, ab25138; 1:16000), mouse anti-LGALS3 (BD Biosciences, 556904; 1:2000), rabbit anti-LGALS8 (Santa Cruz Biotechnology, sc-28254; 1:500) or goat anti-LMNB1 (lamin B1; Santa Cruz Biotechnology, sc-6216; 1:1000) antibodies and matching horseradish peroxidase (HRP)-coupled secondary antibodies (anti-rabbit IgG, Vector Laboratories, PI-1000; anti-mouse IgG, DAKO, P0260; anti-goat IgG, DAKO, P0160; all 1:5000). Bound antibodies were detected with Clarity Western ECL Substrate (Bio-Rad, 170-5061) using a LAS 4000 mini luminescent image analyzer (Fujifilm, Tokyo, Japan).
Immunocytochemistry and confocal microscopy
Transfected or untransfected cells were grown on glass cover slips in 24-well plates and treated as indicated before fixation with 4% paraformaldehyde (Ampliqon, 43226.1000) or ice-cold methanol (specifically indicated).
For immunocytochemistry, cells were submitted to quenching with 50 mM ammonium chloride (Sigma-Aldrich A0171) in DPBS, permeabilized and blocked in 5% goat serum (DAKO, X0907), 1% BSA (Amresco, E531), 0.3% Triton-X-100 (Sigma-Aldrich, T9284) in DPBS with calcium and magnesium (Life Technologies, 14040-091), and stained with primary antibodies that included rabbit antibodies against BAX (Cell Signaling Technology, 2772; 1:350), EEA1 (early endosome antigen 1; Abcam, ab2900; 1:800), LGALS1 (Abcam, ab25138; 1:1000), GOLGA1 (Abcam, ab84340; 1:200), LAMP1 (Abcam, ab24170; 1:1000), PDIA2 (protein disulfide isomerase family A, member 2; Abcam, ab3672; 1:300), and TOMM20 (translocase of outer mitochondrial membrane 20 homolog (yeast); Santa Cruz Biotechnology, sc-11415; 1:1000), mouse antibodies against CTSB (CB59-4B11, kindly provided by Dr. Ekkehard Weber, Martin Luther University Halle-Wittenberg, Germany; 1:400), LGALS3 (BD Biosciences, 556904; 1:50-1:100), GOLGA1 (Life Technologies, A21270; 1:500), HSPA9 (heat shock 70kDa protein 9; Assay Designs, sps-825; 1:100), LAMP2 (H4B4, Developmental Studies Hybridoma Bank; 0.9 µg/ml), and RAB5 (RAB5, member RAS oncogene family; BD Biosciences, 610724; 1:250), and rat antibodies against LGALS3 (anti-Mac-2, kindly provided by Dr. Hakon Leffler, Lund University, Sweden; supernatant 1:2000), and matching Alexa Fluor (AF) 488- or AF594-coupled secondary antibodies (Life Technologies, A21202, A21203, A21206, A21207 and A21471; 1:1000).
Nuclei were labeled with 5 µg/ml Hoechst 33342 and coverslips were mounted with Prolong Gold Antifade mounting medium (Life Technologies, P36930).
Confocal images were acquired with an LSM 700 confocal microscope using the EC Plan-Neofluar 40x/1.30 Oil DIC M27 or Plan-Apochromat 63x/1.40 Oil DIC M27 objective and Zen 2010 software (all equipment and software from Carl Zeiss, Jena, Germany). Pinholes were set so that the section thickness was equal for all channels.
For colocalization analyses, z-stacks (360-nm slices) were acquired. Cell contours (n > 20) were defined manually and green and red thresholds were set up in single channel mode and retained for all samples in an experiment. Manders' coefficients were obtained with the colocalization module of the Zen 2012 (Black Edition) software (Carl Zeiss) using one slice per stack.
Quantification of cells with active BAX and/or LGALS3 puncta was performed by manual counting of 6 fields per sample, which had been randomly selected viewing only the blue channel.
Mini screen with ScanR automated microscopy system
For the mini screen with lysosome-damaging drugs, U2OS-mCherry-LGALS3 cells were plated in black 96-well culture plates with clear bottom (Greiner Bio-One, 655090). After the indicated treatments, cells were fixed in 4% paraformaldehyde. Nuclei were stained with 8 µg/ml Hoechst 33342 and cells were analyzed in a ScanR automated microscopy system with the UPLSAPO 40x/0.9 objective (Olympus, Hamburg, Germany). Images were acquired in an unbiased fashion (>40 fields per sample) after autofocusing on the nuclei using the ScanR acquisition software (version 2.3.0.5, Olympus) and subjected to automated quantification of mCherry-LGALS3 puncta using the ScanR analysis software (version 1.3.0.3, Olympus) as follows: The Hoechst 33342 signal was used to define each nucleus as an individual object and a mask representing the cell area was generated by applying a fixed distance around each nucleus with touching masks being split in the middle between 2 nuclei. Cells on the border of each image were excluded. Within these “cell masks,” fluorescent puncta were counted with the “spot detection” module. Gating based on size and circularity of the nuclei was applied to exclude cell aggregates and extremely condensed cells. On average >600 individual cells (minimum 55) per sample were analyzed. To quantify rounded cells, images from the screen were used for manual counting of a minimum of 40 cells per sample.
Immunohistochemistry of tumor xenografts and mouse mammary glands
MCF7 cells (10 × 106 cells in Geltrex (Life Technologies, A1413202), 120 µl) were inoculated into an axillary mammary fat pad of female FOX CHASE severe combined immunodeficiency (SCID/FOX) mice treated with 0.7 µg/ml estrone (Sigma-Aldrich, E9750) in drinking water starting 5 to 7 d before tumor inoculation. When the tumor diameter reached 5 to 6 mm, animals were treated p.o. (by mouth) with 200 µl vehicle (0.5% methyl cellulose, viscosity 15 cP (Sigma-Aldrich, M7140) in 0.9% NaCl solution) alone or with 100 mg/kg siramesine or terfenadine. After 2 days, mice were sacrificed, and tumors were placed in 10% formalin fixative until embedding in paraffin.
Lactating (10 d) and involuting (10 d lactation + 24 h after removal of pups) mammary glands from C57B6/J wild type mice were fixed, embedded in paraffin and sectioned as described,8 and kindly provided by Dr. Tim Sargeant (University of Cambridge, Cambridge, UK). All animal studies were approved by local authorities and carried out in accordance with international guidelines.
For chromogenic stainings, 4-µm sections were deparaffinized in xylene and blocked with hydrogen peroxide (Merck Millipore, 107209). Antigen retrieval was performed with TE buffer, pH 9 in a microwave. Sections were stained with rabbit anti-LGALS1 (0.1 µg/ml) or rat anti-LGALS3 antibodies (1:100) and matching HRP-coupled secondary antibodies (EnVision Reagent, rabbit, DAKO, K4003, rabbit anti-rat antibodies, DAKO, P0450; 1:1000). Slides were developed with Liquid DAB+ (DAKO, K3468), counterstained with Tissue-Tek Hematoxylin Modified (Sakura, 8707), and analyzed in a Laborlux S light microscope (Leitz, Leica Microsystems, Wetzlar, Germany) with a PL Fluotar 25x/0.60 objective (Leitz).
For fluorescent stainings, 4-µm sections were deparaffinized in xylene and antigen retrieval was performed by boiling in 10 mM trisodium citrate buffer (pH 6.0) containing 0.05% Tween-20 for 10 min. After blocking with 10% goat serum in PBS, sections were stained with rabbit anti-LAMP1 (1:100), rat anti-LGALS3 (1:200), and matching AF488- or AF594-coupled secondary antibodies (1:200). Nuclei were labeled with 2.5 µg/ml Hoechst 33342 and coverslips mounted with Prolong Gold Antifade mounting medium. Z-stacks (1-µm slices) were acquired with an LSM 700 confocal microscope using the Plan-Apochromat 63x/1.40 Oil DIC M27 objective and Zen 2010 software (all equipment and software from Carl Zeiss). Maximum projections were created from the z-stacks using ImageJ analysis software (Fiji).
Live-cell imaging
For live-cell confocal microscopy, cells were plated in 8-well borosilicate chamber slides (Nunc Lab-Tek, Thermo Scientific, 155411) in RPMI 1640 (without Phenol Red) (Life Technologies, 32404-014) with 6% fetal calf serum, 2 mM GlutaMAX (Life Technologies, 35050-038), 23 U/ml penicillin and 23 µg/ml streptomycin. Images were acquired using an LSM 5 DUO inverted confocal microscope with a heated chamber (37°C), the EC Plan-Neofluar 40x/1.30 Oil DIC M27 objective and Zen 2009 software (all equipment and software from Carl Zeiss). Pinholes were set so that the section thickness was equal for all channels. Alternatively, cells were analyzed by automated fluorescence microscopy in the ScanR system using the UPLSAPO 40×/0.9 objective. For this, nuclei were stained with 8 µg/ml Hoechst 33342 before drug treatment.
For confocal or ScanR time series, images were acquired immediately after manual drug addition with 30 or 60 second intervals using autofocusing. Movies from confocal time series were generated with Zen 2009 software (Carl Zeiss) and movies from ScanR images were produced with ImageJ software after generating a 2-channel stack and combined to a split screen with Movie Studio Platinum software (Version 13.0, Sony Creative Software).
For the dextran release assay, cells were loaded with 50 to 100 µg/ml 10-kDa dextran linked to fluorescein, AF488 or AF594 (Life Technologies, D1821, D22910, D22913) for 6 to 24 h, washed and chased for 1 to 2 h before analysis.
C. elegans
C. elegans N2 Bristol strain and qxIs257(Pced-1nuc-1-mcherry)57 reporter strain, kindly provided by Dr. Xiaochen Wang (National Institute of Biological Sciences, Beijing, China), and jaaIs2(Phyp7gfp-lgals3) reporter strain were maintained according to standard protocols.58 The 49.26Phyp7gfp-lgals3 plasmid was created by inserting Phyp7 (BamHI site), GFP (NheI-KpnI sites) and human LGALS3 (KpnI-SacI sites) into the 49.26 vector (generated by Dr. Andrew Fire, Stanford University, CA, USA and kindly provided by Dr. Xiaochen Wang), and injected and integrated into N2 Bristol strain according to standard protocols.
To induce LMP, L4 and adult (L4 + 24 h) worms were cultured in NGM plates at 37°C for 3 h and recovered at 20°C for 3 h and 4 h, respectively. Worms (n > 10) were placed on a 3% agar pad and imaged with an LSM700 confocal microscope using the Plan-Apochromat 63x/1.40 Oil DIC M27 objective. Three randomly selected areas were analyzed by ImageJ software. The survival of the worms was analyzed by responsiveness to picker touch.
Bioinformatics and statistics
LGALS1, LGALS3, LGALS8, and LGALS9 mRNA levels in 44 cell lines and 32 tissues which had been determined by RNA-Seq were extracted from The Human Protein Atlas (www.proteinatlas.org, Version 12, Ensembl version 73.37). LGALS1 and LGALS3 protein levels were compared based on published mass spectrometry data from the NCI-60 cell line panel.33 Nine cell lines were excluded because data for one of the 2 galectins was missing.
Statistical analysis was performed using a 2-tailed, homoscedastic Student t test in order to evaluate the null hypothesis. The cutoff level for statistical significance was set to 5%.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the DCRC animal facility staff (L. Bregnholt Larsen, L. Jørgensen, T. Brøndsted Sten, L. Svane Drifte), L. Bro, M. Grønvig Nielsen, L. Vanderfox, and C. Dinant for technical assistance. We also thank A. Cerami, H. Leffler, J.P. Luzio, F. Randow, T. Sargeant, C. Volbracht, X. Wang, E. Weber, and H. Wodrich for providing valuable research tools, S. Hautaniemi for bioinformatics consultation and R. Pocock for the use of his laboratory.
Funding
This work was supported by the European Research Council grant #340751 (MJ), the Danish National Research Foundation (MJ), the Danish Cancer Society (MJ), the Danish Medical Research Council (BL, MM, and MJ), the Novo Nordisk Foundation (MJ), the Lundbeck Foundation (SA) and the Swedish Research Council (SA).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886-97; PMID:16239905; http://dx.doi.org/ 10.1038/nrc1738 [DOI] [PubMed] [Google Scholar]
- 2.Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci 2013; 126:1905-12; PMID:23720375; http://dx.doi.org/ 10.1242/jcs.091181 [DOI] [PubMed] [Google Scholar]
- 3.Cesen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res 2012; 318:1245-51; PMID:22465226; http://dx.doi.org/ 10.1016/j.yexcr.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 4.Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mole Cell biol 2013; 5:214-26; PMID: 23918283; http://dx.doi.org/ 10.1093/jmcb/mjt022 [DOI] [PubMed] [Google Scholar]
- 5.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27:6434-51.; PMID:18955971; http://dx.doi.org/ 10.1038/onc.2008.310 [DOI] [PubMed] [Google Scholar]
- 6.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/ 10.1038/nrm2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 2011; 13:303-9; PMID:21336304; http://dx.doi.org/ 10.1038/ncb2171 [DOI] [PubMed] [Google Scholar]
- 8.Sargeant TJ, Lloyd-Lewis B, Resemann HK, Ramos-Montoya A, Skepper J, Watson CJ. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nat Cell Biol 2014; 16:1057-68; PMID:25283994; http://dx.doi.org/ 10.1038/ncb3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michallet MC, Saltel F, Flacher M, Revillard JP, Genestier L. Cathepsin-dependent apoptosis triggered by supraoptimal activation of T lymphocytes: a possible mechanism of high dose tolerance. J Immunol 2004; 172:5405-14; PMID:15100281; http://dx.doi.org/ 10.4049/jimmunol.172.9.5405 [DOI] [PubMed] [Google Scholar]
- 10.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 2000; 106:773-81; PMID:10995788; http://dx.doi.org/ 10.1172/JCI9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinser RD, Dolph PJ. Cathepsin proteases mediate photoreceptor cell degeneration in Drosophila. Neurobiol Dis 2012; 46:655-62; PMID:22426392; http://dx.doi.org/ 10.1016/j.nbd.2012.03.004 [DOI] [PubMed] [Google Scholar]
- 12.Luke CJ, Pak SC, Askew YS, Naviglia TL, Askew DJ, Nobar SM. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 2007; 130:1108-19; PMID:17889653; http://dx.doi.org/ 10.1016/j.cell.2007.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 2002; 419:939-44; PMID:12410314; http://dx.doi.org/ 10.1038/nature01108 [DOI] [PubMed] [Google Scholar]
- 14.Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications. Am J Pathol 2001; 159:2045-54; PMID:11733355; http://dx.doi.org/ 10.1016/S0002-9440(10)63056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houseweart MK, Pennacchio LA, Vilaythong A, Peters C, Noebels JL, Myers RM. Cathepsin B but not cathepsins L or S contributes to the pathogenesis of Unverricht-Lundborg progressive myoclonus epilepsy (EPM1). J Neurobiol 2003; 56:315-27; PMID:12918016; http://dx.doi.org/ 10.1002/neu.10253 [DOI] [PubMed] [Google Scholar]
- 16.Matsuda S, Okada N, Kodama T, Honda T, Iida T. A cytotoxic type III secretion effector of Vibrio parahaemolyticus targets vacuolar H+-ATPase subunit c and ruptures host cell lysosomes. PLoS Pathog 2012; 8:e1002803; PMID:22829766; http://dx.doi.org/ 10.1371/journal.ppat.1002803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canbay A, Guicciardi ME, Higuchi H, Feldstein A, Bronk SF, Rydzewski R. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest 2003; 112:152-9; PMID:12865404; http://dx.doi.org/ 10.1172/JCI200317740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lonborg A, et al.. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013; 24:379-93; PMID:24029234; http://dx.doi.org/ 10.1016/j.ccr.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 19.Groth-Pedersen L, Ostenfeld MS, Høyer-Hansen M, Nylandsted J, Jäättelä M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome destabilizing siramesine. Cancer Res 2007; 67:2217-25; PMID:17332352; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3520 [DOI] [PubMed] [Google Scholar]
- 20.Fehrenbacher N, Bastholm L, Kirkegaard-Sorensen T, Rafn B, Bottzauw T, Nielsen C, Weber E, Shirasawa S, Kallunki T, Jäättelä M. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res 2008; 68:6623-33; PMID:18701486; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0463 [DOI] [PubMed] [Google Scholar]
- 21.Brunk UT, Ericsson JL. Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lysosomal membranes. Histochem J 1972; 4:479-91; PMID:4120446; http://dx.doi.org/ 10.1007/BF01011128 [DOI] [PubMed] [Google Scholar]
- 22.Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Høyer-Hansen M, Elling F, Wallach D, Flavell R, Kroemer G, Nylandsted J, et al.. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol 2006; 26:7880-91; PMID:16966373; http://dx.doi.org/ 10.1128/MCB.00716-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, et al.. Galectins: a family of animal beta-galactoside-binding lectins. Cell 1994; 76:597-8; PMID:8124704; http://dx.doi.org/ 10.1016/0092-8674(94)90498-7 [DOI] [PubMed] [Google Scholar]
- 24.Houzelstein D, Goncalves IR, Fadden AJ, Sidhu SS, Cooper DN, Drickamer K, Leffler H, Poirier F. Phylogenetic analysis of the vertebrate galectin family. Mol Biol Evol 2004; 21:1177-87; PMID:14963092; http://dx.doi.org/ 10.1093/molbev/msh082 [DOI] [PubMed] [Google Scholar]
- 25.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer 2005; 5:29-41; PMID:15630413; http://dx.doi.org/ 10.1038/nrc1527 [DOI] [PubMed] [Google Scholar]
- 26.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/ 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell host Microbe 2009; 6:137-49; PMID:19683680; http://dx.doi.org/ 10.1016/j.chom.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 28.Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost MC, Lafont F, et al.. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 2010; 12:530-44; PMID:19951367; http://dx.doi.org/ 10.1111/j.1462-5822.2009.01415.x [DOI] [PubMed] [Google Scholar]
- 29.Ray K, Bobard A, Danckaert A, Paz-Haftel I, Clair C, Ehsani S, Tang C, Sansonetti P, Tran GV, Enninga J. Tracking the dynamic interplay between bacterial and host factors during pathogen-induced vacuole rupture in real time. Cell Microbiol 2010; 12:545-56; PMID:20070313; http://dx.doi.org/ 10.1111/j.1462-5822.2010.01428.x [DOI] [PubMed] [Google Scholar]
- 30.Maier O, Marvin SA, Wodrich H, Campbell EM, Wiethoff CM. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J Virol 2012; 86:10821-8; PMID:22855481; http://dx.doi.org/ 10.1128/JVI.01428-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al.. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013; 32:2336-47; PMID:23921551; http://dx.doi.org/ 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al.. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 2010; 28:1248-50; PMID:21139605; http://dx.doi.org/ 10.1038/nbt1210-1248 [DOI] [PubMed] [Google Scholar]
- 33.Moghaddas Gholami A, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep 2013; 4:609-20; PMID:23933261; http://dx.doi.org/ 10.1016/j.celrep.2013.07.018 [DOI] [PubMed] [Google Scholar]
- 34.Uchimoto T, Nohara H, Kamehara R, Iwamura M, Watanabe N, Kobayashi Y. Mechanism of apoptosis induced by a lysosomotropic agent, L-Leucyl-L-Leucine methyl ester. Apoptosis : Internatl J Programmed Cell Death 1999; 4:357-62; PMID:14634338; http://dx.doi.org/ 10.1023/A:1009695221038 [DOI] [PubMed] [Google Scholar]
- 35.Do KY, Smith DF, Cummings RD. LAMP-1 in CHO cells is a primary carrier of poly-N-acetyllactosamine chains and is bound preferentially by a mammalian S-type lectin. Biochem Biophys Res Commun 1990; 173:1123-8; PMID:2268316; http://dx.doi.org/ 10.1016/S0006-291X(05)80902-7 [DOI] [PubMed] [Google Scholar]
- 36.Inohara H, Raz A. Identification of human melanoma cellular and secreted ligands for galectin-3. Biochem Biophys Res Commun 1994; 201:1366-75; PMID:8024581; http://dx.doi.org/ 10.1006/bbrc.1994.1854 [DOI] [PubMed] [Google Scholar]
- 37.Delacour D, Greb C, Koch A, Salomonsson E, Leffler H, Le Bivic A, Jacob R. Apical sorting by galectin-3-dependent glycoprotein clustering. Traffic 2007; 8:379-88; PMID:17319896; http://dx.doi.org/ 10.1111/j.1600-0854.2007.00539.x [DOI] [PubMed] [Google Scholar]
- 38.Salomonsson E, Carlsson MC, Osla V, Hendus-Altenburger R, Kahl-Knutson B, Oberg CT, Sundin A, Nilsson R, Nordberg-Karlsson E, Nilsson UJ, et al.. Mutational tuning of galectin-3 specificity and biological function. J Biol Chem 2010; 285:35079-91; PMID:20807768; http://dx.doi.org/ 10.1074/jbc.M109.098160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunk UT, Svensson I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep 1999; 4:3-11; PMID:10714269; http://dx.doi.org/ 10.1179/135100099101534675 [DOI] [PubMed] [Google Scholar]
- 40.Ostenfeld MS, Fehrenbacher N, Høyer-Hansen M, Thomsen C, Farkas T, Jäättelä M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res 2005; 65:8975-83; PMID:16204071; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0269 [DOI] [PubMed] [Google Scholar]
- 41.Ostenfeld MS, Høyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P, Kirkegaard-Sorensen T, Nylandsted J, Farkas T, et al.. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008; 4:487-99; PMID:18305408; http://dx.doi.org/ 10.4161/auto.5774 [DOI] [PubMed] [Google Scholar]
- 42.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther 2005; 4:14-22; PMID:15662118; http://dx.doi.org/ 10.4161/cbt.4.1.1505 [DOI] [PubMed] [Google Scholar]
- 43.Thyberg J, Moskalewski S. Role of microtubules in the organization of the Golgi complex. Exp Cell Res 1999; 246:263-79; PMID:9925741; http://dx.doi.org/ 10.1006/excr.1998.4326 [DOI] [PubMed] [Google Scholar]
- 44.Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2001; 2:589-98; PMID:11483992; http://dx.doi.org/ 10.1038/35085008 [DOI] [PubMed] [Google Scholar]
- 45.Rammer P, Groth-Pedersen L, Kirkegaard T, Daugaard M, Rytter A, Szyniarowski P, Høyer-Hansen M, Povlsen LK, Nylandsted J, Larsen JE, et al.. BAMLET activates a lysosomal cell death program in cancer cells. Mol Cancer Ther 2010; 9:24-32; PMID:20053771; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0559 [DOI] [PubMed] [Google Scholar]
- 46.Ellegaard AM, Groth-Pedersen L, Oorschot V, Klumperman J, Kirkegaard T, Nylandsted J, Jäättelä M. Sunitinib and SU11652 Inhibit Acid Sphingomyelinase, Destabilize Lysosomes, and Inhibit Multidrug Resistance. Mol Cancer Ther 2013; 12:2018-30; PMID:23920274; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0084 [DOI] [PubMed] [Google Scholar]
- 47.Jäättelä M, Benedict M, Tewari M, Shayman JA, Dixit VM. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene 1995; 10:2297-305; PMID:7540278. [PubMed] [Google Scholar]
- 48.Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 1999; 18:2330-41; PMID:10228148; http://dx.doi.org/ 10.1093/emboj/18.9.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung YH, Chen LM, Yang JY, Yang WY. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 2013; 4:2111; PMID:23817530; http://dx.doi.org/ 10.1038/ncomms3111 [DOI] [PubMed] [Google Scholar]
- 50.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, et al.. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16(8):1093-107; PMID: 19373242; http://dx.doi.org/ 10.1038/cdd.2009.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Khambu B, Zhang H, Gao W, Li M, Yoshimori T, Yin XM. Autophagy induced by calcium phosphate precipitates targets damaged endosomes. J Biol Chem 2014; 289:11162-74; PMID:24619419; http://dx.doi.org/ 10.1074/jbc.M113.531855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, Urashima T, Oka T, Futai M, Muller WE, et al.. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta 2002; 1572:232-54; PMID:12223272; http://dx.doi.org/ 10.1016/S0304-4165(02)00311-2 [DOI] [PubMed] [Google Scholar]
- 54.Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective AIF relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem 2003; 278:31401-11; PMID:12782632; http://dx.doi.org/ 10.1074/jbc.M301911200 [DOI] [PubMed] [Google Scholar]
- 55.Bosse JB, Ragues J, Wodrich H. Fast generation of stable cell lines expressing fluorescent marker molecules to study pathogen induced processes. Methods Mol Biol 2013; 1064:153-69; PMID:23996256; http://dx.doi.org/ 10.1007/978-1-62703-601-6_11 [DOI] [PubMed] [Google Scholar]
- 56.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1:23-36; PMID:16874023; http://dx.doi.org/ 10.4161/auto.1.1.1495 [DOI] [PubMed] [Google Scholar]
- 57.Guo P, Hu T, Zhang J, Jiang S, Wang X. Sequential action of Caenorhabditis elegans Rab GTPases regulates phagolysosome formation during apoptotic cell degradation. Proc Natl Acad Sci U S A 2010; 107:18016-21; PMID:20921409; http://dx.doi.org/ 10.1073/pnas.1008946107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974; 77:71-94; PMID:4366476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.