

For failing right ventricle (RV) myocardium, α1-adrenergic receptor (α1-AR) inotropic responses were increased. Of the two predominant cardiac α1-AR subtypes (α1A and α1B), increased α1-AR inotropy for failing RV myocardium required the α1A-subtype and was opposed by the α1B-subtype. The α1A-subtype might be a therapeutic target to improve the function of the failing RV.
Keywords: α1-adrenergic, inotropic, right ventricle, myosin regulatory light chain
Abstract
Dysfunction of the right ventricle (RV) is closely related to prognosis for patients with RV failure. Therefore, strategies to improve failing RV function are significant. In a mouse RV failure model, we previously reported that α1-adrenergic receptor (α1-AR) inotropic responses are increased. The present study determined the roles of both predominant cardiac α1-AR subtypes (α1A and α1B) in upregulated inotropy in failing RV. We used the mouse model of bleomycin-induced pulmonary fibrosis, pulmonary hypertension, and RV failure. We assessed the myocardial contractile response in vitro to stimulation of the α1A-subtype (using α1A-subtype-selective agonist A61603) and α1B-subtype [using α1A-subtype knockout mice and nonsubtype selective α1-AR agonist phenylephrine (PE)]. In wild-type nonfailing RV, a negative inotropic effect of α1-AR stimulation with PE (force decreased ≈50%) was switched to a positive inotropic effect (PIE) with bleomycin-induced RV injury. Upregulated inotropy in failing RV occurred with α1A-subtype stimulation (force increased ≈200%), but not with α1B-subtype stimulation (force decreased ≈50%). Upregulated inotropy mediated by the α1A-subtype involved increased activator Ca2+ transients and increased phosphorylation of myosin regulatory light chain (a mediator of increased myofilament Ca2+ sensitivity). In failing RV, the PIE elicited by the α1A-subtype was appreciably less when the α1A-subtype was stimulated in combination with the α1B-subtype, suggesting functional antagonism between α1A- and α1B-subtypes. In conclusion, upregulation of α1-AR inotropy in failing RV myocardium requires the α1A-subtype and is opposed by the α1B-subtype. The α1A subtype might be a therapeutic target to improve the function of the failing RV.
NEW & NOTEWORTHY
For failing right ventricle (RV) myocardium, α1-adrenergic receptor (α1-AR) inotropic responses were increased. Of the two predominant cardiac α1-AR subtypes (α1A and α1B), increased α1-AR inotropy for failing RV myocardium required the α1A-subtype and was opposed by the α1B-subtype. The α1A-subtype might be a therapeutic target to improve the function of the failing RV.
failure of the right ventricle (RV) is a serious and common clinical problem (15, 24); nevertheless, RV failure remains relatively understudied and poorly understood (37). RV dysfunction is a predictor of survival for patients with moderate or advanced heart failure (7, 9) and is strongly related to the prognosis of heart failure patients with pulmonary hypertension (14). Therefore, strategies to improve RV function may be beneficial for improving the survival and functional state of patients with severe RV failure.
Recent studies suggest that in heart failure, a higher level of α1-adrenergic receptor (AR) activation is beneficial (3, 20, 29). In heart failure β-ARs are markedly downregulated; in contrast, α1-AR levels are not decreased (4). Consequently, α1-ARs, which represent ≈10% of all ARs in nonfailing hearts, are increased to ≈25% of ARs in heart failure (29). Thus α1-ARs may play a relatively greater role in heart failure. Consistent with this, in myocardium isolated from failing human hearts, α1-AR mediated inotropy was upregulated and equaled β-AR mediated inotropy (34). Moreover, we reported that in a mouse model of RV failure induced secondary to LV failure, the RV myocardial inotropic response to α1-ARs was upregulated (38).
There are two predominant α1-AR subtypes on cardiac myocytes (α1A and α1B). The goal of this study was to determine the roles of both α1-AR subtypes in upregulation of α1-AR inotropic responses in the failing RV. We used the RV-specific mouse heart failure model of bleomycin-induced pulmonary fibrosis, pulmonary hypertension, and RV failure (17). We found that upregulated α1-AR inotropy in the falling RV is associated exclusively with the α1A-subtype and involves increased Ca2+ transients and increased phosphorylation of cardiac myosin regulatory light chain. In contrast, inotropic responses mediated by the α1B-subtype were not changed compared with nonfailing RV. Therefore, this study demonstrates that the α1A-subtype, but not the α1B-subtype, plays a role in augmenting contraction in RV failure and suggests that the α1A-subtype might be a therapeutic target for improving the function of the failing RV.
METHODS
This institution is accredited by the American Association for the Accreditation of Laboratory Animal Care (Institutional Public Health Service Assurance Number is A3476-01). The study was approved by the Animal Care and Use Subcommittee of the San Francisco Veterans Affairs Medical Center (Protocol 13-013) and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Revised 2011).
Animals and RV injury model.
Adult male wild-type (C57BL/6J; Jackson Laboratory) and α1A-subtype knock-out mice (AKO) (31) were used, age 10–14 wk, body weight ≈ 28 g, at the beginning of the experiment. We used a right ventricle (RV)-specific heart failure model. The fibrogenic antibiotic bleomycin was introduced into the lungs by endotracheal instillation to cause pulmonary fibrosis, pulmonary hypertension, and RV failure (17). In anesthetized mice, a light-guide positioned close to the chest was used to visualize the trachea and to position a catheter to deliver a solution of bleomycin or saline into the trachea. Bleomycin (Sigma-Aldrich, St. Louis, MO) was solubilized in sterile normal saline. Most animals were given a single instillation of 0.075 U of bleomycin in 100 μl saline or saline alone, and animals were harvested after 14–30 days. A subset of animals (reported in Fig. 3A) were studied <2 wk after bleomycin instillation, but the data from this subset were not pooled with all other data obtained >2 wk after bleomycin instillation. Where indicated in the results, a few animals received a lower dose of bleomycin (0.04 U) repeated every 2 wk (up to 8 instillations) (8). The repetitive instillation protocol was suggested to be a better model for idiopathic pulmonary fibrosis. In the present study, similar results were obtained using either protocol.
Fig. 3.

Upregulation of α1-AR inotropy 2 wk after bleomycin instillation. A: summary of the positive inotropic (●) and negative inotropic responses (○) elicited by the α1A-subtype agonist A61603 (A1603) for different samples of RV myocardium from different animals and for various times after a single bleomycin instillation or with biweekly bleomycin instillation (▲). Positive inotropic responses were observed 14 days after initial bleomycin administration. B: summary of inotropic responses to A61603: absolute levels of developed force before (basal) and after addition of A61603. Data from failing RV are divided into nonconverter or converter groups, depending on whether A61603 elicited a negative inotropic effect (NIE) or positive inotropic effect (PIE). **P < 0.01; n = 6–8/group.
In vivo hemodynamics.
Echocardiography was performed on conscious, gently restrained mice using an Acuson S2000 (Siemens) with a 5- to 14-MHz multi-dimension matrix transducer. RV and LV dimensions were measured using two-dimensional-guided M-mode, acquired from five consecutive cardiac cycles in the long axis view. Measurements were taken at midway along the heart. Fractional shortening for each chamber was calculated from the diastolic minus systolic chamber dimension expressed as a percentage of the diastolic dimension. The echocardiographer was blinded to the treatments of all mice.
RV trabecula preparation and measurement of calcium transients.
Mice were anesthetized with 100 mg/kg ip pentobarbital sodium and heparinized (100 U). Hearts were removed and immediately immersed in ice-cold arrest solution containing (in mM) 120 NaCl, 30 KCl, and 0.1 CaCl2 and then perfused through the aorta with a modified Krebs-Henseleit solution containing (in mM) 137 NaCl, 10 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 10 glucose, 20 NaHCO3, 0.2 CaCl2, and 30 2,3,-butanedione monoxime. The perfusate was oxygenated with 95% O2-5% CO2 to give a pH of 7.4 at 22°C. The RV free-wall was removed, and a trabecula that was free-running between the RV wall and tricuspid valve was dissected.
Trabeculae were placed in a muscle chamber (3 × 3 × 15 mm) and mounted on stainless steel pins with the valvular end attached to a micromanipulator and the ventricular end attached to a force transducer (AE-801; Kronex, Oakland, CA). Sarcomeres were observed using a 40× objective, and sarcomere length was assessed using a video-based system (Model 900B; Aurora Scientific, Ontario, Canada). Diastolic sarcomere length was set to 2.1 μm. Trabeculae were equilibrated for 1 h in Krebs-Henseleit solution (5 ml/min) as described above but with 5 mM KCl and no 2,3-butanedione monoxime. The calcium level of the solution was gradually increased up to 1.1–1.5 mM, and trabeculae stimulated to contract at a pacing frequency of 0.5 Hz using platinum wire electrodes at maximal voltage.
To measure Ca2+ transients, trabeculae were loaded with fura-2-AM by superfusion for 1.5 h with a Krebs-Henseleit solution containing 20 μM fura2-AM (Invitrogen) (from a stock solution of 5 mM fura-2-AM, 20% pluronic F-127, dissolved in DMSO), plus three to four drops of antifoam A (Sigma). Trabeculae were then washed with Krebs-Henseleit solution for 15 min, and the calcium level in the bath was increased as described above. This loading protocol increased recorded muscle fluorescence to 3–5 times the autofluorescence of the unloaded muscle. When measured in two experiments, fura-2-AM loading reduced contraction force 0–10%.
Fura-2 fluorescence was measured at an emission wavelength of 510 nm, with the excitation wavelength alternated between 340 nm and 380 nm (Photon Technology International, Edison, NJ). Fluorescence signals at each wavelength were integrated at 100 points/s. Muscle autofluorescence was subtracted before the fura-2 fluorescence ratio was computed. Fura-2 ratio was measured during contractions before and intermittently during the inotropic responses. Fura-2-AM can result in nonspecific loading into multiple cell compartments (which was not determined in this study). The rapid fura-2 fluorescence transient during contraction was used as a measure of the cytosolic Ca2+ transient.
Inotropic responses.
Cardiac α1-ARs were stimulated by addition of a maximal dose (10 μM) of phenylephrine (PE). In wild-type mice, PE stimulates both predominant α1-AR subtypes on cardiac myocytes (α1A and α1B). A maximal dose of the subtype-selective agonist A61603 (100 nM) was used to selectively stimulate the α1A-subtype. In knockout mice lacking the α1A-subtype (AKO), PE was used to selectively stimulate the remaining α1B-subtype. The β-AR antagonist timolol (10 μM) was present in all experiments. Acute inotropic responses were assessed when contraction force stabilized, typically ≈20 min after agonist stimulation. For some experiments, inotropic responses were expressed using the absolute force level per unit area of myocardium (in mN/mm2). For other experiments, the cross-sectional area of the preparation could not be determined (e.g., due to a complex/branched trabecula structure); therefore, inotropic responses were also assessed from the developed force (systolic minus diastolic force) after addition of agonist, expressed relative to the developed force before agonist. Both measures gave similar results.
Phosphorylation status of myosin regulatory light chain.
Measurements of regulatory light chain (RLC) phosphorylation in each trabecula were performed by urea/glycerol-PAGE and immunoblotting, as previously described (22). This method separates phosphorylated RLC from nonphosphorylated RLC, allowing a direct quantitative measure of phosphorylated RLC as a fraction of total RLC. As the separation results from a single phosphate, data may also be calculated as mole of phosphate per mole RLC. After contractions were recorded, a trabecula was immediately placed in 10% ice-cold trichloroacetic acid and 10 mM dithiothreitol. The precipitated trabecula was washed free of acid with three 5-min washes in ethyl ether and resuspended by vigorous agitation in 15 μl of urea sample buffer of 8 M Urea, 20 mM Tris base, 23 mM glycine, 0.2 mM EDTA, and 10 mM dithiothreitol using an orbital shaker (IKA Vibrax VXR) set at 1,400 rpm for 30 min at room temperature. Complete denaturation and solubilization was achieved by addition of urea crystals and prolonged agitation. Protein samples were centrifuged at 10,000 g for 2 min, and 5 μl of supernantant fraction containing denatured proteins was directly loaded onto the glycerol gel system for separation of phosphorylated from nonphosphorylated RLC proteins. Briefly, polyacrylamide gels containing 40% glycerol were pre-electrophoresed for 1 h at 400 V at room temperature in a mini-gel apparatus. Reservoir buffer contained 20 mM Tris base and 23 mM glycine (pH 8.6); thioglycolate and dithiothreitol (2.3 mM each) were included in the upper reservoir. Samples were subjected to electrophoresis for 90 min at 400 V at room temperature, then transferred to a polyvinylidene difluoride membrane for 1 h at 0.3 A at 4°C. After transfer, proteins were fixed onto the polyvinylidene difluoride membrane with 0.4% glutaraldehyde/PBS for 15 min at room temperature. The membrane was then rinsed 3× in PBS and immunoblotted with antibody to cardiac RLC (Enzo Life, F109 3E1). RLC was detected using ECL Plus (Pierce), and fluorescent signal acquired by Storm (GE Healthcare). RLC and p-RLC bands were auto-detected and quantified by ImageQuant TL (GE Healthcare).
Statistical analysis.
Data are presented as means ± SE. Statistical tests (paired and unpaired t-tests, linear regression, and χ2 test) were performed using Prism 6 software (GraphPad Software, La Jolla, CA) with a significance level set at P < 0.05.
RESULTS
Model of bleomycin-induced RV failure.
The RV-specific bleomycin model of RV failure was reported to cause pulmonary fibrosis, pulmonary hypertension, RV hypertrophy, decreased RV ejection fraction, and RV failure within ∼2 wk (17). Consistent with this we found that 2 wk after bleomycin instillation, there was a greater than twofold increase in lung weight relative to body weight, a 55% reduction in RV fractional shortening (Fig. 1A), and high mortality (≈50%). There was a 21% reduction in fractional shortening in the saline-treated group (Fig. 1A), suggesting that instillation of saline into the lung may result in mild injury. However, the reduction in fractional shortening due to bleomycin was considerably greater than the reduction due to saline (P = 0.018). With confirmation of the RV-specificity of this model, LV fractional shortening was not affected (Fig. 1B).
Fig. 1.

Bleomycin (Bleo) model of right ventricular (RV) failure. Echocardiographic assessment of RV and left ventricular (LV) fractional shortening before and 2 wk after bleomycin instillation was completed. A: 2 wk after bleomycin instillation, RV fractional shortening in vivo was reduced to 45% of the value before bleomycin (P = 0.002). After saline instillation, RV fractional shortening in vivo was slightly reduced to ∼80% of initial (P = 0.04). B: LV in vivo fractional shortening was not affected by bleomycin or saline instillation, demonstrating the RV-specific nature of the bleomycin model. *P < 0.05; **P < 0.01; ns, not significant.
Upregulation of α1-AR inotropy in RV failure.
We studied cardiac trabeculae from failing RV (bleomycin-treated) or nonfailing RV (saline-treated) and measured the acute α1-AR inotropic response to PE, a nonselective α1-AR agonist that stimulates both predominant α1-AR subtypes on cardiac myocytes (α1A and α1B). We also measured the inotropic response to the α1A-subtype-selective agonist A61603. All experiments were in the presence of the β-AR blocker timolol.
Figure 2 shows representative recordings of contractions of RV trabeculae with agonist stimulation. For nonfailing RV (saline treated), stimulation of the α1A-subtype by A61603 elicited a negative inotropic effect (NIE) that was identical to that elicited by combined stimulation of both the α1A- plus α1B-subtypes using PE. Thus, for nonfailing RV, stimulation of the α1A-subtype singly, or in combination with the α1B-subtype, elicited a NIE, consistent with our previous reports (25, 39).
Fig. 2.

Increased α1-adrenergic receptor (α1-AR) inotropy in failing RV. A: slow time-base recordings of contractions of RV myocardium from nonfailing and failing RV in response to acute α1-AR stimulation with maximal doses of α1A-subtype-selective agonist A61603 (100 nM) or nonsubtype-selective α1-AR agonist phenylephrine (PE) (10 μM; (in the presence of a β-AR blocker). α1-AR stimulation mediated negative inotropy in nonfailing RV but positive inotropy in failing RV. B: fast time-base recordings of individual contractions of RV myocardium from nonfailing or failing RV (same recordings as A). Force recordings expressed in absolute units are superimposed for contractions before addition of agonists (dotted traces) and after the inotropic responses to the agonists had developed (solid traces).
In contrast, for failing RV there was a switch to a positive inotropic effect (PIE) (Fig. 2). This is consistent with our previous finding of increased α1-AR inotropy in a model of RV failure induced secondary to LV failure (38). Interestingly, the PIE elicited by A61603 (α1A agonist) was greater than the PIE elicited by PE (α1A plus α1B mixed agonist) (discussed below).
The timing of contraction and relaxation were not affected by α1-AR stimulation, as evidenced by the similar time course of contraction and relaxation both before and after agonist (Fig. 2B) and from the data in Table 1 summarizing the time to peak contraction and the relaxation time.
Table 1.
No effect of α1-AR stimulation on the timing of contraction or relaxation of nonfailing or failing RV myocardium
| Time To Peak, ms |
RT50, ms |
|||||
|---|---|---|---|---|---|---|
| Basal | A61603 | PE | Basal | A61603 | PE | |
| Saline (4–5) | 137 ± 1 | 139 ± 1 | 136 ± 4 | 103 ± 6 | 103 ± 6 | 101 ± 6 |
| Bleomycin (3–7) | 137 ± 1 | 149 ± 2 | 138 ± 3 | 94 ± 6 | 93 ± 8 | 112 ± 6 |
Values are means ± SE. Measurements of the time to peak force (interval from the start of a contraction to the peak contraction force) and 50% relaxation time (RT50, interval from the peak contraction force to the time when force had relaxed by 50%) are shown. Nonfailing or failing right ventricle (RV) myocardium was obtained from animals 2 wk after a single tracheal instillation of saline or bleomycin, respectively. For these experiments, α1-adrenergic receptor (α1-AR) stimulation elicited a negative inotropic response in nonfailing RV, but a positive inotropic response in failing RV. However, the timing of contraction and relaxation was not statistically different than before agonist (number/group indicated in parentheses). PE, phenylephrine.
Upregulation of α1-AR inotropy develops 2 wk after bleomycin instillation.
Figure 3A summarizes the inotropic response elicited by A61603 at various times after bleomycin instillation. For RV myocardium studied less than 2 wk after bleomycin instillation, inotropic stimulation with the α1A-subtype agonist A61603 elicited a NIE. However, for RV trabeculae studied more than 2 wk after bleomycin, a PIE elicited by A61603 emerged. Thus the switch in the inotropic response to A61603 from a NIE in nonfailing RV to a PIE after bleomycin treatment required 2 wk to develop. Accordingly, all of the other data presented in this study involved mice more than 2 wk after bleomycin or saline instillation.
Figure 3A also shows that the switch to a PIE elicited by A61603 did not occur in a subset of hearts. Nevertheless, there was a markedly increased lung weight in animals whether α1A-subtype inotropy was increased (converter group) or not (nonconverter group) (see Table 2), suggesting that for some animals, the lack of upregulation of the α1A-subtype inotropic response was not due to a lack of bleomycin-induced lung injury.
Table 2.
Effect of tracheal instillation of bleomycin or saline on body and organ weights
| Percent Change in Body Weight | Lung Weight/Body Weight, mg/g | RV Weight/Body Weight, mg/g | |
|---|---|---|---|
| Saline (23) | 6.4 ± 0.9 | 5.7 ± 0.2 | 1.11 ± 0.03 |
| Bleomycin nonconverter (8) | −9.4 ± 3.4**** | 12.1 ± 1.1**** | 1.22 ± 0.04 |
| Bleomycin converter (12) | −15.0 ± 3.2**** | 13.2 ± 1.4**** | 1.37 ± 0.04****§ |
Values are means ± SE. A single instillation of bleomycin (0.075 U) or saline was given. After at least 2 wk, the effect of bleomycin was assessed on lung weight and on the RV free wall weight. Bleomycin resulted in a decrease in body weight, an increase in lung weight, and increased RV weight. The bleomycin group is divided into 2 subgroups, depending on whether the α1A-subtype inotropic response was switched to a positive inotropic response (converter) or remained a negative inotropic response (nonconverter) (number/group indicated in parentheses).
Significantly different than vehicle (P < 0.0001);
Significantly different that nonconverter (P < 0.05).
Figure 3B summarizes the inotropic response elicited by A61603 for hearts studied at least 2 wk after bleomycin instillation. As described above, bleomycin-treated hearts were divided into converter or nonconverter groups based on whether A61603 elicited a PIE or NIE, respectively. Moreover, inotropic responses assessed using absolute force units (Fig. 3B) were consistent with inotropic responses assessed using relative force units.
Upregulation of α1-AR inotropy in failing RV requires the α1A-subtype.
Figure 4 summarizes the contractile response to stimulation of the α1A-subtype singly by A61603 or in combination with the α1B-subtype using PE. For nonfailing RV, both A61603 and PE elicited a NIE (Fig. 4A) and there was no statistical difference between the responses to these agonists. After bleomycin treatment, ≈60% of animals had a markedly increased α1A-subtype inotropic response than saline-treated controls (converter group, Fig. 4C). For the remainder, the α1A-subtype inotropic response was similar to nonfailing RV myocardium (nonconverter group, Fig. 4B). For RV myocardium from bleomycin-treated animals, the nonconverter group was similar to the nonfailing RV group in having a NIE elicited both by A61603 or PE (Fig. 4B). In contrast, for the converter group, after 2 wk of bleomycin treatment, there was a switch in the α1A-subtype inotropic response to a PIE (Fig. 4C). As noted in Fig. 2, the pooled data in Fig. 4C show that the PIE elicited by the α1A-subtype agonist A61603 was appreciably greater than that induced by combined stimulation of both α1A- and α1B-subtypes with PE. The difference between the inotropic response to A61603 (reflecting the α1A-subtype) versus the inotropic response to PE (reflecting α1A- plus α1B-subtypes) may reflect the inotropic effect mediated by the α1B-subtype alone. The difference between the inotropic response to A61603 minus the response to PE is calculated to be a NIE attributed to the α1B-subtype (Fig. 4C).
Fig. 4.

Upregulation of α1-AR inotropy in failing RV mediated by the α1A-subtype. Summary of inotropic responses to maximal stimulation of the α1A-subtype either singly using A61603 or in combination with the α1B-subtype using PE are shown. Animals were studied >14 days after a single instillation of saline (nonfailing RV) or bleomycin (failing RV). Some animals received biweekly bleomycin instillation (triangles). Individual experimental values are shown, and the pooled means and SE indicated. A: for nonfailing RV, both A61603 and PE elicited a similar NIE. B: after bleomycin instillation, for a subset of animals (nonconverter group), A61603 and PE continued to elicit a NIE. C: in contrast, for some animals there was a conversion to a PIE mediated by A61603 (converter group). The PIE mediated by A61603 was greater than that mediated by PE (P = 0.0173). The difference between the response to A61603 minus the response to PE may reflect a NIE mediated by the α1B-subtype. *P < 0.05.
Taken together, these data suggest that the α1A-subtype mediates a NIE in nonfailing RV, but in failing RV the α1A-subtype inotropic response can be switched to a PIE. In contrast, the α1B-subtype mediates an NIE in nonfailing RV, and the α1B-subtype response is not switched in failing RV. Thus upregulation of α1-AR inotropy in the failing RV involves functional antagonism between the α1-AR subtypes, with a PIE mediated solely by the α1A-subtype and antagonized by a NIE mediated by the α1B-subtype.
To confirm the key role of the α1A-subtype in upregulation of α1-AR inotropy in the failing RV, we used knockout mice lacking the α1A-subtype (AKO). For AKO mice, the predominant remaining α1-AR subtype on cardiac myocytes is the α1B-subtype. Therefore, inotropic stimulation of myocardium from AKO mice using PE would stimulate the remaining α1B-subtype. Figure 5 shows original records of the contractile response of AKO myocardium following stimulation with A61603 or PE. As expected, RV myocardium from AKO mice was unresponsive to the α1A-subtype-selective agonist A61603. For mice 2 wk after tracheal instillation of saline or bleomycin, there was a NIE elicited by PE. The summary of all data (Fig. 6) shows that for AKO myocardium, stimulation of the remaining α1B-subtype using PE resulted in a NIE (force decreased ≈50%) in both nonfailing RV myocardium (Fig. 6A) and failing RV myocardium (Fig. 6B). Figure 6B shows that a NIE elicited by PE was observed for all AKO mice that had been instilled with bleomycin. Thus, for RV myocardium from animals instilled with bleomycin, a PIE in response to PE was observed for most wild-type hearts but not observed for AKO hearts. This difference is statistically significant (P < 0.05, χ2 test). We conclude that the α1B-subtype elicits a NIE, in both nonfailing and failing RV. This is consistent with the data in Fig. 4 suggesting the α1B-subtype has an NIE in both nonfailing and failing RV. Thus upregulation of α1-AR inotropy in the failing RV requires the α1A-subtype. Moreover, in failing RV, the α1B-subtype does not switch to a PIE but elicits a NIE. Together these data suggest that in the failing RV functional antagonism can emerge between the α1A-subtype (that mediates a PIE) and the α1B-subtype (that mediates an NIE). Specifically, when the α1A-subtype is stimulated in combination with the α1B-subtype the inotropic response is lower than when the α1A-subtype is stimulated in the absence of the α1B-subtype.
Fig. 5.
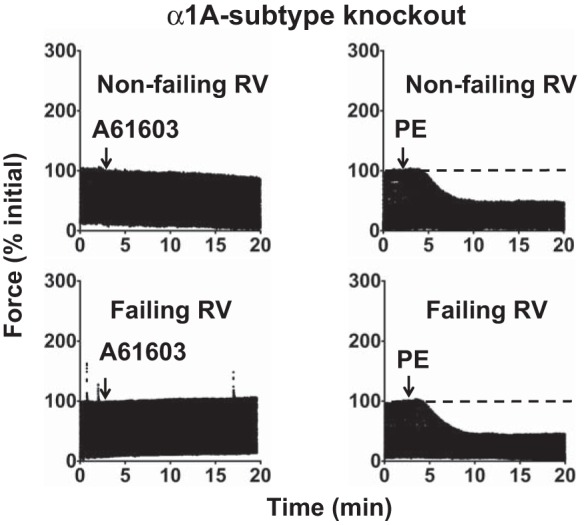
Upregulation of α1-AR inotropy in failing RV requires the α1A-subtype. Slow time-base recordings of contractions of RV myocardium from knockout mice lacking the α1A-subtype (AKO) are shown. RV myocardium was from animals with tracheal instillation of saline (nonfailing) or bleomycin (failing). Shown are acute inotropic responses to α1-AR agonists A61603 (100 nM) or PE (10 μM) in the presence of a β-AR blocker. AKO myocardium was unresponsive to A61603. Stimulation of the remaining α1B-subtype in AKO myocardium using PE elicited a NIE in both nonfailing RV and failing RV.
Fig. 6.
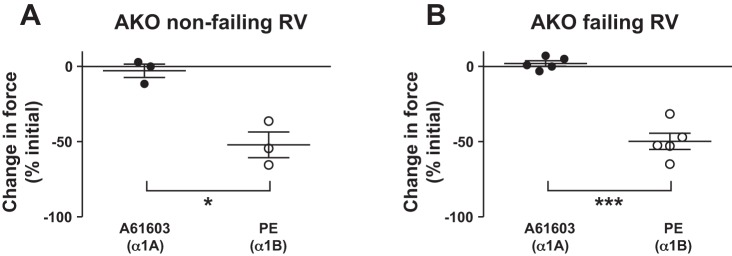
Negative inotropy mediated by the α1B-subtype. A and B: summary of data from all AKO experiments. RV myocardium from AKO mice did not respond to A61603. For AKO mice, stimulation of the remaining α1B-subtype using PE elicited a NIE in all experiments involving nonfailing RV or failing RV. Thus the α1B-subtype mediates a NIE in both nonfailing and failing RV. *P < 0.05; ***P < 0.001.
Mechanisms involved in the switch in α1A-subtype inotropy in failing RV.
The PIE elicited by α1-ARs was suggested to involve increased Ca2+ mobilization (5). We determined the effect of α1-AR inotropic stimulation on the fura-2 Ca2+ transient. A NIE elicited by α1A-subtype stimulation in nonfailing RV was associated with a reduction of the Ca2+ transient (Fig. 7A). In contrast, a PIE elicited by the α1A-subtype in failing RV was associated with an increase of the Ca2+ transient. Figure 7B summarizes that there was a significant positive relationship between changes in the Ca2+ transient and changes in contraction. This suggests that α1A-subtype-mediated effects on Ca2+ handling is a determinant of the contractile response and that an increased Ca2+ transient amplitude contributed to upregulation of α1A-subtype inotropic responses in the failing RV.
Fig. 7.

Relationship between the α1A-subtype inotropic responses versus changes in the Ca2+ transient. A: fast time-base recordings of Fura-2 Ca2+ transients (top) and simultaneously recorded contraction force (bottom) during contractions of RV myocardium from nonfailing RV (left) or failing RV (right). Records are superimposed for contractions before addition of A61603 (dotted traces) and after the inotropic responses to A61603 had developed (solid traces). A decreased Ca2+ transient mediated by A61603 was associated with a negative inotropic response. Conversely, an increased Ca2+ transient mediated by A61603 was associated with a positive inotropic response. B: changes in the amplitude of force- and Ca2+ transients after A61603 were expressed relative to their pre-agonist amplitude (defined as 100%). For all experiments involving myocardium from hearts at least 2 wk after bleomycin or saline instillation into the trachea, following α1A-subtype stimulation, there was a significant positive linear relationship between changes in the amplitude of the Ca2+ transient and changes in the amplitude of contraction (R2 = 0.787, P < 0.0001) (the relationship remained significant after excluding the most positive value). Some animals received biweekly bleomycin instillation (triangles).
The PIE elicited by α1-ARs was also previously suggested to involve increased phosphorylation of myosin RLC, which leads to increased myofilament Ca2+-sensitivity (2). Therefore, for trabeculae that manifested a PIE elicited by A61603, we measured the extent of phosphorylation of cardiac RLC. Figure 8 shows that for nonfailing RV myocardium, the RLC phosphorylation level was ∼50% as previously reported for contracting myocardium (10). Moreover, the RLC phosphorylation level was not changed by A61603 stimulation. This indicates that the NIE elicited by α1A-subtype stimulation in nonfailing RV did not involve decreased RLC phosphorylation, but instead, other mechanisms such as decreased Ca2+ transients may be important. Interestingly, the RLC phosphorylation level for the failing RV (≈15% of total RLC) was substantially lower than nonfailing RV (≈40% of total RLC, P = 0.03). Moreover, for hearts that manifested a PIE in response to α1A-subtype stimulation, there was a marked increase in the level of RLC phosphorylation (P = 0.036). Thus increased RLC phosphorylation may contribute to the PIE elicited by the α1A-subtype in the failing RV.
Fig. 8.

α1A-subtype effect on regulatory light chain (RLC) phosphorylation (p). Immunoblots and quantitative summary of RLC phosphorylation, with or without acute stimulation with A61603 for nonfailing RV and failing RV myocardium that had a positive inotropic response to A61603, are shown. In failing RV, RLC phosphorylation was reduced versus nonfailing RV. In failing RV, RLC phosphorylation was increased by α1A-subtype stimulation.
DISCUSSION
RV failure results in downregulation of β-ARs in humans and multiple experimental models in rodents (30). In contrast, α1-AR inotropic responses are upregulated in failing RV (38). Of the two predominant subtypes of α1-AR on cardiac myocytes (α1A and α1B), the current study indicates that upregulation of α1-AR inotropy in failing RV myocardium is mediated solely by the α1A-subtype. An increased level of contraction mediated by the α1A-subtype might provide inotropic support for the failing RV. Moreover, the α1A-subtype may represent a novel therapeutic target to augment the function of the failing RV. Because the prognosis of heart failure patients with pulmonary hypertension is strongly related to RV dysfunction (14), strategies to improve RV function may be beneficial for improving survival of patients with severe RV failure.
α1-AR signaling in heart failure.
It has been generally believed that α1-ARs, along with all G protein-coupled receptors that signal through Gαq, play a pathogenic role in heart failure. In contrast, this view has been challenged by the suggestion that α1-AR signaling is beneficial in heart failure (3, 20, 29). Consistent with α1-ARs mediating a cardioprotective effect, knockout of α1-ARs prevents normal cardiac growth (28) and worsens cardiomyopathy after pressure overload (27). Furthermore, a large clinical trial (ALLHAT) of an α1-AR antagonist was stopped prematurely because of an increase in heart failure in the α1-AR antagonist-treated group (1). These findings suggest that a higher level of α1-AR activation is cardioprotective in heart failure. Consistent with this, in human heart failure, where β-ARs are downregulated, α1-AR mediated inotropy can equal β-AR mediated inotropy, raising the possibility that a relative increase in α1-AR inotropy has a beneficial effect (34). In the context of the RV, we find that α1-AR inotropic responses are upregulated in the failing RV (38), which might help the failing RV adapt to increased pulmonary pressures.
Cardioprotective role of α1A-subtype signaling.
Previous studies suggest that α1A-subtype signaling has cardioprotective effects. α1A-subtype overexpression enhances contractility without causing hypertrophy (23), protects against pressure-overload-induced dysfunction (11), limits postinfarct cardiomyopathy (12), and protects against ischemic injury (32). Recent studies found that the α1A-subtype mediates a prosurvival effect in cardiac myocytes subjected to multiple pathological conditions (18). Furthermore, α1A-subtype agonist treatment prevents apoptosis in a heart failure model in vivo (6) and prevents cell death and fibrosis and improves in vivo function and survival in multiple heart failure models (26). Consistent with beneficial effects mediated by the α1A-subtype, in the present study we found that the α1A-subtype, but not the α1B-subtype, mediated increased myocardial contraction in the failing RV.
Functional antagonism between α1-AR subtypes in failing RV.
We found that for nonfailing RV, the α1A- and α1B-subtypes both elicited a NIE of similar magnitude when stimulated singly or in combination, consistent with our previous study (25). Nevertheless, in failing RV we found upregulation of α1-AR inotropy and the emergence of functional antagonism between α1A- and α1B-subtypes. In the failing RV, the α1A-subtype inotropic response was switched from a NIE to a PIE in ≈60% of hearts. However, the inotropic response mediated by the α1B-subtype was not observed to be switched and remained as a NIE in both nonfailing and failing RV. Therefore, for the failing RV, the inotropic response to nonsubtype selective α1-AR stimulation might be complex and consist of stimulatory effects mediated by the α1A-subtype that are antagonized by inhibitory effects mediated by the α1B-subtype.
Our findings are consistent with previous studies that suggested that the two predominant cardiac α1-AR subtypes can mediate different inotropic responses in LV myocardium. Previously, the α1A-subtype was linked to a PIE (23), and the α1B-subtype linked to a NIE (16, 33). Furthermore, in rat myocardium, stimulation of the α1A-subtype causes increases of contractions, Ca2+ transients, myofilament response to Ca2+, and intracellular pH (13); however, α1B-subtype stimulation decreases contractions, Ca2+ transients, and pH (13).
Mechanisms contributing to upregulated α1A-subtype response in the failing RV.
In general, our finding that the α1A-subtype elicits a NIE in nonfailing RV but a PIE in failing RV suggests that the signaling pathways linking α1A-subtype receptors to the cellular effectors can be altered in disease.
In the nonfailing RV, stimulation of the α1A-subtype mediated a decrease in both the Ca2+ transient and contraction. This is consistent with our previous studies suggesting a decrease in the amplitude of the Ca2+ transient contributed to a NIE elicited by α1-ARs (5, 25). In contrast, in the failing RV, α1A-subtype stimulation resulted in an increase in the Ca2+ transient and a PIE. This suggests that a switch in the effect of α1A-subtype stimulation on Ca2+ handling, from inhibitory in nonfailing RV to stimulatory in failing RV, contributed to the switch in the inotropic response from a NIE in nonfailing RV to a PIE in failing RV.
Previous studies suggest that phosphorylation of myosin RLC mediates increased myofilament Ca2+ sensitivity and contributes to a PIE mediated by α1-ARs (2, 38, 39). Consistent with this, in the present study we found that in failing RV, the PIE elicited by α1A-subtype stimulation was associated with increased cardiac RLC phosphorylation.
Heterogeneity of α1-AR inotropy after bleomycin treatment.
Previously, we found there was upregulation of α1-AR inotropy in a model of RV failure that was induced secondary to LV failure following myocardial infarction (MI) of the LV (38). However, for hearts subjected to small infarcts we found that the α1-AR inotropic response of RV myocardium was not upregulated, suggesting the existence of an injury threshold for switching of the RV α1-AR inotropic response in heart failure to a PIE (38).
Consistent with our previous study, in the present study, a switch to a PIE elicited by α1-ARs was not observed in all experiments. The duration of RV injury may be a determinant of upregulation of α1-AR inotropy as evidenced by the finding that a PIE elicited by α1-ARs was not observed before 2 wk after bleomycin instillation. Interestingly, the RV injury model of bleomycin-induced pulmonary fibrosis is associated with a high level of mortality within the first 2 wk after bleomycin instillation (17), and before upregulation of α1-AR inotropy develops. Potentially, upregulation of α1-AR inotropy may be protective and contribute to a lower mortality in the period beyond 2 wk after bleomycin instillation.
There was greater RV hypertrophy in hearts that did manifest a switch to a PIE elicited by α1-ARs (Table 2). Potentially, both the increased inotropic response and the greater RV mass might contribute to a beneficial effect on RV function.
Limitations.
The current study found upregulation of myocardial α1A-subtype inotropic responses using cardiac trabeculae studied in vitro. Further study is warranted to search for a functional in vivo correlate. The current study used cardiac trabeculae under in vitro conditions of low temperature and low pacing rate. The level of absolute force development observed was considerably lower than that of previous studies using more physiological conditions (35, 36).
The current study was performed using mouse myocardium. Although, α1-AR levels and regulation in mouse myocardium are similar to those in human (19, 21), future studies should determine whether α1A-subtype signaling is upregulated in RV myocardium from human hearts with RV failure. Further study is needed to determine the significance of our finding upregulation of α1A-subtype inotropy in the failing RV. In this regard, chronic agonist stimulation of the α1A-subtype may be beneficial in the failing heart (6, 26), and upregulation of α1A-subtype function in the failing RV may contribute to a beneficial effect. Finally, the mechanisms mediating upregulation of α1A-subtype inotropy remain unclear. For example, the roles of changes in RLC phosphorylation and Ca2+ transients were not resolved in this study. Moreover, the underlying mechanisms by which the α1A-subtype mediates a decreased Ca2+ transient in nonfailing RV but an increased Ca2+ transient in failing RV need to be defined.
Conclusion
In conclusion, the upregulated α1-AR inotropic response of failing RV is mediated by the α1A-subtype and not the α1B-subtype. The α1A-subtype may represent a novel therapeutic target to augment the function of the failing RV.
GRANTS
This work was supported by Department of Veterans Affairs Merit Review Awards I01BX000740 (to A. J. Baker) and I01BX000593 (to D. H. Lovett), National Heart, Lung, and Blood Institute Grants HL-31113 (to P. C. Simpson) and HL-080536 (to J. T. Stull), and from the Moss Heart Fund (to J. T. Stull).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.M.C., D.H.L., J.T.S., P.C.S., and A.J.B. conception and design of research; P.M.C., G.-Y.W., A.N.C., O.M., P.M.S., D.H.L., J.T.S., and P.C.S. performed experiments; P.M.C., A.N.C., O.M., P.M.S., D.H.L., J.T.S., and A.J.B. analyzed data; P.M.C., D.H.L., P.C.S., and A.J.B. interpreted results of experiments; P.M.C., A.N.C., and A.J.B. prepared figures; P.M.C. and A.J.B. drafted manuscript; P.M.C., J.T.S., P.C.S., and A.J.B. edited and revised manuscript; P.M.C., G.-Y.W., A.N.C., O.M., P.M.S., D.H.L., J.T.S., P.C.S., and A.J.B. approved final version of manuscript.
REFERENCES
- 1.ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive, and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA 283: 1967–1975, 2000. [PubMed] [Google Scholar]
- 2.Andersen GG, Qvigstad E, Schiander I, Aass H, Osnes JB, Skomedal T. α1-AR-induced positive inotropic response in heart is dependent on myosin light chain phosphorylation. Am J Physiol Heart Circ Physiol 283: H1471–H1480, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Baker AJ. Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflügers Arch 466: 1139–1150, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 51: 651–690, 1999. [PubMed] [Google Scholar]
- 5.Chu C, Thai K, Park KW, Wang P, Makwana O, Lovett DH, Simpson PC, Baker AJ. Intraventricular and interventricular cellular heterogeneity of inotropic responses to α1-adrenergic stimulation. Am J Physiol Heart Circ Physiol 304: H946–H953, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dash R, Chung J, Chan T, Yamada M, Barral J, Nishimura D, Yang PC, Simpson PC. A molecular MRI probe to detect treatment of cardiac apoptosis in vivo. Magn Reson Med 66: 1152–1162, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Groote P, Millaire A, Foucher-Hossein C, Nugue O, Marchandise X, Ducloux G, Lablanche JM. Right ventricular ejection fraction is an independent predictor of survival in patients with moderate heart failure. J Am Coll Cardiol 32: 948–954, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 299: L442–L452, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Salvo TG, Mathier M, Semigran MJ, Dec GW. Preserved right ventricular ejection fraction predicts exercise capacity and survival in advanced heart failure. J Am Coll Cardiol 25: 1143–1153, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE, Stull JT. Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo. J Biol Chem 285: 40819–40829, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du XJ, Fang L, Gao XM, Kiriazis H, Feng X, Hotchkin E, Finch AM, Chaulet H, Graham RM. Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J Mol Cell Cardiol 37: 979–987, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Du XJ, Gao XM, Kiriazis H, Moore XL, Ming Z, Su Y, Finch AM, Hannan RA, Dart AM, Graham RM. Transgenic alpha1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc Res 71: 735–743, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Gambassi G, Spurgeon HA, Ziman BD, Lakatta EG, Capogrossi MC. Opposing effects of α1-adrenergic receptor subtypes on Ca2+ and pH homeostasis in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 274: H1152–H1162, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, Arbustini E, Recusani F, Tavazzi L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 37: 183–188, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Greyson CR. Pathophysiology of right ventricular failure. Crit Care Med 36: S57–S65, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Grupp IL, Lorenz JN, Walsh RA, Boivin GP, Rindt H. Overexpression of α1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am J Physiol Heart Circ Physiol 275: H1338–H1350, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am J Physiol Lung Cell Mol Physiol 294: L24–L33, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O'Connell TD. An α1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 115: 763–772, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Jensen B, Swigart PM, Laden M, DeMarco T, Hoopes C, Simpson PC. The alpha-1D Is the predominant alpha-1-adrenergic receptor subtype in human epicardial coronary arteries. J Am Coll Cardiol 54: 1137–1145, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-adrenergic receptors in heart failure: the adaptive arm of the cardiac response to chronic catecholamine stimulation. J Cardiovasc Pharmacol 63: 291–301, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen BC, Swigart PM, De Marco T, Hoopes C, Simpson PC. α1-Adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ Heart Fail 2: 654–663, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamm KE, Hsu LC, Kubota Y, Stull JT. Phosphorylation of smooth muscle myosin heavy and light chains. Effects of phorbol dibutyrate and agonists. J Biol Chem 264: 21223–21229, 1989. [PubMed] [Google Scholar]
- 23.Lin F, Owens WA, Chen S, Stevens ME, Kesteven S, Arthur JF, Woodcock EA, Feneley MP, Graham RM. Targeted α1A-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ Res 89: 343–350, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Markel TA, Wairiuko GM, Lahm T, Crisostomo PR, Wang M, Herring CM, Meldrum DR. The right heart and its distinct mechanisms of development, function, and failure. J Surg Res 146: 304–313, 2008. [DOI] [PubMed] [Google Scholar]
- 25.McCloskey DT, Rokosh DG, O'Connell TD, Keung EC, Simpson PC, Baker AJ. α1-Adrenoceptor subtypes mediate negative inotropy in myocardium from α1A/C-knockout and wild type mice. J Mol Cell Cardiol 34: 1007–1017, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery MD, Chan T, Dash R, Swigart PM, Myagmar BE, Baker AJ, Simpson PC. An α1A adrenergic receptor agonist prevents and treats heart failure. Circulation 130: A20575, 2014. [Google Scholar]
- 27.O'Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC. α1-Adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest 116: 1005–1015, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E, Simpson PC. The α1A/C and α1B adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest 111: 1783–1791, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connell TD, Jensen BC, Baker AJ, Simpson PC. Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev 66: 308–333, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piao L, Fang YH, Parikh KS, Ryan JJ, D'Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation 126: 2859–2869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rokosh DG, Simpson PC. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci USA 99: 9474–9479, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rorabaugh BR, Ross SA, Gaivin RJ, Papay RS, McCune DF, Simpson PC, Perez DM. α1A- but not α1B-adrenergic receptors precondition the ischemic heart by a staurosporine-sensitive, chelerythrine-insensitive mechanism. Cardiovasc Res 65: 436–445, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Ross SA, Rorabaugh BR, Chalothorn D, Yun J, Gonzalez-Cabrera PJ, McCune DF, Piascik MT, Perez DM. The α1B-adrenergic receptor decreases the inotropic response in the mouse Langendorff heart model. Cardiovasc Res 60: 598–607, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Skomedal T, Borthne K, Aass H, Geiran O, Osnes JB. Comparison between alpha-1 adrenoceptor-mediated and beta adrenoceptor-mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther 280: 721–729, 1997. [PubMed] [Google Scholar]
- 35.Stull LB, Hiranandani N, Kelley MA, Leppo MK, Marban E, Janssen PM. Murine strain differences in contractile function are temperature- and frequency-dependent. Pflügers Arch 452: 140–145, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Stull LB, Leppo MK, Marban E, Janssen PM. Physiological determinants of contractile force generation and calcium handling in mouse myocardium. J Mol Cell Cardiol 34: 1367–1376, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114: 1883–1891, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Yeh CC, Jensen BC, Mann MJ, Simpson PC, Baker AJ. Heart failure switches the RV α1-adrenergic inotropic response from negative to positive. Am J Physiol Heart Circ Physiol 298: H913–H920, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GY, McCloskey DT, Turcato S, Swigart PM, Simpson PC, Baker AJ. Contrasting inotropic responses to α1-adrenergic receptor stimulation in left versus right ventricular myocardium. Am J Physiol Heart Circ Physiol 291: H2013–H2017, 2006. [DOI] [PubMed] [Google Scholar]