

Abstract
Aplastic anemia is a rare but life-threatening disorder characterized by cytopenia in at least two of the three blood lineages. A frequent feature of patients with aplastic anemia is that they have shorter telomeres than those of age-matched controls. Testosterone has been used for over half a century in the treatment of aplastic anemia. However, although remissions are frequent following hormone therapy, the molecular mechanism underlying the response to treatment has remained unknown. Here we explored the possibility that the recently described regulation of telomerase activity by sex hormones may be the mechanism responsible. To this end, we used a mouse model of aplastic anemia induced by short telomeres in the bone marrow compartment. We found that testosterone therapy results in telomerase up-regulation, improved blood counts, and a significant extension of life-span of these mice. Importantly, longitudinal follow-up studies revealed longer telomeres in peripheral blood in mice subjected to hormone treatment. Our results demonstrate that testosterone-mediated telomerase activation can attenuate or reverse aplastic anemia disease progression associated with the presence of short telomeres.
Introduction
Aplastic anemia is a rare and potentially fatal bone marrow disease which can develop at any stage of life and is characterized by peripheral cytopenia in at least two of the three blood lineages and marrow hypoplasia.1 Aplastic anemia can be acquired or inherited. The acquired form is mainly caused by T-cell triggered autoimmune processes against the hematopoietic stem cell compartment.2 In contrast, inherited forms of aplastic anemia can be caused by more than 30 mutations in genes involved in biological pathways such as DNA repair, ribosome biogenesis and telomere maintenance.3
Telomeres are specialized nucleoprotein structures located at the end of linear chromosomes.4 They are composed of long stretches of repetitive TTAGGG sequences that are bound by an array of proteins known as shelterins (TRF1, TRF2, TIN2, RAP1, TPP1 and POT1)5 which are important for telomere protection by preventing telomere fusions and telomere fragility. Telomerase is the enzyme capable of de novo synthesis of telomeric repeats. Telomerase activity is high at the blastocyst stage during which telomere length is determined for a given individual.6,7 Thereafter telomeres progressively shorten throughout life due to the down-regulation of telomerase and the loss of telomeric sequences associated with the replication of DNA ends with every cell division. Consequently, cells reaching a critical short telomere length enter into cellular senescence.8–11 In contrast to most somatic cells, adult stem cells retain basal telomerase activity which leads to decelerated telomere attrition in these compartments, although eventually these cells also show telomere shortening associated with the aging process.12 Conversely, impaired telomerase function, for instance by virtue of Tert haploinsufficiency, leads to accelerated telomere shortening in the stem cell compartments which prematurely limits the potential for hematopoietic stem cell proliferation and tissue renewal capacity, leading to the onset of different diseases. Thus, tissues with a high proliferative index such as the hematopoietic system are particularly affected by lower than normal telomerase levels.13 One of the most frequent telomere syndromes is dyskeratosis congenita, clinically defined by a triad of abnormal nails, reticular skin pigmentation, and oral leucoplakia.14,15 The most severe complication however, in ~80% of patients with dyskeratosis congenita, is the development of severe aplastic anemia14,16 (since we exclusively studied effects in the hematopoietic compartment, we will use the term aplastic anemia hereafter). Interestingly, both dyskeratosis congenita and aplastic anemia are characterized by premature telomere shortening. This is caused either by impaired telomere maintenance due to mutations affecting proper telomerase function or, in the case of autoimmune-mediated aplastic anemia, by increased cell turnover and hyperproliferation to compensate for hematopoietic stem cell depletion. Treatment options for aplastic anemia include stem cell transplants and immunosuppressive therapy.17 Interestingly, for more than five decades already, testosterone derivatives such as danazol and oxymetholone have been given to patients with aplastic anemia and such hormone therapies frequently induce remission.18,19 The mechanism underlying the response to treatment in autoimmune-triggered aplastic anemia has remained unclear to date. Interestingly, recent studies demonstrated that sex hormones act on the Tert gene to increase telomerase expression in vitro.20,21 With this in mind, here we set out to test the hypothesis that hormone therapies may induce remissions in patients with aplastic anemia because increased telomerase expression in this setting delays telomere shortening and disease.
To this end, we used a mouse model that faithfully recapitulates the bone marrow phenotype observed in patients with aplastic anemia.22 In this model, which we recently generated, conditional Trf1 gene deletion specifically in the hematopoietic system causes acute telomere uncapping and persistent activation of a DNA damage response at telomeres, leading to fast elimination of those hematopoietic stem cells and progenitor cells lacking Trf1. Because Trf1 deletion is not complete, the remaining non-Trf1 deleted hematopoietic stem cells undergo extra rounds of compensatory proliferation leading to rapid telomere shortening. Thus, incomplete depletion of the stem cell compartment by Trf1 deletion recapitulates the high turnover and hyper-proliferation observed in patients with aplastic anemia of autoimmune origin, as well as the presence of very short telomeres owing to mutations in telomere maintenance genes.
Interestingly, in our mouse model we can experimentally set the frequency of induction of Trf1-mediated stem cell depletion, thus adjusting the rate of progressive telomere shortening and disease onset. In particular, continuous Trf1 deletion leads to rapid telomere shortening and cellular senescence of the progenitor and stem cells in the bone marrow.23 This in turn leads to bone marrow aplasia and pancytopenia.22
Here we used this mouse model to examine whether androgen therapy can increase telomerase activity, decelerate telomere attrition and consequently delay the development of bone marrow failure.
Methods
Study approval
All experiments including mice were done using protocols approved by the CNIO–Instituto de Salud Carlos III (CNIO-ISCIII) Ethics Committee for Research and Animal Welfare (CEIyBA). Mice were treated in accordance with Spanish laws and the guidelines of the Federation of European Laboratory Animal Science Associations (FELASA).
Mice and animal procedures
All mice were bred on a C57/BL6 background and were produced and housed in a specific pathogen-free animal house of the CNIO, Madrid. Trf1lox/lox Mx1-Cre and Trf1lox/lox Mx1-wt mice were generated and bone marrow transplanted as previously described.22,24 Androgen therapy was administered by subcutaneous implantation of a 90-day, slow-release testosterone pellet (Innovative Research of America). For details see the Online Supplementary Material.
Cell culture
Bone marrow cells were isolated from wild-type and Tert+/− mice femora and tibiae. Erythrocytes were lysed with ammonium chloride solution (Stemcell Technologies) for 5 min, washed in phosphate-buffered saline and plated in AlphaMEM supplemented with 5% fetal bovine serum. For the preparation of mouse embryonic fibroblasts (MEF) Tert heterozygous knock-out mice were crossed to yield telomerase wild-type (Tert+/+), heterozygous (Tert+/−) and G1 knock-out mice (Tert−/−). MEF were cultured in Dulbecco’s modified Eagle’s medium without phenol red containing 10% charcoal stripped fetal bovine serum. Estradiol or testosterone (Sigma) was dissolved in MeOH and cells incubated at concentrations as indicated in individual experiments. 4-hydroxytamoxifen was added at a final concentration of 1 μM. For gene expression analyses cells were incubated for 1.5 h prior to harvesting. Long-term culture of MEF was done using the 3T3 protocol.25 MEF were prepared as described elsewhere.26
Quantitative real-time polymerase chain reaction
Total RNA from tissues was extracted with a Qiagen RNeasy Mini kit, according to the manufacturer’s instructions. Before processing, RNA samples were treated with DNaseI. Quantitative real-time polymerase chain reaction was performed using an ABI PRISM 7700 (Applied Biosystems). Primers are listed in Online Supplementary Table S2.
Testosterone enzyme-linked immunosorbent assay
At various time-points, 100 μL of peripheral blood were extracted from the same mice. Serum was isolated and frozen at −80°C until measurement of serum testosterone levels with a testosterone (mouse/rat) enzyme-linked immunosorbent assay kit (MyBioSource, MBS494055) according to the manufacturer’s protocol.
Blood counts
Peripheral blood was drawn from the facial vein (~50 μL) and collected into tubes containing anti-coagulant (EDTA). Blood counts were determined using an Abacus Junior Vet veterinary hematology analyzer.
Histology
Bone marrow and liver samples were fixed in phosphate-buffered 4% formaldehyde, bones were decalcified and embedded in paraffin. Tissue sections (5 μm) were stained with hematoxylineosin. Immunohistochemistry was performed on deparaffinized tissue sections and processed with anti-p19 antibodies (sc-32748, Santa Cruz Biotechnology).
Telomere analysis
Quantitative fluorescence in situ hybridization (Q-FISH) on tissue sections or metaphase spreads was performed as described elsewhere.27 High throughput Q-FISH on peripheral blood leukocytes was done using 120–150 μL of blood, as described previously.28 Details are provided in the Online Supplementary Material.
Statistics
Survival curves were analyzed using the log-rank (Mantel-Cox) test. Time course experiments were analyzed using two-way ANOVA (population doubling assay, high-throughput Q-FISH, testosterone level). The remaining experiments were analyzed using the Student t-test (quantitative polymerase chain reaction, Q-FISH, blood counts, immunohistochemistry). Data are presented as mean ± SEM; P values ≤0.05 were considered statistically significant.
Results
Testosterone and estradiol treatment increases Tert expression in vitro
We first investigated whether treatment with sex hormones (testosterone and estradiol) increases Tert expression in murine cells, as observed in cultured human cells.20,21 To this end, we isolated and cultured whole bone marrow cells and MEF (E13.5). As determined by quantitative reverse transcriptase polymerase chain reaction all cultures showed significantly increased Tert mRNA levels as soon as 1.5 h after incubation with testosterone or estradiol (~2-fold increase) compared to cells treated with MeOH (vehicle) only (Figure 1A,B). The effects of testosterone were dosedependent and the level of Tert induction varied between cell types (Figure 1A,B). Since both testosterone and estradiol could increase Tert expression, and estradiol seemed to be a more potent stimulant, we next tested whether this activation is mediated through the estrogen receptor, as has been described for primary human hematopoietic cells.21 We treated bone marrow cells and MEF simultaneously with testosterone and the estrogen receptor antagonist 4 hydroxytamoxifen and found that this significantly decreased the effects of testosterone in both types of cells suggesting that Tert up-regulation is mainly executed through the estrogen receptor (Figure 1C,D). Because MEF up-regulated Tert in response to testosterone treatment, we next studied primary bone marrow cells and MEF haploin-sufficient (Tert+/−) and null (Tert−/−) for the Tert gene.29 As expected, Tert expression in untreated Tert+/− cells (bone marrow and MEF) was around 50% of that of wild-type MEF (Figure 1E,F). Interestingly, testosterone treatment of Tert+/− cells significantly rescued Tert expression to levels close to those found in the respective wild-type cells. Simultaneous incubation with testosterone and 4 hydroxy tamoxifen abolished this effect (Figure 1E,F). No Tert mRNA was detected in Tert knock-out bone marrow cells or MEF (data not shown). Next, we analyzed the promoter region of Tert and identified, in close proximity to the transcriptional start site, an estrogen receptor response element half-site adjacent to two SP1 sites, a constellation previously shown to regulate gene expression in response to hormone treatment.30,31 In addition, we identified an imperfect palindromic estrogen receptor response element ~2.3 kb upstream from the transcriptional start site (Figure 1G). We did not find an androgen receptor response element consensus sequence within 5 kb upstream of Tert. These findings suggest direct Tert activation via the estrogen receptor in mice.
Figure 1.
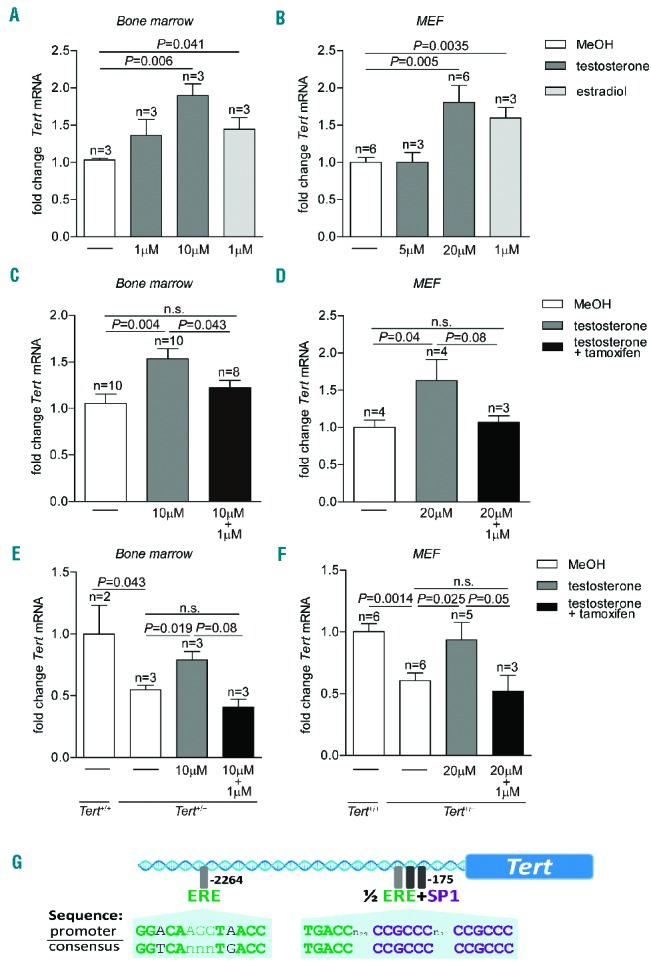
Tert activation by testosterone in vitro. (A) Fold change of Tert mRNA in murine bone marrow cells treated with testosterone or estradiol at indicated concentrations. (B) Fold change of Tert mRNA in MEF treated with testosterone or estradiol at indicated concentrations. (C) Fold change of Tert mRNA in murine bone marrow cells treated with testosterone and testosterone in combination with 1 μM 4-hydroxytamoxifen. (D) Fold change of Tert mRNA in MEF treated with testosterone and testosterone in combination with 1 μM 4-hydroxytamoxifen. (E) Fold change of Tert mRNA in Tert+/+ and Tert+/− bone marrow cells. Tert+/− cells were incubated with testosterone and testosterone in combination with 1 μM 4-hydroxytamoxifen. (F) Fold change of Tert mRNA in Tert+/+ and Tert+/− MEF. Tert+/− cells were incubated with testosterone and testosterone in combination with 1 μM 4-hydroxytamoxifen. All graphs show mean values, error bars indicate SEM, n = number of samples from different mice or MEF clones. A one-sided Student t-test was used for the statistical analysis. P-values are indicated. n.s. = not significant. (G) Illustration of putative binding sites for the estrogen receptor (ERE) and the Sp1 transcription factor in the promoter region of the mouse Tert gene.
Hormone treatment affects telomere length but not proliferation of mouse embryonic fibroblasts
Next, we investigated whether hormone treatment resulted in increased telomere length in MEF and whether this was mediated by telomerase activity. To this end, we incubated Tert wild-type, heterozygous and first generation Tert knock-out MEF (G1) with estradiol or MeOH for four passages (p3–p7). To measure telomere length, we performed telomere-specific Q-FISH analysis on metaphase spreads, which allows quantification of the length of individual telomeres. First, we observed normal telomere function when comparing estradiol-treated with untreated cells as indicated by similar frequencies of telomeric aberrations such as signal free ends and chromosome fusions (neither detected), and multi-telomeric signals (Figure 2A,B). Interestingly, estradiol-treated wild-type MEF showed significantly increased average telomere length as well as decreased abundance of short telomeres compared to MeOH-treated cells (Figure 2C,D). Importantly, this telomere elongation as the result of hormone treatment was mediated by telomerase, as we did not find a significant difference in telomere length between the estradiol-treated and untreated cells when we used G1 Tert knock-out MEF (Figure 2C). No significant differences in telomere length were observed when comparing MeOH with estradiol-treated heterozygous knock-out MEF (Online Supplementary Figure S1).
Figure 2.
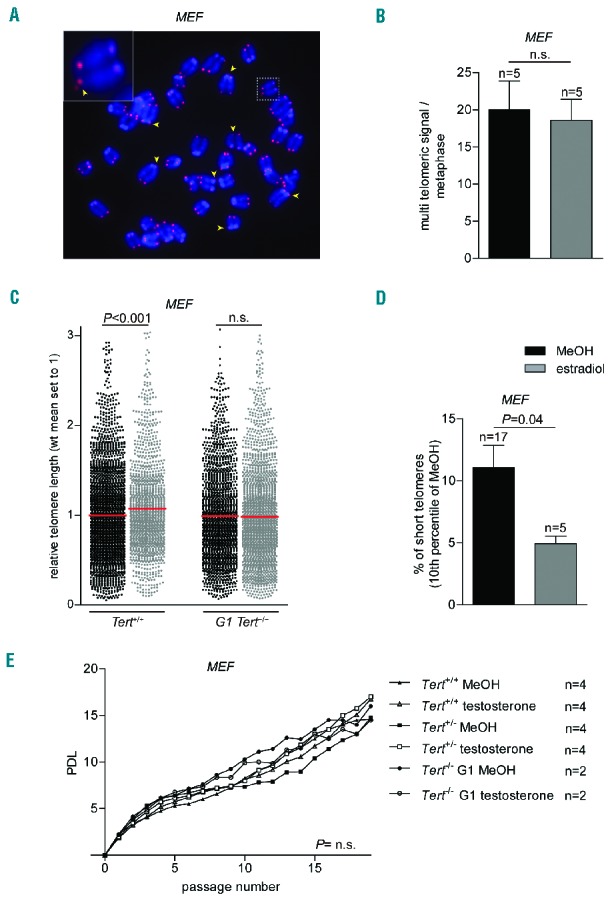
Effects of testosterone and estradiol on MEF. (A) Representative image of a MEF metaphase spread subjected to telomere Q-FISH analysis. DNA is stained with DAPI (blue) and a telomere-specific Cy3-labeled probe (red). Arrows indicate multi-telomeric signals. (B) Number of multitelomeric signals found in metaphases of wild-type MEF treated with 1 μM estradiol or vehicle alone. n = number of metaphases (C) Relative telomere length (arbitrary units of fluorescence) of Tert+/+ or Tert−/− MEF incubated with 1 μM estradiol for four passages. In Tert+/+ n = 1346 telomeres for estradiol and n = 2576 telomeres for methanol treatment. In Tert−/− n = 2041 telomeres for estradiol and n = 1670 telomeres for methanol (MeOH) treatment. (D) Percentage of short telomeres of wild-type MEF treated with 1 μM estradiol or MeOH represented as 10th percentile of telomere length of the MeOH control. (E) Population doubling level (PDL) of wild-type Tert+/+, heterozygous Tert+/− or knock-out Tert−/− MEF treated with or without 20 μM testosterone for 20 passages. n = number of MEF clones for each genotype and treatment. Graphs show mean values, error bars indicate SEM. The Student t-test was used for statistical analysis of the experiments shown in panels (B–D) and two-way ANOVA analysis for that shown in panel (E). P-values are indicated. n.s. = not significant.
To rule out that these effects on telomere length could be mediated by an effect of the hormones on MEF proliferation, we performed a long-term 3T3 assay with Tert wild-type, heterozygous and Tert (G1) knock out MEF in the presence or absence of testosterone. After 20 passages, we could not find any differences in proliferation in any of the three genotypes independently of the hormone treatment (Figure 2E).
In vivo testosterone treatment results in longer telomeres in wild-type mice
To investigate the effects of hormone treatment in vivo, we first implanted mice with 90-day, slow-release testosterone pellets subcutaneously and subsequently performed enzyme-linked immunosorbent assays to check serum testosterone levels at several time-points after implantation of the pellets. We found a more than 20-fold increase in testosterone levels which remained stable over time (Online Supplementary Figure S2A). To determine whether this increased testosterone was biologically active, we tested the well-described erythropoietic stimulating effect of androgens.32,33 Eight weeks after testosterone pellet implantation, we found significantly elevated erythrocyte counts, increased hematocrit, and increased hemoglobin levels in the hormone-treated mice compared with the untreated controls (Online Supplementary Figure S2B–D).
Next, we determined the effects of long-term hormone treatment on telomere length in vivo by performing Q-FISH analysis on tissue sections from mice treated with testosterone for 6 months compared to the untreated controls. We found significantly longer telomeres in bone marrow cells in the testosterone-treated mice compared with non-treated controls (Online Supplementary Figure S2E). Of note, we did not observe significant differences in telomere length between treated and untreated mice in the case of liver (Online Supplementary Figure S2F), in agreement with the lower cell proliferation in this organ than in the bone marrow and the fact that telomerase-dependent telomere elongation occurs in association with cell division.34,35
In vivo testosterone treatment significantly rescues survival in a mouse model of aplastic anemia associated with short telomeres
Next, we set out to investigate the potential benefits of androgen therapy in a mouse model of aplastic anemia induced by short telomeres, which we recently generated.22 In this mouse model, Trf1 deletion leads to rapid telomere shortening and the onset of aplastic anemia. We first generated wild-type irradiated mice, which received either Trf1lox/lox Mx1-Cre or Trf1lox/lox Mx1-wt bone marrow transplants. In this experimental setting, Trf1 deletion can be induced specifically in the bone marrow upon treatment with polyinosinic:polycytidylic acid (pI:pC), which will trigger Cre expression in the Trf1lox/lox Mx1-Cre transplanted bone marrow but not in the Trf1lox/lox Mx1-wt control. We hypothesized that in vivo activation of Tert by androgen therapy can antagonize telomere attrition associated with pI:pC-induced Trf1 deletion and thus prevent or delay the appearance of aplastic anemia and prolong survival in this mouse model.
First, we performed a protocol for long-term induction of Trf1 deletion and induction of telomere shortening previously described by us.22 In detail, bone marrow-transplanted mice were injected twice weekly with pI:pC until they developed signs of aplastic anemia (Figure 3A). Strikingly, androgen therapy in these mice, delivered by subcutaneous implantation of a slow-release pellet of testosterone, significantly improved survival compared with that of testosterone-untreated mice (Figure 3B). No significant differences in average survival were observed when mice were injected three times per week with pI:pC, although some of the testosterone-treated mice survived for double the time of the non-treated controls, suggesting that the level of stem cell depletion and telomere attrition induced by this treatment may be too severe to see the beneficial effects of hormone therapy on average survival in this experimental model (Online Supplementary Figure S3A). As controls to rule out potential adverse effects of pI:pC treatment and testosterone therapy, we treated Trf1lox/lox Mx1-wt mice with pI:pC or Trf1lox/lox Mx1-Cre with testosterone alone, respectively. In both cases, we did not see any adverse effects of the treatment on survival during 200 days after treatment, when all mice were sacrificed (Online Supplementary Figure S3B,C).
Figure 3.
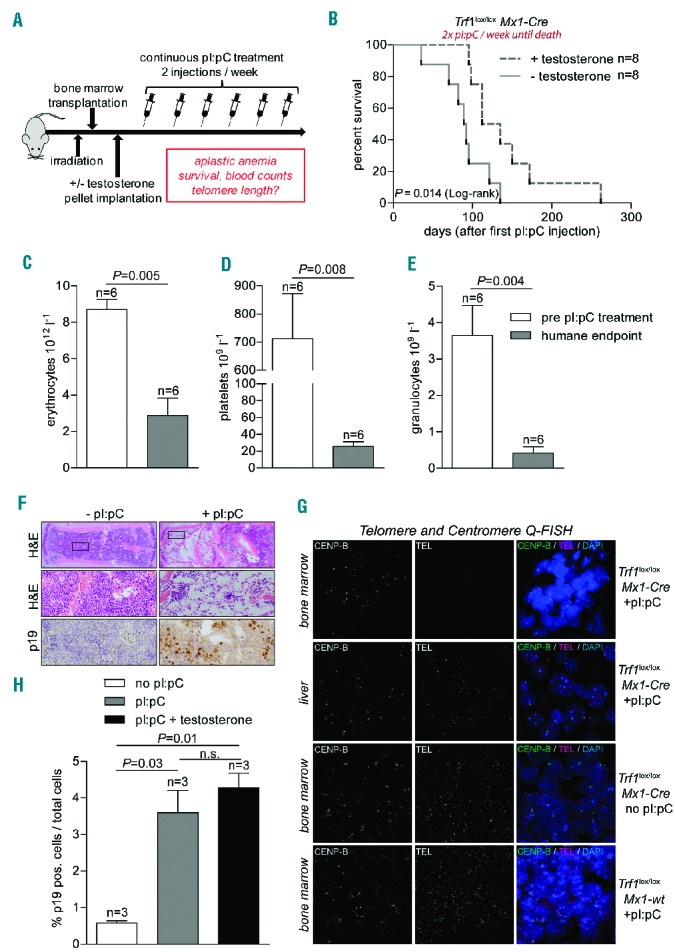
Effects of in vivo androgen therapy during long-term Trf1 deletion. (A) Experimental design. Eight-week old wild-type mice were transplanted with bone marrow (Trf1lox/lox Mx1-Cre). After a latency period of 1 month mice were subcutaneously implanted with a slow-release testosterone pellet. Trf1 deletion was induced by pI:pC at the indicated frequencies. (B) Kaplan-Meier survival curve of mice treated with pI:pC twice weekly until death. In addition, mice were or were not treated with testosterone, as indicated. A log-rank (Mantel-Cox) test was used for statistical analysis. The P-value is depicted. (C) Erythrocyte, (D) platelet and (E) granulocyte counts of mice before pI:pC treatment and after they presented with clear signs of anemia (humane endpoint). (F) Images of bone marrow cross-sectioned and stained with hematoxylin and eosin (H&E) at different magnifications and anti-p19 immunohistochemistry. (G) Representative Q-FISH images of bone marrow and liver cross-sections. Genotypes (Trf1lox/lox Mx1-Cre; Trf1lox/lox Mx1-wt) and pI:pC treatment are indicated on the right. Cross sections were stained with a centromeric probe (CENP-B, Alexa-488) and a telomere-specific probe (TEL-Cy3). Nuclei were stained with DAPI. (H) Percentage of p19-positive cells in bone marrow sections of mice which received the indicated treatment. All bar graphs show mean values, error bars indicate SEM, n = number of mice. A two-sided Student t-test was used for statistical analysis. P-values are indicated.
We next performed full histopathological analyses of mice sacrificed because they had anemic pallor and a poor health status regardless of the androgen treatment. First, analysis of peripheral blood counts confirmed the aplastic anemia phenotype as indicated by drastically lowered erythrocyte, platelet and, granulocyte counts compared to the blood counts of the same mice before pI:pC treatment had been started (Figure 3C–E). Further, histological analysis revealed severe hypoplasia of the bone marrow in mice treated with pI:pC compared with normal bone marrow cellularity in pI:pC-untreated mice (Figure 3F). Next, we determined telomere length in mice showing signs of aplastic anemia; we performed telomere Q-FISH analysis on cross-sections of bone marrow. Strikingly, telomere signals were undetectable in the bone marrow sections of Trf1-deleted mice indicating that telomeres had shortened beyond the detection limit of the Q-FISH technique (i.e., between 500–1000 bps). As a control for the presence of chromosomes, centromeric sequences were detectable in these samples (Figure 3G – top row). In contrast, telomere signals were readily detected in liver samples from the same mice, demonstrating extreme telomere loss specifically in the bone marrow compartment of this experimental model (Figure 3G – second row). As a control, Q-FISH analysis on bone marrow sections from control mice which did not express Cre (Trf1lox/lox Mx1-Cre mice without pI:pC injection or Trf1lox/lox Mx1-wt with pI:pC injection) showed no telomere loss (Figure 3G – third and bottom row). In line with the extremely short telomere phenotype in these mice,23 we noted an increased percentage of cells positive for the senescence marker p19 in bone marrow cross-sections in pI:pC-treated mice (~4%) compared to untreated controls (~0.5%) (Figure 3F,H).
In conclusion, persistent Trf1 deletion resulting from regular administration of pI:pC twice weekly in our experimental model resulted in severe stem cell depletion and extreme telomere shortening in the bone marrow, thus leading to aplastic anemia. Importantly, this process could be significantly attenuated by androgen therapy.
Next, we used a different protocol for Trf1 deletion in our mouse model, in which we first induced deletion of Trf1 by administering pI:pC three times a week for a total of 4 weeks and then gave androgen therapy to some of the mice (Figure 4A). Interestingly, this regime resulted in longer overall survival times upon Trf1 deletion (Figure 4B).
Figure 4.
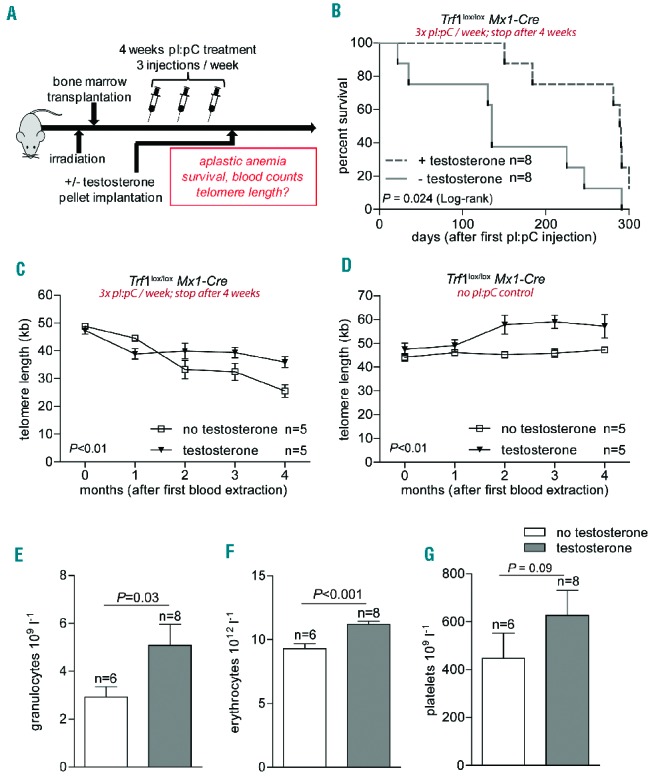
In vivo androgen therapy during moderate Trf1 deletion. (A) Experimental design. Eight-week old wild-type mice were transplanted with bone marrow (Trf1lox/lox Mx1-Cre). After a latency period of 1 month Trf1 deletion was induced by three pI:pC injections per week for a total of 4 weeks. Mice were then subcutaneously implanted with a slow-release testosterone pellet. (B) Kaplan-Meier survival curve of mice treated as detailed above. A log-rank (Mantel-Cox) test was used for statistical analysis. The P-value is depicted. (C) High throughput-Q-FISH longitudinal analysis of telomere length in peripheral blood leukocytes in mice treated with or without testosterone. All mice also received pI:pC injections three times per week for a total period of 4 weeks. (D) High throughput-Q-FISH longitudinal analysis of telomere length in peripheral blood leukocytes in mice not treated with pI:pC and treated or not with testosterone. (E) Granulocyte, (F) erythrocyte and (G) platelet counts 3 months after pI:pC treatment was stopped. Mice given or not given androgen therapy are compared. All graphs show mean values, error bars indicate SEM, n = number of mice. A two-way ANOVA test was used for statistical analysis in (C) and (D); a two-sided Student t-test was used for the experiments shown in (E–G). P-values are indicated.
The longer survival times allowed us to perform longitudinal telomere length analysis on peripheral blood leukocytes and follow telomere dynamics over time in both mouse cohorts.11 As expected, Trf1 deletion resulted in a rapid shortening of telomere length in leukocytes as early as 1 month after the treatment had been started. Importantly, after this initial decrease in telomere length, mice receiving androgen therapy were able to maintain a stable telomere length over time, while telomeres continued to shorten in mice that did not receive androgen therapy (Figure 4C). As controls, mice in which Trf1 deletion was not induced (not treated with pI:pC) showed a stable telomere length throughout the experiment. Interestingly, control mice in which Trf1 deletion was not induced (not treated with pI:pC) but which received androgen therapy, showed a significant elongation of telomeres during the period of therapy (Figure 4D).
In agreement with telomere length maintenance, the Trf1-deleted mice that underwent androgen therapy showed significantly higher granulocyte and erythrocyte counts after 3 months of androgen therapy compared to controls not given androgen therapy (Figure 4E–F). A similar trend was observed for platelet counts (Figure 4G).
In summary, these data support our hypothesis that androgen therapy counteracts telomere attrition in mice with aplastic anemia due to Trf1-induced telomere shortening, thus preventing replicative exhaustion of hematopoietic stem cells and the development of aplastic anemia.
Discussion
Some patients with aplastic anemia have been treated with androgens since the 1950s and in some of them remission was observed.18,36 However, the underlying mechanism for treatment success in some cases but not in others has remained unknown to date. There is growing evidence that sex hormones stimulate telomerase expression.20,21 This is of particular importance for those cases of aplastic anemia presenting with very short telomeres, which in turn may be associated with an exhaustion of hematopoietic stem cells.
The findings described here indicate that treatment with androgens increases Tert expression in mouse bone marrow cells and MEF grown in vitro. In particular, primary MEF treated for several passages with estradiol show increased Tert expression and significantly increased telomere length. Furthermore, we show that this effect is mediated by telomerase, as similarly treated Tert-deficient MEF did not show telomere elongation.
Importantly, we demonstrated here the beneficial effects of androgen therapy in an in vivo mouse model of aplastic anemia produced by short telomeres.22 In this experimental model, androgen therapy improved telomere length maintenance and resulted in a significant delay of aplastic anemia and in an increased survival. We showed that testosterone treatment can slow down telomere erosion caused by continuous depletion of the hematopoietic stem cell compartment. Despite the aforementioned androgen-induced remission in some studies,18,36 no such beneficial effects of androgens were observed in a larger cohort study.37 This discrepancy may be due to different mutation spectra causing the disease in these studies. Our work suggests that aplastic anemia patients with flawed telomere maintenance and/or short telomeres are those who would benefit in particular from androgen-mediated Tert stimulation while the treatment may have no effects in patients with normal telomere length. Interestingly, in this regard, clinical investigations are underway to address the efficacy of danazol (an androgen derivative) treatment for genetic bone marrow disorders (ClinicalTrials.gov Identifier: NCT01441037) such as dyskeratosis congenita.
Of note, our mouse model of aplastic anemia associated with short telomeres due to depletion of hematopoietic stem cells may be of relevance to understand the pathobiology of aplastic anemia associated not only with telomerase mutations but also with mutations in other genes that cause hematopoietic stem cell depletion (e.g. Fanconi anemia38). In this regard we previously demonstrated that accelerated telomere shortening in patients with Fanconi anemia is not caused by dysfunctional telomeres,39 suggesting that hormone-mediated telomerase activation may be used prophylactically in patients with Fanconi anemia to slow down telomere erosion and disease progression. In addition, immune-mediated hematopoietic stem cell depletion is thought to cause telomere shortening and immune-mediated aplastic anemia is currently being treated with immunosuppressive drugs. Our results also suggest that androgen derivatives might provide a useful addition to recent treatment protocols in order to prevent replicative exhaustion as a consequence of the underlying autoimmune process in aplastic anemia.
In summary, several lines of evidence suggest that telomerase activation and telomere elongation can be mediated through the action of sex hormones in vivo. Here, we present the first experimental evidence that testosterone therapy in a mouse model of aplastic anemia produced by short telomeres is beneficial for survival and is associated with longer telomere length in bone marrow and peripheral blood cells. On the basis of our findings, further investigation of hormone therapy in humans with aplastic anemia is warranted. Finally, hormone treatment may be extended to treat and/or prevent other disease manifestations associated with short telomeres and replicative exhaustion.
Acknowledgments
We acknowledge the support of Blasco’s Laboratory which is funded by the Spanish Ministry of Economy and Competitiveness and the Fundación Botín.
Footnotes
The online version of this article has a Supplementary Appendix
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dolberg OJ, Levy Y. Idiopathic aplastic anemia: diagnosis and classification. Autoimmun Rev. 2014;13(4–5):569–573. [DOI] [PubMed] [Google Scholar]
- 2.Nakao S. Immune mechanism of aplastic anemia. Int J Hematol. 1997;66(2):127–134. [DOI] [PubMed] [Google Scholar]
- 3.Dokal I, Vulliamy T. Inherited bone marrow failure syndromes. Haematologica. 2010;95(8):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106(6):661–673. [DOI] [PubMed] [Google Scholar]
- 5.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–2110. [DOI] [PubMed] [Google Scholar]
- 6.Schaetzlein S, Lucas-Hahn A, Lemme E, et al. Telomere length is reset during early mammalian embryogenesis. Proc Natl Acad Sci USA. 2004;101(21):8034–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela E, Schneider RP, Ortega S, Blasco MA. Different telomere-length dynamics at the inner cell mass versus established embryonic stem (ES) cells. Proc Natl Acad Sci USA. 2011;108(37):15207–15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vera E, Bernardes de Jesus B, Foronda M, Flores JM, Blasco MA. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep. 2012;2(4):732–737. [DOI] [PubMed] [Google Scholar]
- 9.Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22(5):654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. [DOI] [PubMed] [Google Scholar]
- 11.Canela A, Vera E, Klatt P, Blasco MA. High-throughput telomere length quantification by FISH and its application to human population studies. Proc Natl Acad Sci USA. 2007;104(13):5300–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiyama E, Hiyama K. Telomere and telomerase in stem cells. Br J Cancer. 2007;96(7): 1020–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359(9324):2168–2170. [DOI] [PubMed] [Google Scholar]
- 14.Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009;23(2):215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de la Fuente J, Dokal I. Dyskeratosis congenita: advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant. 2007;11(6):584–594. [DOI] [PubMed] [Google Scholar]
- 17.Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. [DOI] [PubMed] [Google Scholar]
- 18.Shahidi NT, Diamond LK. Testosterone-induced remission in aplastic anemia of both acquired and congenital types. Further observations in 24 cases. N Engl J Med. 1961;264:953–967. [DOI] [PubMed] [Google Scholar]
- 19.Jaime-Perez JC, Colunga-Pedraza PR, Gomez-Ramirez CD, et al. Danazol as first-line therapy for aplastic anemia. Ann Hematol. 2011;90(5):523–527. [DOI] [PubMed] [Google Scholar]
- 20.Kyo S, Takakura M, Kanaya T, et al. Estrogen activates telomerase. Cancer Res. 1999;59(23):5917–5921. [PubMed] [Google Scholar]
- 21.Calado RT, Yewdell WT, Wilkerson KL, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009;114(11):2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012(15)2990–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8(6):450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez P, Thanasoula M, Munoz P, et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23(17):2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17(299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol. 2005;Chapter 28(Unit 28): 21. [DOI] [PubMed] [Google Scholar]
- 27.Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep. 2000;1(3):244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canela A, Klatt P, Blasco MA. Telomere length analysis. Methods Mol Biol. 2007; 371:45–72. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Snow BE, Hande MP, et al. The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr Biol. 2000;10(22):1459–1462. [DOI] [PubMed] [Google Scholar]
- 30.Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88(2):113–122. [DOI] [PubMed] [Google Scholar]
- 31.Porter W, Wang F, Wang W, Duan R, Safe S. Role of estrogen receptor/Sp1 complexes in estrogen-induced heat shock protein 27 gene expression. Mol Endocrinol. 1996;10(11): 1371–1378. [DOI] [PubMed] [Google Scholar]
- 32.Gurney CW, Fried W. Further studies on the erythropoietic effect of androgens. J Lab Clin Med. 1965;65(775–782. [PubMed] [Google Scholar]
- 33.Fried W, Gurney CW. The erythropoietic-stimulating effects of androgens. Ann N Y Acad Sci. 1968;149(1):356–365. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Sfeir AJ, Zou Y, et al. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell. 2009;138(3):463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcand S, Brevet V, Mann C, Gilson E. Cell cycle restriction of telomere elongation. Curr Biol. 2000;10(8):487–490. [DOI] [PubMed] [Google Scholar]
- 36.Gardner FH. Androgen therapy of aplastic anaemia. Clin Haematol. 1978;7(3):571–585. [PubMed] [Google Scholar]
- 37.Champlin RE, Ho WG, Feig SA, Winston DJ, Lenarsky C, Gale RP. Do androgens enhance the response to antithymocyte globulin in patients with aplastic anemia? A prospective randomized trial. Blood. 1985;66(1):184–188. [PubMed] [Google Scholar]
- 38.Dokal I, Vulliamy T. Inherited aplastic anaemias/bone marrow failure syndromes. Blood Rev. 2008;22(3):141–153. [DOI] [PubMed] [Google Scholar]
- 39.Franco S, van de Vrugt HJ, Fernandez P, Aracil M, Arwert F, Blasco MA. Telomere dynamics in Fancg-deficient mouse and human cells. Blood. 2004;104(13):3927–3935. [DOI] [PubMed] [Google Scholar]