

Background: Thiol dioxygenation is catalyzed by enzymes specific for each substrate.
Results: Kinetic, structural, and spectroscopic data describe an enzyme from P. aeruginosa that is a 3-mercaptopropionate dioxygenase with secondary cysteine dioxygenase activity.
Conclusion: An arginine to glutamine switch and the absence of a cis-peptide bond correlate with substrate preference.
Significance: Characterization of this enzyme deepens our understanding of substrate specificity in thiol dioxygenases.
Keywords: crystal structure, dioxygenase, enzyme kinetics, Mossbauer spectroscopy, Pseudomonas aeruginosa (P. aeruginosa), substrate specificity
Abstract
Thiol dioxygenation is the initial oxidation step that commits a thiol to important catabolic or biosynthetic pathways. The reaction is catalyzed by a family of specific non-heme mononuclear iron proteins each of which is reported to react efficiently with only one substrate. This family of enzymes includes cysteine dioxygenase, cysteamine dioxygenase, mercaptosuccinate dioxygenase, and 3-mercaptopropionate dioxygenase. Using sequence alignment to infer cysteine dioxygenase activity, a cysteine dioxygenase homologue from Pseudomonas aeruginosa (p3MDO) has been identified. Mass spectrometry of P. aeruginosa under standard growth conditions showed that p3MDO is expressed in low levels, suggesting that this metabolic pathway is available to the organism. Purified recombinant p3MDO is able to oxidize both cysteine and 3-mercaptopropionic acid in vitro, with a marked preference for 3-mercaptopropionic acid. We therefore describe this enzyme as a 3-mercaptopropionate dioxygenase. Mössbauer spectroscopy suggests that substrate binding to the ferrous iron is through the thiol but indicates that each substrate could adopt different coordination geometries. Crystallographic comparison with mammalian cysteine dioxygenase shows that the overall active site geometry is conserved but suggests that the different substrate specificity can be related to replacement of an arginine by a glutamine in the active site.
Introduction
The global sulfur cycle involves interconversion of sulfur through a wide range of oxidation states. The most abundant oxidation states of sulfur are −2 as thiol/thiolate (e.g. cysteine or hydrogen sulfide), 0 as elemental sulfur, and +6 as sulfate. Within the sulfur cycle, microbiological transformation dominates the conversion of inorganic sulfur and to some extent organic sulfur (1). Whereas bacteria are very flexible in terms of their sulfur source, animals require dietary intake of sulfur-containing amino acids, such as methionine, which serves as a biosynthetic precursor of cysteine (2). In organisms, sulfur is present in proteins, nucleic acids, sulfate esters of polysaccharides, steroids, phenols, and sulfur-containing coenzymes (3, 4). Therefore, sulfur-dependent enzymology represents an important means of biological sulfur utilization. In particular, two thiol-containing compounds, cysteine and 3-mercaptopropionic acid, are of interest because they are both abundant and are irreversibly removed during their respective metabolic cycle by dioxygenation (2, 5–7).
In mammals, cysteine levels are maintained directly from dietary sources and/or through biosynthesis from methionine (2, 7). High cysteine levels have been implicated in a number of neurodegenerative diseases due to its exitotoxic effect in the nervous system (8–12). Accumulation of cysteine has been attributed to impaired function of cysteine dioxygenase (CDO)5 (9, 13–15), an enzyme catalyzing dioxygenation of cysteine thiolate to sulfinate (CSA) (16–21). Functional CDO occurs widely and has been identified in mammals, lower eukaryotes and in bacteria (21–23). CDO belongs to a family of non-heme mononuclear iron dioxygenases but stands out due to a number of unique characteristics. The iron atom has His3 coordination, and, in mammalian forms, a post-translational modification involving a cysteine-to-tyrosine cross-link is present (24, 25).
3-Mercaptopropionic acid (3-MPA) is a structural analogue of cysteine (Fig. 1), lacking only the amine group. Despite the fact that it is one of the most abundant thiols in wetlands (26) and fresh water lakes (27), most bacteria cannot use 3-MPA as a sole carbon source (28–30). Instead, bacteria are capable of growing on various 3-MPA precursors, which are catabolized to 3-MPA inside the cell (28–31). In bacteria, 3-MPA is a central metabolite in both catabolic and assimilatory sulfur metabolism (5), and although bacteria can form 3-MPA as a metabolite of methionine, homocysteine (32), or dimethylsulfoniopropionate, (31–33), little is known about the dissimilation of 3-MPA. In one bacterium, Variovarax paradoxus, 3-MPA utilization was shown to involve dioxygenation of 3-MPA to 3-sulfinopropionic acid (3-SPA), catalyzed by a CDO homologue, 3-mercaptopropionate dioxygenase (3MDO), which showed high specificity for 3-MPA but not for cysteine (6). Interestingly, 3-MPA acts as an inhibitor of metallo-β-lactamase, an enzyme involved in antibiotic resistance in bacteria (34, 35). In mammals, 3-MPA is toxic by inhibiting the oxidation of fatty acid (36, 37) and the synthesis of the inhibitory neurotransmitter γ-aminobutyric acid, resulting in deformation, seizures, or death of rats (38–40).
FIGURE 1.
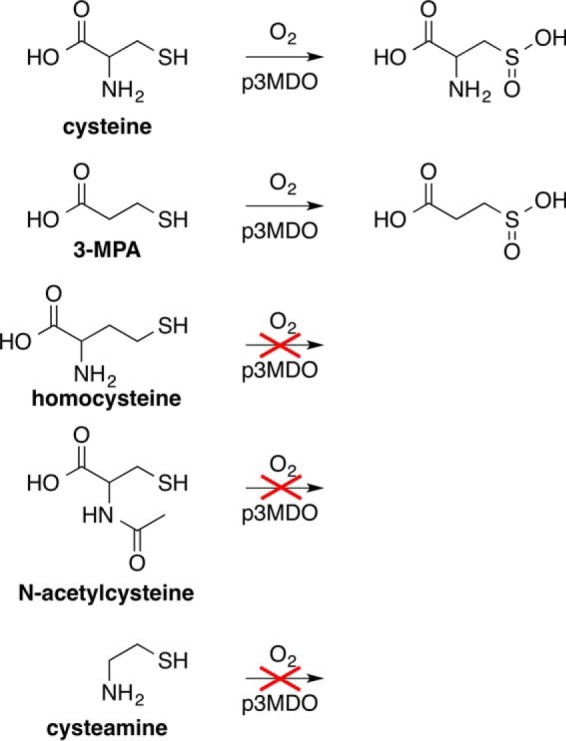
Structural formula of thiol-containing compounds tested as substrates for p3MDO.
The Gram-negative proteobacterium Pseudomonas aeruginosa is an opportunistic pathogen, which can infect plants and animals, including humans. This bacterium has gained importance as a nosocomial pathogen that infects immunocompromised patients and is highly resistant to antibiotic treatment (41). Due to its ability to utilize a wide range of organic matter and cope with different environmental conditions, this organism can colonize many natural environments, including soil, water, and skin. By implication, P. aeruginosa metabolic enzymology illustrates various means of environmental adaptation. Considering that P. aeruginosa colonizes human hosts, it is not surprising that it can utilize cysteine or methionine as a sulfur source (42). Although it has been established that cysteine can be fully reduced to H2S in the course of P. aeruginosa metabolism (43), oxidation of cysteine to CSA through CDO or an equivalent enzyme has not been reported. Similarly, little is known about bacterial enzymes involved in 3-MPA metabolism. Strains from the Pseudomonas family are capable of 3-MPA utilization (28). Conversion of 3-SPA to 3-sulfinopropionyl-CoA in Proteobacteria has been attributed to succinyl-CoA synthetase, (44, 45); however, enzymatic oxidation of 3-MPA to 3-SPA remains to be identified. CDO homologues from bacteria (Ralstonia eutropha, Protein Data Bank accession number 4QMA (formerly 2GM6); Bacillus subtilis, 3EQE) are structurally very similar to mammalian CDO, for which several x-ray crystal structures are available, but the substitution of glutamine in bacterial enzymes for arginine at position 60 of the mammalian active site suggests altered substrate specificity.
In this study, we report identification, classification, structural, kinetic, and spectroscopic characterization of P. aeruginosa thiol dioxygenase (p3MDO). This enzyme is a 3-MPA dioxygenase with secondary cysteine dioxygenase activity. This is the first report of the enzymology of cysteine and 3-MPA dissimilation in P. aeruginosa metabolism and the first example of a CDO homologue that utilizes a second substrate with near stoichiometric coupling to dioxygen consumption.
Experimental Procedures
Protein Sequence Identification, Expression, and Purification
The NCBI reference sequence (NC_002516.2) for P. aeruginosa (strain PAO1) (46) contains a region gi|110645304:2945264–2945869 coding for a hypothetical protein PA2602 (NP_251292.1) (47). A BLAST search (48) of putative bacterial CDO sequence (YP_299237.1) from R. eutropha (strain JMP134) was performed using the SIB-BLAST network service and identified PA2602 as a CDO homologue (Fig. 2). The experimental design for the PCR amplification of the PA2602 coding sequence was optimized using Primer D'Signer software (IBA). DNA sequence coding for CDO was PCR-amplified using Pfu DNA polymerase (Stratagene) and inserted into pPR-IBA1, a Strep-tag affinity-based expression plasmid (IBA) digested with BsaI (Fermentas), according to standard procedures to yield an open reading frame encoding the full PA2602 protein with a C-terminal extension of SAWSHPQFEK. Both strands of the coding region of the P. aeruginosa PA2602 expression construct (pPR-IBA1/PA2602/P. aeruginosa) were fully sequenced at the Genetic Analysis Services at the University of Otago to confirm the absence of adventitious mutations. A molecular mass of 23,746.5 Da was calculated based on the amino acid sequence of the expressed protein (49, 50). PA2602 expression was induced in BL21(DE3)pLysS cells (Novagen) and purified using Strep-tag affinity chromatography according to the manufacturer's protocol (IBA) (51, 52). We employed the following modifications to the standard protein expression/purification procedures: (i) cells were incubated overnight at 18 °C during protein expression; (ii) cell lysis buffer contained 100 mm Tris (pH 8.0), 1.15 m NaCl, 1 mm EDTA, 5 mm Tween 20 (1.5%, v/v), and a Roche complete protease inhibitor; and (iii) cells were lysed using a French pressure cell (model FA-078, SLM Aminco). The purified protein preparations were dialyzed extensively (>109 dilution factor) against buffer compatible with the subsequent experiment. Protein was concentrated on Vivaspin centrifugation concentrators (GE Healthcare) to the appropriate concentrations for the subsequent experiments. As expected, P. aeruginosa CDO homologue preparations resolved into a single band when subjected to SDS-PAGE, revealing the absence of a posttranslational modification present in mammalian CDO (53–56) (Fig. 3). Protein preparations were found to be at least 97.5% pure and contained ∼15% endogenously bound iron as judged by colorimetric assay using ferrozine in a 96-well format as described previously (57). Protein concentrations were determined spectrophotometrically at 280 nm using an extinction coefficient of 29,700 m−1 cm−1 estimated from a bovine serum albumin standard curve quantified by SDS-PAGE densitometry. For anaerobic study protein preparations, corresponding buffers and solvents were made anaerobic by alternating cycles of vacuum and purging with argon gas on a Schlenk line kept on ice slurry. All anaerobic manipulations were performed in a nitrogen atmosphere in a glovebox (Belle Technology; <1 ppm oxygen).
FIGURE 2.

Alignment of the P. aeruginosa CDO homologue with selected family members. Numbering of P. aeruginosa residues is shown at the top, and the extent of each sequence is indicated at the left. Residues of the CDO fingerprint proposed by Stipanuk et al. (23) are shown in white on black. White on gray indicates the possible divergence of the fingerprint in substrate specificity at position 62 in PA2602 (glutamine versus arginine) and the difference in uncross-linked residue at position 95 (glycine or alanine versus cysteine) and at position 168 (arginine versus various). Residues adjacent to and possibly influencing the cross-link are shown in black on light gray. The conserved cupin motif residues are indicated at the bottom. PA2602 from P. aeruginosa PAO1, NP_251292.1, is aligned with characterized 3MDOs (6) (V. paradoxus TBEA6, ACB72254; R. eutropha H16, YP_841375); CDOs (20, 21, 25) (Homo sapiens, NCBI protein sequence accession number BAA12873; R. norvegicus, BAA11925; Streptomyces coelicolor A3(2) (SCO3035), NP_627257; S. coelicolor A3(2) (SCO5772), NP_629897; B. subtilis, NP_390992; Bacillus cereus, NP_832375) and putative CDOs (23) with a putative cysteine-tyrosine cross-link (Vibrio splendidus 12B01, EAP93565.1; Chromobacterium violaceum ATCC, NP_902852.1).
FIGURE 3.
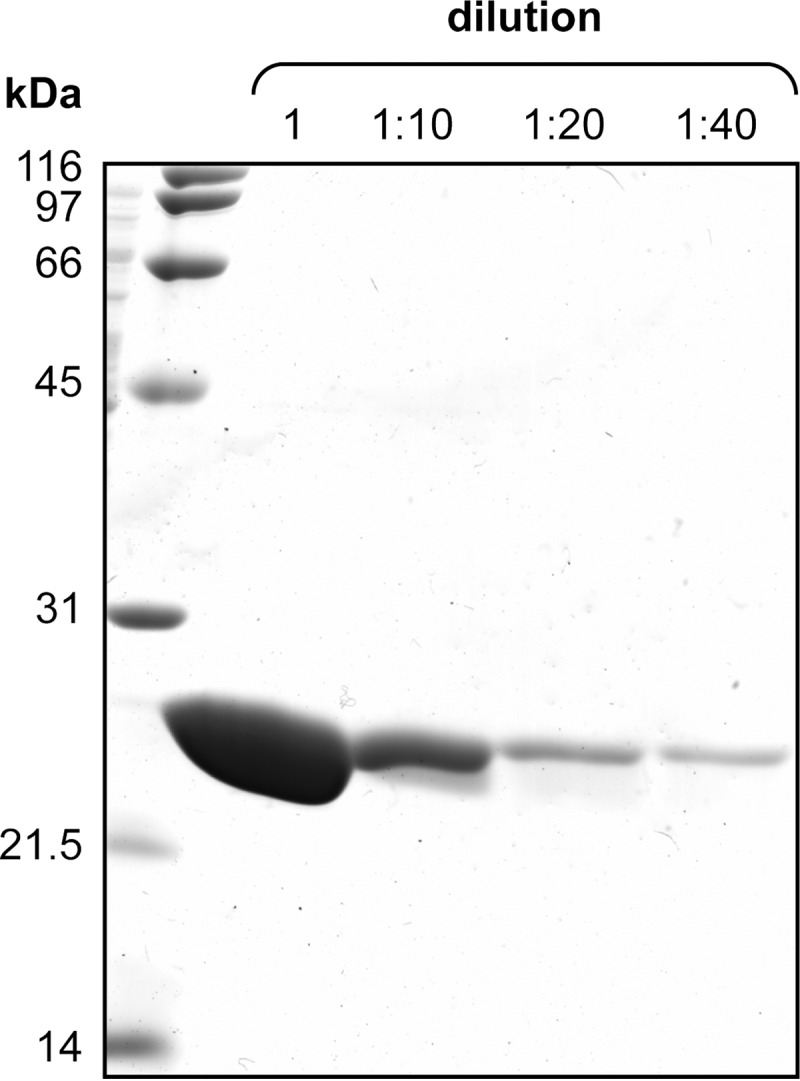
Purified samples of p3MDO were loaded on a 15% (w/v) polyacrylamide gel, resolved by SDS-PAGE, and detected by staining with Coomassie Brilliant Blue dye. A series of p3MDO dilutions (1, 1:10, 1:20, and 1:40) is shown. A 40-fold difference in protein sample loaded illustrates at least 97.5% purity of the protein preparation.
Assessment of PA2602 Protein Expression in P. aeruginosa
P. aeruginosa strain PAO1 was grown in L-broth to late logarithmic phase (A600 ∼3.3), and the cells were collected by centrifugation. Approximately 0.6 g of cell pellet was resuspended in 2.3 ml of 100 mm phosphate buffer, pH 7.5, in the presence of protease inhibitors. Cells were sonicated (SONICS Vibra cell) on ice for 5 min (9 s on, 9 s off, amplitude 38%). The lysed cells were spun down at 30,000 × g at 4 °C for 30 min. Supernatant (100 μl) was mixed with ice-cold acetone (500 μl) and stored at −20 °C for 45 min to precipitate the proteins. After spinning the solution for 30 min at 16,000 × g at room temperature, the supernatant was discarded, and the was pellet air-dried on the bench overnight. The dried pellet was dissolved in SDS-buffer, and proteins were separated by SDS-gel electrophoresis. Comparison with the retention of purified p3MDO on a separate gel and with the use of molecular weight markers revealed a region of interest of about 1 cm between 21.5 and 31 kDa, which was excised from the gel to be measured by mass spectrometry. The proteins were digested “in gel” with trypsin using a robotic work station for automated protein digestion (DigestPro Msi, Intavis AG, Cologne, Germany) essentially following the protocol as described by Shevchenko et al. (58).
Parallel reaction monitoring (PRM) assays were performed by nanoflow ultra-HPLC-coupled TripleTOF 5600+ mass spectrometry (AB Sciex, Framingham, MA). The m/z values of doubly and triply charged precursor ions and their 10 strongest singly charged transitions (m/z of fragment ions) were predicted for seven proteotypic peptides of p3MDO using Skyline software (available from the MacCoss Lab Web site) (59). An aliquot of the sample was first analyzed by an unscheduled PRM assay using the target peptides given in Table 1.
TABLE 1.
Peptides identified by PRM and software-assisted spectrum interpretation using Mascot
| Predicted m/z (charge state)a | Peptide sequence identified by Mascot | Score | Modification |
|---|---|---|---|
| gi|372467082 chain A, crystal structure of cysteine dioxygenase from P. aeruginosa | |||
| 474.5772 (3+) | YQQYLLHVDSR | 36 | |
| 648.8433 (2+) | LEPGEVEALSPR | 85 | |
| 515.5866 (3+) | IGDVHQVSNAFSDR | 40 | |
| 829.4547 (2+) | TSISIHVYGANIGAVR | 48 | |
| 725.6833 (3+) | AVFSAEGEEKPFISGYSNSR | 70 | |
| gi|489228560 isoprenoid biosynthesis protein, partial (P. aeruginosa) | |||
| 755.7236 (3+) | LVTTPAYMLAQSIAEAASGINK | 85 | Oxidation (M) |
| No identification | |||
| 764.3578 (3+) | GAEYSQPYAFDAGGRPHPSGAR | ||
a Predicted m/z (charge state) is the m/z of the strongest charge state predicted by the Skyline software that was used to build the scheduled PRM assay. Score: Mascot ion score at a significance threshold of p < 0.05. An ion score of >56 indicates a significant peptide identification in a search against 48 million sequence entries of the non-redundant NCBI sequence database. All peptide sequences assigned to cysteine dioxygenase were unique for the identified protein.
LC separation of peptides was performed on an Ekspert 400 LC system (Eksigent, AB Sciex) at a flow rate of 500 nl/min using a homemade emitter tip column (75-μm inner diameter PicoTip fused silica tubing (New Objectives, Woburn, MA) packed with 3-μm C-18 beads on a length of 12 cm). The gradient was developed from 2% acetonitrile, 0.2% formic acid in water to 25% acetonitrile, 0.2% formic acid in water over 45 min, followed by an increase to 90% acetonitrile, 0.2% formic acid in water over 15 min. The mass spectrometer was operated in precursor mode with a precursor isolation window of 0.7 units (unit resolution) and an ion accumulation time of 100 ms to allow for the acquisition of both charge states of all seven precursors (14 ions/cycle) within a 2-s cycle time. For precursor fragmentation, rolling collision energy was selected, and all transitions were monitored in parallel by the TOF analyzer in a high resolution fragment ion spectrum. Raw data of the unscheduled PRM were imported into the Skyline software for compound detection and determination of retention times. Skyline was then used to build a scheduled PRM method of only the strongest detected charge state of each precursor within a retention time window of 2 min to be used by the instrument software (Analyst software, AB Sciex). For the scheduled PRM method, the same LC gradient and MS settings were used as for the unscheduled PRM with the exception of an increased ion accumulation time of 150 ms for each precursor and a maximum of 4 precursors/cycle, resulting in a cycle time of 0.9 s.
Raw data were analyzed using Skyline for compound detection and the peak view software (AB Sciex) for the extraction of high resolution fragment ion spectra and peak lists. Peak lists were converted to a Mascot generic format for software-assisted sequence assignment by the database-dependent search engine Mascot (available from the Matrix Science Web site). Spectra were searched against the non-redundant NCBI amino acid sequence database (48,573,147 sequence entries), allowing for the identification of semitryptic peptides with a maximum of three missed cleavage sites and the variable modifications of carboxyamidomethylated cysteine, oxidized methionine, and deamidated asparagine/glutamine. Precursor and fragment ion accuracy were 25 ppm and 0.1 Da, respectively.
Crystallization and Structure Determination
Crystals were grown using protein that had been reconstituted with iron (see below) by both the hanging drop and the sitting drop vapor diffusion methods. To prepare hanging drops, 300 nl of protein at 11 mg/ml in water was mixed with 150 nl of 100 mm HEPES, 100 mm NaCl, 1.6 m (NH4)2SO4, pH 7.5. Crystals grew as extended rhombohedra of ∼100 μm at 16 °C in less than 14 days. Crystals were transferred to a 15% (v/v) solution of glycerol in the well solution and mounted in a loop at −180 °C. Sitting drop crystals were produced in 70-μl reservoirs by mixing 2 μl of protein (11 mg/ml) with 1 μl of mother liquor containing 200 mm sodium acetate, 8% (w/v) PEG 4000, pH 5.7. Crystals grew quickly as extended rhombohedra of ∼500 μm within days at 18 °C, and these were transferred to 25% (v/v) ethylene glycol in well solution and mounted in a loop at −180 °C.
Using crystal A, grown from ammonium sulfate, 180° of data were collected using 1° oscillations at the MX1 and MX2 beamlines of the Australian Synchrotron with a wavelength of 0.9537 Å and an Area Detector Systems Corp. detector. Data were indexed in space group P212121, with unit cell dimensions of 184.38, 63.02, and 35.49 Å, integrated in Mosflm, and scaled using Scala; 5% of reflections were reserved for calculation of the Rfree statistics (Table 2). Programs of the CCP4 suite (Collaborative Computational Project, Number 4) were used for structure solution. Phases were solved by molecular replacement using the R. eutropha structure (Protein Data Bank code 2GM6, modified in Chainsaw) as a search model in Phaser (60). Two monomers were placed in the asymmetric unit. Automated model refinement was performed in Refmac 5 (61), with two TLS groups defined in TLSMD (62) and medium NCS restraints. Models were inspected in COOT (63), and manual adjustment was performed to match chemical expectations, electron density maps, and anomalous electron density maps (this last for iron atoms only). Electron density adjacent to the active site iron was modeled as solvent molecules, including a sulfate ion, in agreement with other CDO structures from crystals prepared in the presence of sulfate and absence of substrate and product. However, the resolution was not sufficient to rule out other explanations, such as bound product (CSA) carried through the purification. A final round of refinement in Refmac5.5 used diffraction data to 2.7 Å but no prior phase information (Table 2).
TABLE 2.
Data collection, reduction, and crystallographic refinement statistics
| Crystal A ((NH4)2SO4) | Crystal B (NaOAc) | Crystal C (NaOAc) | |
|---|---|---|---|
| Resolution range (highest shell) | 48.9–2.70 Å (2.85–2.70 Å) | 47.3–2.50 Å (2.64–2.50 Å) | 47.2–2.14 Å (2.25–2.14 Å) |
| Observed reflections | 112,263 (16,607) | 238,593 (31,848) | 665,020 (92,078) |
| Unique reflections | 16,270 (2,339) | 31,044 (4,391) | 49,221 (6,940) |
| Redundancy | 6.9 (7.1) | 7.7 (7.3) | 13.5 (13.3) |
| Completeness | 99.9% (99.7%) | 99.9% (99.5%) | 99.8% (98.7%) |
| I/σ | 13.9 (4.2) | 16.6 (2.8) | 16.5 (7.7) |
| Rmergea | 0.100 (0.464) | 0.099 (0.664) | 0.114 (0.371) |
| Reflections used in refinement | 16,261 (1,180) | 28,051 (4132) | 45,220 (5,937) |
| Rcrystb | 0.208 (0.283) | 0.183 (0.312) | 0.198 (0.271) |
| Rfreec | 0.283 (0.375) | 0.245 (0.391) | 0.249 (0.332) |
| Deviation from ideal bond lengths | 0.017Å | 0.009Å | 0.009Å |
| Deviation from ideal bond angles | 1.753° | 1.258° | 1.168° |
| Average B factor | 41.64 Å2 | 40.91 Å2 | 26.13 Å2 |
| Macromolecules | 41.64 Å2 | 41.02 Å2 | 25.63 Å2 |
| Iron atoms | 79.04 Å2 | 42.95 Å2 | 26.10 Å2 |
| Solvent | 41.69 Å2 | 37.42 Å2 | 31.31 Å2 |
| Protein Data Bank code | 3USS | 4TLF |
a Rmerge = Σ|Iobs − Iave|/ΣIave.
b Rcryst = Σ|Fobs − Fcalc|/ΣFobs computed over a working set composed of 95% of data.
c Rfree = Σ|Fobs − Fcalc|/ΣFobs computed over a test set composed of 5% of data.
A preliminary data set of 180° at 1° oscillation was collected at MX2 of the Australian Synchrotron from sodium acetate-grown crystal B and indexed in space group P41212, with unit cell dimensions of 66.94, 66.94, 377.35 Å. Data were integrated to 2.5 Å in XDS and scaled using Scala; 5% of reflections were reserved for calculation of the Rfree statistics (Table 2). The structure obtained using crystal A was used for molecular replacement, yielding four monomers in the asymmetric units. A higher resolution data set was collected by the same method from a second sodium acetate-grown crystal (crystal C) and indexed with parameters indistinguishable from those for the preliminary data set from crystal B. The programs of the CCP4 suite and Phenix were used for structure solution, with the refined 2.5 Å structure from crystal B used for molecular replacement. Four monomers were placed in the asymmetric unit. The active site iron and solvent molecules were placed using Phaser-EP (MR-SAD). Phenix Refine and Readyset were used for refinement. The following residues were not included in the final model (chain A: 1–4, 199–211; chain B: 1–4, 198–211; chain C: 1–3, 200–211; chain D: 1–3, 202–211).
Iron Binding by PA2602 Protein
Iron binding was investigated by adding various amounts of ferrous iron sulfate to deoxygenated protein, removing the excess iron by Chelex 100, and then measuring the protein-bound iron by a ferrozine-based assay (57). The data were fitted to the equation (64, 65), y = 0.5(Kapp + E + x) − (0.25(Kapp + E + x)2 − Ex)½, where x is the concentration of added iron, y is the concentration of detected protein-bound iron, E is the concentration of protein competent for iron binding, and Kapp is the apparent dissociation constant for the protein-iron complex.
Cysteine-dependent Oxygen Depletion Activity of PA2602 Protein
Activities upon the addition of cysteine to protein samples were tested by following oxygen depletion with a Clarke-type oxygen electrode (Rank Brothers) as described previously (57). Appropriate control experiments were conducted to confirm that oxygen depletion was not due to cysteine autoxidation under reaction conditions.
Quantitative Assessment of Cysteine Dioxygenase Activity of PA2602 Protein after FeII Enrichment
PA2602 cysteine dioxygenase activity after treatment of the protein with ferrous iron sulfate followed by Chelex was measured using an HPLC-evaporative light scattering detector with a Luna 5 μ hydrophobic interaction liquid chromatography 200 Å column (Phenomenex) as described previously (77, 78). 5–13 μm PA2602 was incubated at 37 °C with 1–21 mm l-cysteine in the presence of 20–400 μm bathocuproinedisulfonic acid (10 mm phosphate buffer, 0–20 mm NaCl, pH 7.5). The reaction was initiated by mixing 5 μl of PA2602 with 845 μl of temperate buffered substrate. The reaction mixture was incubated for 60 min, with aliquots withdrawn at regular time intervals. Both substrate and product showed linear changes over 60 min, and the slopes were used to calculate the respective reaction velocities. The velocities were plotted versus initial cysteine concentrations and were fitted to the Michaelis-Menten equation with Vmax and Km constants using Prism version 5 (GraphPad) software. Vmax was normalized to the PA2602 active site iron concentration to calculate kcat. Alternative reaction conditions of pH 6.5 were also tested; however, the rate of the reaction was slower, and therefore pH 7.5 was used for subsequent kinetic experiments.
Assessment of PA2602 Substrate Specificity
p3MDO (170 μm active site iron) cysteine-specific activity in the presence of 10 mm cysteine was tested for inhibition by 10 mm cysteine analogs (3-MPA, dl-homocysteine, N-acetyl-l-cysteine, or cysteamine) using the HPLC-evaporative light scattering detector assay described above.
Quantitative Assessment of 3-MPA Dioxygenase Activity of PA2602 after FeII Enrichment
A derivatization method (66) based on the reaction between the free-thiol group of 3-MPA and 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellman's reagent) was adapted to quantify 3-MPA. PA2602 (9 μm) was incubated at 37 °C with 0.2–5 mm 3-MPA (100 mm phosphate buffer, pH 7.5). The reaction was initiated by mixing 50 μl of PA2602 with 450 μl of temperate buffered substrate. Reaction aliquots were withdrawn at various time points and processed and analyzed as described previously. Michaelis-Menten parameters were obtained and analyzed in the same way as described for cysteine dioxygenation above.
Mössbauer Spectroscopy
Ferrous 57Fe stock solutions for Mössbauer spectroscopy experiments were prepared as described previously (67). Anaerobic preparations of protein (0.8–1.5 mm) in 100 mm Tris-HCl (pH 8.0) and 50 mm NaCl were iron-saturated anaerobically by exogenously adding ferrous iron in the presence of 1 eq of dithionite, followed by treatment with analytical grade Chelex 100 sodium form, 200–400 mesh (Bio-Rad). Protein samples were frozen in liquid nitrogen in a glovebox (Belle Technology). 57Fe Mössbauer spectra of frozen liquid samples in a custom Teflon sample holder (∼400-μl volume) were recorded on a Mössbauer spectrometer from Science Engineering & Education Co. (Edina, MN) equipped with a closed cycle refrigerator system from Janis Research Co. and Sumitomo Heavy Industries Ltd. and a temperature controller from Lakeshore Cryotronics, Inc. Data were collected in constant acceleration mode in transmission geometry with an applied field of 47 milliteslas parallel to the γ-rays. The zero velocity of the Mössbauer spectra refers to the centroid of the room temperature spectrum of a 25-μm metallic iron foil. Analyses of the spectra were conducted using the WMOSS program (Science Engineering & Education Co.). The Mössbauer spectra of p3MDO·FeII resting state (n = 10), cysteine-saturated (n = 4), and 3-MPA-saturated (n = 2) samples were recorded, and average p3MDO Mössbauer parameters were determined. All spectra shown in this work were fitted using a parameter file containing constrained average p3MDO Mössbauer parameters such that only relative areas of p3MDO·FeII and p3MDO·FeII·substrate species were allowed to vary.
Determination of p3MDO·FeII·Substrate Complex Dissociation Constants
Formation of E·S complex as a function of substrate concentration was followed using Mössbauer spectroscopy, and the substrate-bound fraction was determined assuming that the Lamb-Mössbauer factors of bound and non-bound protein were the same. The fraction bound was plotted against cysteine concentration normalized to active site ferrous iron concentration present in the sample. Consequently, a modified version of the equation describing E·S complex formation under conditions of substrate depletion was derived: y = 0.5(K+1 + x) − (0.25(K+1 + x)2 − x)½, where y = [E·S]/[E], x = [S]/[E], and K = Kd/[E].
Synthesis of the Disodium Salt of 3-SPA
3-SPA was synthesized based on the method by Jollès-Bergeret (68) and a modified version by Schurmann et al. (44). All chemicals for the synthesis were used without further purification from Sigma-Aldrich. Briefly, an intermediate bis-(2-carboxyethyl)sulfone was formed by the condensation of acrylic acid and sodium formaldehyde sulfoxylate in aqueous solution. The intermediate was precipitated through acidification, dried, and then cleaved under basic conditions. The product 3-sulfinopropionate was precipitated as a disodium salt with a large excess of an ethanol/acetone (5:1) mixture with a final yield of 61% after a second cleavage step. Elemental analysis was carried out at the Campbell Micro-analytical Laboratory, University of Otago.
Elemental analysis calcd. for C3H4O4SNa2: C = 19.8; H = 2.2; S = 17.6; Na = 25.3. Found: C = 18.5; H = 2.3; S = 12.4; Na = 23.0; m.p. 308 °C; HRMS (−ve ESI) m/z calcd. For [M-H]−: 136.9914, found: 136.9919; 1H NMR (400 MHz, D2O): δ (ppm) 2.45 (2H, s, H2), 2.55 (2H, s, H3); 13C NMR (125 Mhz, D2O): δ (ppm) 30.05 (C3), 57.23 (C2), 181.04 (C3); IR νmax (ATR, cm−1): 2960 (C-H stretch weak), 1678 (C=O asy. stretch medium), 1420 (C=O sy. stretch strong), 1020, 1014, 975 (S=O stretch strong).
Product Determination by 1H NMR and 13C NMR Analysis
PA2602 (200 μm) was incubated for 30 min in 10 mm phosphate buffer (pH 7.5) at 37 °C in the presence of 10 mm 3-MPA. The solution was diluted 6.5-fold and measured by nuclear magnetic resonance (NMR) using a TSP-d4-capillary for 1H NMR 500-MHz and 13C NMR 125-MHz analysis at 25 °C on a Varian 500-MHz AR NMR spectrometer using VNMRJ 2.3A software. 1H NMR and 13C NMR spectra of 3-SPA dissolved in D2O served as reference spectra for enzymatically produced 3-SPA. Considering the relatively high concentrations of reactants used in this experiment, the possibility of 3-MPA autoxidation to 3-MPA disulfide was investigated. To this end, 1H NMR spectra of room temperature buffered solutions of 3-MPA alone and 3-MPA disulfide produced with H2O2 were measured. Using these spectra allowed estimation of 3-MPA disulfide levels in enzymatic mixtures, which represented less than 1%.
Results
Protein Sequence Identification
BLASTP search (48) of putative bacterial CDO sequence (YP_299237.1) from R. eutropha (strain JMP134) identified 20 P. aeruginosa strains, which each contained a single possible CDO homologue. The strains originated from a variety of habitats, including human hosts (e.g. PAO1), plant (E2), industrial water (ATCC700888), and soil (ATCC14886). The P. aeruginosa CDO homologues exhibited high sequence similarity with variations observed at only three residues: 32, 35, and 42. BLASTX search (48) of P. aeruginosa PAO1 genome (NC_002516.2) using PA2602 nucleotide sequence (gi|110645304:2945264–2945869) revealed only one putative gene coding for CDO homologue. No other related gene was found. Alignment of this homologue with other CDOs, including putative bacterial CDO sequence (YP_299237.1) from R. eutropha, highlighted conserved residues and location of the highly conserved cupin motif (23). Like that of R. eutropha and other bacteria, the P. aeruginosa sequence lacks a cysteine that participates in the cysteine-tyrosine cross-link observed in all mammalian CDOs. Comparison with the previously determined CDO fingerprint motif (21) allowed unambiguous identification of all other key residues within the PA2602 sequence, suggesting a possible thiol dioxygenase function (Fig. 2). Purified, recombinant PA2602 protein was assessed for ligand binding and catalytic ability. As we show below, it is a 3-mercaptopropionate dioxygenase; therefore, we name it p3MDO.
p3MDO Is Expressed in Low Levels in P. aeruginosa
To investigate whether p3MDO is expressed by P. aeruginosa, we initially assayed thiol dioxygenase activity in cell lysate. Selected growth conditions were tried, including media containing disulfides as the sole carbon source. Under all growth conditions tried, no activity was detected. It was therefore decided to use mass spectrometry to determine whether the protein itself is expressed. P. aeruginosa was grown in rich media and lysed, and protein was separated by SDS-PAGE before analysis by mass spectrometry.
Untargeted shotgun tandem mass spectrometry failed to identify peptides of p3MDO, suggesting that the protein is either absent or only present at very low abundance (data not shown). To further assess this, we performed PRM assays on seven predicted proteotypic p3MDO peptides as a more sensitive targeted approach. PRM in conjunction with a Mascot sequence database search identified five p3MDO peptide sequences of the seven recorded peptide spectra with ion scores between 36 and 85 (Fig. 4). The identification is highly significant and specific for p3MDO within the 48 million sequence entries in the non-redundant NCBI sequence database. Fig. 4 shows the raw fragment ion spectrum of the doubly charged precursor at m/z 648.8433 and the Mascot fragment ion assignment for the sequence LEPGEVEALSPR as a representative example of a significant peptide identification. One of the seven fragment ion spectra was assigned to isoprenoid biosynthesis protein from P. aeruginosa (gi|489228560), indicating that a false positive signal and retention time had been considered for the scheduled PRM assay, and another peptide did not match any sequence in the database. The PRM assay clearly confirmed the presence of p3MDO. However, the failure to identify the protein by shotgun LC-MS/MS and the requirement of a highly sensitive targeted approach suggests that the p3MDO protein is present at very low abundance, and this would explain the lack of activity observed.
FIGURE 4.
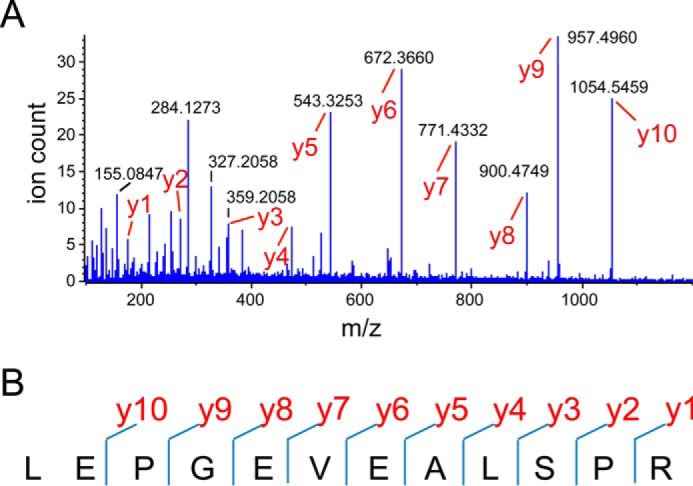
Significant identification of a proteotypic peptide sequence of p3MDO by PRM. A, representative raw spectrum of fragment ions after collision-induced dissociation of the doubly charged precursor at m/z 648.8433. Peaks annotated as y-ions (fragment ions carrying the C terminus) were assigned to fragment ions of the peptide sequence LEPGEVEALSPR by the Mascot software. B, detected sites of peptide fragmentation that confirm the sequence. Only y-ions were identified.
Crystallographic Model
Initial crystallization of p3MDO was achieved from ammonium sulfate at pH 7.5 (crystal A), and a 2.7 Å resolution structure was obtained. Crystallization using sodium acetate conditions similar to those used to crystallize rat CDO (56) produced crystals with a different space group, which diffracted to higher resolutions, 2.5 Å (crystal B) and 2.1 Å (crystal C). Data were complete and moderately redundant, with acceptable merging statistics (Table 2). Refinement yielded good geometry and good agreement with data. The four independent copies in the asymmetric unit are similar (pairwise root mean square deviation of 0.24 to 0.40 Å) and similar to the two copies in our preliminary 3USS structure (pairwise root mean square deviation of 0.27–0.44 Å, crystal A versus either crystal B or C). All monomers in both crystals participate in similar dimers, mediated by a β-strand interaction between Leu189 across the interface and hydrogen bonds between the side chains of Asp68 and Ser69 on opposite molecules. These interactions may influence the orientation of these loops in these crystals. We know of no evidence that the dimer exists under physiological conditions or has biological significance.
Initially, the structure can be compared with that from R. eutropha, with which it shares high identity (62%) and which has been suggested to be a 3MDO (69). The structures are indeed very similar with an overall root mean square deviation of 0.66 Å. This similarity is also true of the active site (Fig. 5A), where identical distances between the iron and the second sphere residues, such as the hydroxyl of Tyr159, are observed (4.1 Å). The only notable difference is the absence of a dioxygen molecule trans to His142 that was modeled in the equivalent position in R. eutropha (4QMA). Although increased electron density was observed at that position, this was modeled as a sulfate in our original structure of p3MDO (crystal A) and in the original analysis of R. eutropha (2GM6). In our subsequent higher resolution structures presented here (crystals B and C), where sulfate was not present, we attempted modeling dioxygen and other molecules in solution but were unable to produce convincing fits in all four chains in the asymmetric unit. Without further supporting evidence, this is modeled conservatively as a water molecule.
FIGURE 5.

Structural comparison of p3MDO (4TLF, blue) with proposed 3MDO from R. eutropha (4QMA, green) and rat CDO (4KWJ, red). Selected conserved side chains are shown with oxygen, nitrogen, sulfur, and iron atoms highlighted in red, blue, yellow, and orange. Shown is a comparison with proposed 3MDO. A, close similarities between the active site of the two enzymes. Slightly larger electron density trans to His142 was modeled as O2 in 3MDO but as water in p3MDO. Comparison with rat CDO (B) shows the overall cupin fold aligned on all backbone atoms. C, active site when the peptide backbones are aligned (iron-bound water molecules have been removed for clarity). D, alignment based on iron, the hydroxyl oxygen of Tyr159, and the δ-N of His157 (the iron-bound waters of only 4TLF are shown). This last alignment clearly shows the difference in the position of Gln62 compared with the corresponding residue of rat CDO, Arg60*. This difference can explain the difference in reactivity of p3MDO toward cysteine and 3-MPA. To aid comparison, the asterisks refer to the rat CDO sequence numbering.
Given the close similarity and lack of kinetic data for R. eutropha 3MDO, comparisons of the bacterial structures offered only limited insights into the structural basis of function. A useful comparison was made with the structure of rat CDO (4KWJ), for which there is considerable crystallographic and kinetic data (70–73). Alignment of the structures based on the backbone confirms the structure similarity suggested by sequence homology (root mean square deviation of 0.86 Å; Fig. 5B). This similarity is also exhibited by the organization of the active site with conserved residues occupying similar positions in both structures, although p3MDO does not contain a cysteine-tyrosine cross-link observed in all mammalian CDO structures (Fig. 5C). The iron, coordinated by three histidines and three waters, has slightly distorted octahedral geometry (176 ± 3, 159 ± 4, and 175 ± 3°; average angles as seen in all four chains). The side chain of His142 occupies an unfavorable rotamer, as it does in all other CDO x-ray crystal structures, suggesting strain induced by the iron coordination. Interestingly, dioxygen is proposed to bind iron at the site trans to this histidine (71).
Surprisingly, superposition of the bacterial and rat CDO structures based on the full polypeptide chain places the catalytic iron in a different position in the two structures, although the relative position of iron, the hydroxyl oxygen of Tyr159, and the δ-N of His157 remain constant (numbering from PA2602). This suggests that a more informative structural comparison is possible if these three centers are aligned. Superposition of these three conserved features (Fig. 5D) displaces the β-strand formed by residues 153–159, demonstrating that the key elements of the bacterial active site are displaced by ∼3 Å relative to the β-barrel, if compared with the mammalian enzyme. This shift correlates with the presence or absence of a cis-peptide bond following the residue C-terminal to Tyr159. All structures of mammalian CDO show both the cysteine-tryrosine cross-link and a serine-proline cis-dipeptide immediately following the tyrosine. Both our structure and the R. eutropha structure (4QMA) lack this sequence, and the corresponding bond is in the trans conformation. The resulting shift of the main chain would open a void, which is filled in the bacterial enzymes by the indole side chain of Trp94, immediately preceding the glycine that replaces the cysteine observed in the mammalian cross-link. This alternate main chain conformation allows the triangle formed by the catalytic iron, the hydroxyl oxygen of Tyr159, and the δ-N of His157 to superimpose closely, with the hydroxyl of Ser153 moved by 2.3 Å. This rotation of the active site relative to the β-barrel results in Gln62 in p3MDO occupying a very different position relative to the corresponding residue Arg60 in the rat structure. Arg60 has been shown in CDO to interact with the carboxyl group of cysteine substrate when cysteine is bound as a chelate to the active site iron via its thiol and amine groups (71).
Iron Binding and Active Site Titration
The relationship between exogenously added iron and iron bound to p3MDO is presented in Fig. 6A. The initial slope of the fit (∼1) reveals stoichiometric binding between iron and p3MDO. The data points were fitted to a quadratic equation (64) to estimate the upper limit (75) of the p3MDO·FeII dissociation constant as 3 ± 1 μm. Currently, the only known enzymes with His3 iron coordination with a reported Kd are CDO, and Acinetobacter johnsonii diketone-cleaving dioxygenase (Dke1). In both cases, the Kd is ∼5 μm, (57, 76), which is comparable with the estimate of the p3MDO affinity for iron presented here.
FIGURE 6.

Affinity and kinetic characterization of p3MDO·FeII complex. Protein (70 μm) was mixed anaerobically with varying concentrations of ferrous sulfate. Unbound iron was removed by Chelex under anaerobic conditions, and the concentration of bound iron was measured using the ferrozine assay. A, the protein-bound iron is plotted versus total iron added and fitted as described under “Experimental Procedures.” B, aliquots of 70 μm p3MDO with various amounts of iron bound were reacted with 10 mm cysteine at 25 °C, and O2 uptake was measured by an oxygen electrode in an air-tight system. The initial velocities of O2 depletion were calculated by linear regression and plotted versus the concentration of protein-bound iron. The best fit line is plotted with a slope of 0.007 s−1.
We then measured the cysteine-dependent oxygen depletion activity by following time-dependent O2 uptake in the presence of p3MDO with increasing concentrations of bound iron. The relationship between protein-bound iron and initial velocities of oxygen consumption exhibited a linear correlation with the slope of the best fit line corresponding to 0.007 ± 0.001 s−1 (Fig. 6B), reflecting the catalytic turnover number per active site bound iron. Activity plateaued when bound iron equaled total enzyme concentration, suggesting a stoichiometric, catalytically competent relationship between p3MDO and iron.
Measurement of P. aeruginosa Thiol Dioxygenase Activities
We tested p3MDO for cysteine dioxygenase activity using an HPLC assay, described previously (77, 78). Incubation reduced the cysteine-associated peak and produced a peak in the HPLC chromatogram with a retention time (∼16 min) identical to that of CSA standards. In addition, samples analyzed by HPLC-evaporative light scattering detection were also analyzed by mass spectrometry (−ve ESI) and showed an m/z peak of 152.0029, which compared well with the calculated profile (152.0023) and the CSA standard (152.0029).
Fidelity of cysteine utilization as a substrate for p3MDO was tested in a number of ways. First, the rate of cysteine depletion and CSA formation could be followed by our HPLC-based assay in the same experiment. The relative rates using 10 mm cysteine at 25 °C (0.005 s−1 CSA formation versus 0.007 s−1 cysteine depletion, n = 2) suggested ∼70% conversion of substrate cysteine into CSA. Importantly, this rate of cysteine depletion mirrors O2 uptake measured using an oxygen electrode described above (0.007 s−1). Finally, we took a Mössbauer sample (774 μm p3MDO) with 10 eq of cysteine added to ensure that all protein has substrate bound and added 1 eq of O2 dissolved in buffer in a sealed system. The amount of CSA produced was determined by HPLC (560 ± 20 μm, n = 9), and this also corresponds to ∼70% use of dioxygen to produce CSA. Although these ratios are dependent on enzyme concentration, pH, and other experimental variables, the data obtained are similar to those for rat CDO with cysteine (72) and strongly support cysteine being a viable substrate for p3MDO, even if not its preferred substrate.
The steady state parameters for cysteine were determined, and Fig. 7 illustrates the dependence and saturation behavior of the rate of CSA formation as a function of cysteine substrate concentration. A summary of Michaelis-Menten kinetic parameters for p3MDO is shown in Table 3. p3MDO exhibited a Km in the millimolar range (of cysteine), which is in accordance with previously published values for other bacterial CDOs (21). However, p3MDO had a significantly slower steady state rate of catalysis than other bacterial CDOs (21).
FIGURE 7.

Michaelis-Menten kinetics of the dioxygenation reaction catalyzed by p3MDO using 3-MPA (open circles) and cysteine (full circles) as substrates measured at 37 °C. Michaelis-Menten constants obtained through fitting (black lines) are reported in Table 2. Error bars, S.D. of at least n = 3 data points.
TABLE 3.
Michaelis-Menten parameters of p3MDO measured at 37 °C
| 3-MPA | Cysteine | |
|---|---|---|
| kcat (s−1) | 0.11 ± 0.02a | 0.021 ± 0.003 |
| Km (mm) | 1.0 ± 0.4 | 8 ± 3 |
| kcat/Km (m−1 s−1) | 110 ± 50 | 3 ± 2 |
a S.E. represents the error associated with the fit to the Michaelis-Menten equation.
The relatively slow rate of cysteine dioxygenation led us to investigate other thiols as potential inhibitors or substrates of the thiol dioxygenase activity of p3MDO. Cysteamine, homocysteine, N-acetyl-l-cysteine, and 3-MPA were chosen. In our HPLC assays, we detected only minor reduction of cysteine dioxygenase activity in the presence of stoichiometric concentrations of N-acetyl-l-cysteine, dl-homocysteine, and cysteamine, 86 ± 15% (n = 6), 79 ± 14% (n = 8), and 75 ± 19% (n = 10), respectively. We previously showed that dl-homocysteine binds to the active site of rat CDO (57). However, the reported extent of inhibition of mammalian CDOs by homocysteine remains ambiguous (17, 19, 25), and very inefficient use of homocysteine as a substrate has recently been reported (80). Comparable inhibition by cysteamine has been reported for other bacterial CDOs (21). N-Acetyl-l-cysteine, homocysteine, and cysteamine dioxygenation product were not observed either in the competition assays or when incubated with P. aeruginosa CDO alone. In contrast, incubation of P. aeruginosa p3MDO with cysteine in the presence of stoichiometric 3-MPA resulted in no detectable levels of CSA within the measured time window. However, HPLC chromatograms revealed a new peak (retention time ∼6 min), which was later assigned to the presence of 3-SPA (see below), suggesting the possibility that 3-MPA not only competed effectively with cysteine for binding in the active site but could be utilized as a substrate for dioxygenase reaction. This is in contrast to bacterial CDOs that showed only a modest (<6%) inhibition of CSA production by 3-MPA and no enzymatic turnover (21).
To confirm the formation of 3-SPA, we synthesized the disodium salt of 3-SPA by an established method (44, 68) and compared 1H and 13C NMR spectra of it and enzymatically produced 3-SPA. This, with reference to published spectra (44, 81, 82), unambiguously showed that p3MDO readily oxidizes 3-MPA. Further evidence was provided by mass spectrometry (−ve ESI), which showed a m/z peak of 136.9918 that compared well with the calculated profile (136.9914) and the 3-SPA standard (136.9919).
An Ellman's reagent-based method was optimized to monitor 3-MPA to 3-SPA conversion. Fig. 7 illustrates the dependence and saturation behavior of the initial rate of 3-MPA substrate utilization as a function of its concentration. The summary of Michaelis-Menten kinetic parameters for p3MDO is shown in Table 3. Our kinetic data show that p3MDO exhibits Km values in the millimolar range for both cysteine and 3-MPA. A 5-fold faster turnover rate of 3-MPA catalysis combined with a lower Km results in a 40-fold higher overall efficiency of dioxygenation of 3-MPA as compared with cysteine. Therefore, in combination with the competition data presented above, we concluded that 3-MPA is the kinetically preferred substrate for p3MDO.
The efficiency of this reaction was also investigated. Time-dependent 1H NMR spectra were taken as described previously for CDO and cysteine (77). The rate of 3-SPA appearance was seen to be ∼95% of 3-MPA disappearance. This relationship was further confirmed by measuring substrate disappearance through Ellman's assay (0.038 ± 0.007 s−1, n = 4) and O2 electrode (0.041 ± 0.008 s−1, n = 4) at 25 °C, with 5 mm 3-MPA (i.e. well above Km). This confirms that both substrates are converted to product during this reaction with 3-MPA.
Substrate Binding to the Active Site
Mössbauer spectroscopy has been successfully applied by us to study active site iron interactions in Rattus norvegicus CDO (57). Iron binding studies suggested a tight binding p3MDO·FeII complex (see above), which allowed anaerobic reconstitution of the active site with exogenously added anaerobic ferrous 57Fe during preparation of protein samples for Mössbauer spectroscopy experiments. Consistent with previous studies on other non-heme mononuclear iron enzymes, the Mössbauer spectrum of p3MDO·FeII exhibited minor heterogeneity, which was substantially removed by adding 1 eq of dithionite, a common treatment of protein samples for Mössbauer spectroscopy. The resulting Mössbauer spectrum of the p3MDO·FeII resting state consists of a single broad quadrupole doublet (Fig. 8A, top spectrum) with parameters (δ = 1.24 mm s−1; ΔEQ = 2.91 mm s−1) typical for high-spin FeII in octahedral coordination.
FIGURE 8.

Determination of p3MDO·FeII·cysteine complex dissociation constant. A, Mössbauer spectra of anaerobic p3MDO resting state (top spectrum) and upon anaerobically added increasing concentrations of cysteine (spectra below). Spectra represent data from two separate titration experiments. Cysteine concentrations were normalized to the concentration of the p3MDO active site ferrous iron present in the sample and are illustrated by the ratios. The spectra can be fitted to two quadrupole doublets employing average Mössbauer parameters (Table 3) for unbound p3MDO·FeII (red line) and substrate-bound p3MDO·FeII·cysteine (blue lines) complexes as described under “Experimental Procedures.” B, similar titration experiment using 3-MPA. The same color scheme is used to label bound and unbound species as for cysteine. C, the relative area occupied by cysteine-bound (full circles) and 3-MPA-bound (open circles) species is plotted versus final thiol concentration (normalized to the active site concentration) and fitted as described under “Experimental Procedures.”
Our kinetic data identified both cysteine and 3-MPA as substrates for p3MDO undergoing the dioxygenation reaction. Therefore, we used Mössbauer spectroscopy to study the thermodynamics of formation of the enzyme substrate complex at 20 °C (Fig. 8). The addition of cysteine led to the gradual conversion of the single broad quadrupole doublet of the p3MDO·FeII resting state (Fig. 8A, top spectrum) to a narrow symmetrical quadrupole doublet (Fig. 5A, bottom spectrum) with Mössbauer parameters (δ = 1.11 mm s−1; ΔEQ = 3.55 mm s−1) also consistent with homogeneous high-spin FeII. Subsequent experiments with resting state (n = 10) and saturating cysteine concentrations (n = 4) allowed unambiguous identification of the p3MDO·FeII·cysteine complex from which average Mössbauer parameters of both species were calculated (Table 4). The reduction in isomer shift and increase in quadrupole splitting observed upon the anaerobic addition of cysteine to p3MDO (Δδ = −0.12 and ΔΔEQ = +0.63 mm s−1) are consistent with coordination of the thiol to iron.
TABLE 4.
Mössbauer parameters of P. aeruginosa thiol dioxygenase measured at 5.2 K
| δ | ΔEQ | ΓL | ΓR | |
|---|---|---|---|---|
| mm s−1 | mm s−1 | mm s−1 | mm s−1 | |
| FeII·p3MDO | 1.23 ± 0.02c | 2.92 ± 0.09 | 0.57a ± 0.04 | 0.80 ± 0.06 |
| FeII·p3MDO·cysteine | 1.11 ± 0.01 | 3.6 ± 0.2 | 0.39a ± 0.01 | ΓL = ΓR |
| FeII·p3MDO·3-MPA | 1.11 ± 0.02 | 2.5 ± 0.3 | 0.90b ± 0.2 | ΓL = ΓR |
a Spectra fitted using a Lorentzian line shape.
b Spectra fitted using Voigt line shape (dithionite was not added).
c S.D. represent errors in parameters calculated using data from at least four independent experiments. n = 10 for FeII·p3MDO parameters.
Similar experiments were carried out with 3-MPA. Interestingly, initial spectra measured in the presence of dithionite used to remove heterogeneity, as described for cysteine above, led to loss of iron from the protein (data not shown). Unable to fully explain this observation yet, we speculate that 3-MPA persulfide is formed in the presence of 3-MPA and dithionite, as we described recently (73), and this species is able to remove iron. The spectra were therefore repeated without pretreatment with dithionite (Fig. 8B) and are thus significantly broader than those shown for cysteine (Fig. 8A). However, the 3-MPA-bound species has the same isomer shift as the cysteine-bound form, suggesting that in both cases, the thiol is coordinated. The change in quadrupole splitting upon binding of 3-MPA is quite different from cysteine (ΔΔEQ = −0.39 mm s−1), and this is attributable to a difference in overall charge of the E·S complexes or different binding geometries or a combination of both effects.
Fitting spectra to a linear combination of spectra with average Mössbauer parameters for resting and E·S complexes allowed quantitative monitoring of p3MDO·FeII·substrate complex formation (Fig. 8). The proportion of substrate-bound species was determined directly from the spectra. Data from several titration experiments exploring different E/S ratios were plotted versus the concentration of cysteine added, normalized to active site iron present, and fitted to a modified version of the quadratic equation (Fig. 8C) (see “Experimental Procedures”). The use of a quadratic equation is warranted due to comparable concentrations of receptor and ligand. We report a dissociation constant for the p3MDO·FeII·cysteine complex in the lower millimolar range (Kd = 1.3 ± 0.1 mm), whereas the p3MDO·FeII·3-MPA shows stoichiometric binding with a upper limit for Kd of 5 μm. In the case of cysteine, the concentration of p3MDO used in the experiments is approximately the same as Kd. This, and the fact the p3MDO·FeII concentration is known to 3 significant figures means that in this case, Kd is quite well defined. In contrast, the stoichiometric binding of 3-MPA means that only an upper limit is defined.
Discussion
In contrast to other non-heme mononuclear iron enzymes, mammalian and bacterial CDOs display a remarkably high specificity for their substrate cysteine. Mammalian ADO, bacterial mercaptosuccinate dioxygenase, and bacterial 3MDO are only able to dioxygenate cysteamine, mercaptosuccinate, and 3-MPA, respectively (6, 83, 84). Although recent work (80) has shown mouse CDO is able to oxidize other substrates, such as l-penicillamine, the coupling between dioxygen consumption and product formation is ≤5%, suggesting that these substrates interact with the enzyme active site and undergo catalysis very differently from the authentic substrate. In this work, we identified and characterized a P. aeruginosa CDO homologue capable of oxidizing two related thiol-containing substrates, cysteine and 3-MPA, to their respective sulfinic acids with a high level of coupling to dioxygen consumption.
There is a high level of sequence identity (98.5%) within the 20 p3MDO homologues identified across 20 strains of P. aeruginosa, with variations occurring only outside the previously identified CDO fingerprint motif (21, 23). Genomic analysis (BLASTX) identified only one copy of the gene coding for the thiol dioxygenase homologue per genome. Mass spectrometry has shown that the protein is indeed expressed by the bacterium, albeit at very low levels. Although we have not identified growth conditions where this enzyme is up-regulated, we have shown that the thiol dioxygenase breakdown pathway is present in P. aeruginosa. The physiological role of p3MDO is unclear. P. aeruginosa was unable to grow with 3MPA as the sole carbon source; the bacteria were able to grow using cysteine as sole carbon source, but a mutation in the p3MDO gene did not prevent growth, showing that p3MDO is not essential for thiol catabolism (data not shown). Whatever the physiological role of this enzyme, it is present in strains inhabiting a diverse spectrum of habitats ranging from human hosts to plants, industrial waters, and soil, indicating its biological importance.
Our structural data provide insight into active site organization in thiol dioxygenases and the role of the cysteine-tyrosine cross-link found in mammalian CDO. Interestingly, the active site geometry is preserved independent of its orientation to the polypeptide backbone: the distances between the iron and hydroxyl of Tyr159, between iron and δ-nitrogen of His157, and between hydroxyl of Tyr159 and δ-nitrogen of His157 are the same in the two structures (numbering from p3MDO). Remarkably, the side chain rings of Tyr159 and His157 exhibit almost perfect superposition despite the fact that p3MDO is unable to be cross-linked. The position of this tyrosine phenol probably depends not only on the presence or absence of cross-link but also on the presence of a serine-proline cis peptide bond in the mammalian enzyme or Trp94 in the bacterial enzyme. This suggests that interpretation of the role of the cross-link and of the functional consequences of its removal must account for this structural feature as well.
Kinetic characterization of p3MDO using both substrates, cysteine and 3-MPA, has shown that 3-MPA is the kinetically preferred substrate, with a 5-fold greater kcat and 8-fold smaller Km. The enzyme is able to utilize both substrates, although inhibition studies show that when both substrates are present, only 3-MPA is oxidized.
Preference for 3-MPA can be rationalized to a certain extent through our structural studies of p3MDO. Previous structural investigations of rat CDO by us (73) and others (71) have shown that Arg60 is integral to cysteine substrate binding. This residue, which reorganizes upon substrate cysteine, forming a sulfur/nitrogen chelate with the ferrous iron in the active site, forms an ionic bond with the carboxyl group of cysteine and holds the substrate in place. Driggers et al. (69) proposed that in the R. eutropha homologue, replacement of this arginine with Gln67, along with the presence of Arg173 (R. eutropha numbering for both), supports 3-MPA substrate specificity. Indeed, structural comparison with p3MDO suggests that Gln62 of the P. aeruginosa p3MDO assumes the role of Arg60 of rat CDO or Gln67 of the R. eutropha enzyme. Significantly, glutamine is shorter than arginine, and Gln62 is situated farther away from the iron atom and thus farther from the site of substrate binding. The active site can therefore accommodate a more elongated structure of 3-MPA bound by its thiol group alone, suggested by our recent structure of 3-MPA-persulfide bound to rat CDO (73). Conversely, because of the weaker interaction of its carboxylate with Gln62, cysteine binding would be expected to be destabilized relative to rat CDO, with its cationic arginine in this position. We do not observe for p3MDO residue Arg168 the potential clash with the amino group of substrate cysteine suggested for Arg173 of the R. eutropha enzyme (69). It is possible that Arg168 of p3MDO interacts favorably with the carboxylate of a cysteine substrate, depending on the conformation of bound substrate. B. subtilis CDO resembles mammalian CDOs in lacking this glutamine/arginine pair and retains the “mammalian” serine-cisproline noted above as lying between Tyr157 and Arg168, despite lacking the adjacent cysteine-tyrosine cross-link. This raises the possibility that the cis dipeptide is correlated with substrate specificity rather than cross-link occurrence.
We investigated the iron site of catalysis and substrate binding using Mössbauer spectroscopy. The resting state, p3MDO·FeII, exhibits a broad asymmetric quadrupole doublet with parameters similar to rat CDO. The broadness in the signal could be due to heterogeneity in the number of water molecules bound to the iron. This idea is supported by crystallographic studies presented here and previously (71, 73) that show either one, two, or three well defined water molecules. Further support comes from the sharp symmetrical quadrupole doublet exhibited by p3MDO·FeII·cysteine. Cysteine binding must replace at least one of the three bound waters, and the lowering of the isomer shift supports thiolate binding to the iron. The geometry of this species is very well defined, as exhibited by significant narrowing of the spectrum upon cysteine binding. In contrast, although 3-MPA also binds via its sulfur, as shown by the lowering of the isomer shift, the broadness of the spectrum is intriguing in light of different dissociation constants described below.
The large differences in the spectra observed upon substrate binding allowed the proportion of cysteine-bound p3MDO to be determined as a function of substrate added in the absence of dioxygen. These conditions preclude catalysis and allow quantification of the enzyme·substrate complex poised for oxygen binding. p3MDO showed stoichiometric binding with 3-MPA, suggesting a low dissociation constant. In contrast, we determined a Kd of 1.3 mm for the p3MDO·FeII·cysteine complex. This is substantially weaker than the low micromolar dissociation constants reported for E·S complexes within other non-heme mononuclear iron enzymes, such as homoprotocatechuate dioxygenase and taurine/α-ketoglutarate dioxygenase (74, 79). It is also very much weaker than rat CDO.6 This weaker binding constant can perhaps be related to Gln62 occupying the position of Arg60 in rat CDO, as discussed above.
In conclusion, dioxygenation occurs at an octahedral FeII ion in p3MDO, but unlike other thiol dioxygenases, p3MDO has significant substrate specificity toward more than one substrate: 3-MPA and cysteine, the latter with 40-fold lower catalytic efficiency but significant efficiency of coupling to dioxygen consumption. The preference for 3-MPA can be explained by a proposed weaker binding of cysteine caused by a weak interaction between Gln62 and the carboxylate of cysteine due to Gln62 being uncharged, shorter, and placed farther from the iron than the equivalent arginine residue in rat CDO. The presence of Arg168 or the lack of a cis-proline following Tyr157 may also favor 3-MPA binding over cysteine. Further studies to more clearly define how these substrates bind are under way.
Author Contributions
G. N. L. J. directed the study. G. N. L. J. and S. M. W. conceived experiments and wrote the paper. E. P. T. and G. N. L. J. designed and analyzed the experiments in Figs. 6 and 8. M. F., E. S., and G. N. L. J. designed and analyzed the experiments in Fig. 7. E. P. T., M. F., and S. M. W. designed and analyzed the experiments in Fig. 5. T. K. designed and analyzed the experiments in Fig. 4. L. W. M. and I. L. L. designed, performed, and analyzed growth studies in P. aeruginosa. S. A. carried out kinetic studies to investigate the efficiency of the reaction. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank M. Thomas, I. Stewart, and J. Lyburn (Department of Chemistry, University of Otago) for help with NMR and MS experiments and A. Steinbüchel and M. Schürmann (University of Münster) for insight into 3-SPA synthesis.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 3USS and 4TLF) have been deposited in the Protein Data Bank (http://wwpdb.org/).
E. P. Tchesnokov and G. N. L. Jameson, unpublished results.
- CDO
- cysteine dioxygenase
- CSA
- cysteine sulfinic acid
- 3-MPA
- 3-mercaptopropionic acid
- 3-SPA
- 3-sulfinopropionic acid
- 3MDO
- 3-mercaptopropionate dioxygenase
- p3MDO
- P. aeruginosa 3-mercaptopropionate dioxygenase
- PRM
- parallel reaction monitoring
- ESI
- electrospray ionization.
References
- 1. Lomans B. P., van der Drift C., Pol A., Op den Camp H. J. M. (2002) Microbial cycling of volatile organic sulfur compounds. Cell. Mol. Life Sci. 59, 575–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stipanuk M. H. (2004) Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 24, 539–577 [DOI] [PubMed] [Google Scholar]
- 3. Schiff J. A. (1979) Pathways of assimilatory sulphate reduction in plants and microorganisms. Ciba Found. Symp. 49–69 [DOI] [PubMed] [Google Scholar]
- 4. Wang L., Chen S., Xu T., Taghizadeh K., Wishnok J. S., Zhou X., You D., Deng Z., Dedon P. C. (2007) Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 3, 709–710 [DOI] [PubMed] [Google Scholar]
- 5. Kiene R. P., Taylor B. F. (1988) Biotransformations of organosulfur compounds in sediments via 3-mercaptopropionate. Nature 332, 148–150 [Google Scholar]
- 6. Bruland N., Wübbeler J. H., Steinbüchel A. (2009) 3-Mercaptopropionate dioxygenase, a cysteine dioxygenase homologue, catalyzes the initial step of 3-mercaptopropionate catabolism in the 3,3-thiodipropionic acid-degrading bacterium Variovorax paradoxus. J. Biol. Chem. 284, 660–672 [DOI] [PubMed] [Google Scholar]
- 7. Stipanuk M. H., Dominy J. E. Jr., Lee J. I., Coloso R. M. (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J. Nutr. 136, 1652S–1659S [DOI] [PubMed] [Google Scholar]
- 8. Janáky R., Varga V., Hermann A., Saransaari P., Oja S. S. (2000) Mechanisms of l-cysteine neurotoxicity. Neurochem. Res. 25, 1397–1405 [DOI] [PubMed] [Google Scholar]
- 9. Townsend D. M., Tew K. D., Tapiero H. (2004) Sulfur containing amino acids and human disease. Biomed. Pharmacother. 58, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olney J. W., Zorumski C., Price M. T., Labruyere J. (1990) l-Cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science 248, 596–599 [DOI] [PubMed] [Google Scholar]
- 11. Jameson G. N. L. (2011) Iron, cysteine and Parkinson's disease. Monatsh. Chem. 142, 325–329 [Google Scholar]
- 12. Jameson G. N. L., Zhang J., Jameson R. F., Linert W. (2004) Kinetic evidence that cysteine reacts with dopaminoquinone via reversible adduct formation to yield 5-cysteinyl-dopamine: an important precursor of neuromelanin. Org. Biomol. Chem. 2, 777–782 [DOI] [PubMed] [Google Scholar]
- 13. Heafield M. T., Fearn S., Steventon G. B., Waring R. H., Williams A. C., Sturman S. G. (1990) Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson's and Alzheimer's disease. Neurosci. Lett. 110, 216–220 [DOI] [PubMed] [Google Scholar]
- 14. Perry T. L., Norman M. G., Yong V. W., Whiting S., Crichton J. U., Hansen S., Kish S. J. (1985) Hallervorden-Spatz disease: cysteine accumulation and cysteine dioxygenase deficiency in the globus pallidus. Ann. Neurol. 18, 482–489 [DOI] [PubMed] [Google Scholar]
- 15. Luchsinger J. A., Tang M. X., Shea S., Miller J., Green R., Mayeux R. (2004) Plasma homocysteine levels and risk of Alzheimer disease. Neurology 62, 1972–1976 [DOI] [PubMed] [Google Scholar]
- 16. Pierce B. S., Gardner J. D., Bailey L. J., Brunold T. C., Fox B. G. (2007) Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry 46, 8569–8578 [DOI] [PubMed] [Google Scholar]
- 17. Chai S. C., Jerkins A. A., Banik J. J., Shalev I., Pinkham J. L., Uden P. C., Maroney M. J. (2005) Heterologous expression, purification, and characterization of recombinant rat cysteine dioxygenase. J. Biol. Chem. 280, 9865–9869 [DOI] [PubMed] [Google Scholar]
- 18. Chai S. C., Bruyere J. R., Maroney M. J. (2006) Probes of the catalytic site of cysteine dioxygenase. J. Biol. Chem. 281, 15774–15779 [DOI] [PubMed] [Google Scholar]
- 19. Yamaguchi K., Hosokawa Y., Kohashi N., Kori Y., Sakakibara S., Ueda I. (1978) Rat liver cysteine dioxygenase (cysteine oxidase): further purification, characterization, and analysis of the activation and inactivation. J. Biochem. 83, 479–491 [DOI] [PubMed] [Google Scholar]
- 20. Simmons C. R., Hirschberger L. L., Machi M. S., Stipanuk M. H. (2006) Expression, purification, and kinetic characterization of recombinant rat cysteine dioxygenase, a non-heme metalloenzyme necessary for regulation of cellular cysteine levels. Protein Expr. Purif. 47, 74–81 [DOI] [PubMed] [Google Scholar]
- 21. Dominy J. E. Jr., Simmons C. R., Karplus P. A., Gehring A. M., Stipanuk M. H. (2006) Identification and characterization of bacterial cysteine dioxygenases: a new route of cysteine degradation for eubacteria. J. Bacteriol. 188, 5561–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasperova A., Kunert J., Horynova M., Weigl E., Sebela M., Lenobel R., Raska M. (2011) Isolation of recombinant cysteine dioxygenase protein from Trichophyton mentagrophytes. Mycoses 54, e456–e462 [DOI] [PubMed] [Google Scholar]
- 23. Stipanuk M. H., Simmons C. R., Karplus P. A., Dominy J. E. Jr. (2011) Thiol dioxygenases: unique families of cupin proteins. Amino Acids 41, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dominy J. E. Jr., Hwang J., Guo S., Hirschberger L. L., Zhang S., Stipanuk M. H. (2008) Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J. Biol. Chem. 283, 12188–12201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye S., Wu X., Wei L., Tang D., Sun P., Bartlam M., Rao Z. (2007) An insight into the mechanism of human cysteine dioxygenase: key roles of the thioether-bonded tyrosine-cysteine cofactor. J. Biol. Chem. 282, 3391–3402 [DOI] [PubMed] [Google Scholar]
- 26. Zhang J. Z., Wang F. Y., House J. D., Page B. (2004) Thiols in wetland interstitial waters and their role in mercury and methylmercury speciation. Limnol. Oceanogr. 49, 2276–2286 [Google Scholar]
- 27. Hu H. Y., Mylon S. E., Benoit G. (2006) Distribution of the thiols glutathione and 3-mercaptopropionic acid in Connecticut lakes. Limnol. Oceanogr. 51, 2763–2774 [Google Scholar]
- 28. Wübbeler J. H., Bruland N., Wozniczka M., Steinbüchel A. (2010) Biodegradation of the xenobiotic organic disulphide 4,4′-dithiodibutyric acid by Rhodococcus erythropolis strain MI2 and comparison with the microbial utilization of 3,3′-dithiodipropionic acid and 3,3′-thiodipropionic acid. Microbiology 156, 1221–1233 [DOI] [PubMed] [Google Scholar]
- 29. Toups M., Wübbeler J. H., Steinbüchel A. (2010) Microbial utilization of the industrial wastewater pollutants 2-ethylhexylthioglycolic acid and iso-octylthioglycolic acid by aerobic Gram-negative bacteria. Biodegradation 21, 309–319 [DOI] [PubMed] [Google Scholar]
- 30. Wübbeler J. H., Raberg M., Brandt U., Steinbüchel A. (2010) Dihydrolipoamide dehydrogenases of Advenella mimigardefordensis and Ralstonia eutropha catalyze cleavage of 3,3′-dithiodipropionic acid into 3-mercaptopropionic acid. Appl. Environ. Microbiol. 76, 7023–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Visscher P. T., Taylor B. F. (1994) Demethylation of dimethylsulfoniopropionate to 3-mercaptopropionate by an aerobic marine bacterium. Appl. Environ. Microbiol. 60, 4617–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kiene R. P., Malloy K. D., Taylor B. F. (1990) Sulfur-containing amino acids as precursors of thiols in anoxic coastal sediments. Appl. Environ. Microbiol. 56, 156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoch D. C. (2002) Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide. Appl. Environ. Microbiol. 68, 5804–5815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goto M., Takahashi T., Yamashita F., Koreeda A., Mori H., Ohta M., Arakawa Y. (1997) Inhibition of the metallo-β-lactamase produced from Serratia marcescens by thiol compounds. Biol. Pharm. Bull. 20, 1136–1140 [DOI] [PubMed] [Google Scholar]
- 35. Siemann S., Clarke A. J., Viswanatha T., Dmitrienko G. I. (2003) Thiols as classical and slow-binding inhibitors of IMP-1 and other binuclear metallo-β-lactamases. Biochemistry 42, 1673–1683 [DOI] [PubMed] [Google Scholar]
- 36. Sabbagh E., Cuebas D., Schulz H. (1985) 3-Mercaptopropionic acid, a potent inhibitor of fatty acid oxidation in rat heart mitochondria. J. Biol. Chem. 260, 7337–7342 [PubMed] [Google Scholar]
- 37. Cuebas D., Beckmann J. D., Frerman F. E., Schulz H. (1985) Mitochondrial metabolism of 3-mercaptopropionic acid. Chemical synthesis of 3-mercaptopropionyl coenzyme A and some of its S-acyl derivatives. J. Biol. Chem. 260, 7330–7336 [PubMed] [Google Scholar]
- 38. Rodríguez de Lores A., Alberici de Canal M., Robiolo B., Mistrorigo de Pacheco M. (1973) The effect of the convulsant 3-mercaptopropionic acid on enzymes of the γ-aminobutyrate system in the rat cerebral cortex. J. Neurochem. 21, 615–623 [DOI] [PubMed] [Google Scholar]
- 39. Löscher W. (1979) 3-Mercaptopropionic acid: convulsant properties, effects on enzymes of the γ-aminobutyrate system in mouse brain and antagonism by certain anticonvulsant drugs, aminooxyacetic acid and gabaculine. Biochem. Pharmacol. 28, 1397–1407 [DOI] [PubMed] [Google Scholar]
- 40. Ding R., Tsunekawa N., Obata K. (2004) Cleft palate by picrotoxin or 3-MP and palatal shelf elevation in GABA-deficient mice. Neurotoxicol. Teratol. 26, 587–592 [DOI] [PubMed] [Google Scholar]
- 41. Prithiviraj B., Weir T., Bais H. P., Schweizer H. P., Vivanco J. M. (2005) Plant models for animal pathogenesis. Cell. Microbiol. 7, 315–324 [DOI] [PubMed] [Google Scholar]
- 42. Vermeij P., Kertesz M. A. (1999) Pathways of assimilative sulfur metabolism in Pseudomonas putida. J. Bacteriol. 181, 5833–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Von Riesen V. L. (1963) Hydrogen sulfide production by Pseudomonas aeruginosa. J. Bacteriol. 85, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schürmann M., Wübbeler J. H., Grote J., Steinbüchel A. (2011) Novel reaction of succinyl coenzyme A (succinyl-CoA) synthetase: activation of 3-sulfinopropionate to 3-sulfinopropionyl-CoA in Advenella mimigardefordensis strain DPN7(T) during degradation of 3,3′-dithiodipropionic acid. J. Bacteriol. 193, 3078–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kapatral V., Bina X., Chakrabarty A. M. (2000) Succinyl coenzyme A synthetase of Pseudomonas aeruginosa with a broad specificity for nucleoside triphosphate (NTP) synthesis modulates specificity for NTP synthesis by the 12-kilodalton form of nucleoside diphosphate kinase. J. Bacteriol. 182, 1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F. S., Hufnagle W. O., Kowalik D. J., Lagrou M., Garber R. L., Goltry L., Tolentino E., Westbrock-Wadman S., Yuan Y., Brody L. L., Coulter S. N., Folger K. R., Kas A., Larbig K., Lim R., Smith K., Spencer D., Wong G. K., Wu Z., Paulsen I. T., Reizer J., Saier M. H., Hancock R. E., Lory S., Olson M. V. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 [DOI] [PubMed] [Google Scholar]
- 47. Winsor G. L., Van Rossum T., Lo R., Khaira B., Whiteside M. D., Hancock R. E., Brinkman F. S. (2009) Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 37, D483–D488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoc H. A. (2005) Protein Identification and Analysis Tools on the ExPASy Server, pp. 571–607, Humana Press, New York: [DOI] [PubMed] [Google Scholar]
- 50. Bjellqvist B., Basse B., Olsen E., Celis J. E. (1994) Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis 15, 529–539 [DOI] [PubMed] [Google Scholar]
- 51. Schmidt T. G. M., Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 52. Skerra A., Schmidt T. G. (2000) Use of the Strep-tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol. 326, 271–304 [DOI] [PubMed] [Google Scholar]
- 53. Bella D. L., Hirschberger L. L., Hosokawa Y., Stipanuk M. H. (1999) Mechanisms involved in the regulation of key enzymes of cysteine metabolism in rat liver in vivo. Am. J. Physiol. 276, E326–E335 [DOI] [PubMed] [Google Scholar]
- 54. Dominy J. E. Jr., Hirschberger L. L., Coloso R. M., Stipanuk M. H. (2006) Regulation of cysteine dioxygenase degradation is mediated by intracellular cysteine levels and the ubiquitin-26 S proteasome system in the living rat. Biochem. J. 394, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kleffmann T., Jongkees S. A. K., Fairweather G., Wilbanks S. M., Jameson G. N. L. (2009) Mass-spectrometric characterization of two posttranslational modifications of cysteine dioxygenase. J. Biol. Inorg. Chem. 14, 913–921 [DOI] [PubMed] [Google Scholar]
- 56. Simmons C. R., Liu Q., Huang Q., Hao Q., Begley T. P., Karplus P. A., Stipanuk M. H. (2006) Crystal structure of mammalian cysteine dioxygenase: a novel mononuclear iron center for cysteine thiol oxidation. J. Biol. Chem. 281, 18723–18733 [DOI] [PubMed] [Google Scholar]
- 57. Tchesnokov E. P., Wilbanks S. M., Jameson G. N. L. (2012) A strongly bound high-spin iron(II) coordinates cysteine and homo-cysteine in cysteine dioxygenase. Biochemistry 51, 257–264 [DOI] [PubMed] [Google Scholar]
- 58. Shevchenko A., Jensen O. N., Podtelejnikov A. V., Sagliocco F., Wilm M., Vorm O., Mortensen P., Shevchenko A., Boucherie H., Mann M. (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. U.S.A. 93, 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 62. Painter J., Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 63. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 64. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 76–108, Wiley-VCH, Inc., New York [Google Scholar]
- 65. Diebold A. R., Neidig M. L., Moran G. R., Straganz G. D., Solomon E. I. (2010) The three-His triad in Dke1: comparisons to the classical facial triad. Biochemistry 49, 6945–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fellner M., Doughty L. M., Jameson G. N. L., Wilbanks S. M. (2014) A chromogenic assay of substrate depletion by thiol dioxygenases. Anal. Biochem. 459, 56–60 [DOI] [PubMed] [Google Scholar]
- 67. Bollinger J. M., Tong W. H., Ravi N., Huynh B. H., Edmonson D. E., Stubbe J. (1994) Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase. 2. kinetics of the excess Fe2+ reaction by optical, EPR, and Moessbauer spectroscopies. J. Am. Chem. Soc. 116, 8015–8023 [Google Scholar]
- 68. Jollés-Bergeret B. (1974) Enzymatic and chemical synthesis of 3-sulfinopropionic acid: analog of succinic acid. Eur. J. Biochem. 42, 349–353 [DOI] [PubMed] [Google Scholar]
- 69. Driggers C. M., Hartman S. J., Karplus P. A. (2015) Structures of Arg- and Gln-type bacterial cysteine dioxygenase homologs. Protein Sci. 24, 154–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Davies C. G., Fellner M., Tchesnokov E. P., Wilbanks S. M., Jameson G. N. L. (2014) The Cys-Tyr cross-link of cysteine dioxygenase changes the optimal pH of the reaction without a structural change. Biochemistry 53, 7961–7968 [DOI] [PubMed] [Google Scholar]
- 71. Driggers C. M., Cooley R. B., Sankaran B., Hirschberger L. L., Stipanuk M. H., Karplus P. A. (2013) Cysteine dioxygenase structures from pH 4 to 9: consistent Cys-persulfenate formation at intermediate pH and a Cys-bound enzyme at higher pH. J. Mol. Biol. 425, 3121–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li W., Blaesi E. J., Pecore M. D., Crowell J. K., Pierce B. S. (2013) Second-sphere interactions between the C93–Y157 cross-link and the substrate-bound Fe site influence the O2 coupling efficiency in mouse cysteine dioxygenase. Biochemistry 52, 9104–9119 [DOI] [PubMed] [Google Scholar]
- 73. Souness R. J., Kleffmann T., Tchesnokov E. P., Wilbanks S. M., Jameson G. B., Jameson G. N. L. (2013) Mechanistic implications of persulfenate and persulfide binding in the active site of cysteine dioxygenase. Biochemistry 52, 7606–7617 [DOI] [PubMed] [Google Scholar]
- 74. Ryle M. J., Padmakumar R., Hausinger R. P. (1999) Stopped-flow kinetic analysis of Escherichia coli taurine/α-ketoglutarate dioxygenase: interactions with α-ketoglutarate, taurine, and oxygen. Biochemistry 38, 15278–15286 [DOI] [PubMed] [Google Scholar]
- 75. Goodrich J. A., Kugel J. F. (2007) Binding and Kinetics for Molecular Biologists, pp. 19–48, Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 76. Leitgeb S., Straganz G. D., Nidetzky B. (2009) Biochemical characterization and mutational analysis of the mononuclear non-haem Fe2+ site in Dke1, a cupin-type dioxygenase from Acinetobacter johnsonii. Biochem. J. 418, 403–411 [DOI] [PubMed] [Google Scholar]
- 77. Siakkou E., Wilbanks S. M., Jameson G. N. L. (2010) Simplified cysteine dioxygenase activity assay allows simultaneous quantitation of both substrate and product. Anal. Biochem. 405, 127–131 [DOI] [PubMed] [Google Scholar]
- 78. Siakkou E., Rutledge M. T., Wilbanks S. M., Jameson G. N. L. (2011) Correlating crosslink formation with enzymatic activity in cysteine dioxygenase. Biochim. Biophys. Acta 1814, 2003–2009 [DOI] [PubMed] [Google Scholar]
- 79. Groce S. L., Miller-Rodeberg M. A., Lipscomb J. D. (2004) Single-turnover kinetics of homoprotocatechuate 2,3-dioxygenase. Biochemistry 43, 15141–15153 [DOI] [PubMed] [Google Scholar]
- 80. Li W., Pierce B. S. (2015) Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch. Biochem. Biophys. 565, 49–56 [DOI] [PubMed] [Google Scholar]
- 81. Katritzky A. R., Akhmedov N. G., Denisko O. V. (2003) H-1 and C-13 NMR spectroscopic study of oxidation of dl-cystine and 3,3′-dithiobis(propionic acid) with hydrogen peroxide in aqueous solution. Magn. Reson. Chem. 41, 37–41 [Google Scholar]
- 82. Nakayama H., Hirami S., Tsuhako M. (2004) Intercalation of mercaptocarboxylic acid into layered double hydroxide accompanied with oxidation of mercapto group. Chem. Lett. 33, 712–713 [Google Scholar]
- 83. Dominy J. E. Jr., Simmons C. R., Hirschberger L. L., Hwang J., Coloso R. M., Stipanuk M. H. (2007) Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem. 282, 25189–25198 [DOI] [PubMed] [Google Scholar]
- 84. Brandt U., Schürmann M., Steinbüchel A. (2014) Mercaptosuccinate dioxygenase, a cysteine dioxygenase homologue, from Variovorax paradoxus strain B4 is the key enzyme of mercaptosuccinate degradation. J. Biol. Chem. 289, 30800–30809 [DOI] [PMC free article] [PubMed] [Google Scholar]