

Abstract
Increasing evidence has emerged that supports an important intersection between 3 fundamental cell biologic pathways in the pathogenesis of inflammatory bowel disease. These include the intersection between autophagy, as revealed by the original identification of ATG16L1 and IRGM as major genetic risk factors for Crohn’s disease, and intracellular bacterial sensing, as shown by the importance of NOD2 in autophagy induction upon bacterial entry into the cell. A pathway closely linked to autophagy and innate immunity is the unfolded protein response, initiated by endoplasmic reticulum stress due to the accumulation of misfolded proteins, which is genetically related to ulcerative colitis and Crohn’s disease (XBP1 and ORMDL3). Hypomorphic ATG16L1, NOD2, and X box binding protein-1 possess the common attribute of profoundly affecting Paneth cells, specialized epithelial cells at the bottom of intestinal crypts involved in antimicrobial function. Together with their functional juxtaposition in the environmentally exposed intestinal epithelial cell, their remarkable functional convergence on Paneth cells and their behavior in response to environmental factors, including microbes, these 3 pathways are of increasing importance to understanding the pathogenesis of inflammatory bowel disease. Moreover, in conjunction with studies that model deficient nuclear factor-κB function, these studies suggest a central role for altered intestinal epithelial cell function as one of the earliest events in the development of inflammatory bowel disease.
Keywords: Inflammatory Bowel Disease, Crohn’s Disease, Ulcerative Colitis, Pathogenesis, Autophagy, Endoplasmic Reticulum Stress, Unfolded Protein Response, Innate Immunity, Genetics
The inflammatory bowel diseases (IBD), Crohn’s disease (CD), and ulcerative colitis (UC) have long been considered to arise from an inappropriate immune response directed toward the commensal microbiota in a genetically susceptible host.1,2 Based on twin experiments, the genetic contribution in CD has been estimated at 50%, and in UC at approximately 20%, with the remainder ascribed to environmental factors; most notably smoking and enteropathogens.1,3,4 These interrelated genetic and environmental factors, which are associated with risk for developing IBD, likely share the common feature that they beneficially or adversely affect the functional relationships between the commensal microbiota, intestinal epithelium, and gut-associated lymphoid tissues.1 Although much remains to be learned about the relevant environmental factors that affect these relationships, significant progress has been made in understanding the immunobiology of IBD and, more recently, its genetic underpinnings. The latter has largely been facilitated by genome-wide association studies (GWAS),5,6 with the identification of >90 genetic loci associated with CD and UC,7–10 but also linkage and candidate gene studies that have allowed, in certain circumstances, for the identification of rare, functionally important variants, including those associated with early-onset IBD.11–14 Despite this dramatic expansion in the number of genetic loci linked to IBD, in CD, a single gene, NOD2 (followed by IL23R and ATG16L1), is associated with by far the largest, albeit limited, fraction of genetic heritability, with the remainder of genes imposing only limited degrees of risk.15 However, the most recent meta-analysis of CD GWAS data has shown that 71 of the common genetic loci discovered so far may account for only 23% of CD’s heritability; although this could be an underestimate.15 Despite this, it is increasingly clear that the identified genetic risk factors appear to be operating within functionally interacting pathways and, as such, likely amplify their individual importance relative to disease pathogenesis. One group of potentially interacting pathways that are rapidly coming to light and have received a significant amount of attention because of the implications that they have for understanding relevant environmental factors is that associated with autophagy, the unfolded protein response (UPR), and intracellular bacterial sensing. This review will summarize the evidence for this in light of recent observations in each of these 3 areas.
Autophagy
The ability of GWAS to generate knowledge about the genetic underpinning of IBD in a hypothesis-free unbiased way was exemplified by the discovery of genes associated with a fundamental biological pathway that had not previously been considered in the context of IBD, namely autophagy.16,17 Initially, a coding variant (T300A) in ATG16L1 was identified in a genome-wide assessment of ~20,000 protein coding variants (nonsynonymous single nucleotide polymorphisms)16 and was later independently identified in subsequent GWAS studies.6,17 Further support for an involvement of autophagy in IBD came from association of IRGM,6,18,19 another gene that had previously been linked to autophagy induction, and LRRK2.8
Macroautophagy (autophagy) refers to a conserved biological process that has evolved as a physiological response to the removal of damaged organelles and micro-organisms (selective) or cellular starvation (nonselective).20,21 Other types of autophagy, such as chaperone and mitochondria-associated autophagy, are not yet linked to IBD but are worthy of mention (for review see references 22 and 23). Autophagy designates the engulfment of cellular macromolecular contents via a double-layered membrane (autophagosome) and its subsequent fusion with lysosomes (lysophagosome) and, consequently, lysosomal degradation.20 This catabolic pathway allows for recycling of proteins and macromolecular components into their basic constituents and the availability of the latter for anabolic processes. It is therefore not surprising that autophagy plays a fundamental role in embryogenesis, immune function, and tissue remodeling and, consequently, a wide variety of disease processes, including, neurodegeneration, cancer, infection, and inflammation among others.21
Several distinct steps that have largely been elucidated in yeast model systems are involved in the autophagy process.20 These involve induction, cargo recognition, vesicle formation, autophagosome-vacuole fusion, and, finally, breakdown of the autophagic contents and the release of their degradation products into the cytosol (Figure 1).20 All cells exhibit a certain low level of basal autophagy, which can be induced under various conditions (ie, induced autophagy). The latter mechanism allows cells to efficiently adapt to various intracellular and extracellular stressors. The serine/threonine protein kinase target of rapamycin plays a central role as an inhibitor of autophagy, and several signaling pathways that regulate autophagy induction mechanistically converge on mammalian target of rapamycin (mTOR).20,24 Upon release of the inhibitory activity provided by mTOR, autophagy is generated by autophagy (Atg) proteins that function in a cascade of modular interactions. The most nascent complex includes Beclin 1, Atg1 (ULK1 and ULK2 in mammals), Atg13, and Atg17 (FIP2000 in mammals)20 that motivate the recruitment of Atg8 (LC3-I) to the isolation membrane via a membrane anchoring, phosphatidylethanolamine modification of Atg8 (LC3-II). Identification of LC3-II recruitment to the isolation membrane is a major means to quantify autophagy (Figure 2). Other proteins involved in the formation of the isolation membrane that initially surrounds the organelles or organisms destined for degradation include Atg 2, 3, 9, and 16. Interestingly, the observation that sirolimus, an mTOR inhibitor, was clinically beneficial in a case report of a CD patient with refractory disease, lends support to the importance of this pathway in human IBD, with the implication that the host’s ability to competently induce autophagy is beneficial.25 However, mTOR inhibitors affect multiple other pathways apart from autophagy, and properly designed clinical trials will be required to address their role in the treatment of CD.26
Figure 1.

Molecules involved in autophagy induction.
Figure 2.
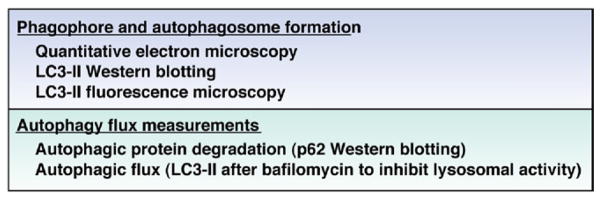
Experimental measures to determine induction of autophagy.
ATG16L1, the mammalian homologue of Atg16, is likely important in linking the isolation membrane to the formation of the autophagosome by virtue of its ability to bind Atg5, a coiled-coil domain containing protein that is important for self-multimerization and shown to be essential for starvation-induced autophagy in yeast (Figure 1).27,28 In addition to Atg5, formation of the double-membraned phagophore as characteristically observed by electron microscopy (Figure 2) requires Atg7, 10, and 12. Ultimately, fusion of the autophagosome with lysosomes to create the auto(phago)lysosome (autolysosome) results in the degradation of the internalized contents, including associated cargo characteristically associated with the autolysosome, such as sequestasome (P62), which can be utilized as a measure of this degradative pathway.29 As autophagy requires lysosomal fusion, it is dependent upon organelle acidification and thus is inhibited by agents such as bafilomycin, which blocks a membrane-associated vacuolar Na+-H+ adenosine triphosphatase. As such, in experimental systems, treatment with bafilomycin results in inhibition of lysosomal function and consequently protection of P62 from degradation and accumulation of LC3-II when autophagy is active; a useful means in experimental settings to monitor cellular autophagy.29
One mechanism by which autophagy is associated with risk for development of CD has recently come to light and involves alterations in the ability of the host to regulate the inflammasome. The inflammasome is generated in response to a variety of inflammatory cytokines (eg, tumor necrosis factor [TNF]) and innate immune signals associated with damage-associated molecular patterns (eg, uric acid) or microbial-associated molecular patterns (eg, lipopolysaccharide) binding to Toll-like receptors (TLR).30 Integration of a priming (eg, TLR or TNF signaling) and a subsequent triggering signal (eg, adenosine triphosphate or uric acid) by the inflammasome activates caspase-1, resulting in cleavage of immature interleukin-1β (IL-1β) and IL-18 into their mature forms and extracellular secretion.30 Using fetal liver from Atg16l1−/− mice to generate chimeric mice with an ATG16L1-deficient hematopoietic system, Saitoh and colleagues showed that innate immune cells from such mice (ie, macrophages) exhibit hyperactive and thus unregulated inflammasome function in response to combined TNF and TLR stimulation.31 Such an observation is consistent with the notion that autophagy is required for removal and regulation of inflammasome function such that hypomorphic ATG16L1 activity leads to exaggerated inflammatory responses secondary to the effects of IL-1β and IL-18. It is interesting to note that IL-1β is also critical for the derivation of naïve T cells into T helper 17 (Th17) cells.32,33 Th17 cells and their related pathways are well known to be an important source of genetic risk in the development of IBD as revealed by polymorphisms in IL23R (encoding the IL-23 receptor β chain), IL12B (encoding IL-12p40 chain of IL-12 and IL-23), JAK2 (encoding Janus-associated kinase 2), and STAT3 (encoding signal transducer associated factor 3) genes.5,8,18 Moreover, polymorphisms have also been identified in NLRP3, encoding Nod-like receptor (NLR), pyrin containing protein 3 (NALP3)/cryopyrin, which is critical for the activation of caspase-1 within the inflammasome,34 and IL18RAP (encoding IL-18 receptor accessory protein).8,35,36 It remains to be determined whether the pathways associated with autophagy, Th17 cells, and IL-1 (and IL-18) are functionally related but worthy of note.
A second functional pathway by which ATG16L1 polymorphisms may confer risk for development of CD is through its effects on Paneth cell function. Paneth cells are specialized epithelial cells located at the base of small intestinal crypts that secrete antimicrobial peptides and inflammatory mediators, and have recently been shown to play an important role in the maintenance of the intestinal stem cell niche.37 Using gene-trap technology to target Atg16l1, mice have recently been generated with hypomorphic (HM) Atg16l1 alleles.38,39 Atg16l1HM mice were shown to develop normally without any evidence of intestinal inflammation.38 However, they exhibited substantial structural abnormalities in Paneth cells, which normally store and secrete abundant amounts of antimicrobial peptides within large intracellular granules.38 Specifically, Paneth cells in Atg16l1HM mice contained a distorted granule compartment with defects in their exocytosis pathway, resulting in decreased granule numbers and diffuse cytoplasmic staining for proteins typically found in granules such as lysozyme.38 A similar distortion was also noted in Atg5ΔIEC mice, wherein the Atg5 allele was specifically deleted in the intestinal epithelium, also implicating a Paneth cell (or at least intestinal epithelial cell)-intrinsic function of Atg16l1HM.38 Interestingly, these structural defects did not translate into a mishandling of a model mucosal pathogen, Listeria monocytogenes, which is consistent with an absence of defects in α-defensin expression in Atg16l1HM-associated Paneth cells.38 In contrast to Atg16l1HM mice, L monocytogenes infection of Nod2−/− mice results in substantially increased systemic translocation of bacteria, in association with decreased α-defensin (cryptdin) expression in Paneth cells before infection (vide infra).40 In addition to these structural alterations, Atg16l1HM Paneth cells also exhibit an aberrant transcriptional profile, with alterations that specifically cluster around peroxisome proliferator of activation receptor signaling, the production of acute-phase reactants and inflammatory mediators (eg, serum amyloid A and TNF), the statin pathway, and adipocytokine signaling, among others.38 The profile of transcriptional changes observed was only detected in Paneth cells and was distinct from that found in thymocytes.38
Remarkably, this Paneth cell phenotype has recently been shown to be entirely dependent on persistent infection with a specific strain of murine norovirus (MNV CR6).39 Specifically, rederivation of Atg16l1HM mice into an MNV-free enhanced specific pathogen-free barrier facility abrogated the structural alterations in Paneth cells that could only be recovered by infection with MNV CR6; however, MNV CR6 infection did not induce intestinal inflammation.39 Infection with a nonpersistent MNV strain, MNV CW3, did not affect Paneth cell morphology.39 The aforementioned alterations in the transcriptional profile observed in Atg16l1HM Paneth cells was also dependent upon MNV CR6 infection, indicating that there might be a fundamental difference how Atg16l1HM Paneth cells respond to viral infection.39 Notably, MNV infection localizes to the lamina propria41 and neither MNV proteins nor RNA could be detected in Paneth cells.39 This suggests that mediators induced by specific types of viral infection act on Paneth cells, which in turn respond differently in the presence or absence of full ATG16L1 function. Although MNV CR6 infection itself did not induce spontaneous intestinal inflammation, MNV CR6-infected Atg16l1HM mice exhibited increased severity in the dextran sodium sulfate-induced colitis model, which could be abrogated by depletion of the commensal flora via broad-spectrum antibiotics.39 Although there is no indication to date that norovirus infection might be a causative factor in human CD, it is interesting that Paneth cells studied in patients homozygous for the risk-conferring ATG16L1T300A variant were shown to exhibit similar structural alterations as compared with those in Atg16l1HM mice.38 This suggests that interrogation of the intestinal virome may provide important insights into the pathogenesis of IBD. Moreover, these studies together show how a phenotypic trait (Paneth cell dysfunction) associated with a genotypic risk factor (ATG16L1) may require the presence of a specific environmental factor (viral infection) to make the host susceptible to inappropriate immune responses to the commensal microbiota. It should also be noted that the absence of spontaneous intestinal inflammation in Atg16l1HM mice, even in the presence of MNV CR6 infection, highlights the importance of other environmental and/or genetic factors in the development of intestinal inflammation, even in the context of abnormal ATG16L1 function. In this context, it is noteworthy that Nod2−/− mice infected with Helicobacter hepaticus, a pathobiont, develop granulomatous ileitis,42 although uninfected Nod2−/− mice exhibit perfectly normal intestinal architecture. These studies further support the concept that a particular genotype is converted into a clinically relevant phenotype only when a specific set of environmental factors are present.
Autophagy Is Regulated by Intracellular Bacterial Sensing Mediated by NOD2
It has recently been recognized that NOD2 exerts an important role in the induction of autophagy associated with the intracellular presence of bacteria.43,44 Because NOD2 polymorphisms account for the vast majority of currently explained genetic heritability of CD, and ATG16L1 is among the genes most strongly associated with CD, autophagy and the intracellular sensing of bacteria may be part of a major shared mechanism in the development of CD.45
NOD2 is a cytosolic pattern recognition receptor that is activated upon binding to muramyl dipeptide (MDP), which derives from peptidoglycan present in gram-positive and gram-negative bacteria.46 MDP-induced NOD2 signaling involves RIP2, TRAF6, and NEMO and hence modulates nuclear factor-κB, but also mitogen-activated protein kinase activation (Figure 3).46 In addition, N-glycolyl–derived MDP from mycobacteria and viral single-stranded RNA can also bind to and activate NOD2. In the case of viral single-stranded RNA, the downstream signaling is distinct from MDP-induced NOD2 activation and involves IRF3 and TRIF.1,47,48 NOD2 and its variants encoded by CD risk-conferring NOD2 variants affect TLR and NF-κB signaling in a complex manner,49 and no comprehensive model would yet allow a full explanation of the mechanistic pathway from which NOD2 variants link to intestinal inflammation in human CD.1 However, several potential but not necessarily mutually exclusive pathways have been shown to exist. These include a role for NOD2 as a negative regulator of TLR signaling and, consequently, regulation of innate immune tolerance to bacterial products,50–52 a direct role of NOD2 in promoting IL-10 expression,53 a T-cell intrinsic role for NOD2 in regulating the production of cytokines by CD4+ T cells in an NF-κB dependent pathway54 and a role for NOD2 in Paneth cell function through direct regulation of antimicrobial peptide (α-defensin) expression.4055, The latter is interesting because it further directs attention to the convergence of multiple pathways on the normal and IBD-associated, pathobiologic functions of intracellular microbial sensing via NOD2, autophagy via ATG16L1 and, as will be discussed here, the unfolded protein response via X box binding protein-1 (XBP1).11 At a higher level, recent evidence suggests that these 3 pathways not only share the common feature that they affect Paneth cells, but that they are likely cross-regulating of each other.
Figure 3.

Signaling pathways induced by NOD2 activation.
The evidence first comes from the discovery that NOD2 functions as a key regulator of autophagy induction. Specifically, NOD2 (and NOD1) activation via their cognate ligands, MDP and C12-iEDAP (a derivative of the natural ligand mesodiaminopimelic acid), respectively, induce the formation of autophagic vesicles in epithelial as well as dendritic cells (DCs).43,44 Using several model pathogens, it could be shown that absent or decreased NOD2 function resulted in impaired autophagic uptake and consequent impaired intracellular bacterial killing.43,44 Remarkably, NOD proteins localized to the bacterial entry sites and mediated the selective recruitment of ATG16L1 to these intracellular locales as deduced from experiments with Shigella flexneri.44 This function was shown to be independent of RIP2 and downstream NF-κB signaling, and involved direct physical contact between NOD2 and ATG16L1.44 Importantly, CD-associated NOD2 variants exhibited impaired autophagy induction and were deficient in their capacity to recruit ATG16L1 to the bacterial entry sites at the plasma membrane, while the biochemical interaction between the two molecules appeared intact.44 Similarly, the CD-associated T300A variant of ATG16L1 exhibited impaired autophagy induction upon MDP stimulation,43 indicating that CD-associated variants of both molecules impair a common functional pathway; intracellular bacterial sensing leading to induction of autophagy and removal of the organism. In DCs, NOD2-induced autophagy was dependent on RIP2 as deduced from knock-down experiments.43 These studies in DCs also revealed important insights into how this impairment in autophagy might connect to alterations in the adaptive immune system. In elegant experiments involving a live attenuated Salmonella typhimurium serovar engineered to express the carboxy-terminal fragment of tetanus toxoid, Cooney et al showed that silencing of NOD2 in infected DCs resulted in decreased activation of autologous CD4+ T cells from tetanus-immune individuals consistent with the role of autophagy in antigen processing and presentation.43 Thus, the inability to properly sense an intracellular microbe is not only associated with diminished autophagic removal, but also likely autophagy-associated processing of the micro-organism for presentation to and activation of an appropriate adaptive immune response. Another recent study corroborated the role of NOD2 in autophagy induction and consequent intracellular bacterial killing, and also suggested that autophagy plays an important role in the uptake and delivery of MDP to its receptor NOD2, and thereby affects NOD2-dependent NF-κB activation.56 In addition, this latter study suggested that the ATG16L1T300A variant may profoundly impair NOD2-dependent antibacterial function in intestinal epithelial cells, but not monocytic cells derived from healthy, genotyped donors.56 These studies highlight cell-and potentially tissue-specific effects of ATG16L1 genotypes in disease pathogenesis.
Endoplasmic Reticulum Stress
Intracellular bacterial sensing and autophagy are now recognized to be interacting pathways in DCs, intestinal epithelial, and Paneth cells.43,44,56 Paneth cells are an appealing cell type to consider in the pathogenesis of IBD in view of their critical role in sensing and regulating the composition of the commensal microbiota, the major driver of the aberrant immune response in IBD,57–59 and the stem cell niche with its implications for barrier repair and cancer.60 To serve both of these functions, the Paneth cell must carry out important secretory functions. This latter property of Paneth cells draws attention to another pathway that has a close mechanistic relationship with autophagy, ie, UPR, as a consequence of endoplasmic reticulum (ER) stress, which is also genetically linked to the pathogenesis of both forms of IBD (CD and UC).11 Although the UPR and autophagy are mechanistically linked with important implications for the pathogenesis of CD as will be discussed, it is interesting to note that only genes involved in the UPR, but not autophagy, have yet been shown to be associated with UC. This is a notable fact that (among other possibilities) could point toward a general difference in the environmental (microbial?) factors that drive CD compared to UC.
ER stress is characteristically induced by the accumulation of misfolded proteins in the ER arising from either primary (genetic) or secondary (environmental) factors that affect the folding of proteins within the ER. Consequently, cells that are reliant on protein secretion for either their basal or induced functions are highly dependent upon a normal UPR. Such cells that have been shown to be especially dependent upon a UPR, and the IRE1/XBP1 branch of the UPR in particular (vide infra), for their function and in many cases survival include a variety of immune cells (plasma cells, plasmacytoid dendritic cells, macrophages, and CD8+ cytolytic T cells)61– 67 and parenchymal cells (hepatocytes, salivary gland epithelial cells, pancreatic acinar cells, and pancreatic endocrine cells).68 –70 Consistent with this, intestinal epithelial cells and especially Paneth cells and goblet cells are highly dependent upon an intact UPR for their normal function,11 with important consequences for IBD and other intestinal conditions.
The UPR as a consequence of ER stress in metazoans involves 3 pathways. Each of these sense events within the ER and link these to the generation of cytosolic transcription factors that direct a distinct transcriptional program that affects the ability of the cell to adapt to ER stress. Such an adaptive response involves the general features of affecting protein translation, the quantity of ER membranes through regulation of lipid metabolism, the protein-folding capabilities through regulation of chaperones, protein quality control associated with the ER-associated degradative pathways, and possibly apoptosis (reviewed in reference 71). The UPR is regulated by glucose-regulated protein 78 (grp78), an ER-associated chaperone (Figure 4). When associated with grp78, the 3 membrane proteins that regulate the UPR (ie, pancreatic ER kinase [PERK], IRE1, and ATF6) are maintained in an inactive state. Binding of unfolded proteins to grp78 releases each of these factors, resulting in characteristic UPR-related behaviors. PERK phosphorylates eIF2α, resulting in cessation of translation except for ATF4, a transcription factor that transactivates CHOP (C/EBP-homologous protein 10) involved in the induction of programmed cell death. ATF6, after migration to the Golgi, undergoes proteolytic cleavage of its cytosolic tail, which is transcriptionally active, by site 1 and site 2 proteases.71 Finally, IRE1 exhibits kinase activity leading to activation of c-Jun N-terminal kinase (JNK) and NF-κB pathways and endoribonuclease activity that is directed at the messenger RNA of XBP1, its only known substrate. Only XBP1 protein translated from spliced XBP1 messenger RNA (XBP1s) is transcriptionally active because it contains a transactivating domain in addition to the DNA binding domain, with only the latter also encoded by the protein translated from unspliced XBP1.71
Figure 4.
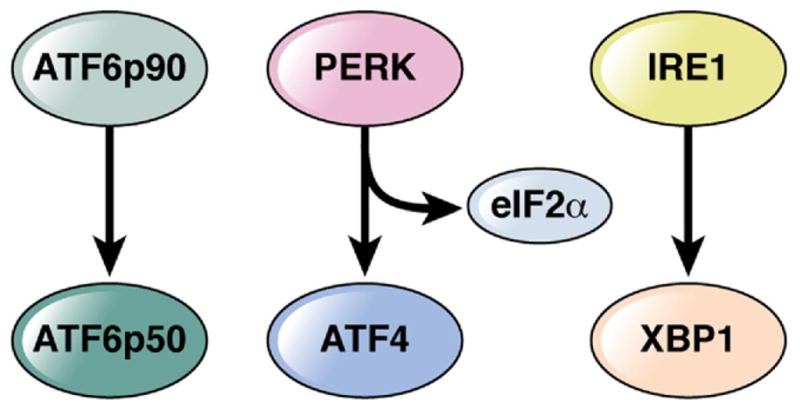
Proximal effectors of the unfolded protein response.
Several primary genetic factors that impose risk for development of IBD affect the UPR. The first to be described is IRE1-regulated XBP1.11 A genetic locus in the vicinity of XBP1 on chromosome 22 was initially implicated as a risk factor for IBD (both UC and CD) based upon several linkage studies.72–74 Based upon this a candidate gene study was performed in 3 cohorts of Northern European descent that, in total, included 4300 IBD and 5300 controls, resulting in the identification of a significant signal in association in both CD and UC confirming the linkage studies.11 More importantly, deep sequencing of the XBP1 gene in 1200 patients and controls identified several functionally hypomorphic rare variants of XBP1 in the IBD, but not control subjects, which were unable to elicit a normal transcriptional response in the setting of ER stress, suggesting that XBP1 conferred risk through the effect of rare variants,11 as recently also shown for the sialic acid acetylesterase gene by Pillai and colleagues.12 Consistent with this hypomorphic function, genetic deletion of Xbp1, specifically in the intestinal epithelium, resulted in spontaneous small intestinal inflammation and increased susceptibility to the colitis associated with dextran sodium sulfate administration in association with a functional and phenotypic impairment in Paneth cells.11 Not only did heterozygotic loss of Xbp1 lead to decreased Paneth cell antimicrobial function in association with spontaneous ileitis, but total loss of XBP1 function as observed in homozygotically deleted mice resulted in a nearly complete absence of Paneth cells and reduction of goblet cells due to apoptosis and spontaneous small intestinal inflammation and increased susceptibility to challenge with L monocytogenes.11 In the absence of XBP1, the epithelium also became hypersensitive toward inflammatory cytokines (eg, TNF) and microbial factors (eg, flagellin) due to increased JNK and NF-κB signaling, consistent with increased IRE1 kinase activity.11 Thus, hypomorphic XBP1 function in the epithelium has 2 consequences, impaired Paneth cell antimicrobial function and hyper-responsiveness to the microbial and inflammatory milieu contained within the intestines potentially leading to spontaneous inflammation.11 These studies with XBP1 also reveal the possibility that IBD may primarily emanate from genetically determined abnormalities within the intestinal epithelium. Other primary genetic abnormalities may also directly result in ER stress and possibly intestinal inflammation. Although not yet linked to human IBD, genetic deletion of IRE1β75 or Site-1 protease (S1P)76 in mice leads to increased susceptibility to dextran sodium sulfate–induced colitis. Multiple different factors associated with the UPR in response to ER stress are associated with either spontaneous intestinal inflammation (XBP1) or sensitivity to intestinal inflammation in response to secondary factors (IRE1β and S1P).
A variety of secondary factors can affect the ability of a cell to properly fold and secrete functional proteins.77 Experimentally, these include a variety of factors that affect the environment of the ER that facilitate protein folding and the ER-associated quality control machinery that ensures the proper sensing and removal of misfolded proteins. Consistent with this, experimental factors that affect the glycosylation of proteins (glucose deprivation), the folding of proteins (calcium deprivation and altered redox balance due to hypoxia), or alterations of protein removal (decreased proteasome function) cause the accumulation of misfolded proteins and ER stress.77 Thus, a variety of genetically determined alterations that either directly affect protein structure or processes involved in protein folding can lend themselves to development of ER stress. The first that deserves mention is ORMDL3, encoding orsomucoid-1-like gene 3, which was linked to the pathogenesis of IBD as a genetic risk factor through GWAS.8,9 Polymorphisms in ORMDL3 have also been recently linked to the pathogenesis of asthma,78,79 primary biliary cirrhosis,80 and insulin-dependent diabetes mellitus.81,82 ORMDL3 is an approximately 15-kDa 2-membrane–spanning ER-resident protein.83 ORMDL3 regulates the uptake of Ca++ from the cytosol to the ER such that deletion of ORMDL1/2 in yeast and silencing in HEK293 cells increases sensitivity to ER stress.83,84 This predicts that hypomorphic ORMDL3 function would impede the reuptake of Ca++ into the ER resulting in protein misfolding. The mechanism by which ORMDL3 is linked to intestinal inflammation remains to be defined. In another example, genetic deletion of anterior gradient gene 2 (Agr2), an ER-resident protein disulfide isomerase that regulates protein folding, in mice results in development of spontaneous enteritis and colitis. Agr2−/− mice exhibit increased ER stress in intestinal epithelium (↑grp78, ↑XBP1s) with no detectable mucus in goblet cells and expansion of Paneth cells before inflammation. Interestingly, a candidate gene study has identified AGR2 variants that are associated with decreased AGR2 messenger RNA expression and confer risk for both CD and UC.85 Finally, alterations in protein structure that result in instability leading to ER stress and, consequently, intestinal inflammation have been described to include those associated with Muc286 and HLA-B27.87,88 In a forward-genetics approach, mice (Winnie and Eeyore) that possess mutations in Muc2 have been described that develop accumulation of abnormal Muc2 in goblet cells and spontaneous colitis in association with severe ER stress.86 It is likely, given results with Xbp1−/− mice,11 that the inflammation is due to both the loss of mucin function and the effects of ER stress within the intestinal epithelium. Similarly, HLA-B27/β2-micro-globulin transgenic rats, the first genetic model of intestinal inflammation to be described87 and dependent upon the presence of the commensal microbiota,89 is now recognized to suffer from a “pathologic” ER stress response that is affiliated with the hematopoietic system.88,90 It is likely that the ER machinery in the rodent is unable to cope with the folding of this complicated human HLA-A molecule. Thus, a number of genetically determined events are now recognized to lead directly to either an unstable protein and/or a disability in ER-associated protein folding resulting in ER stress and potentially intestinal inflammation.
In a similar manner, a variety of secondary environmental factors or processes can directly cause the accumulation of unfolded proteins in the ER and thereby overwhelm the capacity of the UPR to deal with the imposed stress or directly interfere with the components of the UPR itself. The former include inflammation (eg, TNF and hypoxia) and dietary factors (eg, excess iron) with highly secretory cells being the cell type(s) most likely affected (reviewed in reference 77). Consistent with this, ischemia-reperfusion in humans leads to nonphysiologic levels of ER stress, as shown by increased grp78 and XBP1s in association with Paneth cell apoptosis and increased bacterial translocation.91 Microbes are also extremely important environmental stress factors to be considered. These include microbes such as picornavirus EV71,92 Streptomyces species that secrete trierixins that directly impede XBP1 function,93 or Shiga toxigenic Escherichia coli AB5 cytotoxins that degrade grp78,94 an ER-associated chaperone important for protein folding and the sensing of misfolded proteins as discussed here. Moreover, the ER-localized stress response chaperone gp96 is also upregulated in the inflamed intestinal epithelium of CD patients, interestingly on the apical membrane.95 Gp96 was recently reported to have an important role in the invasion of AIEC via recognition of OmpA at the outer membrane of AIEC.95 AIECs have been reported to be present in increased numbers in CD patients compared to healthy controls and are observed to colonize the ileal mucosa of patients with CD.96 –98 In this context, it is remarkable that DCs from donors with hypomorphic NOD2 variants exhibit impaired autophagic uptake as well as impaired intracellular killing of AIEC, leading to an increased intracellular bacterial burden43; potentially linking intracellular bacterial sensing to autophagy and possibly ER stress. Interestingly, IL-10 may protect the cell from excessive ER stress by enabling the UPR.99 These secondary environmental factors and potentially others may explain the common finding of ER stress as defined by increased grp78 and/or XBP1s in the epithelium of human specimens in both CD and UC.11,86,99 Thus, the genetic susceptibility of a host to respond to a variety of secondary environmental factors can determine either the initial development and/or propensity to perpetuate intestinal inflammation.
Autophagy and ER Stress Are Cross-Regulated
Several mediators of the UPR can directly induce autophagosome formation and initiate the process of autophagy. IRE1, a kinase and endoribonuclease directly upstream of XBP1, via its interaction with TRAF2, can induce activation of JNK signaling under conditions of ER stress,100 with JNK directly capable of inducing autophagosome formation via LC3 (Atg8).101,102 Massive IRE1 overactivation has been reported due to hypomorphic XBP1 function, along with increased JNK phosphorylation in small intestinal epithelium of Xbp1-deficient mice.11 Another branch of the UPR that is initiated by PERK and leads to activation of eukaryotic initiation factor 2α (eIF2α) can also directly link ER stress to the induction of autophagy.39,40 Further highlighting the close link that exists between the UPR and autophagy is the observation that Ddit3 (CHOP) and Atf4, both target gene effectors of PERK signaling, enhance the transcription of genes that are essential for autophagy.103
The functional impact of this mechanistic intertwining in the intestinal epithelium between the UPR and autophagy has not yet been specifically studied. However, insights may be gained from other model systems. Amyotrophic lateral sclerosis, a progressive and deadly motoneuron disease, is caused by mutations in superoxide dismutase-1, encoded by SOD1, which trigger its misfolding, abnormal aggregation, and motoneuron dysfunction.104 Transgenic overexpression of human amyotrophic lateral sclerosis-associated SOD1 variants in mice recapitulates human disease in association with unabated ER stress in degenerating motoneurons.105 Contrary to what would have been expected, conditional deletion of Xbp1 in neurons decreased toxicity of the SOD1 variants and ameliorated disease. This was shown to be associated with profound induction of autophagy and a decrease in SOD1 accumulation in the spinal cord.106 The mechanism implicated was by a compensatory autophagic response induced by an augmented UPR secondary to XBP1-deficiency resulting in increased degradation of disease-associated SOD1 protein aggregates. In this context it is remarkable that postmortem specimens of patients with amyotrophic lateral sclerosis exhibit evidence of profound activation of the UPR and autophagy.106 Similarly, accumulation of misfolded dysferlin has been linked to limb girdle muscular dystrophy in humans and a similar disorder in mice that express the mutant protein.107 In the mouse model, activation of eIF2α, downstream of PERK in the UPR, leads to induction of autophagy and protection from the pathologic consequences of misfolded dysferlin.108 A final example demonstrating the linkage between ER stress and autophagy is through studies of genetic Atg7 deficiency in mice.109 Atg7 deficiency is associated with decreased autophagy and increased ER stress in liver,109 resulting in impaired insulin signaling, a previously reported consequence of pathological ER stress in obesity.110 Indeed, Atg7 restoration in obese mice is associated with alleviation of ER stress and consequently normalization of insulin action.109 Together, these studies reveal the existence of a very close cross-regulation between ER stress and autophagy.
Dependence of Innate Immune Responses and the UPR
Recent evidence has emerged showing a close functional linkage between the ability to sustain an effective innate immune response and the UPR.62,111,112 This was originally predicted by the importance of XBP1 in the normal function of plasmacytoid DC.62 However, functional evidence has been provided that shows that the ability of the DC and macrophages to both function and survive, respectively, subsequent to innate immune signaling requires a proficient and regulated UPR. Specifically, TLR4 signaling links to IRE1 and PERK pathways of the UPR via TRIF.66 In the former case, TRIF-mediated, TLR4 signaling in DC leads to IRE1-directed, XBP1 splicing, which is required for the secretion of cytokines during an in vivo infection with Francisella tularensis, the etiologic agent of tularemia.66 In the latter case, TRIF-mediated, TLR4 signaling in macrophages leads to inhibition of eIF2α-directed transcription of ATF4 and consequently CHOP, a factor involved in programmed cell death induction.66 As such, TLR4 signaling not only induces a proper anti-infective response, but also regulates autophagy and the UPR. Whether NLRs such as NOD2 are capable of the same control remains to be elucidated.
Epithelial Function and Concluding Remarks
As detailed here, several lines of evidence highlight the critical importance of altered epithelial function in the context of genetically impaired autophagy, NLR and UPR function. Moreover, there is increasing evidence that these 3 pathways are functionally inter-related and altogether represent a biologically defined, integrated super-pathway acting within intestinal epithelia. This is most emphasized in the common functional impairment that converges upon Paneth cells (but also myeloid cells31,113) due to deficient ATG16L1, NOD2, and XBP1 activity in mice11,38,40 and humans carrying CD-associated NOD255 and ATG16L1 variants.38 How these inter-relationships affect UC relative to CD in view of the limited genotypic evidence for autophagy and NLRs in the former remains to be understood. The critical importance of the intestinal epithelium in mucosal homeostasis is also evident from studies with mice genetically deficient in IKK2 (encoded by Ikbkb) and NEMO (Ikbkg) specifically in the intestinal epithelium.114,115 NEMO-deficient mice develop spontaneous colitis,115 and intestinal epithelial-IKK2 deficiency results in colitis driven by the adaptive immune system due to inappropriate handling of an infestation with Trichuris muris.114 NOD2 expression in the absorptive intestinal epithelium is relatively low, but clearly present,116,117 which suggests that epithelial NOD2 through its interaction with the aforementioned NF-κB pathway and its role in autophagy induction may play an important role in the pathogenesis of CD even beyond its role in Paneth cells. Given the juxtaposition of intestinal epithelial cells to environmental exposures and the reactivity of autophagy, innate immune sensing, and the UPR to environmental factors, it is hoped that the study of these intersecting pathways will reveal important insights into gene-environment interactions as they relate to IBD (Figure 5).
Figure 5.

Relationship between the unfolded protein response, in response to endoplasmic reticulum stress, autophagy, and intracellular bacterial and viral sensing.
Acknowledgments
Funding Work in the authors’ laboratories is supported by National Institutes of Health RO1 grants DK44319, DK51362, DK53056, and DK08819 (R.S.B.), Innsbruck Medical University (MFI 2007-407, A.K.), the Austrian Science Fund and Ministry of Science P21530 and START Y446 (A.K.), the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement no. 260961 (A.K.) and the National Institute for Health Research Cambridge Biomedical Research Centre (A.K.).
Abbreviations used in this paper
- Agr2
anterior gradient gene 2
- AIEC
enteroadherent-invasive Escherichia coli
- Atg
autophagy
- CD
Crohn’s disease
- CHOP
C/EBP homologous protein
- DC
dendritic cell
- eIF2α
eukaryotic initiation factor 2α
- grp78
glucose-regulated protein 78
- GWAS
genome-wide association studies
- HM
hypomorphic
- IBD
inflammatory bowel disease
- IL
interleukin
- JNK
c-Jun N-terminal kinase
- MDP
muramyl dipeptide
- MNV
murine norovirus
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor κB
- NLR
Nod-like receptor
- PERK
pancreatic ER kinase
- S1P
Site-1 protease
- ssRNA
single-stranded RNA
- Th17
T helper 17
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- UC
ulcerative colitis
- UPR
unfolded protein response
- XBP1
X box binding protein-1
- XBP1s
spliced X box binding protein-1
Footnotes
Conflicts of interest The authors disclose no conflicts.
Supplementary Material
Note: The first 50 references associated with this article are available below in print. The remaining references accompanying this article are available online only with the electronic version of the article. Visit the online version of Gastroenterology at www.gastrojournal.org, and at doi:10.1053/j.gastro.2011.02.048.
References
- 1.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen R, Thomson JM, El-Omar EM, et al. The role of infection in the aetiology of inflammatory bowel disease. J Gastroenterol. 2010;45:266–276. doi: 10.1007/s00535-009-0191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lakatos PL. Environmental factors affecting inflammatory bowel disease: have we made progress? Dig Dis. 2009;27:215–225. doi: 10.1159/000228553. [DOI] [PubMed] [Google Scholar]
- 5.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. doi: 10.1146/annurev-genom-082908-150013. [DOI] [PubMed] [Google Scholar]
- 8.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGovern DP, Gardet A, Torkvist L, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke A, Balschun T, Sina C, et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL) Nat Genet. 2010;42:292–294. doi: 10.1038/ng.553. [DOI] [PubMed] [Google Scholar]
- 11.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surolia I, Pirnie SP, Chellappa V, et al. Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature. 2010;466:243–247. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glocker EO, Frede N, Perro M, et al. Infant colitis—it’s in the genes. Lancet. 2010;376:1272. doi: 10.1016/S0140-6736(10)61008-2. [DOI] [PubMed] [Google Scholar]
- 15.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 17.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol. 2010;75:795–800. doi: 10.1111/j.1365-2958.2009.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68:749–763. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 25.Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut. 2008;57:1294–1296. doi: 10.1136/gut.2008.157297. [DOI] [PubMed] [Google Scholar]
- 26.Reinisch W, Panes J, Lemann M, et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn’s disease. Am J Gastroenterol. 2008;103:2284–2292. doi: 10.1111/j.1572-0241.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 27.Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 28.Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 31.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 32.Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009;41:71–76. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhernakova A, Festen EM, Franke L, et al. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. 2008;82:1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 41.Mumphrey SM, Changotra H, Moore TN, et al. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J Virol. 2007;81:3251–3263. doi: 10.1128/JVI.02096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas A, Liu YJ, Hao L, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A. 2010;107:14739–14744. doi: 10.1073/pnas.1003363107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 44.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 45.Klionsky DJ. Crohn’s disease, autophagy, and the Paneth cell. N Engl J Med. 2009;360:1785–1786. doi: 10.1056/NEJMcibr0810347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–290. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strober W, Kitani A, Fuss I, et al. The molecular basis of NOD2 susceptibility mutations in Crohn’s disease. Mucosal Immunol. 2008;1(Suppl 1):S5–S9. doi: 10.1038/mi.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe T, Kitani A, Murray PJ, et al. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe T, Kitani A, Murray PJ, et al. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Hedl M, Li J, Cho JH, et al. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci U S A. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noguchi E, Homma Y, Kang X, et al. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol. 2009;10:471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shaw MH, Reimer T, Sanchez-Valdepenas C, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol. 2009;10:1267–1274. doi: 10.1038/ni.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Homer CR, Richmond AL, Rebert NA, et al. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139:1630–1641. 1641 e1–2. doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaishnava S, Behrendt CL, Ismail AS, et al. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. 2005;6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 59.Salzman NH, Hung K, Haribhai D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 61.Iwakoshi NN, Lee AH, Vallabhajosyula P, et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 62.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 64.Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. 2008;181:5433–5441. doi: 10.4049/jimmunol.181.8.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brunsing R, Omori SA, Weber F, et al. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem. 2008;283:17954–17961. doi: 10.1074/jbc.M801395200. [DOI] [PubMed] [Google Scholar]
- 66.Martinon F, Chen X, Lee AH, et al. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 68.Reimold AM, Etkin A, Clauss I, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 69.Lee AH, Chu GC, Iwakoshi NN, et al. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee AH, Scapa EF, Cohen DE, et al. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 72.Vermeire S, Rutgeerts P, Van Steen K, et al. Genome wide scan in a Flemish inflammatory bowel disease population: support for the IBD4 locus, population heterogeneity, and epistasis. Gut. 2004;53:980–986. doi: 10.1136/gut.2003.034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barmada MM, Brant SR, Nicolae DL, et al. A genome scan in 260 inflammatory bowel disease-affected relative pairs. Inflamm Bowel Dis. 2004;10:513–520. doi: 10.1097/00054725-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Hampe J, Schreiber S, Shaw SH, et al. A genomewide analysis provides evidence for novel linkages in inflammatory bowel disease in a large European cohort. Am J Hum Genet. 1999;64:808–816. doi: 10.1086/302294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bertolotti A, Wang X, Novoa I, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brandl K, Rutschmann S, Li X, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U S A. 2009;106:3300–3305. doi: 10.1073/pnas.0813036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaser A, Blumberg RS. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010;3:11–16. doi: 10.1038/mi.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 79.Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, Invernizzi P, Lu Y, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. 2010;42:658–660. doi: 10.1038/ng.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 83.Hjelmqvist L, Tuson M, Marfany G, et al. ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biol. 2002;3:RESEARCH0027. doi: 10.1186/gb-2002-3-6-research0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cantero-Recasens G, Fandos C, Rubio-Moscardo F, et al. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet. 2010;19:111–121. doi: 10.1093/hmg/ddp471. [DOI] [PubMed] [Google Scholar]
- 85.Zhao F, Edwards R, Dizon D, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2−/− mice. Dev Biol. 2010;338:270–279. doi: 10.1016/j.ydbio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hammer RE, Maika SD, Richardson JA, et al. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099–1112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- 88.Turner MJ, Sowders DP, DeLay ML, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 89.Taurog JD, Richardson JA, Croft JT, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tran TM, Satumtira N, Dorris ML, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172:5110–5119. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 91.Grootjans J, Hodin CM, de Haan JJ, et al. Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia-reperfusion. Gastroenterology. 2011;140:529–539. e3. doi: 10.1053/j.gastro.2010.10.040. [DOI] [PubMed] [Google Scholar]
- 92.Jheng JR, Lau KS, Tang WF, et al. Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell Microbiol. 2010;12:796–813. doi: 10.1111/j.1462-5822.2010.01434.x. [DOI] [PubMed] [Google Scholar]
- 93.Futamura Y, Tashiro E, Hironiwa N, et al. Trierixin, a novel Inhibitor of ER stress-induced XBP1 activation from Streptomyces sp. II. structure elucidation. J Antibiot (Tokyo) 2007;60:582–585. doi: 10.1038/ja.2007.74. [DOI] [PubMed] [Google Scholar]
- 94.Paton AW, Beddoe T, Thorpe CM, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 95.Rolhion N, Barnich N, Bringer MA, et al. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut. 2010;59:1355–1362. doi: 10.1136/gut.2010.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Darfeuille-Michaud A, Neut C, Barnich N, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 97.Martin HM, Campbell BJ, Hart CA, et al. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 98.Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 99.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 100.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 101.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rouschop KM, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 105.Atkin JD, Farg MA, Turner BJ, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 106.Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 108.Fujita E, Kouroku Y, Isoai A, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 109.Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 111.Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaser A, Blumberg RS. Survive an innate immune response through XBP1. Cell Res. 2010;20:506–507. doi: 10.1038/cr.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maeda S, Hsu LC, Liu H, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 114.Zaph C, Troy AE, Taylor BC, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 115.Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 116.Gutierrez O, Pipaon C, Inohara N, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 117.Hisamatsu T, Suzuki M, Reinecker HC, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]