

Abstract
The selectins are transmembrane, Ca2+-dependent lectins that mediate leucocyte rolling on vascular surfaces, the first adhesive step during inflammation and immune surveillance. Leucocytes express L-selectin, activated platelets express P-selectin, and activated endothelial cells express E- and P-selectin. Rolling involves force-regulated, rapidly reversible interactions of selectins with a limited number of glycosylated cell surface ligands. Rolling permits leucocytes to interact with immobilized chemokines that convert β2 integrins to high-affinity conformations, which mediate arrest, post-arrest adhesion strengthening, and transendothelial migration. However, rolling leucocytes also transduce signals through selectin ligands, the focus of this review. These signals include serial activation of kinases and recruitment of adaptors that convert integrins to intermediate-affinity conformations, which decrease rolling velocities. In vitro, selectin signalling enables myeloid cells to respond to suboptimal levels of chemokines and other agonists. This cooperative signalling triggers effector responses such as degranulation, superoxide production, chemokine synthesis, and release of procoagulant/proinflammatory microparticles. In vivo, selectin-mediated adhesion and signalling likely contributes to atherosclerosis, arterial and deep vein thrombosis, ischaemia–reperfusion injury, and other cardiovascular diseases.
Keywords: Neutrophil, Selectin, Integrin, Cell adhesion, Inflammation
1. Selectins: initiators of leucocyte adhesion
In the multistep adhesion cascade, leucocytes tether to and roll along postcapillary venules. They then arrest and crawl through or between endothelial cells into the perivascular space1,2 (Figure 1). This process occurs during constitutive homing of naive lymphocytes to lymph nodes, of haematopoietic stem/progenitor cells to bone marrow, and of T cell precursors to thymus. During inflammation, neutrophils, monocytes, or effector T cells migrate to specific tissues in response to infection or injury. Interactions of selectins with their glycosylated ligands mediate rolling.3 In the original multistep paradigm, selectin-dependent rolling allows leucocytes to encounter chemokines that activate leucocyte integrins, which permit arrest and crawling. However, selectins also transduce signals that modulate leucocyte behaviour, for example, by enabling integrin-dependent reductions in rolling velocities (slow rolling; Figure 1). The multistep paradigm emphasizes interactions of leucocytes with endothelial cells. However, selectins also mediate leucocyte adhesion to activated platelets and to other leucocytes (Figure 1, inset). Thus, selectins initiate multicellular adhesive and signalling events during physiological or pathological inflammation and haemostasis. This review focuses on how selectins mediate signalling as well as adhesion, particularly with regard to neutrophils.
Figure 1.

Multistep leucocyte adhesion cascade. Endothelial selectins initiate tethering and rolling of leucocytes, including neutrophils as depicted here. Signals from engaged selectins and chemokines activate β2 integrins that mediate slow rolling and arrest. Integrins also direct spreading, intraluminal crawling, and migration between or through endothelial cells into perivascular tissues. Inset: Adherent neutrophils polarize to form a leading edge (lamellipodium) and a trailing edge (uropod). The selectin ligand PSGL-1 redistributes to the uropod and captures activated platelets by interacting with P-selectin. Platelet–neutrophil interactions transduce signals that facilitate intraluminal crawling.
2. Selectins and selectin ligands
Each of the three selectins has an N-terminal Ca2+-dependent (C-type) lectin domain, an epidermal growth factor (EGF)-like module, a series of tandem consensus repeats, a transmembrane domain, and a cytoplasmic tail.3 L-selectin is expressed on the surfaces of all leucocytes. Some agonists induce proteolytic release of the ectodomain.4 P-selectin is expressed by megakaryocytes and endothelial cells. It is stored in membranes of α granules of platelets and Weibel–Palade bodies of endothelial cells. Mediators such as thrombin, histamine, or complement induce rapid redistribution of P-selectin to the plasma membrane. TNF-α, IL-1β, or lipopolysaccharide (LPS) increases transcription of mRNA for P-selectin in most mammals, including mice, but not in primates, including humans.5,6 E-selectin is constitutively expressed on the surfaces of venular endothelial cells of bone marrow and skin. In most organs, however, endothelial cells must be stimulated with TNF-α, IL-1β, or LPS to synthesize E-selectin.7 This mechanism of induced expression is conserved in mice and humans.
The selectins are Ca2+-dependent lectins. The minimal recognition determinant is sialyl Lewis x or sLex (NeuAcα2-3Galβ1-4[Fucα1-3]GlcNAcβ1-R), a terminal component of some N- and O-glycans.3 Another posttranslational modification, sulfation, enables certain glycoproteins to serve as physiologically relevant ligands for P- and L-selectin. Thus, P- and L-selectins bind cooperatively to an sLex-capped O-glycan, sulfated tyrosines, and other amino acids near the N terminus of P-selectin glycoprotein ligand-1 (PSGL-1), a homodimeric mucin on leucocytes.3 L-selectin binds to sialylated, fucosylated, and sulfated N- and O-glycans on glycoproteins expressed on endothelial cells of high endothelial venules in lymph nodes.8,9 Sulfation does not enhance binding to E-selectin. Nevertheless, only a limited number of glycoproteins are physiologically relevant ligands for E-selectin, at least on murine neutrophils, where they have been best studied.10 These include PSGL-1, CD44, and E-selectin ligand-1 (ESL-1). Other glycoprotein ligands for E-selectin have been described,10 and glycolipids are candidate ligands for E-selectin on human (but not murine) neutrophils.11 However, additional evidence is required to support their physiological functions.
3. Rolling adhesion through selectin–ligand interactions
In flowing blood, leucocytes roll on vascular surfaces through reversible interactions of selectins with their ligands.3 A cell rolls by forming new adhesive bonds at the leading edge to replace bonds that dissociate at the trailing edge. These bonds are short-lived, usually lasting less than a second. Furthermore, the shear stresses of blood flow exert force on bonds at the trailing edge. Slip bonds have lifetimes that are shortened by force. Catch bonds, which are less intuitive, have lifetimes that are prolonged by force.12 Remarkably, increasing force exerts a triphasic effect on selectin–ligand interactions, first shortening lifetimes (slip bonds), then prolonging lifetimes (catch bonds), and then again shortening lifetimes (slip bonds).12–14 The biological functions of catch bonds are not fully understood. The strongest evidence, from in vitro studies, supports a role for catch bonds in mediating flow-enhanced adhesion by L-selectin.15 At low shear stresses (<0.3 dynes/cm2), leucocytes roll rapidly and irregularly and detach frequently. Higher shear stresses (0.3–1.0 dynes/cm2) enable cells to roll slower and more regularly and detach less frequently. At still higher shear stresses (>1.0 dynes/cm2), cells roll faster and detach more frequently. At low shear stresses, bond lifetimes are very short. As shear stress rises, increasing force prolongs bond lifetimes (catch bonds), causing rolling to become slower and more regular until a shear optimum (∼1.0 dynes/cm2) is reached. Above the shear optimum, force shortens bond lifetimes (slip bonds), causing rolling to become faster and less regular. Catch bonds may prevent agglutination of circulating leucocytes, which express both L-selectin and its ligand, PSGL-1. This is because very little force is applied to L-selectin–PSGL-1 bonds that form during a random contact of circulating leucocytes.15 Because these bond lifetimes are short, they likely dissociate rapidly, preventing stable agglutination. Catch bonds may also prevent leucocyte aggregation during low flow or stasis, as might occur during arterial ischaemia or deep vein thrombosis. However, in vivo studies are required to definitively establish the physiological roles of catch bonds. Models for the structural basis of catch bonds have been reviewed3 and will not detailed here. In brief, force-regulated changes in the relative orientations of the lectin and EGF domains appear to allosterically alter the ligand-binding surface on the lectin domain.
In addition to the intrinsic features of selectin–ligand binding, cellular features modulate the forces applied to adhesive bonds and thus affect rolling. These include the densities of selectins and their ligands and their clustering in membrane domains. Tensile and compressive forces also affect the geometries of rolling cells and the resulting orientations of selectin–ligand bonds.3 For example, rolling neutrophils extrude long membrane tethers at the trailing edge,16,17 and they launch membrane slings over the leading edge that form new bonds with the vascular surface.18
4. Selectin-induced signalling in leucocytes
Selectin-mediated adhesion potentially brings leucocytes in proximity to other agonists. This form of signalling was first supported by studies of myeloid cell adhesion to P-selectin on activated platelets or endothelial cells in vitro.19–22 P-selectin-mediated adhesion enables chemokine- or platelet-activating factor-triggered activation of β2 integrins, which stabilizes adhesion. It also facilitates release of chemokines from adherent leucocytes. Later studies indicated that engagement of selectin ligands (or L-selectin) on leucocytes directly transduces signals. For example, interactions of PSGL-1 with immobilized P-selectin or anti-PSGL-1 mAbs rapidly induce tyrosine phosphorylation of multiple proteins.23 However, such studies, performed under static conditions in vitro, do not address whether selectin-triggered signalling occurs under physiological conditions and if so, whether it exerts biologically relevant effects. To date, the most extensive evidence is for selectin-triggered signals that modulate β2 integrin function on rolling neutrophils.
5. Selectin-induced, integrin-mediated slow rolling of neutrophils
Mild trauma during surgical exteriorization of the murine cremaster muscle mobilizes P-selectin from Weibel–Palade bodies to the endothelial cell surface of postcapillary venules. Leucocytes, mostly neutrophils, roll rapidly on the mobilized P-selectin.24 Injection of TNF-α up-regulates endothelial synthesis of E-selectin. In this setting, neutrophils roll slowly on E-selectin because it is expressed at high densities.25 Slow rolling velocities prolong the transits of neutrophils through venules, providing more opportunities for the cells to encounter endothelial-bound chemokines such as CXCL1.26 Chemokine signals activate β2 integrins, which mediate arrest and then transendothelial migration.27 However, E-selectin ligands engaged during rolling transduce signals that also affect β2 integrin function.28 These signals enable β2 integrins to further reduce rolling velocities by ∼50%, i.e. from ∼10 to ∼5 µm/s. This slower rolling, compared with that on E-selectin in the absence of signalling, results from reversible interactions of β2 integrins, particularly αLβ2, with endothelial cell ligands, particularly intercellular adhesion molecule-1 (ICAM-1). In other words, neutrophils roll slowly by forming and breaking integrin–ligand bonds as well as E-selectin–ligand bonds.
Integrins are transmembrane heterodimers of non-covalently paired α and β subunits.29 Each subunit has multiple domains. Integrins on blood cells must be activated to bind their ligands. Three principal integrin conformations have been described: a bent form with a closed headpiece that has low affinity for ligand, an extended form with a closed headpiece that retains low affinity for ligand, and an extended form with an open headpiece with high affinity for ligand (Figure 2). Transitional conformations also exist.30 Current evidence, primarily obtained with mAbs that bind to particular conformations of human β2 integrins, indicates that selectin signals trigger extension of αLβ2 with the closed headpiece31 (Figure 2). This conformation mediates rolling on ICAM-1. It is often said to have intermediate affinity even though the headpiece, which contains the ligand-binding surface on the αI domain, does not change and retains low affinity for ligand. However, extension does increase encounters of the αI domain with ICAM-1 on apposing cells, which accelerates on-rates. Faster on-rates increase overall affinity. Thus, this conformation can be considered to have intermediate affinity on cell surfaces. In contrast to selectin signals, chemokine signals induce extension of αLβ2 with an open headpiece (Figure 2). Because of its higher affinity for ligand (and longer bond lifetimes), this conformation mediates neutrophil arrest on ICAM-1. Importantly, arrest rapidly triggers integrin outside-in signals by bound ICAM-1 that cause spreading and strengthen adhesion.32
Figure 2.

Integrin activation in rolling neutrophils. Left, signals transmitted through selectin ligands, e.g. PSGL-1 or CD44, convert integrin αLβ2 to an extended conformation that retains a closed headpiece with low affinity for ICAM-1. This conformation decreases rolling velocities. Right, signals transmitted through chemokine receptors, e.g. CXCL1, convert αLβ2 to an extended conformation that has an open headpiece with high affinity for ICAM-1. This conformation causes arrest. Both conformations require recruitment of talin to β2 tails. The extended, high-affinity conformation also requires kindlin and actomyosin-dependent tension.
Extensive information on the neutrophil signalling cascade triggered by selectin interactions has been obtained (Figure 3). The data emerge from flow chamber studies using isolated murine or human leucocytes31,33 or autoperfused murine blood,28 or intravital microscopy of postcapillary venules in the cremaster muscle after challenge with TNF-α.28,33 In flow chambers, rolling velocities are compared on immobilized P- or E-selectin with or without co-immobilized ICAM-1. Blocking mAbs identify the integrin-dependent component of rolling. A chemokine such as CXCL1 may be co-immobilized to trigger integrin-dependent arrest and spreading. In the cremaster muscle, pertussis toxin (PTx) is typically injected to block chemokine signalling through Gαi-coupled receptors on neutrophils. In vivo, selectin signalling results from interactions with E-selectin because of its higher density.28,33 In vitro, P- and E-selectin densities can be adjusted to yield rolling velocities like those in venules.33 Under these conditions, P-selectin signals as effectively as E-selectin. This may be relevant when neutrophils interact with activated platelets, which express P-selectin at high densities.
Figure 3.
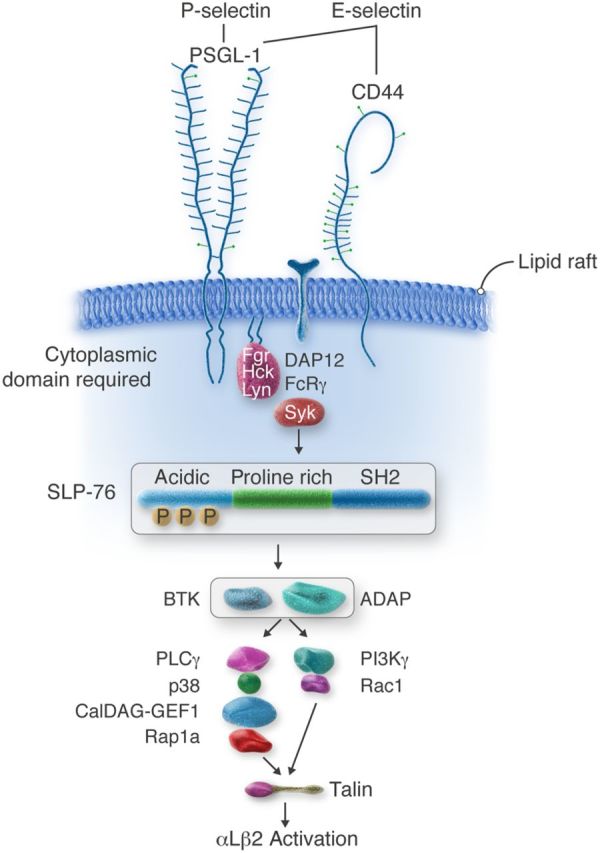
Selectin signalling cascade in neutrophils. Engagement of PSGL-1 or CD44 triggers serial activation of kinases and recruitment of adaptors, resulting in talin-dependent activation of integrin αLβ2 that mediates slow rolling on ICAM-1.
In broad terms, selectin signalling in neutrophils resembles signalling by classical immunoreceptors such as the T-cell receptor, the B-cell receptor, and the Fc receptor. The pathway involves sequential activation of tyrosine kinases and recruitment of adaptors that propagate downstream signals culminating, in this case, in conversion of β2 integrins to a conformation with intermediate affinity, i.e. an extended ectodomain with the closed headpiece.31 P-selectin signals through PSGL-1, the major ligand for P-selectin on murine and human leucocytes.33 E-selectin signals through PSGL-128 or CD44,33 two of the three major ligands for E-selectin on murine neutrophils; the ligands that transduce signals on human neutrophils are less well defined. Chelation or sequestration of cholesterol blocks signalling, indicating that intact lipid rafts are required.33 This is consistent with the well-described assembly of signalling proteins in these membrane domains. Indeed, both PSGL-1 and CD44 are enriched in lipid rafts.34–36 Selectin-mediated adhesion likely clusters PSGL-1 or CD44 to initiate signalling.23,37,38 Slow rolling, however, does not require intact actin filaments in neutrophils.39 Clustering of PSGL-1 or CD44 is more likely when P- or E-selectin is presented in a multivalent form on a cell membrane or surface. Soluble P-selectin can signal when it is dimeric or multimeric, but not when it is monomeric.23,37 Neutrophil adhesion to P-selectin on activated platelets or endothelial cells induces cleavage of the ectodomain of P-selectin.40–42 Modestly elevated levels of circulating soluble P-selectin are present in patients with thrombotic or inflammatory disorders43 and in mice expressing P-selectin without the cytoplasmic domain.44 The elevated soluble P-selectin has been postulated to be proinflammatory and prothrombotic.44–46 However, the levels are well below the dissociation constant for binding of monomeric soluble P-selectin to PSGL-1.47 If the shed ectodomain of P-selectin remains monomeric in plasma, its role as a signalling protein remains unclear.
The earliest known signalling event is activation (tyrosine phosphorylation) of Src family kinases (SFKs). Murine neutrophils express three SFKs: Fgr, Hck, and Lyn. Deficiency of Fgr or both Hck and Lyn blocks signalling.33,48 How selectin binding triggers activation of SFKs is not understood. Neutrophils from knockin mice that express PSGL-1 lacking the cytoplasmic domain (ΔCD PSGL-1) do not signal in response to P-selectin.35 CD44-deficient neutrophils expressing ΔCD PSGL-1 do not signal in response to E-selectin.33 These data demonstrate a critical signalling role for the PSGL-1 cytoplasmic domain. How it does so is not known. ΔCD PSGL-1 still associates with lipid rafts, indicating that PSGL-1 does not require its cytoplasmic tail to concentrate in these membrane domains.35 The PSGL-1 cytoplasmic tail was reported to bind to Nef-associated factor, thereby recruiting phosphatidylinositol-3-kinase δ (PI3Kδ) to activate β2 integrins.49 As discussed below, however, PI3Ks are dispensable for slow rolling, and PI3Ks can be activated by downstream complexes. Thus, the PSGL-1 tail probably acts more proximally in the signalling cascade. One possibility is that it binds directly to SFKs, as does the cytoplasmic domain of CD44 in some cells.50
Neutrophil SFKs activated by selectin engagement phosphorylate the immunoreceptor tyrosine-based activation motifs (ITAMs) in the cytoplasmic domains of DAP12 and FcRγ,48 which also concentrate in lipid rafts.51 The phosphorylated ITAMs serve as docking sites for spleen tyrosine kinase (Syk), which activates and recruits other kinases and adaptors as part of a multiprotein complex.28 These include the key adaptor SLP-76, the Tec kinase Btk, and the adaptor ADAP.33,52,53 In turn, this complex activates further downstream mediators. One pathway involves serial activation of phospholipase C γ, p38 MAPK, and a Ca2+ and diacylglycerol-activated guanine nucleotide exchange factor (CalDAG-GEF1), which activates the GTPase Rap1a.52,54 Activated Rap1a, probably through effectors,55 recruits talin1 to the membrane. A second pathway may involve activation of PI3Kγ and Rac1 to recruit talin1.52,56 Talin1 (see below) then directly activates integrin αLβ2.
Although the major components of the signalling cascade are well established, some remain to be confirmed by a second laboratory, and some are the subject of debate. For example, one group reports that PSGL-1 is the sole signalling ligand for E-selectin,28 whereas another group reports that either PSGL-1 or CD44 is sufficient to transduce signals from E-selectin.33 The first group reports that a novel complex of PSGL-1 with L-selectin is responsible for signalling.57 This may represent only a small subset of the two proteins since the great majority of L-selectin is not in lipid rafts.35 Whether PI3Kγ contributes to selectin-induced signalling is also controversial.33 Additional studies are required to address these discrepancies.
A final common step in integrin activation is binding of talin to the cytoplasmic tail of the β subunit.58,59 Talin1 is the predominant isoform expressed in haematopoietic cells, including neutrophils. Talin is a large cytoplasmic protein with a head domain and a rod domain.60 The head domain binds to β tails, and the rod domain binds to β tails, actin, and other proteins. Binding of the talin head domain to β tails destabilizes interactions between the α and β tails and transmembrane domains, inducing allosteric activation of the integrin ectodomain.58,59 As mentioned earlier, selectin signals induce extension of the αLβ2 ectodomain that retains a closed headpiece with low affinity for ICAM-1.31 This intermediate-affinity conformation mediates slow rolling but not arrest. Talin1-deficient neutrophils are defective both in selectin-triggered, αLβ2-mediated slow rolling and in chemokine-triggered, αLβ2-mediated arrest and spreading.61 Notably, selectin-triggered, αLβ2-mediated slow rolling does not require intact actin filaments or actomyosin tension.39 This suggests that talin1 bound to β2 tails need not simultaneously bind to the cytoskeleton to support slow rolling (Figure 2). Chemokine signals induce extension of the αLβ2 ectodomain with an open headpiece with high affinity for ICAM-1.31 Importantly, both intact actin filaments and actomyosin tension are required for chemokine-induced arrest and spreading.39 This is consistent with the notion that simultaneous talin interactions with integrins and the cytoskeleton further separate the α and β subunits and stabilize the open headpiece of the ectodomain (Figure 2).
Kindlins, another group of cytoplasmic adaptors, bind to a different region of integrin β tails.59 Kindlin-3 is the most abundant kindlin in haematopoietic cells, including neutrophils. In response to selectin or chemokine signals, kindlin-3-deficient neutrophils extend the αLβ2 ectodomain, but do not open the headpiece. As a result, they manifest αLβ2-mediated slow rolling but not arrest.61 Kindlins increase the clustering of talin-activated integrins.62 This stabilizes the open headpiece and promotes outside-in signalling. Outside-in signalling causes extensive cytoskeletal rearrangements that promote spreading and strengthen adhesion.32 Transitions from integrin-dependent arrest to integrin-dependent outside-in signalling occur rapidly and are often difficult to distinguish. The dispensability of kindlin-3 for selectin-triggered, αLβ2-mediated slow rolling implies that selectin signalling, unlike chemokine signalling, may not recruit kindlins to β2 tails (Figure 2). Other mediators may contribute to integrin activation. Thus, neutrophils rolling on E-selectin in TNF-α-stimulated venules stimulate PSGL-1-dependent release of myeloid-related proteins 8 and 14 from the cytosol. These proteins, acting in an autocrine loop, engage Toll-like receptor 4 on neutrophils to enhance β2 integrin-dependent slow rolling and arrest.63
How might selectin signalling facilitate neutrophil recruitment? First, β2 integrin-dependent slow rolling may increase encounters with immobilized chemokines that induce neutrophil arrest. In vivo, however, blocking β2 integrins increases rolling velocities by only two-fold;28,33 that is, neutrophils still roll relatively slowly and probably remain able to interact efficiently with endothelial-bound chemokines. Alternatively, selectin signalling may prime neutrophils to respond to lower levels of chemokines. Under static conditions in vitro, neutrophil or monocyte adhesion to P- or E-selectin enables effector responses to otherwise suboptimal levels of a conventional agonist such as a platelet-activating factor or a chemokine.20–22,37 These responses include integrin activation, superoxide production, or synthesis of chemokines. Whether such priming occurs during leucocyte rolling in vivo is less clear. Indeed, selectin signalling under flow is rapidly reversible.28 Blocking selectin signalling by deleting or inhibiting a key signalling protein such as Syk has a minimal effect on neutrophil recruitment in several models of acute inflammation. Similarly, blocking chemokine signalling by disabling Gαi receptors with PTx only partially inhibits recruitment. In contrast, blocking both pathways markedly inhibits recruitment. Such data have been interpreted to indicate that selectin signalling and chemokine signalling cooperate to maximize neutrophil recruitment during acute inflammation.28,33,48,52,54 However, these studies employed inhibitors or gene knockouts that impair integrin outside-in signalling as well as PSGL-1- or CD44-triggered inside-out signalling. Like selectin signalling, ligand binding to chemokine-activated β2 integrins triggers a signalling cascade that resembles that of classical immunoreceptors.32 Indeed, most of the components are shared, including SFKs, Syk, and SLP-76. Therefore, determining the physiological importance of selectin-triggered activation of β2 integrins will require methods to block signalling through selectin ligands without impairing integrin outside-in signalling. As noted earlier, knockin mice expressing PSGL-1 lacking the cytoplasmic domain (ΔCD PSGL-1) were made to address this issue.35 The limitation of these mice, however, is that the density of PSGL-1 on leucocyte surfaces is reduced by ∼90%. This is due to loss of an export signal in the cytoplasmic domain, which impairs transport of newly synthesized PSGL-1 from the endoplasmic reticulum to the Golgi apparatus.64 In vitro, the limitation of reduced ΔCD PSGL-1 surface density on neutrophils can be overcome by adjusting selectin densities to match bond numbers and rolling velocities with those of neutrophils from wild-type mice.33,35 In vivo, selectin densities cannot be adjusted between genotypes. Therefore, it is difficult to determine whether signalling defects in the knockin mice are due to reduced PSGL-1 levels, loss of the cytoplasmic domain, or both. Mutations of the cytoplasmic domain that impair signalling without altering PSGL-1 expression may overcome this limitation.
6. Signalling during platelet–myeloid cell interactions
P-selectin-mediated adhesion of activated platelets to PSGL-1 on myeloid cells can trigger signalling in both cell types.65 In vitro, adhesion is rapidly stabilized by interactions of β2 integrins, particularly αMβ2, with glycoprotein Ibα and other ligands on platelets.66–68 Stable adhesion requires activation of SFKs.69,70 As for neutrophil slow rolling and arrest on endothelial cells, it is difficult to determine whether SFK activation results from signalling through PSGL-1, outside-in signalling through ligand-bound β2 integrins, or both. Close cell contact immediately brings both platelets and myeloid cells into contact with other mediators. Platelets express chemokines that activate integrins and induce synthesis of chemokines and other mediators in myeloid cells.22 Activated neutrophils secrete CTG, which activates platelets through protease-activated receptor 4.71,72 Thus, selectin-initiated adhesion triggers bidirectional signalling in platelets and myeloid cells. Feedback-loop signal amplification likely induces other effector responses. For example, platelet–neutrophil interactions trigger formation of neutrophil extracellular traps,73 and platelet–monocyte interactions trigger expression of tissue factor;74 both exacerbate venous thrombosis.75 However, it remains difficult to distinguish the contributions of selectin-mediated adhesion and selectin-mediated signalling in vivo, where chemokines, proteases, and other mediators are also present.
Although the multistep adhesion model usually emphasizes adhesion of leucocytes to endothelial cells, platelets also interact with neutrophils as they adhere to the endothelium (Figure 1). Cells that arrest in venules rapidly polarize in response to chemokine signals. Intraluminal crawling is mediated by integrin αMβ2 that redistributes to the leading edge, or lamellipodium.1 The other end of the polarized cells forms a protruding domain, or uropod. PSGL-1 and several other glycoproteins are redistributed to this domain.76–78 In vivo, most uropods are oriented away from the endothelial cell surface.79 Remarkably, the concentrated PSGL-1 in the uropods serves as an efficient platform to capture circulating activated platelets that express P-selectin79 (Figure 1, inset). The adhesion of the activated platelets to the uropods is typically transient, yet these interactions induce signals that facilitate the redistribution of integrin αMβ2 to the leading edge, increase crawling velocities, and reduce the time required for the crawling neutrophil to migrate out of the venule79 (Figure 1, inset). In mice expressing ΔCD PSGL-1, activated platelets still adhere to polarized neutrophils, but neutrophil migration is impaired.79 This is consistent with impaired signalling due to loss of the PSGL-1 cytoplasmic domain. As noted above, however, decreased signalling could also result from less stable adhesion due to the marked decrease in ΔCD PSGL-1 density on the neutrophils.35 Notably, integrin αMβ2 redistributed to the leading edge captures unactivated platelets,78 probably through interactions with glycoprotein Ibα. This process is less efficient in neutrophils with reduced expression of ESL-1.78 As for PSGL-1 or CD44, distinguishing a direct signalling role of ESL-1 from an indirect role in juxtacrine adhesion/signalling requires further investigation. Nevertheless, these studies establish a major contribution of platelets to acute inflammation in physiological and pathological states.
7. Selectins and human disease
Over the past 25 years, selectins, particularly P-selectin, have been shown to contribute to pathological inflammation and thrombosis in many preclinical disease models, including atherosclerosis,80–82 ischaemia–reperfusion injury,83 arterial thrombosis,84 and deep vein thrombosis.75,85 Patients with inflammatory and thrombotic disorders typically have higher levels of activated platelets displaying P-selectin, platelet–leucocyte aggregates, and soluble P-selectin in peripheral blood.43,86 Although these findings have led to intense interest in the development of selectin inhibitors as drugs, testing such drugs in human trials is still in the early stages. In an early study, patients with ST-segment elevation myocardial infarction were randomized to receive a placebo or a soluble form of PSGL-1 fused to the Fc portion of human IgG1 (a P-selectin antagonist) intravenously as an adjunct to standard thrombolytic therapy.87 The trial was terminated prematurely due to the lack of efficacy in ST-segment elevation, left ventricular function, clinical outcome, and other endpoints. In a more encouraging phase 2 trial, patients with non-ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention were randomized to receive a placebo or a humanized anti-P-selectin mAb to block P-selectin binding to PSGL-1.88 Relative to placebo, the antibody decreased plasma troponin I to levels that approached statistical significance. These disparate results illustrate the challenges in introducing a new drug for cardiovascular disorders in which established drugs, e.g. thrombolytics and anti-platelet agents, have already improved outcomes, and they suggest a need for careful design of larger clinical trials.
Patients with sickle cell anaemia develop injury to many organs that is consistent with repeated episodes of ischaemia–reperfusion injury.89 Murine models of sickle cell anaemia have established that sickled red cells obstructing small vessels trigger pathological inflammation and thrombosis that involves adhesive interactions among red cells, platelets, and the endothelium. E-selectin binding to neutrophils triggers adhesion to sickle red cells.78 Furthermore, P-selectin on activated platelets and endothelial cells binds directly to ligands on sickle red cells as well as to PSGL-1 on neutrophils.90 Blockade of P- or E-selectin decreases these adhesive interactions and improves microcirculatory flow in sickle cell mice.91–94 In a recent phase 2 trial, intravenous infusion of a small-molecule selectin inhibitor reduced the time for resolution of acute, painful vasoocclusive crisis in patients with sickle cell disease.95 Although this did not reach statistical significance, secondary analysis revealed a statistically significance reduction in opioid analgesic use. These results suggest that a larger phase 3 trial is warranted. A separate phase 2 trial is examining whether chronic administration of a humanized anti-P-selectin mAb will prevent or reduce vasoocclusive complications (https://clinicaltrials.gov/ct2/show/NCT01895361).
These initial clinical trials suggest that selectin antagonists may be effective in some cardiovascular disorders, as predicted from the many preclinical studies. Given the inherent risks in extrapolating data from animal models to humans, careful selection of clinical targets and proper trial design will be essential for further progress.
8. Conclusion
The function of selectins as adhesion receptors in models of inflammation, thrombosis, and immune responses is well established. Early clinical trials suggest promise for drugs that inhibit selectin–ligand interactions. Selectins also trigger signals in leucocytes by engaging ligands such as PSGL-1 and CD44. These signals are sufficient for some responses, notably conversion of β2 integrins to an intermediate-affinity conformation that mediates slow rolling. In vitro, selectin signals are usually insufficient to induce responses such as superoxide production, synthesis of tissue factor, secretion of cytokines, or formation of neutrophil extracellular traps. However, selectin signals enable suboptimal signals from other agonists, e.g. chemokines or lipid autacoids, to generate such responses. Whether cooperative signalling occurs in vivo requires further investigation. Insights from such studies may lead to new drug targets for thrombotic and inflammatory diseases.
Conflict of interest: The author holds equity interest in Selexys Pharmaceuticals Corporation.
Funding
Research in the author's laboratory was supported by National Institutes of Health grants HL034363 and HL085607.
References
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7:678–689. [DOI] [PubMed] [Google Scholar]
- 2.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 2010;11:366–378. [DOI] [PubMed] [Google Scholar]
- 3.McEver RP, Zhu C. Rolling cell adhesion. Annu Rev Cell Dev Biol 2010;26:363–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahn J, Walcheck B, Migaki GI, Jutila MA, Kishimoto TK. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell 1998;92:809–818. [DOI] [PubMed] [Google Scholar]
- 5.Yao L, Setiadi H, Xia L, Laszik Z, Taylor FB, McEver RP. Divergent inducible expression of P-selectin and E-selectin in mice and primates. Blood 1999;94:3820–3828. [PubMed] [Google Scholar]
- 6.Liu Z, Miner JJ, Yago T, Yao L, Lupu F, Xia L, McEver RP. Differential regulation of human and murine P-selectin expression and function in vivo. J Exp Med 2010;207:2975–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev 1999;79:181–213. [DOI] [PubMed] [Google Scholar]
- 8.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol 2004;22:129–156. [DOI] [PubMed] [Google Scholar]
- 9.Mitoma J, Bao X, Petryanik B, Schaerli P, Gauguet JM, Yu SY, Kawashima H, Saito H, Ohtsubo K, Marth JD, Khoo KH, von Andrian UH, Lowe JB, Fukuda M. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol 2007;8:409–418. [DOI] [PubMed] [Google Scholar]
- 10.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011;118:6743–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nimrichter L, Burdick MM, Aoki K, Laroy W, Fierro MA, Hudson SA, Von Seggern CE, Cotter RJ, Bochner BS, Tiemeyer M, Konstantopoulos K, Schnaar RL. E-selectin receptors on human leukocytes. Blood 2008;112:3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature 2003;423:190–193. [DOI] [PubMed] [Google Scholar]
- 13.Sarangapani KK, Yago T, Klopocki AG, Lawrence MB, Fieger CB, Rosen SD, McEver RP, Zhu C. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J Biol Chem 2004;279:2291–2298. [DOI] [PubMed] [Google Scholar]
- 14.Wayman AM, Chen W, McEver RP, Zhu C. Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys J 2010;99:1166–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yago T, Wu J, Wey CD, Klopocki AG, Zhu C, McEver RP. Catch bonds govern adhesion through L-selectin at threshold shear. J Cell Biol 2004;166:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramachandran V, Williams M, Yago T, Schmidtke DW, McEver RP. Dynamic alterations of membrane tethers stabilize leukocyte rolling on P-selectin. Proc Natl Acad Sci USA 2004;101:13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidtke DW, Diamond SL. Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J Cell Biol 2000;149:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, Groisman A, Ley K. ‘Slings’ enable neutrophil rolling at high shear. Nature 2012;488:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorant DE, Patel KD, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Coexpression of GMP-140 and PAF by endothelium stimulated by histamine or thrombin: a juxtacrine system for adhesion and activation of neutrophils. J Cell Biol 1991;115:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorant DE, Topham MK, Whatley RE, McEver RP, McIntyre TM, Prescott SM, Zimmerman GA. Inflammatory roles of P-selectin. J Clin Invest 1993;92:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-α secretion. J Clin Invest 1995;95:2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest 1996;97:1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hidari KI-PJ, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem 1997;272:28750–28756. [DOI] [PubMed] [Google Scholar]
- 24.Ley K, Bullard DC, Arbonés ML, Bosse R, Vestweber D, Tedder TF, Beaudet AL. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. J Exp Med 1995;181:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunkel EJ, Ley K. Distinct phenotype of E-selectin-deficient mice—E-selectin is required for slow leukocyte rolling in vivo. Circ Res 1996;79:1196–1204. [DOI] [PubMed] [Google Scholar]
- 26.Jung U, Norman KE, Scharffetter-Kochanek K, Beaudet AL, Ley K. Transit time of leukocytes rolling through venules controls cytokine- induced inflammatory cell recruitment in vivo. J Clin Invest 1998;102:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lefort CT, Ley K. Neutrophil arrest by LFA-1 activation. Front Immunol 2012;3:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced aLb2 integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 2007;26:773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol 2007;25:619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol 2012;24:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 2010;116:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 2009;27:339–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, Coggeshall KM, McEver RP. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin αLβ2-mediated slow leukocyte rolling. Blood 2010;116:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005;106:1604–1611. [DOI] [PubMed] [Google Scholar]
- 35.Miner JJ, Xia L, Yago T, Kappelmayer J, Liu Z, Klopocki AG, Shao B, McDaniel JM, Setiadi H, Schmidtke DW, McEver RP. Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood 2008;112:2035–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neame SJ, Uff CR, Sheikh H, Wheatley SC, Isacke CM. CD44 exhibits a cell type dependent interaction with triton X-100 insoluble, lipid rich, plasma membrane domains. J Cell Sci 1995;108:3127–3135. [DOI] [PubMed] [Google Scholar]
- 37.Ma YQ, Plow EF, Geng JG. P-selectin binding to P-selectin glycoprotein ligand-1 induces an intermediate state of aMb2 activation and acts cooperatively with extracellular stimuli to support maximal adhesion of human neutrophils. Blood 2004;104:2549–2556. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Yago T, Zhang N, Abdisalaam S, Alexandrakis G, Rodgers W, McEver RP. Cytoskeletal regulation of CD44 membrane organization and interactions with E-selectin. J Biol Chem 2014;289:35159–35171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shao B, Yago T, Coghill PA, Klopocki AG, Mehta-D'souza P, Schmidtke DW, Rodgers W, McEver RP. Signal-dependent slow leukocyte rolling does not require cytoskeletal anchorage of P-selectin glycoprotein ligand-1 (PSGL-1) or integrin αLβ2. J Biol Chem 2012;287:19585–19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michelson AD, Barnard MR, Hechtman HB, MacGregor H, Connolly RJ, Loscalzo J, Valeri CR. In vivo tracking of platelets: circulating degranulated platelets rapidly lose surface P-selectin but continue to circulate and function. Proc Natl Acad Sci USA 1996;93:11877–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dole VS, Bergmeier W, Patten IS, Hirahashi J, Mayadas TN, Wagner DD. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb Haemost 2007;98:806–812. [PubMed] [Google Scholar]
- 42.Bodary PF, Homeister JW, Vargas FB, Wickenheiser KJ, Cudney SS, Bahrou KL, Ohman M, Rabbani AB, Eitzman DT. Generation of soluble P- and E-selectins in vivo is dependent on expression of P-selectin glycoprotein ligand-1. J Thromb Haemost 2007;5:599–603. [DOI] [PubMed] [Google Scholar]
- 43.Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J 2003;24:2166–2179. [DOI] [PubMed] [Google Scholar]
- 44.Hartwell DM, Mayadas TN, Berger G, Frenette PS, Rayburn H, Hynes RO, Wagner DD. Role of P-selectin cytoplasmic domain in granular targeting in vivo and in early inflammatory responses. J Cell Biol 1998;143:1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.André P, Hartwell D, Hrachovinová I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci USA 2000;97:13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woollard KJ, Kling D, Kulkarni S, Dart AM, Jackson S, Chin-Dusting J. Raised plasma soluble P-selectin in peripheral arterial occlusive disease enhances leukocyte adhesion. Circ Res 2006;98:149–156. [DOI] [PubMed] [Google Scholar]
- 47.Ushiyama S, Laue TM, Moore KL, Erickson HP, McEver RP. Structural and functional characterization of monomeric soluble P-selectin and comparison with membrane P-selectin. J Biol Chem 1993;268:15229–15237. [PubMed] [Google Scholar]
- 48.Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med 2008;205:2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, Huo Y, Zhu X, Plow EF, Chen M, Geng JG. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol 2007;8:882–892. [DOI] [PubMed] [Google Scholar]
- 50.Thorne RF, Legg JW, Isacke CM. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J Cell Sci 2004;117:373–380. [DOI] [PubMed] [Google Scholar]
- 51.Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol 2005;26:208–214. [DOI] [PubMed] [Google Scholar]
- 52.Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood 2010;115:3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Block H, Herter JM, Rossaint J, Stadtmann A, Kliche S, Lowell CA, Zarbock A. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J Exp Med 2012;209:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, Van Aken H, Wagner DD, Laudanna C, Ley K, Zarbock A. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur J Immunol 2011;41:2074–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem 2009;284:5119–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood 2013;121:2301–2310. [DOI] [PubMed] [Google Scholar]
- 57.Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, Lefort C, Fisher CI, Buscher K, Gelschefarth B, Urzainqui A, Gerke V, Ley K, Zarbock A. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med 2013;210:2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol 2011;10:10. [DOI] [PubMed] [Google Scholar]
- 59.Ye F, Snider AK, Ginsberg MH. Talin and kindlin: the one-two punch in integrin activation. Front Med 2014;8:6–16. [DOI] [PubMed] [Google Scholar]
- 60.Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Ann Rev Biophys 2009;38:235–254. [DOI] [PubMed] [Google Scholar]
- 61.Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, Critchley DR, Ginsberg MH, Fassler R, Ley K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 2012;119:4275–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-Friede A, Shattil SJ, Ruppert R, Moser M, Fassler R, Ginsberg MH. The mechanism of kindlin-mediated activation of integrin αIIbβ3. Curr Biol 2013;23:2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pruenster M, Kurz ARM, Chung K-J, Cao-Ehlker X, Bieber S, Nussbaum CF, Bierschenk S, Eggersmann TK, Rohwedder I, Heinig K, Immler R, Moser M, Koedel U, Gran S, McEver RP, Vestweber D, Verschoor A, Leandersson T, Chavakis T, Roth J, Vogl T, Sperandio M. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun 2015;6:6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miner JJ, Shao B, Wang Y, Chichili GR, Liu Z, Klopocki AG, Yago T, McDaniel JM, Rodgers W, Xia L, McEver RP. Cytoplasmic domain of P-selectin glycoprotein ligand-1 facilitates dimerization and export from the endoplasmic reticulum. J Biol Chem 2011;286:9577–9586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, Totani L, Piccardoni P, Vestweber D, de Gaetano G, Cerletti C. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood 1999;93:876–885. [PubMed] [Google Scholar]
- 66.Cerletti C, Evangelista V, De Gaetano G. P-selectin-b-integrin cross-talk: a molecular mechanism for polymorphonuclear leukocyte recruitment at the site of vascular damage. Thromb Haemost 1999;82:787–793. [PubMed] [Google Scholar]
- 67.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the β2-integrin CD11b/CD18. Blood 1996;88:146–157. [PubMed] [Google Scholar]
- 68.Simon DI, Chen ZP, Xu H, Li CQ, Dong JF, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, López JA. Platelet glycoprotein Iba is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 2000;192:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piccardoni P, Sideri R, Manarini S, Piccoli A, Martelli N, De Gaetano G, Cerletti C, Evangelista V. Platelet/polymorphonuclear leukocyte adhesion: a new role for SRC kinases in Mac-1 adhesive function triggered by P-selectin. Blood 2001;98:108–116. [DOI] [PubMed] [Google Scholar]
- 70.Evangelista V, Manarini S, Coller BS, Smyth SS. Role of P-selectin, beta2-integrins, and Src tyrosine kinases in mouse neutrophil-platelet adhesion. J Thromb Haemost 2003;1:1048–1054. [DOI] [PubMed] [Google Scholar]
- 71.Selak MA, Chignard M, Smith JB. Cathepsin G is a strong platelet agonist released by neutrophils. Biochem J 1988;251:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L, Varki A, McEver RP. Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood 2011;118:4015–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 2010;107:15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost 2005;3:1590–1596. [DOI] [PubMed] [Google Scholar]
- 75.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012;209:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruehl RE, Moore KL, Lorant DE, Borregaard N, Zimmerman GA, McEver RP, Bainton DF. Leukocyte activation induces surface redistribution of P-selectin glycoprotein ligand-1. J Leukoc Biol 1997;61:489–499. [DOI] [PubMed] [Google Scholar]
- 77.Serrador JM, Nieto M, Alonso-Lebrero JL, Del Pozo MA, Calvo J, Furthmayr H, Schwartz-Albiez R, Lozano F, González-Amaro R, Sánchez-Mateos P, Sánchez-Madrid F. CD43 interacts with moesin and ezrin and regulates its redistribution to the uropods of T lymphocytes at the cell-cell contacts. Blood 1998;91:4632–4644. [PubMed] [Google Scholar]
- 78.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 2009;15:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nacher M, Pitaval C, Radovanovic I, Fukui Y, McEver RP, Filippi MD, Lizasoain I, Ruiz-Cabello J, Zarbock A, Moro MA, Hidalgo A. Neutrophils scan for activated platelets to initiate inflammation. Science 2014;346:1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. Combined role of P- and E-selectins in atherosclerosis. J Clin Invest 1998;102:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med 2000;191:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation 2008;117:3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thiagarajan RR, Winn RK, Harlan JM. The role of leukocyte and endothelial adhesion molecules in ischemia–reperfusion injury. Thromb Haemost 1997;78:310–314. [PubMed] [Google Scholar]
- 84.Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, Celi A, Croce K, Furie BC, Furie B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med 2003;197:1585–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wakefield TW, Myers DD, Henke PK. Role of selectins and fibrinolysis in VTE. Thromb Res 2009;123(Suppl 4):S35–S40. [DOI] [PubMed] [Google Scholar]
- 86.Totani L, Evangelista V. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol 2010;30:2357–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mertens P, Maes A, Nuyts J, Belmans A, Desmet W, Esplugas E, Charlier F, Figueras J, Sambuceti G, Schwaiger M, Mortelmans L, Van de Werf F. Recombinant P-selectin glycoprotein ligand-immunoglobulin, a P-selectin antagonist, as an adjunct to thrombolysis in acute myocardial infarction. The P-Selectin Antagonist Limiting Myonecrosis (PSALM) trial. Am Heart J 2006;152:125 e121–128. [DOI] [PubMed] [Google Scholar]
- 88.Tardif JC, Tanguay JF, Wright SS, Duchatelle V, Petroni T, Gregoire JC, Ibrahim R, Heinonen TM, Robb S, Bertrand OF, Cournoyer D, Johnson D, Mann J, Guertin MC, L'Allier PL. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-ST-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol 2013;61:2048–2055. [DOI] [PubMed] [Google Scholar]
- 89.Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am 2014;28:181–198. [DOI] [PubMed] [Google Scholar]
- 90.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood 2001;98:1955–1962. [DOI] [PubMed] [Google Scholar]
- 91.Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am J Physiol Heart Circ Physiol 2004;286:H1608–H1614. [DOI] [PubMed] [Google Scholar]
- 92.Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, Diwan BA, Mohandas N, Cheung AT. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood 2004;104:3378–3385. [DOI] [PubMed] [Google Scholar]
- 93.Gutsaeva DR, Parkerson JB, Yerigenahally SD, Kurz JC, Schaub RG, Ikuta T, Head CA. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood 2011;117:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010;116:1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Telen MJ, Wun T, McCavit TL, De Castro LM, Krishnamurti L, Lanzkron S, Hsu LL, Smith WR, Rhee S, Magnani JL, Thackray H. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015;125:2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]