

Abstract
Background
Ataxia with oculomotor apraxia type 1 is an autosomal-recessive neurodegenerative disorder characterized by a childhood onset of slowly progressive cerebellar ataxia, followed by oculomotor apraxia and a severe primary motor peripheral axonal motor neuropathy. Ataxia with oculomotor apraxia type 1 is caused by bi-allelic mutations in APTX (chromosome 9p21.1).
Case presentation
Our patient has a clinical presentation that is typical for ataxia with oculomotor apraxia type 1 with no particularly severe phenotype. Multiplex Ligation-dependent Probe Amplification analysis resulted in the identification of a homozygous deletion of all coding APTX exons (3 to 9). SNP array analysis using the Illumina Infinium CytoSNP-850 K microarray indicated that the deletion was about 62 kb. Based on the SNP array results, the breakpoints were found using direct sequence analysis: c.-5 + 1225_*44991del67512, p.0?. Both parents were heterozygous for the deletion. Homozygous complete APTX deletions have been described in literature for two other patients. We obtained a sample from one of these two patients and characterized the deletion (156 kb) as c.-23729_*115366del155489, p.0?, including the non-coding exons 1A and 2 of APTX. The more severe phenotype reported for this patient is not observed in our patient. It remains unclear whether the larger size of the deletion (156 kb vs 62 kb) plays a role in the phenotype (no extra genes are deleted).
Conclusion
Here we described an ataxia with oculomotor apraxia type 1 patient who has a homozygous deletion of the complete coding region of APTX. In contrast to the patient with the large deletion, our patient does not have a severe phenotype. More patients with deletions of APTX are required to investigate a genotype-phenotype effect.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0213-y) contains supplementary material, which is available to authorized users.
Keywords: APTX, Ataxia with oculomotor apraxia type 1, Deletion, Homozygous, MLPA, SNP array analysis, Breakpoint mapping, Genetic testing
Background
Ataxia with oculomotor apraxia type 1 (AOA1) is an autosomal-recessive neurodegenerative disorder mainly characterized by a childhood onset of slowly progressive cerebellar ataxia, oculomotor apraxia, dysarthria, limb dysmetria, motor and sensory axonal neuropathy [1–3]. Clinical symptoms can also include dystonia, chorea, optic atrophy and cognitive impairment. AOA1 is caused by homozygosity or compound heterozygosity for mutations in APTX, the gene that encodes the protein Aprataxin [4, 5]. Aprataxin is a member of the histidine triad (HIT) superfamily and plays a role in DNA-single-strand break repair [6–12]. The pathological mechanism leading to the neurodegenerative phenotype, as observed for AOA1 patients, is still unknown.
In the current report we describe the extensive clinical and molecular genetic testing of an AOA1 patient who is homozygous for a complete APTX deletion rather than compound heterozygous for point mutations in APTX as is normally found in AOA1 patients. We also introduce the Leiden Open (source) Variation Database for APTX mutations.
Case presentation
Case report
Our index patient is the first child of healthy non-consanguineous Moroccan parents. He has two healthy brothers and parents refer no family history of interest. He was born after a normal gestation and both delivery and neonatal period were unremarkable. He started walking around 12 months and his parents considered his language development normal during the first years. At age 3 years, his parents started to notice abnormal ocular movements, walking disorder with frequent falls and language difficulties. He was clinically diagnosed with cerebellar ataxia. Cranial Computed Tomography and cranial Magnetic Resonance imaging (MRI) scans showed cerebellar atrophy (for MRI scan see Additional file 1). At age 6 years the child was referred to the Neuropaediatrics unit of the Sabadell university hospital in Spain. Neurological exam showed oculomotor apraxia, ataxic gait and dysmetria. Furthermore, he also presented distal limb dystonia, bilateral spontaneous Babinski and rotulian and achillean hyporeflexia. Blood levels of albumin, cholesterol, immunoglobulins, alpha-fetoprotein and carcinoembryonic antigen were all normal. Vitamin E levels at age 7 years, however, were low (repeated measurements: 1.3 μg/ml and 0.4 μg/ml; normal range 3–15 μg/ml). Because of the low vitamin E levels, molecular testing of the gene involved in vitamin E deficiency, TTPA, was performed [13]. No abnormalities were observed. Vitamin E levels returned to normal (11.7 μg/ml) after oral vitamin E treatment. Results of the Brainstem Auditory Evoked Response (BAER) test and the ophthalmologic and cardiological assessments were all normal. Electromyography (EMG) at age 7 years showed a discrete reduction of the sural sensory nerve action potential (SNAP) amplitude. At age 9 years the EMG results were in agreement with sensitive axonal polyneuropathy. No abnormalities were found in the gene involved in Ataxia-telangiectasia, ATM [14]. Finally, molecular testing of APTX was performed.
APTX sequencing and MLPA
All seven coding exons (exons 3 to 9B) and exon/intron boundaries of APTX (NM_175073.2, isoform a, GRCh build 37 (UCSC hg19, February 2009)) were screened using direct sequence analysis (primers available upon request). Although we have previously used this setup to test for mutations in APTX (n = 158 index patients tested, 17 different APTX variants found (for 9 novel APTX variants see Additional file 2), also see http://www.lovd.nl/aptx, in total 45 APTX sequence variants), we were unable to amplify all exons, raising the possibility that APTX was not present on both alleles (homozygous deletion) of our patient. Therefore, the SALSA Multiplex Ligation-dependent Probe Amplification (MLPA) kit P316 (MRC Holland, Amsterdam, The Netherlands) was used to detect large deletions in APTX. MLPA analysis was performed according to manufacturer’s instructions. MLPA products were run on an ABI 3730XL automated sequencer (Applied Biosystems, Foster City, CA, USA) and data was analyzed using Genemarker software version 2.4 (Softgenetics, State College, PA, USA). MLPA analysis resulted in the identification of a deletion of exons 3 to 9. The non-coding exons 1 and 2 were not deleted. The peak ratio was clearly zero instead of 0.5, indicating that the deletion was homozygous and not heterozygous. The MLPA results were in agreement with the sequence analysis findings.
SNP array analysis
SNP array analysis was performed to confirm the MLPA finding and to further characterize the deletion. The Illumina Infinium CytoSNP-850 K microarray was used in combination with the Illumina platform and the Nexus Copy Number 7.0 software (BioDiscovery, El Segundo, CA, USA) according to standard protocols of the manufacturer. A deletion of 62 kb between positions 32.932.194 and 32.994.500 on chromosome chr9p21.1 (NCBI 37; UCSC hg19) was found (see Fig. 1a). Both the Log2ratio and the b-allele frequency (not shown in Fig. 1) indicate that the deletion is present in homozygous form. The deletion is in agreement with the MLPA results.
Fig. 1.
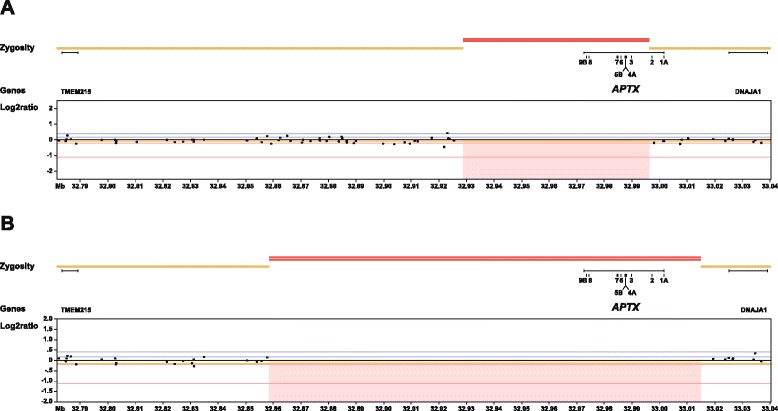
Results SNP array analysis. Indicated in the figure are the zygosity (heterozygous: yellow bars; homozygous: red bars) and Log2Ratio (reds square: deleted region based on Log2Ratio) tracks, the locations of APTX and surrounding genes on chromosome 9p21.1 (NCBI 37; UCSC hg19), and the exon numbering of APTX for our index patient (a) and the reference sample of Dr. Yoon et al. (b)
Breakpoint mapping
Breakpoint mapping of the deletion was performed to characterize the exact breakpoints of the deletion. Based on the SNP array results, the breakpoints were found using direct sequence analysis. The exact nomenclature of the APTX deletion is c.-5 + 1225_*44991del67512, p.0?. A breakpoint specific PCR was developed (primers available upon request, for more details see reference [15]), confirming the homozygous occurrence of the deletion in the index patient. Both parents were heterozygous for the c.-5 + 1225_*44991del67512, p.0? deletion and were therefore identified as carriers of AOA1.
Discussion
Most of the AOA1 patients are homozygous or compound heterozygous for a pathogenic point mutation in APTX. In the present report we describe a AOA1 patient who has a homozygous deletion of the complete coding region of APTX. Homozygous APTX deletions have been previously reported in a Tunisian family and a Pakistani patient [16, 17]. These deletions were found using southern blotting, after sequence fragments of all exons failed to amplify. Confirmation and characterization with more modern techniques like MLPA analysis and array analysis was not performed. Because their molecular analyses also lack breakpoint mapping we could not check whether our APTX deletion is the exactly the same deletion. We were however able to obtain a DNA sample of the patient of Yoon et al. SNP array analysis and breakpoint mapping, performed as described above, indicated a 156 kb APTX deletion,
c.-23729_*115366del155489, p.0? (APTX, exon 01A tm 09B), including the two non-coding APTX exons 1A and 2 (also see Fig. 1b).
The more severe phenotype reported for the patient of Yoon et al. [17], including fast deterioration and cognitive impairment, is not observed in our patient. It remains unclear whether the larger size of the deletion (156 kb vs 62 kb) plays a role in the phenotype. No extra genes are deleted, however, the two non-coding APTX exons 1 and 2B are deleted in the patient of Yoon et al. (also see Fig. 1). The function of these two exons is not studied. It is possible that this region contains regulatory elements that play in role in the transcription of (distance) genes, other than APTX, that might explain the more severe phenotype. Furthermore, a possible role of the different APTX transcripts in the AOA1 phenotype is not studied to date. Even though the predominant transcript in human tissues, NM_175073.2, contains both non-coding exons, several APTX transcripts exist that lack exons 1 and/or 2B [18]. Finally, a third option explaining the difference in phenotype between our patient and the patient of Yoon et al. is coincidence rather than a genetic feature. So far, genotype-phenotype correlations were only studied for a limited set of APTX point mutations [11, 12, 19]. More patients with deletions of APTX are required to investigate a genotype-phenotype effect. We were unfortunately unable to contact Amouri et al. to request a DNA sample [16]. Therefore, the size of the APTX deletion is unknown for their patient, neither do we know whether the two non-coding APTX exons are deleted in their patient. This is especially unsatisfactory because the typical AOA1 clinical presentation of our patient is similar to that reported for the patient of Amouri et al. [16].
To our knowledge, a database with APTX genetic variants was not available when writing this report. Because databases can be helpful in classifying variants we have launched a APTX database in LOVD (Leiden Open Variation Database) format [20]. All our mutations, including the total APTX deletion, and some well-known variants from literature were deposited in this database (accessible at http://www.lovd.nl/aptx). We will continue to update this database with new variants and encourage other APTX diagnostic labs to do the same.
Conclusions
In conclusion, here we described the extensive clinical and genetic analysis of a AOA1 patient homozygous for a complete deletion of the coding exons of APTX. Our patient has a typical AOA1 clinical presentation without a severe phenotype. More patients with clearly characterized APTX deletions are required to investigate a genotype-phenotype effect.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal
Acknowledgements
The authors would like to thank the index patient and his parents for participating in this case report. In addition, the authors thank Tom de Vries Lentsch of the Clinical Genetics Department of Erasmus Medical Center in Rotterdam for technical assistance with the figure.
Abbreviations
- AOA1
Ataxia with oculomotor apraxia type 1
- BAER
Brainstem auditory evoked response
- EMG
Electromyography
- GTC
Genotyping console
- MLPA
Multiplex ligation-dependent probe amplification
- LOH
Loss of heterozygosity
- LOVD
Leiden open variation database
- SNAP
Sensory nerve action potential
- QC
Quality control
Additional files
MRI scan of index patient(white arrow). (PDF 307 kb)
Novel APTX variants. (PDF 65 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MG and CE carried out the clinical characterization of the index patient and his parents. RvdG, GB, RvdH and PE performed the molecular genetic analyses. GY provided a reference sample. RvM and AvdO performed the interpretation of the molecular genetic data and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Rick van Minkelen, Phone: 31 107043197, Email: r.vanminkelen@erasmusmc.nl.
Miriam Guitart, Email: MGuitart@tauli.cat.
Conxita Escofet, Email: CEscofet@tauli.cat.
Grace Yoon, Email: grace.yoon@utoronto.ca.
Peter Elfferich, Email: p.elfferich@erasmusmc.nl.
Galhana M. Bolman, Email: g.bolman@erasmusmc.nl
Robert van der Helm, Email: r.vanderhelm@erasmusmc.nl.
Raoul van de Graaf, Email: r.vandegraaf@erasmusmc.nl.
Ans M.W. van den Ouweland, Email: a.vandenouweland@erasmusmc.nl
References
- 1.Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126(Pt 12):2761–72. doi: 10.1093/brain/awg283. [DOI] [PubMed] [Google Scholar]
- 2.Tranchant C, Fleury M, Moreira MC, Koenig M, Warter JM. Phenotypic variability of aprataxin gene mutations. Neurology. 2003 Mar 11;60(5):868-70. [DOI] [PubMed]
- 3.Castellotti B, Mariotti C, Rimoldi M, Fancellu R, Plumari M, Caimi S, et al. Ataxia with oculomotor apraxia type1 (AOA1): novel and recurrent aprataxin mutations, coenzyme Q10 analyses, and clinical findings in Italian patients. Neurogenetics. 2011;12(3):193–201. [DOI] [PubMed]
- 4.Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nature genetics. 2001;29(2):184–8. [DOI] [PubMed]
- 5.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189–93. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 6.Hirano M, Yamamoto A, Mori T, Lan L, Iwamoto TA, Aoki M, et al. DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann Neurol. 2007;61(2):162–74. doi: 10.1002/ana.21078. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds JJ, El-Khamisy SF, Katyal S, Clements P, McKinnon PJ, Caldecott KW. Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia 1. Mol Cell Biol. 2009;29(5):1354–62. doi: 10.1128/MCB.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443(7112):713–6. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, Tada M, Igarashi S, Koyama A, Date H, Yokoseki A, et al. Aprataxin, causative gene product for EAOH/AOA1, repairs DNA single-strand breaks with damaged 3'-phosphate and 3'-phosphoglycolate ends. Nucleic Acids Res. 2007;35(11):3797–809. doi: 10.1093/nar/gkm158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris JL, Jakob B, Taucher-Scholz G, Dianov GL, Becherel OJ, Lavin MF. Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum Mol Genet. 2009;18(21):4102–17. doi: 10.1093/hmg/ddp359. [DOI] [PubMed] [Google Scholar]
- 11.Mosesso P, Piane M, Palitti F, Pepe G, Penna S, Chessa L. The novel human gene aprataxin is directly involved in DNA single-strand-break repair. Cell Mol Life Sci. 2005;62(4):485–91. doi: 10.1007/s00018-004-4441-0. [DOI] [PubMed] [Google Scholar]
- 12.Gueven N, Becherel OJ, Kijas AW, Chen P, Howe O, Rudolph JH,et al. Aprataxin, a novel protein that protects against genotoxic stress. Hum Mol Genet. 2004;13(10):1081–93. [DOI] [PubMed]
- 13.Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet. 1995;9(2):141–5. [DOI] [PubMed]
- 14.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 15.van den Ouweland AM, Elfferich P, Lamping R, van de Graaf R, van Veghel-Plandsoen MM, Franken SM, et al. Identification of a large rearrangement in CYLD as a cause of familial cylindromatosis. Fam Cancer. 2011;10(1):127–32. doi: 10.1007/s10689-010-9393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amouri R, Moreira MC, Zouari M, El Euch G, Barhoumi C, Kefi M, et al. Aprataxin gene mutations in Tunisian families. Neurology. 2004;63(5):928–9. doi: 10.1212/01.WNL.0000137044.06573.46. [DOI] [PubMed] [Google Scholar]
- 17.Yoon G, Westmacott R, MacMillan L, Quercia N, Koutsou P, Georghiou A, et al. Complete deletion of the aprataxin gene: ataxia with oculomotor apraxia type 1 with severe phenotype and cognitive deficit. J Neurol Neurosurg Psychiatry. 2008;79(2):234–6. [DOI] [PubMed]
- 18.Sano Y, Date H, Igarashi S, Onodera O, Oyake M, Takahashi T, et al. Aprataxin, the causative protein for EAOH is a nuclear protein with a potential role as a DNA repair protein. Ann Neurol. 2004;55(2):241–9. doi: 10.1002/ana.10808. [DOI] [PubMed] [Google Scholar]
- 19.Yokoseki A, Ishihara T, Koyama A, Shiga A, Yamada M, Suzuki C, et al. Genotype-phenotype correlations in early onset ataxia with ocular motor apraxia and hypoalbuminaemia. Brain. 2011;134(Pt 5):1387–99. doi: 10.1093/brain/awr069. [DOI] [PubMed] [Google Scholar]
- 20.Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT, et al. LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–63. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]