

Abstract
The Brain and Body Donation Program (BBDP) at Banner Sun Health Research Institute (http://www.brainandbodydonationprogram.org) started in 1987 with brain-only donations and currently has banked more than 1600 brains. More than 430 whole-body donations have been received since this service was commenced in 2005. The collective academic output of the BBDP is now described as the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND). Most BBDP subjects are enrolled as cognitively normal volunteers residing in the retirement communities of metropolitan Phoenix, Arizona. Specific recruitment efforts are also directed at subjects with Alzheimer’s disease, Parkinson’s disease and cancer. The median age at death is 82. Subjects receive standardized general medical, neurological, neuropsychological and movement disorders assessments during life and more than 90% receive full pathological examinations by medically licensed pathologists after death. The Program has been funded through a combination of internal, federal and state of Arizona grants as well as user fees and pharmaceutical industry collaborations. Subsets of the Program are utilized by the US National Institute on Aging Arizona Alzheimer’s Disease Core Center and the US National Institute of Neurological Disorders and Stroke National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders. Substantial funding has also been received from the Michael J. Fox Foundation for Parkinson’s Research. The Program has made rapid autopsy a priority, with a 3.0-hour median postmortem interval for the entire collection. The median RNA Integrity Number (RIN) for frozen brain and body tissue is 8.9 and 7.4, respectively. More than 2500 tissue requests have been served and currently about 200 are served annually. These requests have been made by more than 400 investigators located in 32 US states and 15 countries. Tissue from the BBDP has contributed to more than 350 publications and more than 200 grant-funded projects.
Keywords: aging, Alzheimer’s disease, autopsy, biobank, biospecimen, brain bank, cancer, freeze-thaw, Parkinson’s disease, pathology, post-mortem interval, RNA
INTRODUCTION
Purpose and mission
The purpose of brain banks, and tissue, biospecimen or biobanks in general, is at least twofold. First, autopsy and neuropathological examination allow an accurate diagnosis and hence accurate clinicopathological correlation studies. As clinical syndromes generated by most diseases are not sufficiently specific regarding the underlying pathological processes, pathological examination of diseased tissue is often the only means by which a definite diagnosis can be attained. For neurodegenerative diseases, as biopsies are generally not done, neuropathological examination after death has an even more critical role. Accurate diagnoses ensure that clinical data acquired during life may be appropriately matched to a particular diagnostic condition, providing, over time and with sufficient numbers of studied subjects, a reliable categorization that may be used to collect and enhance knowledge regarding etiology, pathogenesis, epidemiology, biomarkers, clinical characteristics, clinical diagnosis, prognosis and treatment effects.
The second major purpose of biobanks is to provide pathologically characterized control and diseased tissue to basic scientists, enabling them to discover the underlying molecular mechanisms specific to each disease and to design appropriate therapeutic interventions. A detailed understanding of the genetic and molecular processes of disease pathogenesis, obtained by comparative study of diseased and non-diseased tissue, remains the major approach to finding such interventions. It is noteworthy that for both Alzheimer’s and Parkinson’s diseases, the existing approved pharmacotherapies are all based on molecular alterations identified wholly or partially from study of post-mortem brains more than 30 years ago.1
The study population and research site
The primary population under study consists of the retirement communities of northwest greater Phoenix, especially Sun City, Sun City West and Sun City Grand. A profile of these communities has been constructed using data from the US Census Bureau (2010) and from the files of the Sun Cities Area Historical Society. The combined population of Sun City, Sun City West and Sun City Grand is approximately 80 000, with a median age of about 70 years. Sun City, established in 1960, was the first large, planned retirement community in the United States. Sun City West and Sun City Grand followed, in 1978 and 1996, respectively. Due to a lack of densification and new land availability, the populations of all three communities have stopped increasing. The population of Sun City has been maintained at about 40 000 for more than 4 decades, suggesting that a continued influx of new retirees will maintain the populations of all of the communities. However, multiple new retirement communities have been developed in the immediate vicinity and throughout the metropolitan Phoenix area and many Brain and Body Donation Program (BBDP) study subjects now reside in these areas as well. Additionally, a subset of subjects is recruited from both metropolitan Phoenix and metropolitan Tucson through neurologists associated with the Arizona Alzheimer’s Disease Core Center.
Although migration of retirees to the region occurred initially in three major waves, coinciding with the establishment and marketing of housing developments in Sun City, Sun City West and Sun City Grand, the residents are all very similar, being composed overwhelmingly of elderly, well-educated, Caucasian (greater than 90%), middle and upper income individuals originating most commonly from Midwestern US states but with considerable influx from Northeast and Western states as well. The average residency time in Sun City is 12.6 years. As there is very little outmigration, the turnover is mostly due to deaths. Of the original Sun City population, 90% were replaced by a second generation of retirees by 1990.
Banner Sun Health Research Institute
Banner Sun Health Research Institute (BSHRI) is an affiliate of Banner Health, a non-profit, regional healthcare provider. Originally named the Institute for Biogerontology Research and then Sun Health Research Institute, BSHRI was conceived and initiated by Sun Health, a community owned and operated healthcare provider that began in 1965 to serve the needs of Sun City. Sun Health grew to provide healthcare to not only the original Sun City but also Sun City West and Sun City Grand as well as northwest greater Phoenix in general, all of which have experienced rapid growth of both retirement and non-retirement populations. Services provided by Sun Health included medical insurance, two acute care hospitals, extended and hospice care, outpatient clinics and Alzheimer care residences. Due to the increasing economic difficulties associated with providing healthcare to a predominantly elderly, Medicare-dependent population, Sun Health merged with Banner Health (http://www.bannerhealth.com) in 2008. Banner Health is larger and serves a more age-diversified population with 36 000 employees and 25 hospitals.
Sun Health Research Institute was established in 1986 with funds from Sun Health operations revenue and charitable contributions to the Sun Health Foundation. The focus of the Institute’s scientific work has been, since its inception, Alzheimer’s disease, with substantial Parkinson’s disease research beginning in 1996. Since 1998, the Institute has been part of the Arizona Alzheimer’s Consortium, a state-funded alliance that also includes Banner Alzheimer’s Institute, the University of Arizona, Barrow Neurological Institute, the Mayo Clinic Arizona, Arizona State University and Midwestern University. The Consortium was awarded a US National Institute on Aging (NIA) Alzheimer’s Disease Core Center (ADCC) in 2001 and a state-funded Arizona Parkinson’s Disease Consortium (APDC) was initiated in 2002. Mayo Clinic and BSHRI together were funded in 2011 by the US National Institute of Neurological Disorders and Stroke (NINDS) to form the National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders.
Origin and evolution of the program
The BBDP has been in existence since 1987 and has enrolled more than 3100 subjects over that time, which constitutes more than 2% of the current combined populations of the surrounding retirement communities. Of these, more than 1600 subjects have expired and their brains have been collected and stored. Whole-body donation was started in 2005, with more than 430 subjects currently accrued. Presently there are about 900 living subjects enrolled. All subjects have volunteered specifically for the Program and are highly motivated, with an annual drop-out rate of less than 3%. Recruitment relied initially on hospital staff, who obtained consents from family members of individuals who died while admitted. Recruitment gradually became entirely done on a prospective basis, primarily through public speaking events and tours of the Institute given by Institute staff to community groups and the general public, although interactions of the population with physicians and nursing staff belonging to the Sun Health and Banner Health provider networks, and relatively frequent local media releases, also contribute. Eligibility criteria for the Program stipulate that subjects must reside in the greater Phoenix metropolitan region (except for about 8% of subjects that have been enrolled for the Arizona NIA ADCC through the University of Arizona in Tucson). Recruitment is directed at subjects with a clinical diagnosis of Alzheimer’s disease (AD), Parkinson’s disease (PD) or cancer or who are free of other major neurological conditions. Subjects must be free of hazardous infectious diseases and must consent to annual clinical assessments. Additionally, applicants’ private medical records are reviewed to exclude confounding diseases and hazardous infectious disease conditions including HIV, hepatitis B or C, Creutzfeldt-Jakob disease and other infectious encephalopathies. In the absence of private medical records review, the enrolling physician must complete a questionnaire responding to the presence or absence of a history of these conditions. All enrolled subjects or legal representatives sign an Institutional Review Board-approved informed consent form allowing both clinical assessments during life, several options for brain and/or bodily organ donation after death, and usage of donated biospecimens for approved future research. A separate section requests the subject to allow or disallow DNA isolation and storage as well as genetic testing.
Between 1987 and 1995 brain donors did not receive formal neuropsychological testing. Their mental status was determined by requisitioning medical records from their primary care physicians, neurologists, psychologists and psychiatrists, and through telephone interviews with family members and caregivers, both at the time of enrollment and in the immediate post-mortem period. In 1996 a clinical psychologist was hired and from then onward, a standardized neuropsychological assessment has been administered to more than 75% of enrolled subjects (more than 90% of subjects since 2005). Also since 1996, most donors have received a standardized neurological evaluation tailored to detect overt and incipient movement disorders. A neuropathologist (TGB) was hired in 1997; prior to this time post-mortem diagnosis was performed by a retired general pathologist. In 2000, the clinical operations of the Program were greatly expanded, with the hiring of a cognitive neurologist, another neuropsychologist and associated support staff, while currently participating personnel include multiple professional staff including neurologists, neuropsychologists, psychiatrists, psychometrists, coordinators, nurses and schedulers. The collective academic output of the BBDP is now described as the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND).
Consideration of sampling issues
Subjects enrolled in the BBDP are not an accurate representative sample of the entire US elderly population, in that they lack racial diversity and are more highly educated, have higher incomes and live longer than the average US citizen. Although the study population lacks the diversity of the general US population, there are advantages of using a more homogeneous group. The use of homogeneous populations with minimal genetic and environmental variability decreases the subject number required to attain adequate statistical power. This has been recognized by many researchers and has been used, for example, by groups utilizing isolated populations to study inheritance.2–6 However, studying a more homogeneous group allows the possibility of missing disease characteristics resulting from alleles that may be enriched in some ethnic groups but not in others.
Autopsy studies, because of the requirement for subjects to volunteer for something that many people will not consider due to personal or religious feelings, cannot ever be considered to constitute a truly randomized population sample. At the very least, then, autopsy studies will be composed of a subset of the population that is more likely to volunteer for altruistic causes. This “volunteer bias” has been studied, and it is suggested, for example, that volunteers generally come from a higher socioeconomic background than non-volunteers.7–9 Volunteer bias may apply differentially to normal and diseased subjects, as normal subjects are almost always true volunteers while diseased subjects are often enrolled by family members after they have lost independent decision-making capacity. Despite these considerations, one study has shown that autopsied subjects do not differ appreciably from those that are not autopsied10 but this may differ with locale and methods of subject selection.
The BBDP is a longitudinal clinicopathological study of AD, PD and normal aging. The normal subjects are volunteers that are recruited from and reside in the surrounding community while the AD and PD subjects are recruited both from the community and from neurologists’ offices. While AD is so common that a population-based study of elderly subjects will obtain sufficient AD cases, PD is much less common, affecting only about 1% of the population over age 65, and so tens of thousands of normal older subjects would have to be autopsied in order to obtain an adequate number for clinical or post-mortem tissue studies. Other neurodegenerative diseases are even less common and for these, population-based sampling would yield negligible case numbers. Although the BBDP is not primarily concerned with studying conditions other than AD or PD, it is necessary to have adequate numbers of subjects with other neurodegenerative diseases so that a “disease control” group is available. For this reason, directed recruitment is necessary, and the study design of the BBDP is necessarily case-control in nature.
Governmental regulation of tissue donation after death
In the United States, as in much of the world, all medical research on human subjects is ethically and legally guided by the Declaration of Helskinki (http://www.wma.net/en/30publications/10policies/b3/). Other documents of historical importance include the Nuremburg Code and Declaration of Geneva. The principles of the Declaration of Helsinki are honored in the US legal system within the Code of Federal Regulations (CFR),11 where they are summarized as the Common Rule. The Common Rule is administered by the US Office of Human Research Protection, which exists to ensure that all human subject research will adhere to the written standards. The health information of human subjects is protected by the Health Insurance Portability and Accountability Act of 1996 (HIPAA). The BBDP performs annual standardized clinical assessments and has obtained the approval of our Institutional Review Board for these and all other aspects of the Program, including the informed consent and protocol. However, the Common Rule applies only to living human subjects and therefore the US federal government does not directly regulate research performed on deceased human subjects but rather places responsibility for such regulation on individual US states.12 Each US state has their own unique set of laws applicable to research usage of tissue from deceased subjects but there is commonality in that 48 states have adopted the most recent form of the Uniform Anatomical Gift Act (UAGA). The original UAGA, enacted in 1968, was directed at increasing the availability of donor organs for transplantation, but the 2006 revision (http://www.uniformlaws.org/ActSummary.aspx?title=Anatomical%20Gift%20Act%20(2006) expressly states that tissue donation may be made for the purposes of transplantation, therapy, research or education.13 An individual may sign an informed consent for post-mortem tissue donation while still alive, or this consent may be given after death by the spouse, adult children, parents, adult siblings, grandparents or legal representative, in hierarchical order. As rapid autopsy is a major objective for the BBDP, consent for autopsy and tissue donation is always obtained well prior to the subject’s death and subjects who are not able to come into our clinic for assessments are not accepted for enrollment. An important part of the UAGA is that a consent given during life does not need to be confirmed or re-acquired after death, as it would be almost impossible to rapidly obtain written consents from family members in these circumstances. Also, the UAGA prevents others, even family members, from revoking permission for tissue donation after death when the deceased individual had given their informed consent while alive. A summary of the UAGA on the official website (see above) states,“. . . there is no reason to seek consent from the donor’s family because the family has no legal right to revoke the gift. The practice of procurement organizations seeking affirmation even when the donor has clearly made a gift results in unnecessary delays in procuring organs and the occasional reversal of the donor’s wishes.” While the UAGA has cleared up many of the uncertainties and state-to-state variability that previously existed, US domestic law still does not provide explicit guidance on the handling or ownership of post-mortem human tissue.13
Clinical assessment
Standardized general medical, neurological and movement examinations as well as cognitive assessments and sleep and autonomic symptom questionnaires are administered annually to most subjects. Olfactory testing is performed every third year (Table 1). Standardized neuropsychological exams began in 1996, movement exams in 1997 and general neurological and extended cognitive exams in 2001. It is the objective to give all these assessments to all subjects regardless of whether they are considered normal controls or have dementia or parkinsonism. In the most recent 5-year period, at BSHRI alone there have been 2543 general neurological exams on 887 subjects, 2646 movement exams on 904 subjects, 2694 neuropsychological tests on 930 subjects, 2410 sleep questionnaires on 882 subjects, 2408 autonomic questionnaires on 881 subjects and 1120 (University of Pennsylvania Smell Identification Test (UPSIT) on 945 subjects. Clinical diagnostic classification is performed after each annual assessment, at a consensus conference attended by neurologists, psychiatrists and neuropsychologists. A final clinicopathological diagnosis is assigned after death, after review of all standardized clinical data, the most recent private medical records and neuropathological examination findings.
Table 1.
Clinical assessment batteries
| Neurological and functional battery | Neuropsychological battery | Movement disorders battery |
|---|---|---|
| General medical history, general medical examination | Rey Auditory Verbal Learning Test | UPDRS parts I–IV |
| Medication history | Stroop | Hoehn and Yahr scale |
| Substance usage (tobacco, ethanol) | Trails A and B | Timed tapping test14,15 |
| General neurological examination | Controlled Oral Word Association | Purdue Pegboard Test15,16 |
| MMSE | Judgment of Orientation | Tremor rating |
| GDS | Clock drawing | Restless legs syndrome score |
| FAST | Digits Forward and Backward | Mayo Clinic sleep questionnaire17 |
| Montreal Cognitive Assessment | Geriatric and Hamilton Depression Scales | SCOPA autonomic questionnaire18 |
| Mattis Dementia Scale | UPSIT smell test19 |
GDS, Global Deterioration Scale; FAST, Functional Assessment Testing; CDR, Clinical Dementia Rating Scale; MMSE, Mini Mental State Examination; UPDRS, Unified Parkinson’s Disease Rating Scale.
Although it is a requirement of the Program that donors consent to annual standardized clinical assessments, and considerable efforts are expended to have every subject assessed, some die without a standardized assessment. There are various reasons why not all subjects are assessed. Some subjects are in a terminal phase of illness when they register with the Program and die or become incapacitated before an assessment can be done, while others have difficulty scheduling an appointment for various reasons. A common impediment is a lack of someone to bring them in if they are not able to drive themselves. Home visits are done to help rectify this but these are limited as they add significant cost due to travel time. Some donors had enrolled in the Program prior to the time that standardized clinical assessments became mandatory (about 1997) and are therefore allowed to remain as brain-only donors. For normal control cases that die without a standardized BBDP clinical assessment, or whose last BBDP assessment was more than 18 months prior to death, an effort is made to obtain detailed information through telephone interviews with informants. These are standardized interviews that cover basic and instrumental activities of daily living, symptoms of cognitive impairment, movement abnormalities, and questions relating to sleep and autonomic functions. Questions are drawn from published scales such as the Clinical Dementia Rating Scale, Neuropsychiatric Inventory and Functional Activities Questionnaire.
Private medical records are obtained for all subjects and these are reviewed in a standardized manner. Generally the records from the two most recent years are obtained at initial enrollment and again after death. Of 1188 subjects with completed neuropathological examinations since 1997 (medically-licensed neuropathological examinations began in 1997), only 16 have no private medical records available. The median time span covered, from earliest to latest record date, is 70 months (5.8 years). After an initial “training period” in which all medical diagnoses were recorded, a total of 498 separate diagnoses were retained and a translation query written to condense all known synonyms for a given condition to a single “approved health condition”. We have found that review of private medical records is essential to gaining a full understanding of the subject’s overall medical history. The records receive multiple reviews by support staff, a neuropsychologist, a psychiatrist, a neurologist and the neuropathologist to ensure accuracy. Subjects also fill out medical history questionnaires at BBDP visits, and by comparing these to information obtained from review of private medical records, it is evident that self-report of medical conditions consistently under-reports health conditions, as compared to private medical records review.
A narrative summary of the clinical history is written after the subject’s death and is included in the neuropathology report, using all available clinical information, including results of BBDP standardized assessments, subject health questionnaires and private medical records.
Flow of subjects through the BBDP
The flow of subjects through the BBDP over the most recent 5-year period is illustrated in Figure 1. On August 1, 2009 there were 1009 living subjects enrolled in the BBDP. This initial input of subjects was supplemented by 511 new subjects recruited over the following 5 years, giving a total of 1520 subjects that were enrolled in the BBDP over this time period (“total subject input” in Fig. 1). Five years later, 896 subjects were still living and still enrolled while 199 had dropped out of the Program and 425 had died. Of those who died, four were not autopsied because the BBDP was not notified at the time of death. Of those autopsied, 357 (84%) had had one or more standardized BBDP clinical assessments during life.
Fig 1.
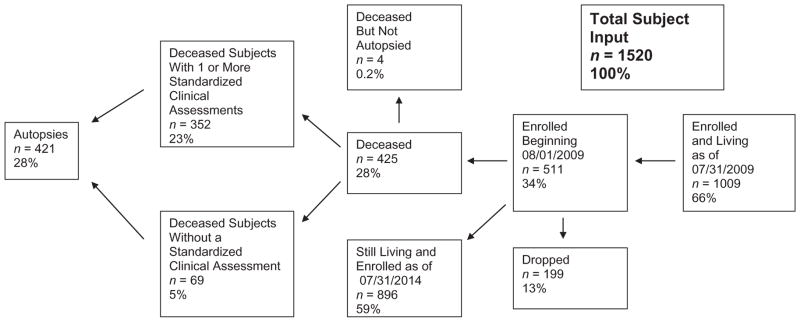
Autopsied subjects of the Brain and Body Donation Program (BBDP) 08/01/2009–07/30/2014. Percentages are calculated with respect to total subject input. Of autopsied subjects, 84% had one or more standardized clinical assessments.
Post-mortem operations
Dedication to short post-mortem interval (PMI)
Since the inception of the Program, the overriding organizing principle has been that the organ must be removed and processed as rapidly as possible. The median PMI, defined as the time elapsing between death and the start of organ removal, for all 1611 of our autopsied subjects, is 3.0 hours. There is a moderately extensive literature on the relationship between PMI and organ tissue quality, which will not be exhaustively summarized here, but it is apparent that deterioration of molecular entities after death varies widely depending on what is being measured. Highly volatile energy storage molecules such as ATP disappear within minutes,20 catecholamines drop precipitously14,15,21,22 and phosphorylated protein concentrations (“phosphoproteome”) fluctuate widely16 within the first few hours while high-molecular-weight DNA may be stable for up to 20 days after death.17 Previous research draws conflicting conclusions about the postmortem stability of RNA in organ tissue. Many report that post-mortem RNA is stable, using measures of global RNA integrity.18,19,23–32 Studies of various individual RNA species report mixed results with some indicating stability post-mortem30,33–39 while others have found associations between PMI and expression of various individual RNA transcripts.32,40–51 Variability in post-mortem preservation of different mRNA transcripts may be due to differing tertiary structures or normal physiological half-lives, which range from minutes to days. For example, c-fos mRNA has a half-life of 15 min while that for globin mRNA is 50 h.21
We analyzed52 total RNA extracted from frozen cerebellar cortex from 79 deceased BBDP subjects (see Fig. 2). The PMI was significantly correlated with overall RNA quality measures including RNA Integrity Number (RIN) (r = −0.34, P = 0.002) and RNA quantitative yield (r = −0.25, P = 0.02). Additionally, we determined the expression of 89 genes using a PCR-based gene expression array. A greater proportion of genes had decreased rather than increased expression with increasing PMI (65/89 vs 20/89; P < 0.0001). Of these, transcripts from the genes ADAM9, LPL, PRKCG and SERPINA3 had significantly decreased expression with increasing PMI (P < 0.01). No individual gene transcripts had significantly increased expression with increasing PMI. Broniscer et al.,53 using a RIN of 7.5 as a definition of “minimally acceptable RNA”, found that, of samples with a PMI of less than 5 h, 70% had acceptable RNA quality, as compared with only 21% of the samples from longer PMI autopsies. From a systematic sample of more than 865 BBDP subjects, the median RIN for organ tissue is 8.9, while for 329 samples from a variety of bodily organs and tissues, the median RIN is 7.4. For organ samples, more than 85% have a RIN greater than 7.5.
Fig 2.
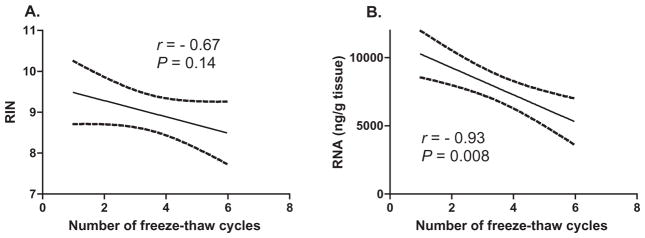
Correlation between the number of thawing cycles and (A) RNA Integrity Number (RIN) and (B) RNA yield, measured as the concentration (ng/μL) per tissue weight (g). Dashed lines show outlines of 95% confidence intervals.
These results and the brief literature review presented here indicate that a short PMI clearly allows optimal use of the resource but it is evident that all post-mortem studies must evaluate the influence of PMI as it is dependent on the specific molecular forms under investigation.
On-call autopsy teams
The maintenance of a rapid autopsy program requires sufficient staffing to prevent “burnout”. We are not aware of any other autopsy program that has had a similarly consistently short PMI. Most rapid autopsy programs fail because of understaffing that leads to burnout and resignation. Our experience has led to the conclusion that it is advisable to limit on-call periods for any one individual to no more than 4 months per year. The minimum number of persons required for each callout is two, for both safety and efficiency reasons. At present, we have four rotating on-call teams, each team consisting of two trained dissectors (one each for the brain and body, working in parallel) and two assistants. The trained dissectors are all BBDP employees and are on call every third month, while the assistants, derived from volunteers amongst the entire BSHRI scientific, technical and administrative staff, generally serve for 1–3 months per year. Trained dissectors and assistants are paid a bonus for each callout. As another strategy to avoid on-call team burnout, we continually optimize procedures to minimize tissue processing time. Generally we complete all procedures in 3–4 h and the total time a team member spends on a call, including transportation to and from their home, is between 3 and 5 h. To expedite organ processing, we always have made preparations for 2–3 autopsies into the future, prelabeling all containers and bags. Having a permanent dedicated autopsy suite enables all tools and equipment to be laid out ready for use.
Time-of-death telephone protocol and cadaver transport
When subjects registered into the Brain Donation Program die, their caregivers or family members, whether they are living at their own home (most subjects who die at home are registered in an approved hospice program) or at a care institution, follow the instructions given to them at the time of acceptance into the Program. The instructions are simple and require only that the caregiver telephone the Institute as soon as possible after death. If the call is after-hours, it is transferred to the operator at one of Banner Health’s acute care hospitals. The on-call autopsy team leader is then paged by the hospital operator and calls in to receive the contact information. The team leader then calls the contact, usually an employee of the care institution or hospice program, and conducts a short interview to ascertain the subject’s identity, confirm that they are registered in the Program, and record the subject’s current charted medical history conditions. The team leader also asks whether the subject has specific hazardous infectious diseases (see later section on safety for details). Subjects are transported to the BBDP morgue by a contracted commercial cadaver transport company. These transportation companies normally serve funeral homes on a 24-h basis and therefore are ideally suited for serving a rapid autopsy program. All charges are borne by the Program; donors and their families are not charged for any aspect relating to their clinical assessments or autopsy.
Blood and CSF draw, scalp and temporalis muscle sampling
Prior to removing the brain, CSF is drawn from the lateral ventricles and heart blood is drawn from the left ventricle by transthoracic puncture, using 30 mL disposable polyethylene syringes fitted with 8 cm long, 18 gauge needles. The CSF is ejected into 15 mL disposable polyethylene tubes for centrifugation while the blood is converted to serum by introduction into standard serum separator vacuum tubes (7 mL) prior to centrifugation. Although a small fraction of blood is also taken into EDTA vacuum tubes (4 mL each) for preparation of plasma, this is not directly comparable to plasma from living subjects as extensive post-mortem coagulation removes many clotting factors and other proteins. About 20–30 mL of CSF, 5–10 mL of serum and 4 mL of EDTA-treated serum are generally obtained. A 2–3 mL sample of blood serum is sent to a commercial clinical pathology laboratory for syphilis, HIV and hepatitis A, B and C serology. Remaining blood serum as well as CSF is centrifuged and supernatants from both blood serum and CSF are aliquotted into 0.5 mL polyethylene microcentrifuge tubes and stored frozen at −80°C.
Analysis of post-mortem CSF has been criticized but offers considerable potential for identifying predictive and diagnostic biomarkers. Cerebrospinal fluid is contiguous with the brain interstitial fluid and thus reflects brain chemistry.54 Therefore, it is likely that neurodegenerative abnormalities in the brain will ultimately produce a characteristic biochemical signature that will be most evident in the CSF. However, a critical deficiency with most CSF tests is that they have rarely been evaluated against the gold standard for diagnosis, which is neuropathological examination of the brain at autopsy. Evaluation of new biomarkers using, as a gold standard, a clinical neurological diagnosis, cannot, by definition, be shown to improve on this. Therefore the numerous CSF studies that claim 80–90% sensitivity and specificity as compared to the clinical diagnosis of AD are misleading in that they actually represent only at most 70% accuracy with respect to the true neuropathological diagnosis.55 The usage of post-mortem CSF in conjunction with neuropathological diagnosis is therefore a valuable and unique approach to biomarker evaluation.
However, it is recognized that post-mortem CSF changes56–59 must be distinguished from changes due to disease. We have conducted extensive studies on postmortem CSF60–66 using Western blot, ELISA, proteomic and metabolomic methodologies and have generally obtained results similar to those published by other groups who have used CSF drawn from living subjects who later died, were autopsied and neuropathologically diagnosed. Perhaps because our CSF studies have been from subjects with very short PMI, we have not found a correlation between measured CSF biomarkers and PMI,61 although we have found that phosphorylated tau protein appears to be depleted in the post-mortem period, suggesting that phosphorylated proteins in the CSF, as in the brain,16 may be especially vulnerable to post-mortem changes. It has often been stated that lumbar CSF has significant differences from ventricular CSF but a relatively recent comprehensive proteomics study found significant concentration differences in only two proteins.67 In our opinion, the benefits obtained by having precise neuropathological diagnoses for subjects greatly outweighs the disadvantages conferred by the presence of any minor post-mortem changes, and post-mortem CSF is thus an ideal medium for biomarker discovery. As with any post-mortem tissue study, all results must be correlated with PMI and possibly confounding peri-mortem factors as biochemical entities are heterogeneous in their behavior with respect to these. Our post-mortem CSF is stored in polypropylene tubes, as recommended by recent studies and organizations.68
Also prior to brain removal, and prior to CSF draw, samples are taken of temporalis muscle and scalp. Many neurological diseases can be diagnosed or studied in muscle or skin and therefore we fix and/or freeze these samples along with the brain tissue. We have published results for Aβ concentrations in temporalis muscle samples69 and have used scalp samples to look for both AD changes (unpublished negative results to date) and Lewy-type synucleinopathy (also negative findings to date).70
Brain removal, gross dissection and initial tissue processing
Brain removal is accomplished in the standard fashion with an oscillating electric saw. As some of our investigators regularly culture glial and vascular cells from the postmortem brain tissue,71–76 when this is being done, extra steps may be taken to minimize bacterial contamination of brain tissue, including washing of the scalp with an antibacterial solution, using sterile gloves and wiping down the cutting board with 70% ethanol. The brain is removed with as much cervical spinal cord as possible. Following brain removal, the pituitary gland is dissected from the sella turcica.
Gross neuropathologic examinations on brain external surfaces, coronal cerebral slices and parasagittal cerebellar slices may be performed by the neuropathologist during working hours but as most autopsies occur outside of working hours, all brains are photographed at the time of removal, whole and after slicing, with a digital camera and dedicated software that transmits the images directly to a computer hard-drive. By reviewing the digital images, the neuropathologist is then able to perform a gross description on brains removed and processed after-hours.
The details of the brain dissection are as follows. The olfactory bulbs and tracts as well as the pineal and pituitary glands are removed and the cerebellum and brainstem are severed from the cerebrum by a transverse cut across the rostral pons, keeping the entire substantia nigra (SN) with the coronally sectioned cerebral slices. The leptomeninges are stripped from both cerebrum and cerebellum for storage as research material.77 The cerebellum is separated from the brainstem by severing the cerebellar peduncles. Each hemicerebellum is sliced into 4–5 segments in the parasagittal plane. The brainstem is sliced into left and right halves. The cerebrum is sliced into 1 cm segments in the coronal plane. The left side slices are used for immersion fixation while the right side slices are rapidly frozen between sheets of dry ice (20 × 20 × 3 cm).
This method of brain processing differs from the conventional neuropathological examination, in which the whole or half of the brain is immersed in formaldehyde for 7–14 days before slicing. Although it has been argued that fresh-slicing detracts from the neuropathological gross exam, we feel that it is essential to slice the brain fresh, to enable both rapid freezing and optimum fixation. Freezing an intact hemisphere slowly on dry ice or by simply placing it in an ultralow-temperature freezer may result in severe ice-crystal artifact,78 rendering the tissue unsuitable for cryostat-section histological examination, which has become increasingly important as a method for molecular studies, especially those utilizing confocal laser-capture microscopic analysis. Also, the long-entrenched idea that rapid freezing in liquid nitrogen or similar fluids is essential for molecular biological studies is simplistic and incorrect. In fact, ultra-rapid freezing may cause membrane damage while slow freezing in iso-osmotic cyroprotectant is optimal for synaptosomal preparations.21 Our experience has shown that freezing rapidly between sheets of dry ice gives morphologically intact tissue suitable for a wide range of molecular biological procedures, including Western blot, ELISA, Northern blot, in situ hybridization, gene microarray, proteomics and RT-PCR.
Similarly, fixing the brain whole or as an intact hemisphere is not optimal as this results in extreme fixation gradients across the tissue, with brain surfaces becoming over-fixed while interior regions are under-fixed. These fixation gradients make even semi-quantitative immunohistochemical (IHC) evaluation very difficult, although a minority of epitopes are tolerant to this. While perfusion-fixation of the post-mortem brain through the circle of Willis (the entire cerebrum or one hemisphere can be fixed this way) is the optimal fixation method,79 this is time-consuming and we have found that fixation of 1 cm-thick slices for 24–48 h at 4°C is a reasonable compromise, although still subject to a fixation gradient.
To aid in the production of uniformly thick fresh brain slices for freezing and fixation, we have developed a brain slicing apparatus (jig) that holds the brain together while being sliced. With the assistance of a retired engineer living in Sun City and a contracted fabrication company (AvTek, Inc, Phoenix, AZ, USA), we have enabled its semiautomated duplication by entering the 3-dimensional plans for the apparatus onto a computer.
Fixation is carried out for 2 days at 4°C with a commercial formalin preparation containing 4% formaldehyde. Following fixation, diagnostic tissue blocks are taken from 28 brain regions (Table 2) for embedding in paraffin wax; additionally, eight large (about 3.5 × 5 cm) tissue blocks, representing all cerebral lobes as well as cerebellum, following cryoprotection in 2% dimethyl sulfoxide/20% glycerol, are sectioned at 40 μm and 80 μm on a sledge-type freezing microtome. These thick sections are superior to standard autopsy brain paraffin-embedded sections for both silver stains and immunohistochemistry, due to their relatively light fixation,79,80 thickness (allowing full visualization of 3-D structure), structural integrity (allowing processing as free-floating sections, which improves antibody access and washing) and lack of exposure to hot paraffin wax.81 These large-format sections also offer the opportunity for grading of cerebral white matter rarefaction and an extensive survey for microscopic infarctions. Pre-cut free-floating 40 μm and 80 μm sections, as well as unsectioned fixed wet brain blocks, are stored in 0.1 mol/L phosphate buffer with 0.1% sodium azide, in a refrigerator at 4°C (sections) or at room temperature (fixed wet brain blocks). Sections and tissue stored in this manner are excellent for IHC studies even after years of storage.
Table 2.
Brain and spinal cord regions sampled for diagnostic histopathology and inventory
| Brain Region | Paraffin6 μm sections | Frozen 40 and 80 μm sections |
|---|---|---|
| Superior frontal gyrus | No | Yes |
| Middle frontal gyrus | Yes | Yes |
| Precentral gyrus | Yes | No |
| Postcentral gyrus | Yes | No |
| Superior parietal lobule | No | Yes |
| Inferior parietal lobule | Yes | No |
| Anterior cingulate gyrus | Yes | Yes |
| Superior temporal gyrus | No | Yes |
| Middle temporal gyrus | Yes | Yes |
| Inferior temporal gyrus | No | Yes |
| Fusiform gyrus | Yes | Yes |
| Parahippocampal gyrus at amygdala | Yes | Yes |
| Parahippocampal gyrus at head hip. | No | Yes |
| Parahippocampal gyrus at lat.genic. | Yes | Yes |
| Hippocampus at head | No | Yes |
| Hippocampus at body | Yes | Yes |
| Occipital cortex: primary | Yes | Yes |
| Occipital cortex: association | Yes | Yes |
| Cerebral white matter: all lobes | No | Yes |
| Caudate nucleus: head | No | Yes |
| Caudate nucleus: body | Yes | Yes |
| Putamen: head | No | Yes |
| Globus pallidus | Yes | Yes |
| Substantia innominata | Yes | Yes |
| Thalamus: anterior | Yes | Yes |
| Thalamus: midpoint | Yes | Yes |
| Hypothalamus | Yes | Yes |
| Mammillary body | Yes | No |
| Subthalamic nucleus | Yes | Yes |
| Substantia nigra | No | Yes |
| Midbrain tegmentum | No | Yes |
| Pons:anterior | Yes | No |
| Pons: midpoint | Yes | Yes |
| Cerebellum: vermis | Yes | No |
| Cerebellum: dentate nucleus | Yes | Yes |
| Cerebellum: hemispheric cortex | Yes | Yes |
| Medulla: anterior | Yes | No |
| Medulla: posterior | Yes | No |
| Olfactory bulb and tract | Yes | No |
| Spinal cord: cervical | Yes | No |
| Spinal cord: thoracic† | Yes | No |
| Spinal cord; lumbar† | Yes | No |
| Spinal cord: sacral† | Yes | No |
Only from whole-body autopsies.
Diagnostic stains
Sections from paraffin blocks are cut at 6 μm and stained with HE. A standard set of brain sections is immunohistochemically stained for phosphorylated α-synuclein70,82 to identify Lewy bodies and Lewy-related neurites (see Fig. 3). Sections from the large frozen blocks are stained with Campbell-Switzer, Gallyas83 and Thioflavine S methods for plaques, tangles and other inclusions (see Fig. 3). While some investigators have criticized the usage of any stains other than molecularly specific IHC stains,84 we continue to use this set of stains to maintain consistency and standardization over many years of operation. The Braak neurofibrillary staging method was initially described using the Gallyas stain85 while the Thal amyloid staging method was initially described with the Campbell-Switzer stain.86 Both Braak and Thal have shown that the Campbell-Switzer silver stain very closely corresponds with IHC methods for Aβ.87–90 Previous and subsequent studies have also supported that IHC is not always or necessarily more sensitive than all other methods.91–95 We have demonstrated the equivalency of Campbell-Switzer and IHC methods for Aβ in an imaging-to-autopsy study of an amyloid imaging agent, F18 florbetapir,96,97 where cortical amyloid standardized uptake value ratio correlated with similar strength to plaque densities estimated with both Campbell-Switzer (Spearman rho = 0.76; P < 0.0001) and Aβ IHC (Spearman rho = 0.71; P < 0.001). Additionally, the validity and accuracy of the Campbell-Switzer method has also been established through strong correlations with autoradiographic binding of F18 florbetapir98 in post-mortem human brain sections (Spearman rho = 0.95; P < 0.0001).
Fig. 3.

Photomicrographs illustrating the histological methods used for investigating neurodegenerative diseases. Frames A–D are 40 μm large format sections stained with Campbell-Switzer silver stain to show all types of senile plaques. Frames E–H are 40 μm large format sections stained with Gallyas silver stain, showing both neurofibrillary changes (neurofibrillary tangles, dystrophic neurites and neuropil threads) and neuritic plaques. Frames I–M show, in 40 μm large format sections, changes of progressive supranuclear palsy, including neurofibrillary tangles in the substantia nigra (I), pontine nuclei (K) and dentate nucleus (L, M), and tufted astrocytes (J). Frame J was obtained from a section stained with the Gallyas method while I and K–M were from sections stained with an immunoperoxidase method for phosphorylated tau protein (AT8 antibody). Frames N and O show, in 40 μm large format sections, astrocytic plaques in a subject with corticobasal degeneration. Frames P–R show changes of Pick’s disease, including a chromatolytic “Pick cell” (P, H, E paraffin section), Pick bodies in the dentate granule cell layer (Q, 40 μm large format section stained with the Campbell-Switzer method) and oligodendroglial coiled bodies (R, 40 μm large format section stained with the Gallyas method). Frame S shows argyrophilic grains in a 40 μm large format section stained with the Gallyas method. Section T is a low-magnification image of the amgydala and adjacent temporal lobe, in a subject with dementia with Lewy bodies, from a 40 μm large format section stained with an immunoperoxidase method for phosphorylated α-synuclein. Frames U–Y are from subjects with Parkinson’s disease (PD) or dementia with Lewy bodies (DLB), stained with an immunoperoxidase method for phosphorylated α-synuclein. Frames U, V and X are from paraffin sections (U – olfactory bulb; V – locus ceruleus; X – submandibular gland). Frame W is from an 80 μm large format tangential section of the lower esophagus. Frame Y is from a retinal whole-mount from a PD subject. Frame Z is from a 40 μm large format section, from a subject with frontotemporal lobar dementia, stained with an immunoperoxidase method for phosphorylated TDP-43.
The 80 μm sections are stained with HE to detect and grade periventricular cerebral white matter rarefaction (leukoaraiosis)77,99 and microinfarctions (see Fig. 4). With these large, thick sections, we have found that HE is superior to Luxol Fast Blue for detection of white matter changes. Additional IHC procedures are used as needed, including those for phosphorylated tau protein to detect atypical tauopathies,100,101 ubiquitin and phosphorylated TDP-43102 to detect intraneuronal inclusions of fronto-temporolobar dementia (see Fig. 3).
Fig 4.

Photographs and photomicrographs illustrating vascular lesions. Frame A shows cross-sections of the circle of Willis from cognitively normal subjects with relatively normal arteries (above) as compared to late-onset Alzheimer’s disease subjects with relatively severe atherosclerotic stenosis (below). Frames B–F are from 40 μm large format sections. Frame B shows periventricular white matter rarefaction in an HE-stained section of the frontal lobe. Frame C shows a microinfarct in the cerebral cortex while frame D shows a microscopic focus of cerebellar cortical sclerosis, both from HE-stained sections. Frame E shows amyloidotic capillaries in the primary visual cortex, stained with the Campbell-Switzer silver stain. Frame F shows a larger amyloidotic blood vessel with dyshoric diffuse perivascular amyloid, stained with the thioflavin S method.
Semi-quantitative assessment of histopathological lesions
Histopathological scoring is performed blinded to clinical and neuropathological diagnosis. Amyloid plaque and neurofibrillary tangle density are graded and staged at standard sites in frontal, temporal, parietal and occipital cortices as well as hippocampus and entorhinal cortex, based on the aggregate impression from the 80 μm sections stained with thioflavin S, Campbell-Switzer and Gallyas methods. The total plaque score, considering all types of plaques (cored, neuritic and diffuse) together, is predominantly derived from the Campbell-Switzer stain while the Gallyas and thioflavine S stains are especially useful for estimating neuritic plaque densities. All three stains show neurofibrillary changes and therefore this score is estimated after viewing slides stained with all three. Both total and neuritic plaque densities are rated as none, sparse, moderate and frequent, using the published CERAD (Consortium to Establish a Registry for Alzheimer Disease) templates.103 Conversion of the descriptive terms to numerical values give scores of 0–3 for each area, with a maximum score of 15 for all five areas combined. Neurofibrillary tangle abundance and distribution is also graded in these thick sections, again using the CERAD templates for this, while the original Braak protocol83 is used for estimating the topographical distribution of neurofibrillary change. We are currently engaged in retrospectively rating a subset of past cases for Thal amyloid phase,86 and re-classifying the same subset according to NIA-AA (National Institute on Aging-Alzheimer’s Association) criteria for AD.104,105
Paraffin sections stained with IHC for phosphorylated α-synuclein70,82 are graded for density of Lewy-type synucleinopathy, considering together both neuronal perikaryal inclusions and neuropil fibers (0–4) according to templates published by the Dementia with Lewy Bodies Consortium.106 Ten standard brain regions are assessed, including olfactory bulb, anterior medulla, anterior and mid-pons, midbrain with SN, amgydala, anterior cingulate gyrus and three neocortical regions (middle frontal gyrus, middle temporal gyrus, inferior parietal lobule). A summary brain score of all 10 regions is recorded to give an overall brain load estimate, with the highest possible score being 40. The topographical distribution of Lewy-type synucleinopathy is classified using the Unified Staging System for Lewy Body Disorders.70
Assessment of the nigrostriatal dopaminergic system
Accurate counting of neurons within defined brain regions requires the methods of unbiased morphometry107,108 and a complete sampling of the brain region of interest. For cases with clinical parkinsonism, as well as many control cases, we have serially sectioned the SN in a subset of subjects, to allow researchers to perform unbiased estimates of neuron numbers. The entire left side of the SN was serially sectioned at 40 μm on a sliding freezing microtome, with all sections collected and stored appropriately. There are 146 cases that have been serially sectioned in this manner. These include 36 controls, 10 with incidental Lewy body disease (ILBD), 58 with PD, nine with dementia with Lewy bodies (DLB), 10 with AD with Lewy bodies (ADLB) and 19 with progressive supranuclear palsy (PSP). However, serial sectioning is very time-consuming and there has been very little demand for sets of these serial sections. To allow correlations of nigrostriatal depletion with clinical and tissue measures, we have semi-quantitative estimates (none, mild, moderate, severe) of SN pigmented neuron loss, at a standard level, available for virtually all cases. We also have measured striatal tyrosine hydroxylase (TH) concentrations by ELISA in 205 cases and the objective is to have this biochemical data available for all the relevant cases (e.g. all Lewy body disorders, normal controls and a selection of non-Lewy body neurodegenerative cases). We have also used this ELISA data to validate our semi-quantitative SN pigmented neuron loss scores; the correlation, in 205 cases, between the neuron loss scores and the striatal TH concentrations is very strong and highly significant (Spearman rho = 0.90; P < 0.0001). The correlation of striatal TH with SN Lewy-type synucleinopathy density score is also strong and significant (Spearman rho = 0.94; P < 0.0001). Our TH ELISA method has been previously published.109
Assessment of vascular lesions
Circle of Willis atherosclerosis is graded by gross visual external inspection, as previously published.110,111 The extent of atheromatous involvement is rated as none, mild, moderate or severe, using a schematic template. The method was validated by comparison with detailed computerized cross-sectional measurements of arterial lumen narrowing, performed on 54 cases in a separately published study. The correlation coefficient between the two methods was highly significant (Spearman rho = 0.64; P < 0.0001), indicating that the gross visual grading method corresponds reasonably well to the degree of atherosclerotic narrowing of the arterial lumen. The inter-rater reliability of the method was assessed in a separate subset of 50 cases, by grading each specimen on two separate occasions. With 40 of the cases, the same grade was assigned by both observers, while in 10 cases, the score differed by one grading unit. The Spearman correlation coefficient between the two observations was 0.92 (P < 0.0001).
Cerebral white matter rarefaction77,97,112–116 is graded in frontal, temporal, parietal and occipital lobes on the large 40 or 80 μm sections stained with HE. The method is analogous to that used by neuroradiologists to grade leukoaraiosis or “small vessel disease” in the cerebral white matter with MRI. Rarefaction restricted to the immediate periventricular region, occupying 25% or less of the centrum semi-ovale, is termed “mild”, while “moderate” is used when this extends to between 25% and 50%, with “severe” being reserved for rarefaction involving more than 50%. A summary score is obtained by adding the scores from all lobes, with a maximum score of 12.
Cerebral amyloid angiopathy (CAA) is graded on the same thick sections, primarily with the thioflavin S stain, and is a semi-quantitative estimate of the density of involved blood vessels, again by analogy with the CERAD templates. The extent of vascular circumferential amyloid deposition is not recorded. Each region receives a score of 0–3, with a maximum total score of 12.
Brain infarcts are classified by estimated age (acute, subacute, old or chronic), location and size (see Table 3). Centrum ovale infarcts are defined as those that are restricted to the centrum ovale; if the infarct involves both cerebral cortex and centrum ovale, it is classified as a cerebral cortex infarct. Deep nuclei infarcts include those of the basal ganglia, thalamus, subthalamic regions and hypothalamus. Infratentorial infarcts are those involving the brainstem and/or cerebellum. A volume is estimated for each infarct, and summary figures are recorded for brain subdivisions as well as for the entire brain. Foci of cerebellar cortical sclerosis are recorded as infarcts and are generally microscopic in size class. Acute ischemic changes are not counted as infarcts but are separately noted. It is recognized that the actual numbers of microscopic infarcts in a brain are likely to be up to 1000 times higher than what is found with standard brain sampling.117 Infarcts are very common in the BBDP population, with 43% of all subjects having one or more autopsy-confirmed infarcts. Prevalences for several subtypes of infarct are given in Table 3.
Table 3.
Categorization and prevalence of brain infarctions by estimated time of occurrence prior to death, brain location and size
| Age of infarct | Brain location of infarct | Size class of cortical infarct |
|---|---|---|
| Acute (n = 101; 12%) | Any location (n = 521; 43%) | Microinfarct (n = 213; 18%) |
| Subacute or chronic (n = 430; 36%) | Cerebral cortex (n = 314; 26%) | Lacunar (1 cc or less) (n = 49; 4%) |
| Centrum ovale (n = 92; 8%) | Small (1–27 cc) (n = 97; 8%) | |
| Deep nuclei (n = 282; 23%) | Large (more than 27 cc) (n = 58; 5%) | |
| Infratentorial (n = 235; 20%) |
Infarcts are also each assigned an estimated volume. Infarcts within the hippocampal formation and amygdala are classified with cerebral cortex infarcts. Centrum ovale infarcts are defined as those that are restricted to the centrum ovale; if the infarct involves both cerebral cortex and centrum ovale, it is classified as a cerebral cortex infarct. Deep nuclei infarcts include those of the basal ganglia, thalamus, subthalamic regions and hypothalamus. Infratentorial infarcts are those involving the brainstem and/or cerebellum. Number and percentage of subjects with each infarct type are indicated in parentheses. Microscopic infarcts are those not identified grossly and generally average about 0.1 cc in volume.
Correlation of neurodegenerative and vascular lesions with cognition
Cognitive impairment in the elderly is usually multifactorial. All of the significant causes have not yet been identified, although neuritic plaque density, Braak tangle stage and Lewy-type synucleinopathy load have all been found to be independently significant factors in most studies.118 A brief analysis of several possible influences in the BBDP population is presented in Table 4. The analysis includes 693 subjects aged 70 or over, for whom Mini Mental State Examination (MMSE) score, total white matter rarefaction (WMR) score, CERAD neuritic plaque density, Braak stage, CAA total score, number of cortical microinfarcts and total brain load for Lewy-type synucleinopathy are all available. All tissue measures had a significant inverse correlation with MMSE score except the number of cortical microinfarcts. Using logistic regression analysis and including age as a covariable, only neuritic plaque density, Braak stage, Lewy-type synucleinopathy brain summary score and total white matter rarefaction score were significant independent predictors of a lower MMSE score. However, it is recognized that the presence of multicolinearity between these variables, and the overwhelming influence of neurofibrillary change, may obscure significant relationships without the employment of more sophisticated analysis methods. In particular, others have reported that the number of microscopic infarcts is associated with a greater risk of dementia.119,120
Table 4.
Relationship of some neurodegenerative and vascular measures with MMSE score, from 693 BBDP subjects
| Measurement | Mean (SD) | Correlation with MMSE | OR for lower MMSE |
|---|---|---|---|
| Age | 84.9 (6.7) | — | — |
| MMSE | 18.6 (9.5) | — | — |
| Neuritic plaque density | 2.1 (1.2) | −0.47 (P < 0.0001) | 1.3 (P = 0.002) |
| Braak stage | 4.1 (1.3) | −0.56 (P < 0.0001) | 2.1 (P < 0.0001) |
| Lewy type synucleinopathy brain summary score | 7.8 (11.2) | −0.18 (P < 0.0001) | 1.02 (P = 0.02) |
| White matter rarefaction score | 4.2 (3.4) | −0.19 (P < 0.0001) | 1.08 (P = 0.02) |
| Amyloid angiopathy score | 3.4 (3.5) | −0.31 (P < 0.0001) | 1.00 (P = 0.88) |
| Number of cortical microinfarcts | 0.84 (2.9) | −0.04 (P = 0.25) | 0.99 (P = 0.87) |
Correlations were performed with the Spearman method. Logistic regression analysis provided odds ratios (OR) for each factor’s association with a lower MMSE score (<24), corrected for the influence of all other factors. MMSE, Mini Mental State Examination.
Diagnosis of neurodegenerative disorders
Diagnostic criteria have been amply discussed in the literature and will not be reviewed here. Published clinicopathological consensus criteria105,106,121–126 are used when available, incorporating clinical determinations of cognitive status and the presence or absence of other neurological signs as well as pertinent medical history. The diagnoses of cases coming to autopsy in the BBDP since 1997, when full neuropathological examinations first became a standard feature, are given in Table 5. The frequency of different conditions reflects both local prevalence and recruiting priorities, which have been focused on normal elderly controls, AD and PD. However, noteworthy are the relatively high frequencies of non-Alzheimer’s dementias, particularly DLB, vascular dementia (VaD), PSP and hippocampal sclerosis, all of which have often come to autopsy as clinically diagnosed undifferentiated dementia or AD. A recent publication is more indicative of the true incidence of these conditions in the BBDP population, as it is based on the conversion rates of BBDP subjects that were initially normal at their BBDP clinical assessments.127 Also notable is the frequent co-existence of more than one major neuropathological condition in the same subject,97,102,114,127,128 in agreement with such findings at other centers.129
Table 5.
Neuropathological diagnoses, 1997–2014, from 1173 autopsies
| Neuropathological diagnosis | n | % |
|---|---|---|
| Normal† | 238 | 20 |
| Alzheimer’s disease | 665 | 57 |
| Dementia with Lewy bodies | 107 | 9 |
| Parkinson’s disease | 170 | 14 |
| Vascular dementia | 110 | 9 |
| Progressive supranuclear palsy | 80 | 6.8 |
| Hippocampal sclerosis | 64 | 5 |
| Dementia lacking distinctive histology | 13 | 1.1 |
| Multiple system atrophy | 8 | 0.7 |
| Frontotemporal lobar degeneration with TDP-43 | 18 | 1.5 |
| Motor neuron disease | 12 | 1.0 |
| Corticobasal degeneration | 8 | 0.7 |
| Pick’s disease | 5 | 0.4 |
| Neurofibrillary tangle predominant dementia | 5 | 0.4 |
| Huntington’s disease | 2 | 0.2 |
| Multiple major neurodegenerative diagnoses | 349 | 37 |
Earlier autopsies before 1997 did not receive a full neuropathological examination. As more than one condition is often present in a single subject, the sum of the percentages exceeds 100. The percentage of subjects with multiple major clinicopathological conditions excludes the normal subjects from the denominator. Subjects listed as “multiple major neurodegenerative diagnoses have more than one of the listed conditions below.
No major clinical neurological diagnosis.
Genotyping and DNA banking
Since the discovery of the molecular genetic cause of Huntington’s disease,130 technological advances in DNA sequencing have led to the identification of multiple genetic risk factors for neurodegenerative diseases. DNA has been routinely prepared from lightly fixed cerebellar cortex from more than 95% of autopsied subject since 1997.131,132 We have found this method more convenient than using fresh-frozen tissue although the method is generally unsuitable for brain tissue fixed in formalin for the standard interval of 10 days to 2 weeks; in these cases we use fresh-frozen cerebellum. The apolipoprotein E (apoE) genotype has been determined for more than 95% of subjects since 1997. Quality control studies are periodically done, consisting of having a series of apoE-genotyped cases repeated by another laboratory. Isolated DNA remaining after apoE genotyping is stored for future studies. Screening for other mutations or polymorphisms (Table 6) is performed when family history, clinical features and/or histopathology indicate increased risk for a particular condition, or when a particular study is funded. For highly penetrant mutations causing early-onset neurological dysfunction, disclosure of test results is offered, through a certified genetic counselor.133–136
Table 6.
Genetic mutations and polymorphisms: results for autopsied cases
| Genetic polymorphism or mutation | No. screened | No. positive (%) |
|---|---|---|
| Apolipoprotein E –ε4 | 1302 | 524 (40) |
| LRRK2 – G2019S polymorphism screened | 237 | 3 (1.3) |
| Glucocerebrosidase (GBA) – all exons screened | 247 | 18 (7.3) |
| Tau (MAPT) – screen of exons 1 and 9–13 | 45 | 3 (1.3) |
| Presenilin 1 – mutation screen | 7 | 4 (57) |
| Progranulin (GRN) – screen of exons 1–13 | 31 | 4 (12.9) |
| C9orf72 – expansion screen | 21 | 5 (24) |
| TDP43 (TARDBP) | 1 | 0 |
| Valosin-containing protein (VCP) | 1 | 0 |
| Charged multivesicular body protein 2B (CHMP2B) | 1 | 0 |
Tissue quality assessment
Post-mortem tissue quality is affected by PMI, pre-mortem (agonal) conditions and post-mortem (pre-analytical) factors. Agonal status impacts tissue quality, as fever, ischemia-hypoxia and acidosis are deleterious to many molecules of interest.14,21,28,36,47,137,138 However, agonal status is difficult to determine in elderly subjects, who usually die in a nursing home without close medical supervision. The pH of CSF and/or brain tissue has been used as a surrogate marker for tissue quality.20 Some studies have reported that when the agonal state, as inferred from clinical data, is expected to have been detrimental, for example in cases of prolonged mechanical ventilation, the pH is low (e.g. pH 5.5–6.0), while in cases of sudden death it is higher, closer to normal (e.g. 7.0–7.4).137,139 Some enzyme activities and measures of RNA integrity have correlated with post-mortem pH, but not in all studies or for all subtypes of these molecules.14,28,35,140,141 Two comprehensive studies may have resolved the variability in prior reports, as it was found that only pH values under about 6.5–6.8 were associated with decreased RNA integrity.28,140 We have found that postmortem CSF pH did not correlate with viable cell yield in post-mortem glial cultures.142 A surprising result was that PMI was significantly correlated (inversely) with CSF pH (R = −0.37, P = 0.01), in agreement with one other group143 but in disagreement with several other reports.35,140,144 As CSF or brain pH is only an indirect marker of tissue quality, we have chosen instead to measure RNA integrity as a direct marker of tissue quality. Currently we have measured RNA RIN on frozen cerebellar samples of 848 cases. As mentioned, the median RIN for brain tissue is 8.9, while for 329 samples from a variety of bodily organs and tissues, the median RIN is 7.4. We also measure RNA quantitative yield; the mean for 863 cases in cerebellar cortex is 124.2 ng/μL. This effort is continuing, as the objective is to have RIN and RNA yield estimates from every brain collected since 1997.
Study of freeze-thaw effects and CSF pH on tissue RNA integrity
We have long suspected that most of the molecular degradation that occurs in banked tissue is due to repeated freeze-thaw cycles, which occur due to improper handling during dissection for tissue retrieval or due to freezer malfunctions. Previous research has indicated that thawing and refreezing of frozen brain tissue may be injurious to RNA because of damage to cellular membranes, including those of lysosomes containing ribonuclease.25,43,44 However, these investigations did not provide a quantitative estimate of RNA loss. An extensive literature search revealed only a single study that systematically addressed quantitative RNA changes due to repeated thaw-refreeze effects. This study was performed on surgically removed tonsillar tissue.145
We have systematically investigated the effects of thawing and refreezing on RNA integrity, by deliberately thawing and freezing small samples of brain tissue over varying time intervals and temperatures. Multiple frozen cerebellar samples (25–30 mg) from a single subject with initially high RIN were taken from an ultralow temperature freezer (−80 C), placed in chilled polyethylene microcentrifuge tubes and subjected to 1–6 thaw-refreeze cycles. For each cycle of thawing and subsequent refreezing, the tubes were removed from the freezer and placed in a tube rack on the lab bench at room temperature (20°C). After 30 min, the tubes were refrozen by placing back in the ultralow temperature freezer. Correlation of RIN and RNA quantitative yield with number of freeze-thaw cycles (Fig. 2) showed a trend for decreasing RIN with increasing number of freeze-thaw events but this did not reach the significance level (r = −0.67; P = 0.14). In contrast, increasing number of freeze-thaw cycles resulted in a significant decline in RNA yield (r = 0.93, P < 0.008). The expression of 89 genes was determined using a PCR-based array (RT2 ProfilerTM PCR Array: Human Alzheimer’s Disease, SABiosciencesTM, Frederick, MD, USA) containing genes pertinent to AD. A greater proportion of genes had decreased rather than increased transcript concentrations with increasing cycle number (80/89 vs 5/89; chi square P < 0.0001). Significantly decreased transcript concentrations occurred for 21 individual genes with increasing cycle number even after correction for total RNA concentration (unpaired, two-tailed t-tests, P < 0.001). In conclusion, we determined that RNA degrades progressively with increasing number of thaw-refreeze cycles and that RIN does not appear to be as sensitive as RNA yield as a marker of RNA degradation related to repeated thaw-refreeze cycles.
When dissecting frozen tissue samples for use by researchers, the tissue must not be allowed to thaw. We dissect on dry ice with heavy razor blades (Thompson tissue slicers), letting the tissue warm slightly (to about –20°C) to avoid shattering.
To avoid loss of tissue due to freezer malfunction, multiply-redundant protective systems are required. Our freezers are currently protected by two separate temperature-sensitive alarm systems that communicate temperature range perturbations by online access, email, text and telephone. Additionally, a diesel generator provides power in the event of electrical failure, and banks of CO2 tanks are connected to allow backup, automatic cooling when triggered by out-of-range freezer temperatures. A disaster plan is evolving to meet challenges posed by local or regional environmental, geological or other exigencies.
Cryostat sections for biochemical study of small, important brain regions
One of the limitations of brain banking is that some of the most important brain regions are very small and just one or two studies requiring frozen tissue may entirely deplete the frozen tissue from that region. Examples of these small yet vital regions are the entorhinal cortex, hippocampus and SN. To overcome this limitation and distribute tissue from these regions to greater numbers of researchers, we have been sectioning these brain areas on a cryostat and providing researchers with samples of 10–20 cryostat sections per case. This has been found sufficient to allow five or six Western blot analyses for proteins of interest and/or numerous RNA analyses for studies of gene expression. Conservatively, we estimate that this increases utilization of these small brain regions by five-fold or greater, while preserving large subject sample sizes with available tissue from these critical areas.
Whole-body autopsies and tissue collection
Between 1987 and 2004, the Program limited autopsies and tissue collection to the brain, cervical spinal cord, occipital scalp and temporalis muscle. In 2005 whole-body donation was offered to all enrolled subjects and to newly recruited subjects. Many brain donors chose to become whole-body donors and whole-body donation has become the option of choice, making up 67% of all autopsies over the most recent 5-year period. We have had more than 430 whole-body donations, with a median PMI of 3.1 h and a median RIN of 7.4. A list of the 42 tissue sites collected is given in Table 7. Although this expansion was undertaken primarily in the interest of pursuing research in diseases of other organ systems, it has been increasingly recognized that medically significant interactions exist between the brain and the body, and studies of our banked bodily tissue have made significant contributions to the understanding of neurodegenerative disease.
Table 7.
Bodily tissue sites collected. All are available as both fixed and frozen samples
| Organ system | Tissue site |
|---|---|
| Cardiovascular | |
| Aorta, thoracic | |
| Aorta, abdominal | |
| Heart, left ventricle | |
| Heart, right ventricle | |
| Genitourinary | |
| Kidney | |
| Bladder | |
| Prostate gland | |
| Uterus | |
| Cervix | |
| Vagina | |
| Endocrine | |
| Adrenal gland | |
| Thyroid gland | |
| Ovary | |
| Testis | |
| Respiratory | |
| Larynx | |
| Bronchus, primary | |
| Lung | |
| Diaphragm | |
| Musculoskeletal | |
| Bone, rib | |
| Muscle, psoas | |
| Muscle, temporalis | |
| Gastrointestinal | |
| Submandibular gland | |
| Esophagus, upper | |
| Esophagus, lower | |
| Stomach, fundus | |
| Duodenum | |
| Jejunum | |
| Ileum | |
| Colon, transverse | |
| Colon, sigmoid | |
| Rectum | |
| Liver | |
| Gallbladder | |
| Pancreas | |
| Other | |
| Breast | |
| Nerve, vagus | |
| Nerve, sciatic | |
| Skin, abdominal | |
| Skin, scalp | |
| Spleen | |
| Lymph node, peribronchial | |
| Mesentery |
Several established risk factors for atherosclerosis, including hypertension, hypercholesterolemia, diabetes, cigarette smoking and the apoE-ε4 allele have also been found to be risk factors for the development of dementia.146–148 However, the relationship between dementia, AD, non-AD dementias and atherosclerotic vascular disease (AVD) is complex and not completely understood. Also, while elevated midlife AVD risk factors appear to increase the probability of dementia that begins in late life,149–151 once dementia begins, these same AVD risk factors appear to diminish, perhaps due to molecular or cellular changes associated with specific dementing diseases, to decreased caloric intake and/or increased caloric expenditure, or to reduced survival of those with high AVD risk factors.152–158 Studies using BBDP data and tissue have contributed to this still-evolving body of knowledge, as we have found that elderly AD subjects have increased measures of intracranial atherosclerosis77,99,110,159–164 but decreased atherosclerosis of the coronary arteries.165 Additionally, another finding of the latter study was that mean heart weights were significantly lower in AD subjects as was the prevalence of clinically diagnosed cardiovascular disorders.165 Clinical alterations of intracardiac flow parameters have also been found to be altered in BBDP subjects.166
The divergent results for intracranial and coronary atherosclerosis are perhaps not so surprising considering that the natural history, severity and risk factors differ, in many ways, for coronary, carotid and intracranial atherosclerosis. Severe coronary artery disease may exist in the relative absence of intracranial atherosclerosis, and vice versa.167,168 Serum cholesterol is a significant risk factor for coronary and carotid atherosclerosis but has been reported to be less significant or even not significant for intracranial atherosclerosis.168–172 Similarly, the apoE-ε4 allele has been more strongly linked with coronary and carotid atherosclerosis than with intracranial atherosclerosis, where our own studies110,111 as well as three independent studies173–175 have found no link. Hypertension is more strongly associated with cerebral than with coronary atherosclerosis. Aging is the strongest risk factor and is common to all three sites.167 Autopsy studies have indicated that the earliest affected site in a US population was the carotid arteries, followed by the coronary and intracranial arteries. The typical age of onset of clinical cerebrovascular disease is 10–15 years later than that of clinically manifest coronary artery disease.167 The site-specific variability of AVD is probably due to both genetic and environmental factors, as there are considerable geographic and racial influences.167 There are multiple longitudinal studies176–180 indicating that there is progressive weight loss in people with dementia, and that this probably begins even prior to dementia diagnosis. Therefore, it seems likely that weight loss plays a major role in the apparent reversal of AVD risk factors, as well as clinically manifest cardiovascular disease, in elderly dementia subjects.
Over the last few years we have collected body mass index (BMI) measures on most BBDP subjects. Of 468 AD subjects and 196 non-demented elderly controls, both the means of repeated BMI measures and the means for final BMI are significantly lower in AD subjects, by about 1 point (mean of all BMIs for AD subjects = 24.2; for control = 25.1; P = 0.008). The final recorded BMI during life is about 1 point lower, for both AD and control subjects, than the mean of all recorded BMIs, supporting a downward trend during the final years of life. The generally lower BMI in AD subjects is a potential explanation and answer to the question posed more than 25 years ago by Wolf-Klein,157 “Are Alzheimer’s patients healthier?” The overwhelming importance of BMI to human health in general needs no review here and it is expected that many future studies will examine BMI in relation to many other clinical and pathological measures in the BBDP population.
In 2006, the Michael J. Fox Foundation for Parkinson’s Research (MJFF) funded the Arizona Parkinson’s Disease Consortium and the BBDP, expressly for the purpose of simultaneously mapping the presence of Lewy-type synucleinopathy in both the central and peripheral nervous systems (PNS). The project resulted in a new staging system applicable to all Lewy body disorders, the Unified Staging System for Lewy Body Disorders,70 and a published PNS survey181 that provided anatomic pathology data for use in clinicopathological correlations of non-motor signs and symptoms of PD and other Lewy body disorders. Additional focused projects have extended this initial anatomic survey with much more detail, especially in the pharynx,182–184 gastrointestinal tract185 and retina.186 Currently, collaborating investigators are conducting extended studies of synucleinopathy in skin.
Of potentially direct importance to neurodegenerative disease clinical management, peripheral tissue sites and organs could be biopsied to improve clinical diagnostic accuracy. For both AD and PD, the clinical diagnostic accuracy is much lower than what most clinicians have assumed it to be.55,187 This has probably been a factor in the high failure rate of phase 2 and 3 clinical trials.188 The sponsorship of the MJFF also enabled a search for a peripheral diagnostic biopsy site. The lower esophagus and submandibular gland were identified as the peripheral sites with the highest Lewy-type synucleinopathy prevalence and a subsequent autopsy study indicated that needle biopsy of the submandibular gland would probably be feasible, sensitive and specific.189 The MJFF funded a small clinical trial in which 9/12 long-duration, clinically diagnosed PD subjects were found to be positive for Lewy-type synucleinopathy on needle biopsy.190 We are currently near the end of a second MJFF-sponsored trial, involving 25 early (signs and symptoms of less than 5 years duration) PD subjects as well as 10 age-similar control subjects. This should indicate whether submandibular needle gland biopsy will be useful for identifying early-stage PD subjects. If so, the selection of early PD subjects for clinical trials could be significantly improved, allowing lower subject numbers, lower trial costs, more trials and a higher probability of trial success, should an effective agent be tested.
For BBDP bodily tissue, just as for brain, medically licensed pathologists perform a microscopic diagnostic examination so that all tissue is confidently classified as normal or abnormal, and the type of abnormality is recorded in a standardized manner. We are currently preparing a manuscript comparing bodily autopsy diagnoses in AD and control subjects. The most common clinical and autopsy diagnoses are those expected of this age group (Table 8). Of autopsy diagnoses, aortic atherosclerosis is virtually universal in elderly BBDP individuals, and is often severe, with endothelial ulcerations and focal calcifications. About 85% of all BBDP subjects aged 65 or over have coronary atherosclerosis. Other very common diagnoses are cardiomegaly (66%), calcification of the mitral (35%) and aortic (67%) valves, myocardial infarction (50%), acute bronchopneumonia (30%), hepatic atrophy (19%), hepatic steatosis (13%) and renal atrophy and/or nephrosclerosis (63%).
Table 8.
Common medical history and autopsy (anatomical pathology) diagnoses for Brain and Body Donation Program whole-body autopsies
| Medical history diagnosis | % | Autopsy diagnoses | % |
|---|---|---|---|
| Metabolic | |||
| Type II diabetes | 24 | Hepatic atrophy | 19 |
| Hypertension | 76 | Hepatic steatosis | 13 |
| Dyslipidemia | 55 | Thyroiditis | 9 |
| Hypothyroidism | 35 | Thyroid goiter | 7 |
| Osteoporosis | 35 | Testicular atrophy | 23 |
| Cardiovascular | |||
| Atrial fibrillation | 25 | Cardiomegaly | 66 |
| Cardiac arrhythmia, other | 30 | Coronary atherosclerosis | 85 |
| Congestive heart failure | 52 | Myocardial infarction | 50 |
| Coronary artery disease | 42 | Aortic valve calcification | 67 |
| Myocardial infarction | 20 | Mitral valve calcification | 35 |
| Gastrointestinal | |||
| Gastroesophageal reflux disease | 58 | Gastritis | 13 |
| Diverticular disease | 27 | Diverticulosis | 38 |
| Genitourinary | |||
| Benign prostatic hypertrophy or hyperplasia | 38 | Renal atrophy or nephrosclerosis | 63 |
| Renal failure | 24 | Cystitis | 27 |
| Other | |||
| Arthritis, degenerative | 59 | Acute bronchopneumonia | 30 |
| Bone fractures | 18 | Emphysema | 16 |
| Chronic obstructive lung disease | 27 | Cholecystectomy | 17 |
| Stroke | 22 | Hysterectomy | 49 |
| Depression | 49 | Mastectomy | 12 |
Percentages for gender-specific conditions are for the involved gender only. See Figure 3 for malignant neoplasms.
A major objective since the expansion to whole-body donation has been to assemble a unique and powerful tissue bank for cancer research and so we have expended additional effort to recruit donors with cancer. In 2002, the National Dialogue on Cancer, convened to understand why the “war on cancer” was falling short of expectations, concluded that of the 10 most important roadblocks to finding cures for cancer, the single most critical one was inadequate availability of “high-quality, highly characterized human tissues for translational research” (US National Cancer Institute Office of Biorepositories and Biospecimen Research). The need for better access to high-quality tissue has been widely cited by other documents, including the Genomics and Personalized Medicine Act of 2007, the Department of Health and Human Services’ Personalized Health Care Report (2007) and the President’s Council of Advisors on Science and Technology: Priorities for Personalized Medicine (2008). This theme has been repeatedly emphasized in subsequent years.191–193
There are several reasons for poor availability of cancer tissue.192 Cancer tissue is collected at the time of therapeutic surgical interventions, such as biopsy, or at the time of death, by autopsy. Biopsy is only done on individuals with disease and therefore normal control tissues are generally not obtained. A major objective of cancer research is to understand the molecular causes of metastasis, but meta-static deposits are rarely biopsied. Additionally, biopsy tissue is often completely used for diagnostic purposes or is insufficient in quantity to constitute a shared resource.
Both diseased and normal control tissue could potentially be obtained at autopsy. Usually, autopsy tissue is not suitable for high-quality molecular research due to autolytic changes resulting from a long PMI. The BBDP therefore is capable of fulfilling this critical need, as the PMI from our rapid autopsies is comparable to the elapsed time between surgical removal and processing, and the RNA quality is also similar.194 Although our efforts toward cancer biobanking are still relatively new, we have made a significant contribution to prostate cancer research. We served a US National Cancer Institute-funded multi-institutional project by providing non-cancerous prostate tissue from more than 50 BBDP subjects for use as controls in gene expression studies of prostate cancer diagnostic and prognostic indicators.195 Other projects have demonstrated the value of our metastatic cancer sample availability, through the genomic analysis of a primary site as well as several different metastatic sites in samples from the same patient.196 Additional studies have included genomic analysis of a BBDP case of cholangiocarcinoma197 and comparison of the molecular genetics of lung adenocarcinoma with normal lung tissue samples from more than 50 BBDP subjects.198 Figure 5 shows our current availability of cancer tissue samples.
Fig 5.

Frozen cancer tissue types available. All have detailed clinical and pathological data. Many have both primary site and metastatic tissue samples.
BBDP website and database
Since 1998, the Institute has developed a sophisticated database and database management protocols, engineered by a contracted professional industrial database consultant. The clinical and pathology components of the BBDP database were designed together and are completely integrated. The database is a customized elaboration of Microsoft Access and My Sequel programs, takes up more than 775 MB and is composed of 135 tables or matrices with more than 6800 separate data elements or fields. These collectively contain data pertaining to basic demographic information, medical history, results of standardized clinical assessments and, for deceased subjects, data obtained from gross and microscopic examinations, tissue inventory and records of tissue requests and shipments. The database contains multiple forms to facilitate data entry and to generate more than 100 different standing data reports. The database is also used to ensure that scheduling of clinical assessments is done at appropriate intervals. The US National Alzheimer’s Coordinating Center (NACC) Uniform Data Set and the Neuropathology Data Set are subsets and we are currently devising export queries to allow deposition of selected data in the NINDS Parkinson’s Disease Biomarker Project Data Management Resource. Numerous additional fields include in-house grading methods for plaques, tangles, Lewy-type synucleinopathy, infarcts and white matter rarefaction, as well as fixed tissue quality, RIN and RNA yield, and current inventory, with more than 400 fields for fixed tissue, frozen tissue, CSF and blood serum.
The database is backed up automatically every day on two servers administered by Banner Health Information Services, located at two sites about 5 miles apart. Storage of data in both locations ensures that structural damage to one server (e.g. fire) does not result in irretrievable loss. Both servers store daily copies of databases for the preceding 30 days; older copies are sent to a commercial server contracted for this purpose. Storage of older database copies allows the recovery of an intact database in the event of inadvertent errors in database operation, hardware malfunction or software malfunction.
The BBDP database has been managed by informal groups that have included the database designer, a statistician, the directors of the clinical and neuropathology cores and the major database operators. This arrangement has worked very well as it includes the individuals who use the database day-to-day, the physician-scientists that collect the data, and the database designer, who can make changes to the database as necessary.
Confidentiality of human subject data in the database is compliant with US HIPAA statutes and is assured by multiple safeguards which restrict access to personal identifying information such as name, date of birth, place of birth, place of residence, date of death, address, email address and phone number. Data entry is restricted to as few personnel as possible. Access to the database requires a unique user name and password, and all transactions are tracked and stored in an electronic log, allowing identification of inappropriate or incorrect usage by any individual.
Access to the Banner Health local area network is protected by a firewall maintained by Banner Health Information Services. Paper copies of subject records are kept in locked filing cabinets in the offices of the major database operators. Banner Sun Health Research Institute has been issued a Certificate of Confidentiality by the US Secretary of Health and Human Services, protecting human subjects’ data from court subpoena.
The BBDP has a website (http://www.brainandbodydonationprogram.org) with a public section and a private section. The private section is secure (https://www.brainandbodydonationprogram.org) and is accessible only after a user registers and is approved. Potential users are encouraged to read the information on the public side, to gain an overall understanding of the Program and a general idea of the tissue inventory. Media items featuring the BBDP, as well as a list of publications that have used BBDP tissue, are posted on the public side. Once approved as a user, restricted documents may be obtained, including a table of user fee charges and a special subset of the BBDP database designed to be user-friendly with the aid of a detailed database dictionary. Researchers may use the website database to help them select cases for tissue studies, or may use it solely for data studies, subject to the conditions on the posted data usage agreement. Tissue requests may be made on the website, and customized email support is available and recommended so that BBDP personnel may match the project needs with the most appropriate tissue and subject types.
Biospecimen allocation procedure
Requests for biological materials are granted on the basis of scientific merit. Requests for Lewy body disorder-focused projects are reviewed by an NINDS-appointed committee, as part of our NINDS-funded National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders. Investigators must first establish appropriate scientific credentials through the website by providing a recent biosketch or curriculum vitae. Requests for biosamples funded through the NIA ADCC are compliant with the NIA Biospecimens Best Practices policy. Small, pilot tissue requests by researchers from not-for-profit organizations are granted free of charge, while larger requests and all requests from for-profit organizations, are assessed a user fee, from tables posted on the BBDP website. The user fee is to recover unfunded BBDP costs, including those related to subject recruitment, longitudinal clinical assessment, autopsy, neuropathological and anatomical pathology tissue characterization, BBDP personnel time assisting with study design, case selection, database searches, retrieval and/or dissection, packaging, shipping, continuing correspondence and institutional overhead charges. Banner Sun Health Research Institute is a not-for-profit organization and fees are used only to meet the costs of operating the BBDP.
We believe that, since governments world-wide often do not provide sufficient funding for comprehensive brain banking, brain banks need to seek non-governmental funding. Most US National Institutes of Health tissue resources use cost recovery or user fees to supplement their funding and our experience shows that reasonable cost recovery fees do not impair usage of the resource. Diversification of funding sources will help protect brain banks from the periodic funding cuts that are common to all government-supported science. Our Program generally receives less than 10% of its operating costs from user fees, as the majority of the funding has come from a combination of federal grants, state of Arizona grants, grants from disease-specific private organizations, including the MJFF, internal institutional overhead support and philanthropic donations from individuals in the surrounding communities.
Distribution of biospecimens to researchers and type of projects served
Over the most recent 5 year period (07/01/2009–06/30/2014), 1035 separate tissue distributions were made. Currently, more than 200 requests are served each year. Since the Program began, more than 2500 requests have been served, coming from more than 400 investigators located in 32 US states and 15 countries. Tissue and/or data from the BBDP has contributed to more than 350 publications and more than 200 grant-funded projects.
The following are types of projects served in the past and likely to be served in the future. Examples of publications are given for each category. For a full list of publications supported by BBDP tissue, please visit our website, http://www.brainandbodydonationprogram.org.
Global gene expression and genome-wide association studies (GWAS)
This technology has gone through several iterations and will likely continue to do so as the technology advances. The studies are increasingly done by large government-funded consortia as well as large pharmaceutical companies. Genome-wide association studies use DNA and do not generally require high-quality tissue, but they are increasingly being paired with global gene expression profiling, which does require a minimum RNA quality. Initially, GWAS were done mostly with clinically diagnosed subjects but there is an increasing trend to use subjects that have had neuropathological diagnoses due to the increased diagnostic accuracy that allows a smaller subject number and lower study cost. The technology is continually advancing, with “deep sequencing” having recently become economically viable. BBDP tissue has been shared with multiple global gene expression and GWAS projects that have provided critical information relating to AD, PD, PSP, pancreatitis and prostate cancer.199–235
MicroRNA profiling
MicroRNA profiling is a relatively new field but the BBDP has supported several such projects recently, some of which have published results.60,236,237
Profiling of gene-specific DNA methylation
Epigenetic changes are increasingly being scrutinized and, of several studies supported, a few have now published their results.238,239
Studies using proteomic technology
Proteins vary widely in their post-mortem stability and therefore short PMI material is crucial for proteomics. As mentioned, BSHRI scientists and collaborators have published several articles61–65 on the CSF proteomics of AD and PD, as well as proteomic analyses of AD white matter112,113 and a “synucleinomics” profile of PD cortex.240 Proteomic studies of cerebral cortex, other brain regions and even laser-captured single cells will probably become more common and will be complementary to global gene expression and GWAS.
Studies using metabolomic technology
This technology is also still relatively new, but a recent publication supported by the BBDP has investigated the metabolomic profile of PD CSF.66 As many small molecules are very volatile, short PMI material will be advantageous to these types of studies.
Preclinical development of diagnostics and therapeutics
The BBDP has collaborated with multiple pharmaceutical industry investigators, supplying tissue or CSF from diseased and control subjects that has been used to aid the development of diagnostic and therapeutic agents. In particular, we have done extensive collaborative work toward tissue-based verification of amyloid binding imaging agents97,98,241–246 and the biochemical effects of anti-amyloid therapy in human brain.247–251
Independent investigator-initiated studies
Many of the study types listed above involve multi-center, technology-driven collaborations, but the great majority of projects that have used BBDP tissue have been single-investigator, hypothesis-driven studies, and we expect that these will continue to be the most frequent users. Some areas of concentration are as follows: studies using living cells cultured from rapidly autopsied donors;73–75,223,252–260 molecular analysis of α-synuclein;261–264 the nigrostriatal dopaminergic system;109,265–268 the basal forebrain cholinergic system;269–272 neuroinflammation;229,255,273–290 and molecular changes relating to Aβ in AD.69,116,291–310
Clinicopathological correlation studies
The clinical data generated by the BBDP offers excellent opportunities for clinicopathological correlation studies, which have been the foundation upon which our knowledge of disease has been based. These investigations are essential if we are to learn more about disease progression, subtypes of disease, improvement of diagnostic accuracy and selection of subjects for clinical trials. Work by BBDP investigators has led to numerous publications about clinical and neuropathological features of essential tremor, restless legs syndrome and rapid eye movement sleep behavior disorder,311–320 electrophysiological investigations of Lewy body disorders,321–330 mild cognitive impairment and functional decline in AD and PD,331–336 clinical and neuropathological findings associated with ILBD109,321,337 and other Lewy body disorders70,114,115,127,224,242,267,269,338,339 and diagnostic methods.187,189,190 Our clinical data is now available for collaborative studies to investigators world-wide; see our website for details (http://www.brainandbodydonationprogram.org).
Protection of autopsy personnel and tissue users
Health risks to individuals exposed to human tissue are minimized by the use of a number of precautionary measures. The process begins with screening of medical records at the time of enrollment and repeated screens with each clinical visit and at the time of death, the latter by a standardized telephone questionnaire. Those individuals with a medical history of a serious infectious pathogen are excluded from enrollment or autopsy. This includes individuals with active or carrier status for hepatitis B, hepatitis C, HIV, syphilis, tuberculosis, Creutzfeldt-Jakob disease and acute or chronic active CNS infections of any type. As BSHRI is a private research organization, not a diagnostic autopsy service, it is our option to refuse to perform an autopsy, and ethical considerations support the exclusion of serious infectious conditions to protect autopsy and research personnel. Only seven BBDP subjects have been rejected for the discovery of a probable or confirmed hazardous infectious disease at the time of death, reflecting the efficacy of our earlier screening procedures and the low rate of these infectious diseases in our study population. Three of these rejections were for high risk of Creutzfeldt-Jakob disease, determined through the peri-mortem telephone interview, and one of the three was later confirmed by the National Prion Disease Surveillance Laboratory at Case Reserve University in Cleveland. A fourth rejection was for sepsis with methicillin-resistant Staphylococcus aureus, while an additional three subjects were found after postmortem blood testing to be carrying hepatitis C, despite negative clinical ante-mortem medical records screening. The frozen tissue from these subjects was destroyed.
Staff involved in brain removal and tissue processing are encouraged to have hepatitis A and B vaccination; during procedures involving exposure to fresh or frozen human tissue, staff wear protective clothing including hoods or caps, eye protection, masks, gowns, gloves and boots. If potentially infectious contamination of an individual occurs, such as splashing of fresh tissue fluids into the eyes or into a wound, the individual receives appropriate first aid measures at the scene and then is taken to a nearby Emergency Room. All Institute staff must complete, upon first hiring and annually afterwards, a formal training program in laboratory safety, including biohazard safety precautions.
The future of autopsy and brain banking
The usage of autopsies has been in decline throughout the world over the last 50 years. In Japan, post-mortem imaging has become a common substitute for autopsy,340 and it has been stated in some recent publications that biomarkers measured in living subjects “. . . could replace autopsy confirmation of AD plaque and tangle pathology as the ‘gold standard’ for the diagnosis of AD in the near future”.68 We believe that this prediction may indeed come true, as amyloid imaging has been shown, through autopsy-confirmed studies, to have a high level of accuracy for the diagnosis of neuropathologically significant AD, at least in the setting of demented subjects.341 Imaging agents for tau or synuclein pathology have not yet been autopsy-confirmed but there are multiple groups working on these. However, amyloid imaging ligands are still insensitive to sparse cortical amyloid loads and it is likely that any tau or synuclein ligands will also lack the sensitivity and specificity of an autopsy with comprehensive neuropathological examination. In our opinion, autopsy and brain banks will become more important, rather than less important, in the future, as new biomarkers will continue to require autopsy validation, as has been done for amyloid imaging agents,96,342 although it is possible that brain biopsy may also help meet this need.343,344 Due to the complexity of brain disease in the elderly, with resultant heterogeneity and co-existence of major pathologies,97,114,243,345 the expansion of our understanding of these conditions will depend almost entirely on the careful study of the post-mortem human brain, in conjunction with increasing attention to the detailed collection of ante-mortem health information and genomic studies. Finally, human brain tissue will continue to serve, as it has in the past, as the most important resource for discovery of new molecularly based therapies.1
Acknowledgments
We have deep admiration and respect for the volunteer subjects who have made the BBDP possible through their altruistic and generous spirits. The BBDP has received financial support from the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research.
References
- 1.Beach TG. Alzheimer’s disease and the “Valley of Death”: not enough guidance from human brain tissue? J Alzheimers Dis. 2013;33 (Suppl 1):S219–S233. doi: 10.3233/JAD-2012-129020. [DOI] [PubMed] [Google Scholar]
- 2.Peschken CA, Esdaile JM. Rheumatic diseases in North America’s indigenous peoples. Semin Arthritis Rheum. 1999;28:368–391. doi: 10.1016/s0049-0172(99)80003-1. [DOI] [PubMed] [Google Scholar]
- 3.Johanneson B, Steinsson K, Lindqvist AK, et al. A comparison of genome-scans performed in multicase families with systemic lupus erythematosus from different population groups. J Autoimmun. 1999;13:137–141. doi: 10.1006/jaut.1999.0305. [DOI] [PubMed] [Google Scholar]
- 4.Lio D, Pes GM, Carru C, et al. Association between the HLA-DR alleles and longevity: a study in Sardinian population. Exp Gerontol. 2003;38:313–317. doi: 10.1016/s0531-5565(02)00178-x. [DOI] [PubMed] [Google Scholar]
- 5.Mathews CA, Reus VI, Bejarano J, et al. Genetic studies of neuropsychiatric disorders in Costa Rica: a model for the use of isolated populations. Psychiatr Genet. 2004;14:13–23. doi: 10.1097/00041444-200403000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Adachi T, Inanami O, Sato A. Nitric oxide (NO) is involved in increased cerebral cortical blood flow following stimulation of the nucleus basalis of Meynert in anesthetized rats. Neurosci Lett. 1992;139:201–204. doi: 10.1016/0304-3940(92)90552-i. [DOI] [PubMed] [Google Scholar]
- 7.Gustavsson JP, Asberg M, Schalling D. The healthy control subject in psychiatric research: impulsiveness and volunteer bias. Acta Psychiatr Scand. 1997;96:325–328. doi: 10.1111/j.1600-0447.1997.tb09924.x. [DOI] [PubMed] [Google Scholar]
- 8.Mandel FS, Weiner M, Kaplan S, Pelcovitz D, Labruna V. An examination of bias in volunteer subject selection: findings from an in-depth child abuse study. J Trauma Stress. 2000;13:77–88. doi: 10.1023/A:1007772931154. [DOI] [PubMed] [Google Scholar]
- 9.Sofuoglu M, Dudish-Poulsen S, Nicodemus KK, Babb DA, Hatsukami DK. Characteristics of research volunteers for inpatient cocaine studies: focus on selection bias. Addict Behav. 2000;25:785–790. doi: 10.1016/s0306-4603(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 10.Lim A, Kukull W, Nochlin D, et al. Cliniconeuropathological correlation of Alzheimer’s disease in a community-based case series. J Am Geriatr Soc. 1999;47:564–569. doi: 10.1111/j.1532-5415.1999.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 11.Allen MJ, Powers ML, Gronowski KS, Gronowski AM. Human tissue ownership and use in research: what laboratorians and researchers should know. Clin Chem. 2010;56:1675–1682. doi: 10.1373/clinchem.2010.150672. [DOI] [PubMed] [Google Scholar]
- 12.Nelkin D, Andrews L. Do the dead have interests? Policy issues for research after life. Am J Law Med. 1998;24:261–291. [PubMed] [Google Scholar]
- 13.Kurtz SF, Strong CWGD. The 2006 revised Uniform Anatomical Gift Act – A law to save lives. Health Lawyers News. 2007 Feb;:44–49. [Google Scholar]
- 14.Spokes EG. An analysis of factors influencing measurements of dopamine, noradrenaline, glutamate decarboxylase and choline acetylase in human postmortem brain tissue. Brain. 1979;102:333–346. doi: 10.1093/brain/102.2.333. [DOI] [PubMed] [Google Scholar]
- 15.Spokes EG, Koch DJ. Post-mortem stability of dopamine, glutamate decarboxylase and choline acetyltransferase in the mouse brain under conditions simulating the handling of human autopsy material. J Neurochem. 1978;31:381–383. doi: 10.1111/j.1471-4159.1978.tb12477.x. [DOI] [PubMed] [Google Scholar]
- 16.Oka T, Tagawa K, Ito H, Okazawa H. Dynamic changes of the phosphoproteome in postmortem mouse brains. PLoS ONE. 2011;6:e21405. doi: 10.1371/journal.pone.0021405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ludes B, Pfitzinger H, Mangin P. DNA fingerprinting from tissues after variable postmortem periods. J Forensic Sci. 1993;38:686–690. [PubMed] [Google Scholar]
- 18.Morrison MR, Griffin WS. The isolation and in vitro translation of undegraded messenger RNAs from human postmortem brain. Anal Biochem. 1981;113:318–324. doi: 10.1016/0003-2697(81)90083-x. [DOI] [PubMed] [Google Scholar]
- 19.Perrett CW, Marchbanks RM, Whatley SA. Characterisation of messenger RNA extracted postmortem from the brains of schizophrenic, depressed and control subjects. J Neurol Neurosurg Psychiatry. 1988;51:325–331. doi: 10.1136/jnnp.51.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravid R, Van Zwieten EJ, Swaab DF. Brain banking and the human hypothalamus – factors to match for, pitfalls and potentials. Prog Brain Res. 1992;93:83–95. doi: 10.1016/s0079-6123(08)64565-3. [DOI] [PubMed] [Google Scholar]
- 21.Hynd MR, Lewohl JM, Scott HL, Dodd PR. Biochemical and molecular studies using human autopsy brain tissue. J Neurochem. 2003;85:543–562. doi: 10.1046/j.1471-4159.2003.01747.x. [DOI] [PubMed] [Google Scholar]
- 22.Palmer AM, Lowe SL, Francis PT, Bowen DM. Are post-mortem biochemical studies of human brain worthwhile? Biochem Soc Trans. 1988;16:472–475. doi: 10.1042/bst0160472. [DOI] [PubMed] [Google Scholar]
- 23.Leonard S, Logel J, Luthman D, Casanova M, Kirch D, Freedman R. Biological stability of mRNA isolated from human postmortem brain collections. Biol Psychiatry. 1993;33:456–466. doi: 10.1016/0006-3223(93)90174-c. [DOI] [PubMed] [Google Scholar]
- 24.Cummings TJ, Strum JC, Yoon LW, Szymanski MH, Hulette CM. Recovery and expression of messenger RNA from postmortem human brain tissue. Mod Pathol. 2001;14:1157–1161. doi: 10.1038/modpathol.3880451. [DOI] [PubMed] [Google Scholar]
- 25.Yasojima K, McGeer EG, McGeer PL. High stability of mRNAs postmortem and protocols for their assessment by RT-PCR. Brain Res Brain Res Protoc. 2001;8:212–218. doi: 10.1016/s1385-299x(01)00119-2. [DOI] [PubMed] [Google Scholar]
- 26.Preece P, Cairns NJ. Quantifying mRNA in postmortem human brain: influence of gender, age at death, postmortem interval, brain pH, agonal state and inter-lobe mRNA variance. Brain Res Mol Brain Res. 2003;118:60–71. doi: 10.1016/s0169-328x(03)00337-1. [DOI] [PubMed] [Google Scholar]
- 27.Preece P, Virley DJ, Costandi M, et al. An optimistic view for quantifying mRNA in post-mortem human brain. Brain Res Mol Brain Res. 2003;116:7–16. doi: 10.1016/s0169-328x(03)00208-0. [DOI] [PubMed] [Google Scholar]
- 28.Li JZ, Vawter MP, Walsh DM, et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13:609–616. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- 29.Tomita H, Vawter MP, Walsh DM, et al. Effect of agonal and postmortem factors on gene expression profile: quality control in microarray analyses of postmortem human brain. Biol Psychiatry. 2004;55:346–352. doi: 10.1016/j.biopsych.2003.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ervin JF, Heinzen EL, Cronin KD, et al. Postmortem delay has minimal effect on brain RNA integrity. J Neuropathol Exp Neurol. 2007;66:1093–1099. doi: 10.1097/nen.0b013e31815c196a. [DOI] [PubMed] [Google Scholar]
- 31.Popova T, Mennerich D, Weith A, Quast K. Effect of RNA quality on transcript intensity levels in microarray analysis of human post-mortem brain tissues. BMC Genomics. 2008;9:91. doi: 10.1186/1471-2164-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durrenberger PF, Fernando S, Kashefi SN, et al. Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J Neuropathol Exp Neurol. 2010;69:70–81. doi: 10.1097/NEN.0b013e3181c7e32f. [DOI] [PubMed] [Google Scholar]
- 33.Sajdel-Sulkowska EM, Majocha RE, Salim M, Zain SB, Marotta CA. The postmortem Alzheimer brain is a source of structurally and functionally intact astrocytic messenger RNA. J Neurosci Methods. 1988;23:173–179. doi: 10.1016/0165-0270(88)90189-6. [DOI] [PubMed] [Google Scholar]
- 34.Gilmore JH, Lawler CP, Eaton AM, Mailman RB. Postmortem stability of dopamine D1 receptor mRNA and D1 receptors. Brain Res Mol Brain Res. 1993;18:290–296. doi: 10.1016/0169-328x(93)90092-4. [DOI] [PubMed] [Google Scholar]
- 35.Kingsbury AE, Foster OJ, Nisbet AP, et al. Tissue pH as an indicator of mRNA preservation in human postmortem brain. Brain Res Mol Brain Res. 1995;28:311–318. doi: 10.1016/0169-328x(94)00219-5. [DOI] [PubMed] [Google Scholar]
- 36.Johnston NL, Cervenak J, Shore AD, Torrey EF, Yolken RH. Multivariate analysis of RNA levels from postmortem human brains as measured by three different methods of RT-PCR. Stanley Neuropathology Consortium. J Neurosci Methods. 1997;77:83–92. doi: 10.1016/s0165-0270(97)00115-5. [DOI] [PubMed] [Google Scholar]
- 37.Mathern GW, Pretorius JK, Kornblum HI, et al. Altered hippocampal kainate-receptor mRNA levels in temporal lobe epilepsy patients. Neurobiol Dis. 1998;5:151–176. doi: 10.1006/nbdi.1998.0200. [DOI] [PubMed] [Google Scholar]
- 38.Schramm M, Falkai P, Tepest R, et al. Stability of RNA transcripts in post-mortem psychiatric brains. J Neural Transm. 1999;106:329–335. doi: 10.1007/s007020050162. [DOI] [PubMed] [Google Scholar]
- 39.Miller CL, Diglisic S, Leister F, Webster M, Yolken RH. Evaluating RNA status for RT-PCR in extracts of postmortem human brain tissue. Biotechniques. 2004;36:628–633. doi: 10.2144/04364ST03. [DOI] [PubMed] [Google Scholar]
- 40.Johnson SA, Morgan DG, Finch CE. Extensive postmortem stability of RNA from rat and human brain. J Neurosci Res. 1986;16:267–280. doi: 10.1002/jnr.490160123. [DOI] [PubMed] [Google Scholar]
- 41.Lukiw WJ, Wong L, McLachlan DR. Cytoskeletal messenger RNA stability in human neocortex: studies in normal aging and in Alzheimer’s disease. Int J Neurosci. 1990;55:81–88. doi: 10.3109/00207459008985953. [DOI] [PubMed] [Google Scholar]
- 42.Burke WJ, O’Malley KL, Chung HD, Harmon SK, Miller JP, Berg L. Effect of pre- and postmortem variables on specific mRNA levels in human brain. Brain Res Mol Brain Res. 1991;11:37–41. doi: 10.1016/0169-328x(91)90018-s. [DOI] [PubMed] [Google Scholar]
- 43.Ragsdale DS, Miledi R. Expressional potency of mRNAs encoding receptors and voltage-activated channels in the postmortem rat brain. Proc Natl Acad Sci U S A. 1991;88:1854–1858. doi: 10.1073/pnas.88.5.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross BM, Knowler JT, McCulloch J. On the stability of messenger RNA and ribosomal RNA in the brains of control human subjects and patients with Alzheimer’s disease. J Neurochem. 1992;58:1810–1819. doi: 10.1111/j.1471-4159.1992.tb10057.x. [DOI] [PubMed] [Google Scholar]
- 45.Eastwood SL, Burnet PW, McDonald B, Clinton J, Harrison PJ. Synaptophysin gene expression in human brain: a quantitative in situ hybridization and immunocytochemical study. Neuroscience. 1994;59:881–892. doi: 10.1016/0306-4522(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 46.Pardue S, Zimmerman AL, Morrison-Bogorad M. Selective postmortem degradation of inducible heat shock protein 70 (hsp70) mRNAs in rat brain. Cell Mol Neurobiol. 1994;14:341–357. doi: 10.1007/BF02088715. [DOI] [PubMed] [Google Scholar]
- 47.Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC. The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett. 1995;200:151–154. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- 48.Castensson A, Emilsson L, Preece P, Jazin EE. High-resolution quantification of specific mRNA levels in human brain autopsies and biopsies. Genome Res. 2000;10:1219–1229. doi: 10.1101/gr.10.8.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bauer M, Gramlich I, Polzin S, Patzelt D. Quantification of mRNA degradation as possible indicator of postmortem interval – a pilot study. Leg Med (Tokyo) 2003;5:220–227. doi: 10.1016/j.legalmed.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Barrachina M, Castano E, Ferrer I. TaqMan PCR assay in the control of RNA normalization in human post-mortem brain tissue. Neurochem Int. 2006;49:276–284. doi: 10.1016/j.neuint.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 51.Dumitriu A, Moser C, Hadzi TC, et al. Post-mortem interval influences á-synuclein expression in Parkinson disease brain. J Park Dis. 2012 doi: 10.1155/2012/614212. Epub 2012 March 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell Tissue Bank. 2011;12:311–318. doi: 10.1007/s10561-010-9210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broniscer A, Baker JN, Baker SJ, et al. Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer. 2010;116:4632–4637. doi: 10.1002/cncr.25405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng PP, Luider TM, Pieters R, et al. Identification of tumor-related proteins by proteomic analysis of cerebrospinal fluid from patients with primary brain tumors. J Neuropathol Exp Neurol. 2003;62:855–862. doi: 10.1093/jnen/62.8.855. [DOI] [PubMed] [Google Scholar]
- 55.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71:266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burgess JA, Lescuyer P, Hainard A, et al. Identification of brain cell death associated proteins in human post-mortem cerebrospinal fluid. J Proteome Res. 2006;5:1674–1681. doi: 10.1021/pr060160v. [DOI] [PubMed] [Google Scholar]
- 57.Lescuyer P, Allard L, Zimmermann-Ivol CG, et al. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics. 2004;4:2234–2241. doi: 10.1002/pmic.200300822. [DOI] [PubMed] [Google Scholar]
- 58.Dayon L, Hainard A, Licker V, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 59.Finehout EJ, Franck Z, Relkin N, Lee KH. Proteomic analysis of cerebrospinal fluid changes related to postmortem interval. Clin Chem. 2006;52:1906–1913. doi: 10.1373/clinchem.2006.070508. [DOI] [PubMed] [Google Scholar]
- 60.Burgos K, Malenica I, Metpally R, et al. Profiles of Extracellular miRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE. 2014;9:e94839. doi: 10.1371/journal.pone.0094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roher AE, Maarouf CL, Sue LI, Hu Y, Wilson J, Beach TG. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers. 2009;14:493–501. doi: 10.3109/13547500903108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maarouf CL, Andacht TM, Kokjohn TA, et al. Proteomic analysis of Alzheimer’s disease cerebrospinal fluid from neuropathologically diagnosed subjects. Curr Alzheimer Res. 2009;6:399–406. doi: 10.2174/156720509788929318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maarouf CL, Beach TG, Adler CH, et al. Cerebrospinal fluid biomarkers of neuropathologically diagnosed Parkinson’s disease subjects. Neurol Res. 2012;34:669–676. doi: 10.1179/1743132812Y.0000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maarouf CL, Beach TG, Adler CH, et al. Quantitative appraisal of ventricular cerebrospinal fluid biomarkers in neuropathologically diagnosed Parkinson’s disease cases lacking Alzheimer’s disease pathology. Biomark Insights. 2013;8:19–28. doi: 10.4137/BMI.S11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castano EM, Roher AE, Esh CL, Kokjohn TA, Beach T. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol Res. 2006;28:155–163. doi: 10.1179/016164106X98035. [DOI] [PubMed] [Google Scholar]
- 66.LeWitt PA, Li J, Lu M, Beach TG, Adler CH, Guo L. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov Disord. 2013;28:1653–1660. doi: 10.1002/mds.25555. [DOI] [PubMed] [Google Scholar]
- 67.Simonsen AH, Bech S, Laursen I, et al. Proteomic investigations of the ventriculo-lumbar gradient in human CSF. J Neurosci Methods. 2010;191:244–248. doi: 10.1016/j.jneumeth.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 68.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 2011;121:597–609. doi: 10.1007/s00401-011-0808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuo YM, Kokjohn TA, Watson MD, et al. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol. 2000;156:797–805. doi: 10.1016/s0002-9440(10)64947-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Little JP, Simtchouk S, Schindler SM, et al. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci. 2014;60:88–96. doi: 10.1016/j.mcn.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 72.Lue LF, Kuo YM, Beach T, Walker DG. Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol Neurobiol. 2010;41:115–128. doi: 10.1007/s12035-010-8106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer’s disease and other neurodegenerative diseases. J Neurosci Res. 2005;81:412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- 74.Lue LF, Brachova L, Walker DG, Rogers J. Characterization of glial cultures from rapid autopsies of Alzheimer’s and control patients. Neurobiol Aging. 1996;17:421–429. doi: 10.1016/0197-4580(96)00006-1. [DOI] [PubMed] [Google Scholar]
- 75.Lue LF, Walker DG. Modeling Alzheimer’s disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- 76.Rogers J, Lue LF, Walker DG, et al. Elucidating molecular mechanisms of Alzheimer’s disease in microglial cultures. Ernst Schering Res Found Workshop. 2002;39:25–44. doi: 10.1007/978-3-662-05073-6_3. [DOI] [PubMed] [Google Scholar]
- 77.Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 78.Vonsattel JP, Aizawa H, Ge P, et al. An improved approach to prepare human brains for research. J Neuropathol Exp Neurol. 1995;54:42–56. doi: 10.1097/00005072-199501000-00006. [DOI] [PubMed] [Google Scholar]
- 79.Beach TG, Tago H, Nagai T, Kimura H, McGeer PL, McGeer EG. Perfusion-fixation of the human brain for immunohistochemistry: comparison with immersion-fixation. J Neurosci Methods. 1987;19:183–192. doi: 10.1016/s0165-0270(87)80001-8. [DOI] [PubMed] [Google Scholar]
- 80.Leong AS, Gilham PN. The effects of progressive formaldehyde fixation on the preservation of tissue antigens. Pathology. 1989;21:266–268. doi: 10.3109/00313028909061071. [DOI] [PubMed] [Google Scholar]
- 81.Pollard K, Lunny D, Holgate CS, Jackson P, Bird CC. Fixation, processing, and immunochemical reagent effects on preservation of T-lymphocyte surface membrane antigens in paraffin-embedded tissue. J Histochem Cytochem. 1987;35:1329–1338. doi: 10.1177/35.11.3309048. [DOI] [PubMed] [Google Scholar]
- 82.Beach TG, White CL, Hamilton RL, et al. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008;116:277–288. doi: 10.1007/s00401-008-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 84.Cummings BJ. Plaques and tangles: searching for primary events in a forest of data. Neurobiol Aging. 1997;18:358–362. doi: 10.1016/s0197-4580(97)00049-3. [DOI] [PubMed] [Google Scholar]
- 85.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 86.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 87.Braak H, Braak E, Bohl J, Lang W. Alzheimer’s disease: amyloid plaques in the cerebellum. J Neurol Sci. 1989;93:277–287. doi: 10.1016/0022-510x(89)90197-4. [DOI] [PubMed] [Google Scholar]
- 88.Braak H, Braak E, Ohm T, Bohl J. Alzheimer’s disease: mismatch between amyloid plaques and neuritic plaques. Neurosci Lett. 1989;103:24–28. doi: 10.1016/0304-3940(89)90479-5. [DOI] [PubMed] [Google Scholar]
- 89.Braak H, Braak E, Kalus P. Alzheimer’s disease: areal and laminar pathology in the occipital isocortex. Acta Neuropathol. 1989;77:494–506. doi: 10.1007/BF00687251. [DOI] [PubMed] [Google Scholar]
- 90.Braak H, Braak E. Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49:215–224. [PubMed] [Google Scholar]
- 91.Akiyama H, Tago H, Itagaki S, McGeer PL. Occurrence of diffuse amyloid deposits in the presubicular parvopyramidal layer in Alzheimer’s disease. Acta Neuropathol. 1990;79:537–544. doi: 10.1007/BF00296114. [DOI] [PubMed] [Google Scholar]
- 92.Halliday G, Flowers D, Baum L. Analysis of staining methods for different cortical plaques in Alzheimer’s disease. Acta Neuropathol. 1994;87:174–186. doi: 10.1007/BF00296188. [DOI] [PubMed] [Google Scholar]
- 93.Rosenwald A, Reusche E, Ogomori K, Teichert HM. Comparison of silver stainings and immunohistology for the detection of neurofibrillary tangles and extracellular cerebral amyloid in paraffin sections. Acta Neuropathol. 1993;86:182–186. doi: 10.1007/BF00334887. [DOI] [PubMed] [Google Scholar]
- 94.Vallet PG, Guntern R, Hof PR, et al. A comparative study of histological and immunohistochemical methods for neurofibrillary tangles and senile plaques in Alzheimer’s disease. Acta Neuropathol. 1992;83:170–178. doi: 10.1007/BF00308476. [DOI] [PubMed] [Google Scholar]
- 95.He Y, Duyckaerts C, Delaere P, Piette F, Hauw JJ. Alzheimer’s lesions labelled by anti-ubiquitin antibodies: comparison with other staining techniques. A study of 15 cases with graded intellectual status in ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol. 1993;19:364–371. doi: 10.1111/j.1365-2990.1993.tb00453.x. [DOI] [PubMed] [Google Scholar]
- 96.Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–678. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 97.Dugger BN, Clark CM, Serrano G, et al. Neuropathologic heterogeneity does not impair florbetapir-positron emission tomography postmortem correlates. J Neuropathol Exp Neurol. 2014;73:72–80. doi: 10.1097/NEN.0000000000000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi SR, Schneider JA, Bennett DA, et al. Correlation of amyloid PET ligand florbetapir F 18 binding with Abeta aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord. 2012;26:8–16. doi: 10.1097/WAD.0b013e31821300bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kalback W, Esh C, Castano EM, et al. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease. Neurol Res. 2004;26:525–539. doi: 10.1179/016164104225017668. [DOI] [PubMed] [Google Scholar]
- 100.Arai T, Ikeda K, Akiyama H, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001;101:167–173. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 101.Evidente VG, Adler CH, Sabbagh MN, et al. Neuropathological findings of PSP in the elderly without clinical PSP: possible incidental PSP? Parkinsonism Relat Disord. 2011;17:365–371. doi: 10.1016/j.parkreldis.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnold SJ, Dugger BN, Beach TG. TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol. 2013;126:51–57. doi: 10.1007/s00401-013-1110-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 104.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 105.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 107.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- 108.Gundersen HJ, Bagger P, Bendtsen TF, et al. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 109.Beach TG, Adler CH, Sue LI, et al. Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol. 2008;115:445–451. doi: 10.1007/s00401-007-0313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beach TG, Wilson JR, Sue LI, et al. Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol (Berl) 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- 111.Roher AE, Esh C, et al. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- 112.Roher AE, Maarouf CL, Malek-Ahmadi M, et al. Subjects harboring presenilin familial Alzheimer’s disease mutations exhibit diverse white matter biochemistry alterations. Am J Neurodegener Dis. 2013;2:187–207. [PMC free article] [PubMed] [Google Scholar]
- 113.Castano EM, Maarouf CL, Wu T, et al. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem Int. 2013;62:145–156. doi: 10.1016/j.neuint.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dugger BN, Adler CH, Shill HA, et al. Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord. 2014;20:525–529. doi: 10.1016/j.parkreldis.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Choi SA, Evidente VG, Caviness JN, et al. Are there differences in cerebral white matter lesion burdens between Parkinson’s disease patients with or without dementia? Acta Neuropathol. 2010;119:147–149. doi: 10.1007/s00401-009-0620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Roher AE, Weiss N, Kokjohn TA, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- 117.Westover MB, Bianchi MT, Yang C, Schneider JA, Greenberg SM. Estimating cerebral microinfarct burden from autopsy samples. Neurology. 2013;80:1365–1369. doi: 10.1212/WNL.0b013e31828c2f52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kovari E, Gold G, Herrmann FR, et al. Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke. 2004;35:410–414. doi: 10.1161/01.STR.0000110791.51378.4E. [DOI] [PubMed] [Google Scholar]
- 120.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42:722–727. doi: 10.1161/STROKEAHA.110.595082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 122.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 124.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 125.Dickson DW. Required techniques and useful molecular markers in the neuropathologic diagnosis of neurodegenerative diseases. Acta Neuropathol. 2005;109:14–24. doi: 10.1007/s00401-004-0950-z. [DOI] [PubMed] [Google Scholar]
- 126.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dugger BN, Hentz JG, Adler CH, et al. Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol. 2014;73:244–252. doi: 10.1097/NEN.0000000000000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sabbagh MN, Sandhu SS, Farlow MR, et al. Correlation of clinical features with argyrophilic grains at autopsy. Alzheimer Dis Assoc Disord. 2009;23:229–233. doi: 10.1097/WAD.0b013e318199d833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jellinger KA, Attems J. Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm. 2014 doi: 10.1007/s00702-014-1288-x. Epub ahead of print August 5. [DOI] [PubMed] [Google Scholar]
- 130.Gusella JF, Macdonald ME. Huntington’s disease. Semin Cell Biol. 1995;6:21–28. doi: 10.1016/1043-4682(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 131.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 132.Beach TG, Sue L, Scott S, et al. Hippocampal sclerosis dementia with tauopathy. Brain Pathol. 2003;13:263–278. doi: 10.1111/j.1750-3639.2003.tb00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Meiser B, Dunn S. Psychological impact of genetic testing for Huntington’s disease: an update of the literature. J Neurol Neurosurg Psychiatry. 2000;69:574–578. doi: 10.1136/jnnp.69.5.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bruni AC, Conidi ME, Bernardi L. Genetics in degenerative dementia: current status and applicability. Alzheimer Dis Assoc Disord. 2014;28:199–205. doi: 10.1097/WAD.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 135.Le BI. Genetics of frontotemporal lobar degeneration: an up-date and diagnosis algorithm. Rev Neurol (Paris) 2013;169:811–819. doi: 10.1016/j.neurol.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 136.Fong JC, Karydas AM, Goldman JS. Genetic counseling for FTD/ALS caused by the C9ORF72 hexanucleotide expansion. Alzheimers Res Ther. 2012;4:27–36. doi: 10.1186/alzrt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Perry EK, Perry RH, Tomlinson BE. The influence of agonal status on some neurochemical activities of postmortem human brain tissue. Neurosci Lett. 1982;29:303–307. doi: 10.1016/0304-3940(82)90334-2. [DOI] [PubMed] [Google Scholar]
- 138.Morrison-Bogorad M, Zimmerman AL, Pardue S. Heat-shock 70 messenger RNA levels in human brain: correlation with agonal fever. J Neurochem. 1995;64:235–246. doi: 10.1046/j.1471-4159.1995.64010235.x. [DOI] [PubMed] [Google Scholar]
- 139.Hardy JA, Wester P, Winblad B, Gezelius C, Bring G, Eriksson A. The patients dying after long terminal phase have acidotic brains; implications for biochemical measurements on autopsy tissue. J Neural Transm. 1985;61:253–264. doi: 10.1007/BF01251916. [DOI] [PubMed] [Google Scholar]
- 140.Stan AD, Ghose S, Gao XM, et al. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yates CM, Butterworth J, Tennant MC, Gordon A. Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. J Neurochem. 1990;55:1624–1630. doi: 10.1111/j.1471-4159.1990.tb04948.x. [DOI] [PubMed] [Google Scholar]
- 142.Beach TG, Sue LI, Walker DG, et al. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Catts VS, Catts SV, Fernandez HR, Taylor JM, Coulson EJ, Lutze-Mann LH. A microarray study of post-mortem mRNA degradation in mouse brain tissue. Brain Res Mol Brain Res. 2005;138:164–177. doi: 10.1016/j.molbrainres.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 144.Karkela JT. Critical evaluation of postmortem changes in human autopsy cisternal fluid. Enzymes, electrolytes, acid-base balance, glucose and glycolysis, free amino acids and ammonia. Correlation to total brain ischemia. J Forensic Sci. 1993;38:603–616. [PubMed] [Google Scholar]
- 145.Botling J, Edlund K, Segersten U, et al. Impact of thawing on RNA integrity and gene expression analysis in fresh frozen tissue. Diagn Mol Pathol. 2009;18:44–52. doi: 10.1097/PDM.0b013e3181857e92. [DOI] [PubMed] [Google Scholar]
- 146.Nash DT, Fillit H. Cardiovascular disease risk factors and cognitive impairment. Am J Cardiol. 2006;97:1262–1265. doi: 10.1016/j.amjcard.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 147.Wolozin B, Bednar MM. Interventions for heart disease and their effects on Alzheimer’s disease. Neurol Res. 2006;28:630–636. doi: 10.1179/016164106X130515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kivipelto M, Helkala EL, Laakso MP, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ. 2001;322:1447–1451. doi: 10.1136/bmj.322.7300.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Launer LJ, Ross GW, Petrovitch H, et al. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging. 2000;21:49–55. doi: 10.1016/s0197-4580(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 151.Skoog I, Lernfelt B, Landahl S, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 152.Astarita G, Jung KM, Berchtold NC, et al. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE. 2010;5:e12538. doi: 10.1371/journal.pone.0012538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bergmann C, Sano M. Cardiac risk factors and potential treatments in Alzheimer’s disease. Neurol Res. 2006;28:595–604. doi: 10.1179/016164106X130498. [DOI] [PubMed] [Google Scholar]
- 154.Mielke MM, Zandi PP, Sjogren M, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005;64:1689–1695. doi: 10.1212/01.WNL.0000161870.78572.A5. [DOI] [PubMed] [Google Scholar]
- 155.Rosano C, Newman AB. Cardiovascular disease and risk of Alzheimer’s disease. Neurol Res. 2006;28:612–620. doi: 10.1179/016164106X130407. [DOI] [PubMed] [Google Scholar]
- 156.Sanderson M, Wang J, Davis DR, Lane MJ, Cornman CB, Fadden MK. Co-morbidity associated with dementia. Am J Alzheimers Dis Other Demen. 2002;17:73–78. doi: 10.1177/153331750201700210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wolf-Klein GP, Siverstone FA, Brod MS, et al. Are Alzheimer patients healthier? J Am Geriatr Soc. 1988;36:219–224. doi: 10.1111/j.1532-5415.1988.tb01804.x. [DOI] [PubMed] [Google Scholar]
- 158.Irina A, Seppo H, Arto M, Paavo R, Sr, Hilkka S. beta-amyloid load is not influenced by the severity of cardiovascular disease in aged and demented patients. Stroke. 1999;30:613–618. doi: 10.1161/01.str.30.3.613. [DOI] [PubMed] [Google Scholar]
- 159.Roher AE, Debbins JP, Malek-Ahmadi M, et al. Cerebral blood flow in Alzheimer’s disease. Vasc Health Risk Manag. 2012;8:599–611. doi: 10.2147/VHRM.S34874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Roher AE, Garami Z, Tyas SL, et al. Transcranial doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer’s disease. Alzheimers Dement. 2011;7:445–455. doi: 10.1016/j.jalz.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Roher AE, Esh C, Rahman A, Kokjohn TA, Beach TG. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke. 2004;35:2623–2627. doi: 10.1161/01.STR.0000143317.70478.b3. [DOI] [PubMed] [Google Scholar]
- 162.Roher AE, Esh C, Kokjohn T, Sue L, Beach T. Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology. 2005;65:974. doi: 10.1212/wnl.65.6.974. [DOI] [PubMed] [Google Scholar]
- 163.Roher AE, Esh C, Kokjohn TA, et al. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- 164.Roher AE, Tyas SL, Maarouf CL, et al. Intracranial atherosclerosis as a contributing factor to Alzheimer’s disease dementia. Alzheimers Dement. 2011;7:436–444. doi: 10.1016/j.jalz.2010.08.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Beach TG, Maarouf CL, Brooks RG, et al. Reduced clinical and postmortem measures of cardiac pathology in subjects with advanced Alzheimer’s Disease. BMC Geriatr. 2011;11:3. doi: 10.1186/1471-2318-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Belohlavek M, Jiamsripong P, Calleja AM, et al. Patients with Alzheimer disease have altered transmitral flow: echocardiographic analysis of the vortex formation time. J Ultrasound Med. 2009;28:1493–1500. doi: 10.7863/jum.2009.28.11.1493. [DOI] [PubMed] [Google Scholar]
- 167.Puddu P, Puddu GM, Bastagli L, Massarelli G, Muscari A. Coronary and cerebrovascular atherosclerosis: two aspects of the same disease or two different pathologies? Arch Gerontol Geriatr. 1995;20:15–22. doi: 10.1016/0167-4943(94)00600-c. [DOI] [PubMed] [Google Scholar]
- 168.Reed DM. The paradox of high risk of stroke in populations with low risk of coronary heart disease. Am J Epidemiol. 1990;131:579–588. doi: 10.1093/oxfordjournals.aje.a115542. [DOI] [PubMed] [Google Scholar]
- 169.Postiglione A, Nappi A, Brunetti A, et al. Relative protection from cerebral atherosclerosis of young patients with homozygous familial hyper-cholesterolemia. Atherosclerosis. 1991;90:23–30. doi: 10.1016/0021-9150(91)90240-4. [DOI] [PubMed] [Google Scholar]
- 170.Kuramoto K, Ueda S, Matsushita S, Suzuki Y, Matsumoto Y, Iijima T. Cholesterol, atherosclerosis and cerebro-cardiovascular complications in 3,236 elderly autopsy cases. Nippon Ronen Igakkai Zasshi. 1991;28:188–193. doi: 10.3143/geriatrics.28.188. [DOI] [PubMed] [Google Scholar]
- 171.Konishi M, Komachi Y, Iso H, et al. Secular trends in atherosclerosis of coronary arteries and basal cerebral arteries in Japan. The Akita pathology study. Arteriosclerosis. 1990;10:535–540. doi: 10.1161/01.atv.10.4.535. [DOI] [PubMed] [Google Scholar]
- 172.Tanaka K, Masuda J, Imamura T, et al. A nation-wide study of atherosclerosis in infants, children and young adults in Japan. Atherosclerosis. 1988;72:143–156. doi: 10.1016/0021-9150(88)90075-5. [DOI] [PubMed] [Google Scholar]
- 173.Honig LS, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology. 2005;64:494–500. doi: 10.1212/01.WNL.0000150886.50187.30. [DOI] [PubMed] [Google Scholar]
- 174.Kosunen O, Talasniemi S, Lehtovirta M, et al. Relation of coronary atherosclerosis and apolipoprotein E genotypes in Alzheimer patients. Stroke. 1995;26:743–748. doi: 10.1161/01.str.26.5.743. [DOI] [PubMed] [Google Scholar]
- 175.Traykov L, Rigaud AS, Caputo L, et al. Apolipoprotein E phenotypes in demented and cognitively impaired patients with and without cerebrovascular disease. Eur J Neurol. 1999;6:415–421. doi: 10.1046/j.1468-1331.1999.640415.x. [DOI] [PubMed] [Google Scholar]
- 176.White H, Pieper C, Schmader K. The association of weight change in Alzheimer’s disease with severity of disease and mortality: a longitudinal analysis. J Am Geriatr Soc. 1998;46:1223–1227. doi: 10.1111/j.1532-5415.1998.tb04537.x. [DOI] [PubMed] [Google Scholar]
- 177.White H, Pieper C, Schmader K, Fillenbaum G. Weight change in Alzheimer’s disease. J Am Geriatr Soc. 1996;44:265–272. doi: 10.1111/j.1532-5415.1996.tb00912.x. [DOI] [PubMed] [Google Scholar]
- 178.Wang PN, Yang CL, Lin KN, Chen WT, Chwang LC, Liu HC. Weight loss, nutritional status and physical activity in patients with Alzheimer’s disease. A controlled study. J Neurol. 2004;251:314–320. doi: 10.1007/s00415-004-0316-4. [DOI] [PubMed] [Google Scholar]
- 179.Barrett-Connor E, Edelstein SL, Corey-Bloom J, Wiederholt WC. Weight loss precedes dementia in community-dwelling older adults. J Am Geriatr Soc. 1996;44:1147–1152. doi: 10.1111/j.1532-5415.1996.tb01362.x. [DOI] [PubMed] [Google Scholar]
- 180.Gu Y, Scarmeas N, Cosentino S, et al. Change in body mass index before and after Alzheimer’s disease onset. Curr Alzheimer Res. 2014;11:349–356. doi: 10.2174/1567205010666131120110930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Beach TG, Adler CH, Sue LI, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mu L, Sobotka S, Chen J, et al. Altered pharyngeal muscles in Parkinson disease. J Neuropathol Exp Neurol. 2012;71:520–530. doi: 10.1097/NEN.0b013e318258381b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mu L, Sobotka S, Chen J, et al. Parkinson disease affects peripheral sensory nerves in the pharynx. J Neuropathol Exp Neurol. 2013;72:614–623. doi: 10.1097/NEN.0b013e3182965886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mu L, Sobotka S, Chen J, et al. Alpha-synuclein pathology and axonal degeneration of the peripheral motor nerves innervating pharyngeal muscles in Parkinson disease. J Neuropathol Exp Neurol. 2013;72:119–129. doi: 10.1097/NEN.0b013e3182801cde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Annerino DM, Arshad S, Taylor GM, Adler CH, Beach TG, Greene JG. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol. 2012;124:665–680. doi: 10.1007/s00401-012-1040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Beach TG, Carew J, Serrano G, et al. Phosphorylated alpha-synuclein-immunoreactive retinal neuronal elements in Parkinson’s disease subjects. Neurosci Lett. 2014;571:34–38. doi: 10.1016/j.neulet.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83:406–412. doi: 10.1212/WNL.0000000000000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Beach TG. Alzheimer’s disease and the “valley of death”: not enough guidance from human brain tissue? J Alzheimers Dis. 2013;33:S219–S233. doi: 10.3233/JAD-2012-129020. [DOI] [PubMed] [Google Scholar]
- 189.Beach TG, Adler CH, Dugger BN, et al. Submandibular Gland Biopsy for the Diagnosis of Parkinson Disease. J Neuropathol Exp Neurol. 2013;72:130–136. doi: 10.1097/NEN.0b013e3182805c72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Adler CH, Dugger BN, Hinni ML, et al. Submandibular gland needle biopsy for the diagnosis of Parkinson disease. Neurology. 2014;82:858–864. doi: 10.1212/WNL.0000000000000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hewitt RE. Biobanking: the foundation of personalized medicine. Curr Opin Oncol. 2011;23:112–119. doi: 10.1097/CCO.0b013e32834161b8. [DOI] [PubMed] [Google Scholar]
- 192.Massett HA, Atkinson NL, Weber D, et al. Assessing the need for a standardized cancer HUman Biobank (caHUB): findings from a national survey with cancer researchers. J Natl Cancer Inst Monogr. 2011;2011:8–15. doi: 10.1093/jncimonographs/lgr007. [DOI] [PubMed] [Google Scholar]
- 193.Myles R, Massett HA, Comey G, Atkinson N, Allsop D, Compton C. Stakeholder research on biospecimen needs and reactions to the development of a national cancer human biobank by the National Cancer Institute. J Natl Cancer Inst Monogr. 2011;2011:16–23. doi: 10.1093/jncimonographs/lgr008. [DOI] [PubMed] [Google Scholar]
- 194.Rudloff U, Bhanot U, Gerald W, et al. Biobanking of human pancreas cancer tissue: impact of ex-vivo procurement times on RNA quality. Ann Surg Oncol. 2010;17:2229–2236. doi: 10.1245/s10434-010-0959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jia Z, Rahmatpanah FB, Chen X, et al. Expression changes in the stroma of prostate cancer predict subsequent relapse. PLoS ONE. 2012;7:e41371. doi: 10.1371/journal.pone.0041371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arora S, Korn RL, Lenkiewicz E, et al. Clonal evolution of a case of treatment refractory maxillary sinus carcinoma. PLoS ONE. 2012;7:e45614. doi: 10.1371/journal.pone.0045614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Liu SV, Lenkiewicz E, Evers L, et al. Genomic analysis and selected molecular pathways in rare cancers. Phys Biol. 2012;9:065004. doi: 10.1088/1478-3975/9/6/065004. [DOI] [PubMed] [Google Scholar]
- 198.Mikse OR, Blake DC, Jr, Jones NR, et al. FOXO3 encodes a carcinogen-activated transcription factor frequently deleted in early-stage lung adenocarcinoma. Cancer Res. 2010;70:6205–6215. doi: 10.1158/0008-5472.CAN-09-4008. [DOI] [PubMed] [Google Scholar]
- 199.Hoglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hamilton G, Killick R, Lambert JC, et al. Functional and genetic analysis of haplotypic sequence variation at the nicastrin genomic locus. Neurobiol Aging. 2012;33:1848.e1–13. doi: 10.1016/j.neurobiolaging.2012.02.005. Epub 2012 Mar 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jun G, Vardarajan BN, Buros J, et al. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Arch Neurol. 2012;69:1270–1279. doi: 10.1001/archneurol.2012.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Miyashita A, Koike A, Jun G, et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE. 2013;8:e58618. doi: 10.1371/journal.pone.0058618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309:1483–1492. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Coppola G, Chinnathambi S, Lee JJ, et al. Evidence for a role of the rare p. A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21:3500–3512. doi: 10.1093/hmg/dds161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haplo-insufficiency in patients with the common 1477C-->T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 206.Chen-Plotkin AS, Martinez-Lage M, Sleiman PM, et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol. 2011;68:488–497. doi: 10.1001/archneurol.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Van DV, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.van Blitterswijk M, Mullen B, Heckman MG, et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging. 2014;35:2421–2427. doi: 10.1016/j.neurobiolaging.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.van Blitterswijk M, Mullen B, Nicholson AM, et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014;127:397–406. doi: 10.1007/s00401-013-1240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.van Blitterswijk M, Baker MC, Dejesus-Hernandez M, et al. C9ORF72 repeat expansions in cases with previously identified pathogenic mutations. Neurology. 2013;81:1332–1341. doi: 10.1212/WNL.0b013e3182a8250c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12:978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Holton P, Ryten M, Nalls M, et al. Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk Loci. Ann Hum Genet. 2013;77:85–105. doi: 10.1111/ahg.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jia Z, Wang Y, Sawyers A, et al. Diagnosis of prostate cancer using differentially expressed genes in stroma. Cancer Res. 2011;71:2476–2487. doi: 10.1158/0008-5472.CAN-10-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pio R, Jia Z, Baron VT, Mercola D. Early growth response 3 (Egr3) is highly over-expressed in non-relapsing prostate cancer but not in relapsing prostate cancer. PLoS ONE. 2013;8:e54096. doi: 10.1371/journal.pone.0054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44:1349–1354. doi: 10.1038/ng.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dumitriu A, Latourelle JC, Hadzi TC, et al. Gene expression profiles in Parkinson disease prefrontal cortex implicate FOXO1 and genes under its transcriptional regulation. PLoS Genet. 2012;8:e1002794. doi: 10.1371/journal.pgen.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Latourelle JC, Dumitriu A, Hadzi TC, Beach TG, Myers RH. Evaluation of Parkinson disease risk variants as expression-QTLs. PLoS ONE. 2012;7:e46199. doi: 10.1371/journal.pone.0046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zou F, Chai HS, Younkin CS, et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLoS Genet. 2012;8:e1002707. doi: 10.1371/journal.pgen.1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zheng B, Liao Z, Locascio JJ, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Berchtold NC, Sabbagh MN, Beach TG, Kim RC, Cribbs DH, Cotman CW. Brain gene expression patterns differentiate mild cognitive impairment from normal aged and Alzheimer’s disease. Neurobiol Aging. 2014;35:1961–1972. doi: 10.1016/j.neurobiolaging.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 224.Stamper C, Siegel A, Liang WS, et al. Neuronal gene expression correlates of Parkinson’s disease with dementia. Mov Disord. 2008;23:1588–1595. doi: 10.1002/mds.22184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liang WS, Reiman EM, Valla J, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008;105:4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liang WS, Dunckley T, Beach TG, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics. 2008;33:240–256. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Liang WS, Dunckley T, Beach TG, et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics. 2007;28:311–322. doi: 10.1152/physiolgenomics.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Liang WS, Dunckley T, Beach TG, et al. Neuronal gene expression in non-demented individuals with intermediate Alzheimer’s Disease neuropathology. Neurobiol Aging. 2008;31:549–566. doi: 10.1016/j.neurobiolaging.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Parachikova A, Agadjanyan MG, Cribbs DH, et al. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–1833. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Weeraratna AT, Kalehua A, Deleon I, et al. Alterations in immunological and neurological gene expression patterns in Alzheimer’s disease tissues. Exp Cell Res. 2007;313:450–461. doi: 10.1016/j.yexcr.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dunckley T, Beach TG, Ramsey K, et al. Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging. 2006;27:1359–1371. doi: 10.1016/j.neurobiolaging.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Coon KD, Valla J, Szelinger S, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6:194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 234.Webster JA, Myers AJ, Pearson JV, et al. Sorl1 as an Alzheimer’s disease predisposition gene? Neurodegener Dis. 2007;5:60–64. doi: 10.1159/000110789. [DOI] [PubMed] [Google Scholar]
- 235.Webster JA, Gibbs JR, Clarke J, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Finch N, Carrasquillo MM, Baker M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Burgos KL, Javaherian A, Bomprezzi R, et al. Identification of extracellular miRNA in human cerebrospinal fluid by next-generation sequencing. RNA. 2013;19:712–722. doi: 10.1261/rna.036863.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chouliaras L, Mastroeni D, Delvaux E, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging. 2013;34:2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kellie JF, Higgs RE, Ryder JW, et al. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep. 2014;4:5797. doi: 10.1038/srep05797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts Braak neurofibrillary stage and clinicopathological Alzheimer’s disease: implications for amyloid imaging. J Alzheimers Dis. 2012;28:869–876. doi: 10.3233/JAD-2011-111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Dugger BN, Serrano GE, Sue LI, et al. Presence of striatal amyloid plaques in Parkinson’s disease dementia predicts concomitant Alzheimer’s disease: usefulness for amyloid imaging. J Parkinsons Dis. 2012;2:57–65. doi: 10.3233/JPD-2012-11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Serrano GE, Sabbagh MN, Sue LI, et al. Positive florbetapir PET amyloid imaging in a subject with frequent cortical neuritic plaques and frontotemporal lobar degeneration with TDP43-positive inclusions. J Alzheimers Dis. 2014;42:813–821. doi: 10.3233/JAD-140162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 245.Thompson PW, Ye L, Morgenstern JL, et al. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. J Neurochem. 2009;109:623–630. doi: 10.1111/j.1471-4159.2009.05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ye L, Velasco A, Fraser G, et al. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J Neurochem. 2008;105:1428–1437. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kokjohn TA, Roher AE. Antibody responses, amyloid-beta peptide remnants and clinical effects of AN-1792 immunization in patients with AD in an interrupted trial. CNS Neurol Disord Drug Targets. 2009;8:88–97. doi: 10.2174/187152709787847315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Maarouf CL, Daugs ID, Kokjohn TA, et al. The biochemical aftermath of anti-amyloid immunotherapy. Mol Neurodegener. 2010;5:39. doi: 10.1186/1750-1326-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Patton RL, Kalback WM, Esh CL, et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Roher AE, Cribbs DH, Kim RC, et al. Bapineuzumab alters abeta composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapy. PLoS ONE. 2013;8:e59735. doi: 10.1371/journal.pone.0059735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Roher AE, Maarouf CL, Daugs ID, et al. Neuropathology and amyloid-beta spectrum in a bapineuzumab immunotherapy recipient. J Alzheimers Dis. 2011;24:315–325. doi: 10.3233/JAD-2011-101809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Walker DG, Dalsing-Hernandez JE, Lue LF. Human postmortem brain-derived cerebrovascular smooth muscle cells express all genes of the classical complement pathway: a potential mechanism for vascular damage in cerebral amyloid angiopathy and Alzheimer’s disease. Microvasc Res. 2008;75:411–419. doi: 10.1016/j.mvr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–956. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 254.Lue LF, Rydel R, Brigham EF, et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 255.Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem Int. 2001;39:333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- 256.Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging. 2001;22:957–966. doi: 10.1016/s0197-4580(01)00306-2. [DOI] [PubMed] [Google Scholar]
- 257.Li R, Shen Y, Yang LB, Lue LF, Finch C, Rogers J. Estrogen enhances uptake of amyloid beta-protein by microglia derived from the human cortex. J Neurochem. 2000;75:1447–1454. doi: 10.1046/j.1471-4159.2000.0751447.x. [DOI] [PubMed] [Google Scholar]
- 258.Hjelm BE, Salhia B, Kurdoglu A, et al. In vitro-differentiated neural cell cultures progress towards donor-identical brain tissue. Hum Mol Genet. 2013;22:3534–3546. doi: 10.1093/hmg/ddt208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hjelm BE, Rosenberg JB, Szelinger S, et al. Induction of pluripotent stem cells from autopsy donor-derived somatic cells. Neurosci Lett. 2011;502:219–224. doi: 10.1016/j.neulet.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Truran S, Franco DA, Roher AE, et al. Adipose and leptomeningeal arteriole endothelial dysfunction induced by beta-amyloid peptide: a practical human model to study Alzheimer’s disease vasculopathy. J Neurosci Methods. 2014;235C:123–129. doi: 10.1016/j.jneumeth.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Prasad K, Beach TG, Hedreen J, Richfield EK. Critical role of truncated alpha-synuclein and aggregates in Parkinson’s disease and incidental Lewy body disease. Brain Pathol. 2012;22:811–825. doi: 10.1111/j.1750-3639.2012.00597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Lue LF, Walker DG, Adler CH, et al. Biochemical increase in phosphorylated alpha-synuclein precedes histopathology of Lewy-type synucleinopathies. Brain Pathol. 2012;22:745–756. doi: 10.1111/j.1750-3639.2012.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Walker DG, Lue LF, Adler CH, et al. Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol. 2013;240:190–204. doi: 10.1016/j.expneurol.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Henderson-Smith A, Chow D, Meechoovet B, et al. SMG1 identified as a regulator of Parkinson’s disease-associated alpha-synuclein through siRNA screening. PLoS ONE. 2013;8:e77711. doi: 10.1371/journal.pone.0077711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Beach TG, Walker DG, Sue LI, Newell A, Adler CC, Joyce JN. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol. 2004;63:329–337. doi: 10.1093/jnen/63.4.329. [DOI] [PubMed] [Google Scholar]
- 266.Beach TG, Sue LI, Walker DG, et al. Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol (Berl) 2007;114:419–424. doi: 10.1007/s00401-007-0250-5. [DOI] [PubMed] [Google Scholar]
- 267.Joyce JN, Ryoo HL, Beach TB, et al. Loss of response to levodopa in Parkinson’s disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 2002;955:138–152. doi: 10.1016/s0006-8993(02)03396-6. [DOI] [PubMed] [Google Scholar]
- 268.Joyce JN, Ryoo H, Gurevich EV, Adler C, Beach T. Ventral striatal D(3) receptors and Parkinson’s Disease. Parkinsonism Relat Disord. 2001;7:225–230. doi: 10.1016/s1353-8020(00)00060-2. [DOI] [PubMed] [Google Scholar]
- 269.Adler CH, Hentz JG, Joyce JN, Beach T, Caviness JN. Motor impairment in normal aging, clinically possible Parkinson’s disease, and clinically probable Parkinson’s disease: longitudinal evaluation of a cohort of prospective brain donors. Parkinsonism Relat Disord. 2002;9:103–110. doi: 10.1016/s1353-8020(02)00012-3. [DOI] [PubMed] [Google Scholar]
- 270.Beach TG, Sue LI, Scott S, Sparks DL. Neurofibrillary tangles are constant in aging human nucleus basalis. Alzheimers Rep. 1998;1:375–380. [Google Scholar]
- 271.Beach TG, Kuo YM, Spiegel K, et al. The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:308–313. doi: 10.1093/jnen/59.4.308. [DOI] [PubMed] [Google Scholar]
- 272.Potter PE, Rauschkolb PK, Pandya Y, et al. Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer’s disease. Acta Neuropathol. 2011;122:49–60. doi: 10.1007/s00401-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Rogers J, Li R, Mastroeni D, et al. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27:1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 276.Strohmeyer R, Shen Y, Rogers J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Brain Res Mol Brain Res. 2000;81:7–18. doi: 10.1016/s0169-328x(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 277.Styren SD, Civin WH, Rogers J. Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer’s disease brain. Exp Neurol. 1990;110:93–104. doi: 10.1016/0014-4886(90)90054-v. [DOI] [PubMed] [Google Scholar]
- 278.Tooyama I, Sato H, Yasuhara O, et al. Correlation of the expression level of C1q mRNA and the number of C1q-positive plaques in the Alzheimer Disease temporal cortex. analysis of C1q mrna and its protein using adjacent or nearby sections. Dement Geriatr Cogn Disord. 2001;12:237–242. doi: 10.1159/000051265. [DOI] [PubMed] [Google Scholar]
- 279.Webster S, Lue LF, Brachova L, et al. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging. 1997;18:415–421. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- 280.Webster S, Bonnell B, Rogers J. Charge-based binding of complement component C1q to the Alzheimer amyloid beta-peptide. Am J Pathol. 1997;150:1531–1536. [PMC free article] [PubMed] [Google Scholar]
- 281.Webster S, Bradt B, Rogers J, Cooper N. Aggregation state-dependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem. 1997;69:388–398. doi: 10.1046/j.1471-4159.1997.69010388.x. [DOI] [PubMed] [Google Scholar]
- 282.Webster S, Rogers J. Relative efficacies of amyloid beta peptide (A beta) binding proteins in A beta aggregation. J Neurosci Res. 1996;46:58–66. doi: 10.1002/(SICI)1097-4547(19961001)46:1<58::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 283.Webster S, Glabe C, Rogers J. Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem Biophys Res Commun. 1995;217:869–875. doi: 10.1006/bbrc.1995.2852. [DOI] [PubMed] [Google Scholar]
- 284.Yermakova AV, Rollins J, Callahan LM, Rogers J, O’Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol. 1999;58:1135–1146. doi: 10.1097/00005072-199911000-00003. [DOI] [PubMed] [Google Scholar]
- 285.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–1088. [PubMed] [Google Scholar]
- 286.Lue LF, Walker DG, Brachova L, et al. Involvement of microglial receptor for advanced glycation endproducts (rage) in alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 287.Lue LF, Yan SD, Stern DM, Walker DG. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord. 2005;4:249–266. doi: 10.2174/1568007054038210. [DOI] [PubMed] [Google Scholar]
- 288.Beach TG, Sue LI, Walker DG, et al. Marked microglial reaction in normal aging human substantia nigra: correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 2007;114:419–424. doi: 10.1007/s00401-007-0250-5. [DOI] [PubMed] [Google Scholar]
- 289.Nakahara H, Konishi Y, Beach TG, Yamada N, Makino S, Tooyama I. Infiltration of T lymphocytes and expression of icam-1 in the hippocampus of patients with hippocampal sclerosis. Acta Histochem Cytochem. 2010;43:157–162. doi: 10.1267/ahc.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Morimoto K, Horio J, Satoh H, et al. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients compared to the brains of non-demented patients with and without increasing AD pathology. J Alzheimers Dis. 2011;25:59–76. doi: 10.3233/JAD-2011-101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tharp WG, Lee YH, Greene SM, Vincellete E, Beach TG, Pratley RE. Measurement of altered AbetaPP isoform expression in frontal cortex of patients with Alzheimer’s disease by absolute quantification real-time PCR. J Alzheimers Dis. 2012;29:449–457. doi: 10.3233/JAD-2011-111337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Konishi Y, Beach T, Sue LI, Hampel H, Lindholm K, Shen Y. The temporal localization of frame-shift ubiquitin-B and amyloid precursor protein, and complement proteins in the brain of non-demented control patients with increasing Alzheimer’s disease pathology. Neurosci Lett. 2003;348:46–50. doi: 10.1016/s0304-3940(03)00567-6. [DOI] [PubMed] [Google Scholar]
- 293.Caselli RJ, Walker D, Sue L, Sabbagh M, Beach T. Amyloid load in nondemented brains correlates with APOE e4. Neurosci Lett. 2010;473:168–171. doi: 10.1016/j.neulet.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kane MD, Lipinski WJ, Callahan MJ, et al. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci. 2000;20:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE. Isolation, chemical characterization, and quantitation of A beta 3- pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun. 1997;237:188–191. doi: 10.1006/bbrc.1997.7083. [DOI] [PubMed] [Google Scholar]
- 296.Kuo YM, Webster S, Emmerling MR, De Lima N, Roher AE. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of Abeta peptides of Alzheimer’s disease. Biochim Biophys Acta. 1998;1406:291–298. doi: 10.1016/s0925-4439(98)00014-3. [DOI] [PubMed] [Google Scholar]
- 297.Kuo YM, Kokjohn TA, Kalback W, et al. Amyloid-beta peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem Biophys Res Commun. 2000;268:750–756. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 298.Kuo YM, Kokjohn TA, Beach TG, et al. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem. 2001;276:12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- 299.Kuo YM, Emmerling MR, Bisgaier CL, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Commun. 1998;252:711–715. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- 300.Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Roher AE, Baudry J, Chaney MO, Kuo YM, Stine WB, Emmerling MR. Oligomerizaiton and fibril asssembly of the mayloid-beta protein. Biochim Biophys Acta. 2000;1502:31–43. doi: 10.1016/s0925-4439(00)00030-2. [DOI] [PubMed] [Google Scholar]
- 302.Van Vickle GD, Esh CL, Kokjohn TA, et al. Presenilin-1 280Glu-->Ala mutation alters C-terminal APP processing yielding longer abeta peptides: implications for Alzheimer’s disease. Mol Med. 2008;14:184–194. doi: 10.2119/2007-00094.VanVickle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Kokjohn TA, Van Vickle GD, Maarouf CL, et al. Chemical characterization of pro-inflammatory amyloid-beta peptides in human atherosclerotic lesions and platelets. Biochim Biophys Acta. 2011;1812:1508–1514. doi: 10.1016/j.bbadis.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kuo YM, Emmerling MR, Lampert HC, et al. High levels of circulating Abeta42 are sequestered by plasma proteins in Alzheimer’s disease. Biochem Biophys Res Commun. 1999;257:787–791. doi: 10.1006/bbrc.1999.0552. [DOI] [PubMed] [Google Scholar]
- 305.Maarouf CL, Daugs ID, Spina S, et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol Neurodegener. 2008;3:20. doi: 10.1186/1750-1326-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Maarouf CL, Daugs ID, Kokjohn TA, et al. Alzheimer’s disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS ONE. 2011;6:e27291. doi: 10.1371/journal.pone.0027291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Roher AE, Kokjohn TA, Esh C, et al. The human amyloid-beta precursor protein770 mutation V717F generates peptides longer than amyloid-beta-(40–42) and flocculent amyloid aggregates. J Biol Chem. 2004;279:5829–5836. doi: 10.1074/jbc.M311380200. [DOI] [PubMed] [Google Scholar]
- 308.Decourt B, Gonzales A, Beach TG, et al. BACE1 levels by APOE genotype in non-demented and Alzheimer’s post-mortem brains. Curr Alzheimer Res. 2013;10:309–315. doi: 10.2174/1567205011310030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Han P, Tang Z, Yin J, et al. Pituitary adenylate cyclase-activating polypeptide protects against beta-amyloid toxicity. Neurobiol Aging. 2014;35:2064–2071. doi: 10.1016/j.neurobiolaging.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 310.Han P, Liang W, Baxter LC, et al. Pituitary adenylate cyclase-activating polypeptide is reduced in Alzheimer disease. Neurology. 2014;82:1724–1728. doi: 10.1212/WNL.0000000000000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Boeve BF, Silber MH, Ferman TJ, et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013;14:754–762. doi: 10.1016/j.sleep.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Adler CH, Hentz JG, Shill HA, et al. Probable RBD is increased in Parkinson’s disease but not in essential tremor or restless legs syndrome. Parkinsonism Relat Disord. 2011;17:456–458. doi: 10.1016/j.parkreldis.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Adler CH, Shill HA, Beach TG. Essential tremor and Parkinson’s disease: lack of a link. Mov Disord. 2011;26:372–377. doi: 10.1002/mds.23509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Shill HA, Adler CH, Hentz JG. Purkinje cell loss in essential tremor. Mov Disord. 2014;29:496–500. doi: 10.1002/mds.25845. [DOI] [PubMed] [Google Scholar]
- 315.Shill HA, Adler CH, Beach TG. Pathology in essential tremor. Parkinsonism Relat Disord. 2012;18 (Suppl 1):S135–S137. doi: 10.1016/S1353-8020(11)70042-6. [DOI] [PubMed] [Google Scholar]
- 316.Shill HA, Adler CH, Beach TG, et al. Brain biochemistry in autopsied patients with essential tremor. Mov Disord. 2012;27:113–117. doi: 10.1002/mds.24004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Shill HA, De LV, Samanta J, Stacy M. Motor learning in essential tremor. Mov Disord. 2009;24:926–928. doi: 10.1002/mds.22479. [DOI] [PubMed] [Google Scholar]
- 318.Shneyder N, Adler CH, Hentz JG, et al. Autonomic complaints in patients with restless legs syndrome. Sleep Med. 2013;14:1413–1416. doi: 10.1016/j.sleep.2013.08.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Symanski C, Shill HA, Dugger B, et al. Essential tremor is not associated with cerebellar Purkinje cell loss. Mov Disord. 2014;29:496–500. doi: 10.1002/mds.25845. [DOI] [PubMed] [Google Scholar]
- 320.Shill HA, Adler CH, Sabbagh MN, et al. Pathologic findings in prospectively ascertained essential tremor subjects. Neurology. 2008;70:1452–1455. doi: 10.1212/01.wnl.0000310425.76205.02. [DOI] [PubMed] [Google Scholar]
- 321.Caviness JN, Adler CH, Hentz JG, et al. Incidental Lewy body disease: electrophysiological findings suggesting pre-clinical Lewy body disorders. Clin Neurophysiol. 2011;122:2426–2432. doi: 10.1016/j.clinph.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Caviness JN, Sabbagh M, Connor D, et al. Quantitative electroencephalography can distinguish between Parkinson’s disease patient groups classified according to clinical cognitive dysfunction and neuropsychometric performance. Neurology. 2006;66 (5 Suppl 2):A210–A211. [Google Scholar]
- 323.Caviness JN, Adler CH, Beach TG, Wetjen KL, Caselli RJ. Small-amplitude cortical myoclonus in Parkinson’s disease: physiology and clinical observations. Mov Disord. 2002;17:657–662. doi: 10.1002/mds.10177. [DOI] [PubMed] [Google Scholar]
- 324.Caviness JN, Smith BE, Clarke SJ, et al. Motor unit number estimates in idiopathic Parkinson’s disease. Parkinsonism Relat Disord. 2002;8:161–164. doi: 10.1016/s1353-8020(01)00007-4. [DOI] [PubMed] [Google Scholar]
- 325.Caviness JN, Lue LF, Beach TG, et al. Parkinson’s disease, cortical dysfunction, and alpha-synuclein. Mov Disord. 2011;26:1436–1442. doi: 10.1002/mds.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Caviness JN, Adler CH, Sabbagh MN, Connor DJ, Hernandez JL, Lagerlund TD. Abnormal corticomuscular coherence is associated with the small amplitude cortical myoclonus in Parkinson’s disease. Mov Disord. 2003;18:1157–1162. doi: 10.1002/mds.10525. [DOI] [PubMed] [Google Scholar]
- 327.Caviness JN, Adler CH, Caselli RJ, Hernandez JL. Electrophysiology of the myoclonus in dementia with Lewy bodies. Neurology. 2003;60:523–524. doi: 10.1212/wnl.60.3.523. [DOI] [PubMed] [Google Scholar]
- 328.Caviness JN, Hentz JG, Evidente VG, et al. Both early and late cognitive dysfunction affects the electroencephalogram in Parkinson’s disease. Parkinsonism Relat Disord. 2007;13:348–354. doi: 10.1016/j.parkreldis.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 329.Caviness JN, Shill HA, Sabbagh MN, Evidente VG, Hernandez JL, Adler CH. Corticomuscular coherence is increased in the small postural tremor of Parkinson’s disease. Mov Disord. 2006;21:492–499. doi: 10.1002/mds.20743. [DOI] [PubMed] [Google Scholar]
- 330.Klassen BT, Hentz JG, Shill HA, et al. Quantitative EEG as a predictive biomarker for Parkinson disease dementia. Neurology. 2011;77:118–124. doi: 10.1212/WNL.0b013e318224af8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Sabbagh MN, Lahti T, Connor DJ, et al. Functional ability correlates with cognitive impairment in Parkinson’s disease and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;24:327–334. doi: 10.1159/000108340. [DOI] [PubMed] [Google Scholar]
- 332.Sabbagh MN, Cooper K, DeLange J, et al. Functional, global and cognitive decline correlates to accumulation of Alzheimer’s pathology in MCI and AD. Curr Alzheimer Res. 2010;7:280–286. doi: 10.2174/156720510791162340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sabbagh MN, Silverberg N, Bircea S, et al. Is the functional decline of Parkinson’s disease similar to the functional decline of Alzheimer’s disease? Parkinsonism Relat Disord. 2005;11:311–315. doi: 10.1016/j.parkreldis.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 334.Adler CH. Mild cognitive impairment in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15 (Suppl 3):S81–S82. doi: 10.1016/S1353-8020(09)70787-4. [DOI] [PubMed] [Google Scholar]
- 335.Caviness JN, Driver-Dunckley E, Connor DJ, et al. Defining mild cognitive impairment in Parkinson’s disease. Mov Disord. 2007;22:1272–1277. doi: 10.1002/mds.21453. [DOI] [PubMed] [Google Scholar]
- 336.Sabbagh MN, Shah F, Reid RT, et al. Pathologic and nicotinic receptor binding differences between mild cognitive impairment, Alzheimer disease, and normal aging. Arch Neurol. 2006;63:1771–1776. doi: 10.1001/archneur.63.12.1771. [DOI] [PubMed] [Google Scholar]
- 337.Adler CH, Connor DJ, Hentz JG, et al. Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord. 2010;25:642–646. doi: 10.1002/mds.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Caviness JN, Adler CH, Beach TG, Wetjen KL, Caselli RJ. Myoclonus in Lewy body disorders. Adv Neurol. 2002;89:23–30. [PubMed] [Google Scholar]
- 339.Damian A, Adler CH, Hentz JG, et al. Autonomic function, as self-reported on the SCOPA-autonomic questionnaire, is normal in essential tremor but not in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:1089–1093. doi: 10.1016/j.parkreldis.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Okuda T, Shiotani S, Sakamoto N, Kobayashi T. Background and current status of postmortem imaging in Japan: short history of “Autopsy imaging (Ai)”. Forensic Sci Int. 2013;225:3–8. doi: 10.1016/j.forsciint.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 341.Beach TG, Schneider JA, Sue LI, et al. Theoretical impact of Florbetapir (18F) amyloid imaging on diagnosis of alzheimer dementia and detection of preclinical cortical amyloid. J Neuropathol Exp Neurol. 2014;73:948–953. doi: 10.1097/NEN.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Leinonen V, Koivisto AM, Savolainen S, et al. Amyloid and tau proteins in cortical brain biopsy and Alzheimer’s disease. Ann Neurol. 2010;68:446–453. doi: 10.1002/ana.22100. [DOI] [PubMed] [Google Scholar]
- 344.Rinne JO, Wong DF, Wolk DA, et al. [(18)F]Flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid beta detection in living subjects with normal pressure hydrocephalus: pooled analysis of four studies. Acta Neuropathol. 2012;124:833–845. doi: 10.1007/s00401-012-1051-z. [DOI] [PubMed] [Google Scholar]
- 345.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]