

Abstract
Vilazodone is a selective serotonin reuptake inhibitor and a 5-HT1A receptor partial agonist that is approved for treatment of major depressive disorder in adults in the USA and Mexico. The efficacy, safety, and tolerability of vilazodone for generalized anxiety disorder (GAD) were investigated in a clinical trial (NCT01766401 ClinicalTrials.gov). Participants (18–70 years, inclusive) who met Diagnostic and Statistical Manual of Mental Disorders, 4th ed., text revision, criteria for GAD were randomized (1 : 1) to placebo or flexible-dose vilazodone (20–40 mg/day) for 8 weeks of double-blind treatment. Primary and secondary efficacy parameters were changes from baseline to week 8 in Hamilton Rating Scale for Anxiety and Sheehan Disability Scale total scores, respectively. Analysis was based on a mixed-effects model for repeated measures approach on the intent-to-treat population. The intent-to-treat population comprised 395 patients (placebo=197, vilazodone=198); 77% completed the study. The least squares mean difference in change from baseline to week 8 in the Hamilton Rating Scale for Anxiety total score was statistically significant for vilazodone versus placebo [−1.50 (−2.96, −0.04), P=0.0438]. The mean change from baseline to week 8 in the Sheehan Disability Scale total score for vilazodone versus placebo was not statistically significant. Adverse events were reported in 60% of placebo-treated and 83% of vilazodone-treated patients. This was a positive clinical trial of 20–40 mg/day vilazodone versus placebo in the treatment of GAD.
Keywords: antidepressant, generalized anxiety disorder, 5-HT1A receptor partial agonist, major depressive disorder, selective serotonin reuptake inhibitor, vilazodone
Introduction
Generalized anxiety disorder (GAD) is a condition of excessive and persistent worry about future events, in which patients have a distorted perception of risks and threats, particularly pertaining to the health, security, and welfare of themselves and their immediate family members (Allgulander, 2012). GAD, which tends to run a waxing and waning course in nonclinical samples (Angst et al., 2009) and a prolonged course in primary care (Rodriguez et al., 2006), is associated with key physical and psychological symptoms including restlessness, somatic symptoms, difficulty concentrating, and disturbed sleep (American Psychiatric Association, 2013). Because the use of different diagnostic criteria and cultural variability affect the diagnosis of GAD globally, data on the disorder worldwide are not standardized nor widely available. Worldwide epidemiology estimates for GAD vary extensively, with a survey of mental disorders in 15 countries finding the prevalence of GAD to be in the range of 1–22% (World Health Organization, 2001). The 12-month prevalence of GAD in the USA and Europe is similar, with rates estimated at 2.9% among US adults (Kessler et al., 2012) and 1.7–3.4% (depending on age) among individuals in the EU (Wittchen et al., 2011) in the community setting. The lifetime morbid risk, indicating the proportion of people who have GAD plus the proportion who will eventually develop it, is estimated at 9% of the US population (Kessler et al., 2012).
GAD, which often occurs comorbidly with major depression, is as seriously impairing as a major depressive episode (Wittchen, 2002). Functional and occupational impairments associated with GAD can lead to diverse issues including poor health-related quality of life, overutilization of medical resources, excess medical costs, and low work productivity (Revicki et al., 2012). Agents from various drug classes are used to treat GAD, but as many as 50% of patients have inadequate response (Buoli et al., 2013), constituting a considerable unmet medical need.
Vilazodone is a selective serotonin reuptake inhibitor (SSRI) and 5-HT1A receptor partial agonist approved by the US Food and Drug Administration (FDA) for the treatment of major depressive disorder (MDD) in adults. The efficacy of vilazodone in MDD was established in two short-term, double-blind, placebo-controlled phase III trials (NCT00285376 and NCT00683592; Rickels et al., 2009; Khan et al., 2011). Two positive phase IV clinical trials (NCT01473394 and NCT01473381; Croft et al., 2014; Mathews et al., 2015) have recently added additional evidence to the foundation of support for vilazodone in MDD. The recommended dose for vilazodone is 20–40 mg/day (Vilazodone, 2015), which is the dose level being evaluated for the treatment of GAD. Safety and tolerability findings in MDD were supported in a 1-year, open-label trial of 40 mg/day vilazodone (NCT00644358; Robinson et al., 2011). Vilazodone was generally well tolerated in all trials; common adverse events (AEs), including diarrhea, nausea, vomiting, and insomnia, were generally transient in nature and considered mild in severity (Laughren et al., 2011; Liebowitz et al., 2011).
Pharmacotherapeutic efficacy in the treatment of GAD and similar conditions has been demonstrated by agents from various drug classes including serotonin–norepinephrine reuptake inhibitors (SNRIs; duloxetine, venlafaxine), benzodiazepines (alprazolam, diazepam, lorazepam), and SSRIs (citalopram, escitalopram, paroxetine, sertraline), in addition to the 5-HT1A receptor partial agonist, buspirone, and the anticonvulsant, pregabalin (Bandelow et al., 2012; Baldwin et al., 2014). Evidence of efficacy for both SSRIs and buspirone suggests that vilazodone, with its proposed combined SSRI and 5-HT1A receptor partial agonist mechanism of action, may have anxiolytic potential that could be effective in treating GAD. In the two pivotal studies on vilazodone in MDD (Rickels et al., 2009; Khan et al., 2011) and in a pooled post-hoc analysis of patients with anxious depression from those studies (Thase et al., 2014), improvement in anxiety symptoms was demonstrated by statistically significant differences in favor of vilazodone over placebo in the mean change from baseline to week 8 in total score on the Hamilton Rating Scale for Anxiety (HAMA; Hamilton, 1959).
Anxiolytic potential for vilazodone has also been suggested by results from phase IV studies on MDD. In one study (Mathews et al., 2015), the decrease in the mean HAMA total score from baseline to week 8 was greater for 20 and 40 mg/day vilazodone than for placebo, but the difference was not statistically significant; in the other study (Croft et al., 2014), a statistically significant difference in the mean HAMA total score reduction for 40 mg/day vilazodone versus placebo was observed at week 4 and persisted until the end of treatment. Collectively, these findings encouraged additional investigation of vilazodone for the treatment of patients with GAD. The objective of the current study was to evaluate the efficacy, safety, and tolerability of 20–40 mg/day vilazodone relative to placebo in adult patients with GAD.
Methods
The study was conducted at 30 US study centers between January 2013 and January 2014 in full compliance with FDA guidelines for Good Clinical Practice and the ethical principles of the Declaration of Helsinki. The protocol was approved by each site’s institutional review board, and all patients provided written informed consent.
Study design
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, flexible-dose study of vilazodone in adult patients with GAD. The overall study duration was 10 weeks, consisting of a 1-week screening period, an 8-week double-blind treatment period, and a 1-week double-blind down-taper period. All patients were eligible to enter the double-blind down-taper if it was considered medically appropriate by the investigator.
Following screening, eligible patients were randomized (1 : 1) to identically appearing pills of placebo or 20–40 mg/day vilazodone, to be taken once daily with food. All patients who were randomized to vilazodone received 10 mg/day during week 1 and 20 mg/day during week 2. At the end of weeks 2 and 4, patients with inadequate response and no significant tolerability issues could have their dose increased to 40 mg/day; patients with adequate response continued taking 20 mg/day. Patients were blinded to all dose increases, and no increases were allowed after the end of week 4.
Patients were randomized by computer-generated numbers; investigators and patients were blinded to the allocation of study drug throughout treatment and down-taper. The blind was maintained through a secured randomization code list and was broken only in the case of emergency. Removing the blind for any reason disqualified a patient from further participation.
Patients
Male and female outpatients who were between 18 and 70 years of age (inclusive) and who met the Diagnostic and Statistical Manual of Mental Disorders, 4th ed. – text revision (DSM-IV-TR; American Psychiatric Association, 2000) criteria for GAD were included. At screening, patients had a HAMA total score of 20 or higher, HAMA item 1 (anxious mood) score of 2 or higher, HAMA item 2 (tension) score of 2 or higher, Clinical Global Impressions–Severity of Illness (CGI-S; Guy, 1976) score of 4 or higher, and 17-item Hamilton Depression Rating Scale (HAMD17; Hamilton, 1960) total score of 17 or lower. BMI between 18 and 40 kg/m2 (inclusive) was required, and female patients of childbearing potential were required to have a negative serum β-human chorionic gonadotropin pregnancy test. Patients had normal physical examination, clinical laboratory, and ECG findings, or abnormal results that were judged to be not clinically significant.
Patients were excluded if they had a DSM-IV-TR-based Axis I diagnosis other than GAD within 6 months; secondary diagnoses of comorbid social anxiety disorder and/or specific phobias were allowed. In addition, patients were excluded if they had a lifetime history of meeting the criteria for various other psychiatric disorders (e.g. bipolar disorder, psychotic disorder, depressive episode with psychotic or catatonic features), suicide risk, substance abuse within 6 months, nonresponse to an adequate trial (≥8 weeks at an adequate dose based on approved package insert) of two or more SSRIs or SNRIs for GAD treatment, or intolerance/hypersensitivity to vilazodone, SNRIs, or SSRIs. Psychoactive drugs or required concomitant treatments with prohibited medications were prohibited; eszopiclone, zopiclone, zaleplon, or zolpidem could be continued for insomnia. Concurrent medical conditions that might have interfered with the conduct of the study, confounded the interpretation of study results, or endangered the patient’s well-being were also exclusionary.
Efficacy and safety assessments
Efficacy was assessed using several measures at various weeks corresponding to study visits. The primary outcome was the HAMA score, which was assessed at weeks −1 (screening), 0 (baseline), 1, 2, 4, 6, and 8 (all study visits). The secondary outcome was the Sheehan Disability Scale score (SDS; Sheehan et al., 1996), which was assessed at weeks 0, 2, 4, 6, and 8. Additional outcome measures comprised HAMD17 (weeks −1, 0, 8), CGI-S (all study visits), and the CGI-Improvement Scale (CGI-I; Guy, 1976; weeks 1, 2, 4, 6, 8). Safety was assessed on the basis of AE reports (coded by MedDRA, version 16.1), physical examination, clinical laboratory and vital sign measures, ECG findings, the Columbia-Suicide Severity Rating Scale (C-SSRS; Posner et al., 2011; all study visits and down-taper), and the Changes in Sexual Functioning Questionnaire (CSFQ; Clayton et al., 1997; weeks 0, 4, 8).
Statistical analyses
The safety population consisted of all randomized patients who took one or more dose of the double-blind study drug; the intent-to-treat (ITT) population consisted of all patients in the safety population who had a baseline HAMA assessment and one or more postbaseline HAMA assessments. Efficacy and safety analyses were based on the ITT and safety populations, respectively; all statistical tests were two-sided hypothesis tests performed at the 5% level of significance for main effects, and confidence intervals were two-sided 95% confidence intervals.
The primary efficacy parameter was change from baseline to week 8 in HAMA total score. The prespecified primary analysis was carried out using a mixed-effects model for repeated measures (MMRM), with treatment group, pooled study center, visit, and the treatment group×visit interaction as fixed effects, and the baseline value and the baseline value×visit interaction as covariates; the analysis was based on observed cases without imputation of missing values. An unstructured covariance matrix was used to model the covariance of within-patient scores; the Kenward–Roger approximation (Kenward and Roger, 1997) was used to estimate denominator degrees of freedom. Two prospectively defined sensitivity analyses were carried out on the primary efficacy parameter: a pattern-mixture model analysis based on non-future-dependent missing value restrictions (Kenward et al., 2003), which tested for violations of the missing at random missingness assumption, and a last observation carried forward (LOCF) analysis, which is a more conservative imputation method than MMRM. Both sensitivity analyses were based on an analysis of covariance model, with treatment group and pooled study center as factors and the baseline HAMA total score as a covariate.
The secondary efficacy parameter was change from baseline to week 8 in SDS total score. Analysis was carried out using an MMRM approach that was similar to the approach used for the primary efficacy parameter on a modified ITT population consisting of patients with evaluable assessments on all three individual SDS domain items (work/school, social life, family life). The SDS total score was calculated as the sum of the scores of the individual domain items. A prespecified LOCF sensitivity analysis was also carried out. To control the overall familywise type I error rate using multiple comparisons when testing the primary and secondary efficacy parameters, the fixed sequence testing procedure was applied; analyses of the secondary efficacy parameter were carried out inferentially only if the null hypothesis for the primary efficacy parameter was rejected.
Additional efficacy parameters included change from baseline to week 8 in the HAMA psychic anxiety and somatic anxiety subscales (Hamilton, 1959), HAMA items 1 and 2, SDS domain items (work/school, social life, family life), HAMD17 score, and CGI-S score. The CGI-I score at week 8 and the rates of response on the HAMA (≥50% improvement from baseline) and CGI-I (score ≤2) scales were also evaluated. The HAMA psychic anxiety subscale comprised items 1 (anxious mood), 2 (tension), 3 (fears), 4 (insomnia), 5 (intellectual), 6 (depressed mood), and 14 (anxious behavior at interview). The HAMA somatic anxiety subscale comprised items 7 [somatic (muscular)], 8 [somatic (sensory)], 9 (cardiovascular symptoms), 10 (respiratory symptoms), 11 (gastrointestinal symptoms), 12 (genitourinary symptoms), and 13 (autonomic symptoms).
Rates of response on the HAMA and CGI-I scales were analyzed using a generalized linear mixed model with random intercept and fixed terms of treatment group, visit, treatment-by-visit interaction, and baseline score. Other additional efficacy parameters were analyzed using an MMRM approach that was similar to that used for the primary efficacy parameter; for the CGI-I score, the baseline CGI-S score was used as an explanatory variable. Analyses of additional efficacy outcomes were not controlled for multiple comparisons. Safety analyses included the number and percentage of patients with AEs recorded in response to a nonleading question; descriptive statistics were used to evaluate change from baseline in laboratory values and vital signs.
Results
Double-blind treatment was completed by 77% of patients (Fig. 1). The difference in the rate of discontinuations for vilazodone-treated patients (28%) compared with placebo-treated patients (19%) was statistically significant (P=0.0329), as was the difference in discontinuations due to AEs (vilazodone=11%, placebo=4%, P=0.0061).
Fig. 1.
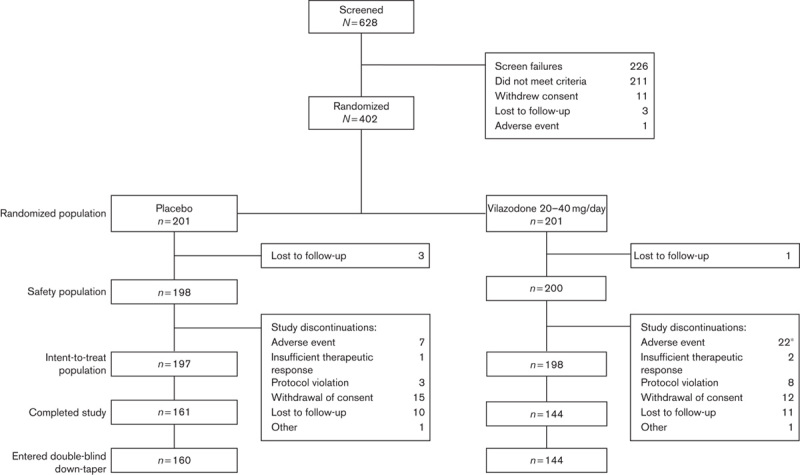
Patient disposition. *P<0.05 versus placebo (Fisher’s exact test).
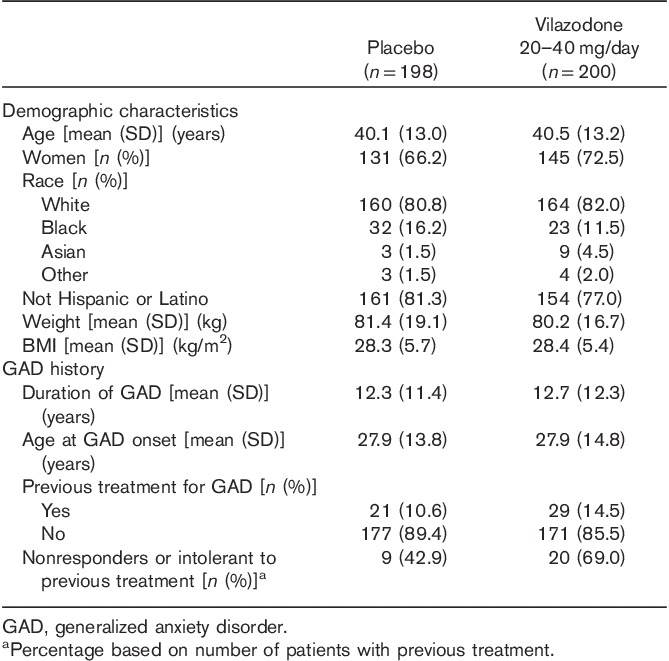
Baseline demographic characteristics and GAD history were similar in the vilazodone-treatment and placebo-treatment groups (Table 1); the mean age of the patients was ~40 years, and the majority of patients were white (81%) and female (69%). Most patients had not received previous treatment for GAD; of the patients who had received prior GAD treatment, 43% of placebo-treated patients and 69% of vilazodone-treated patients reported nonresponse or intolerance to one or more treatment. Other psychiatric disorders in addition to GAD were reported in 31% of patients in the placebo-treatment group and 35% of patients in the vilazodone-treatment group. Mood disorders, which were the most frequently reported psychiatric comorbidities, were reported in 25% of patients overall and at similar rates in the two treatment groups. The mean baseline scores were similar between groups on most efficacy measures; small but statistically significant differences were observed in the HAMA total score (placebo=24.9, vilazodone=25.9, P=0.0040) and somatic anxiety subscale score (placebo=10.3, vilazodone=11.0, P=0.0165). Per protocol, all patients had a CGI-S score of 4 (moderately ill) or higher at baseline; the majority of patients were considered moderately ill (65%). The mean baseline HAMD17 scores (∼13 in both groups) suggested a nondepressed or mildly clinically depressed patient population (Zimmerman et al., 2013).
Table 1.
Demographic characteristics and GAD history (safety population)
Analysis of efficacy
Significantly greater improvement was seen for vilazodone-treated patients compared with placebo-treated patients on the primary efficacy parameter, change from baseline to week 8 in the HAMA total score (Table 2, Fig. 2); the effect size was 0.22. In prespecified sensitivity analyses, the primary MMRM analysis was supported by pattern-mixture model analysis; on the basis of LOCF analysis, the difference between vilazodone and placebo was not statistically significant (data not shown). Lack of statistical significance with the more conservative LOCF analysis does not diminish the robustness of the primary results, as this study was powered to detect differences using MMRM analysis; the ability to detect treatment differences generally requires a larger sample size when using an LOCF analysis relative to an MMRM analysis.
Table 2.
Primary and secondary efficacy (ITT population)
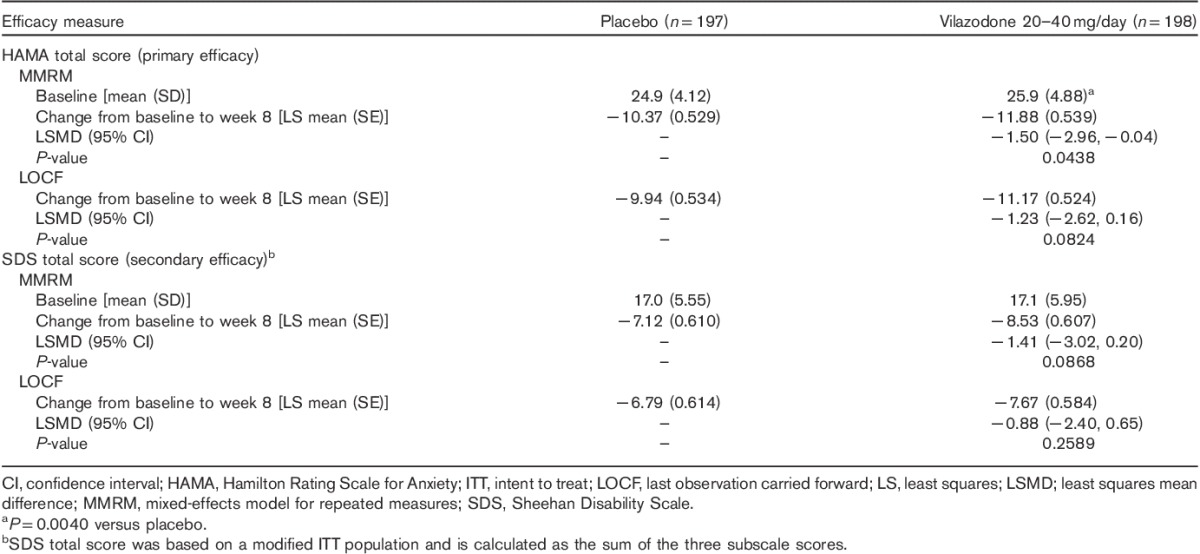
Fig. 2.
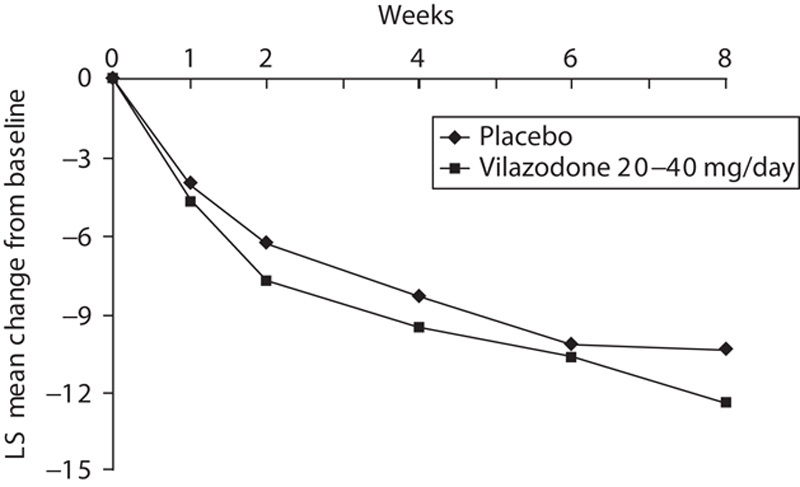
HAMA least squares mean change from baseline (ITT population, MMRM). HAMA, Hamilton Rating Scale for Anxiety; ITT, intent to treat; MMRM, mixed-effects model for repeated measures; LS, least squares. *P<0.05 for 20–40 mg/day vilazodone versus placebo.
On the secondary efficacy parameter, change from baseline to week 8 in the SDS total score, mean decreases in the score, which indicate improvement on the scale, were observed in both vilazodone and placebo groups. The difference between groups was not statistically significant (Table 2).
Additional efficacy parameters are presented in Table 3. Statistically significant improvement in favor of vilazodone versus placebo was observed on the HAMA psychic anxiety subscale, SDS work/school item, and HAMD17 total score. The differences between groups were not statistically significant for other additional efficacy parameters.
Table 3.
Additional efficacy parameters (intent to treat population)

Safety and tolerability
Extent of exposure
The mean treatment duration was 50.4 days for the placebo-treatment group and 47.4 days for the vilazodone-treatment group; patient-years of exposure (total treatment duration in days divided by 365.25) were 27.3 for placebo and 26.0 for vilazodone. At the end of the double-blind treatment period, the final daily vilazodone dose was 40 mg for 63% of patients and 20 mg for 33% of patients.
Adverse events
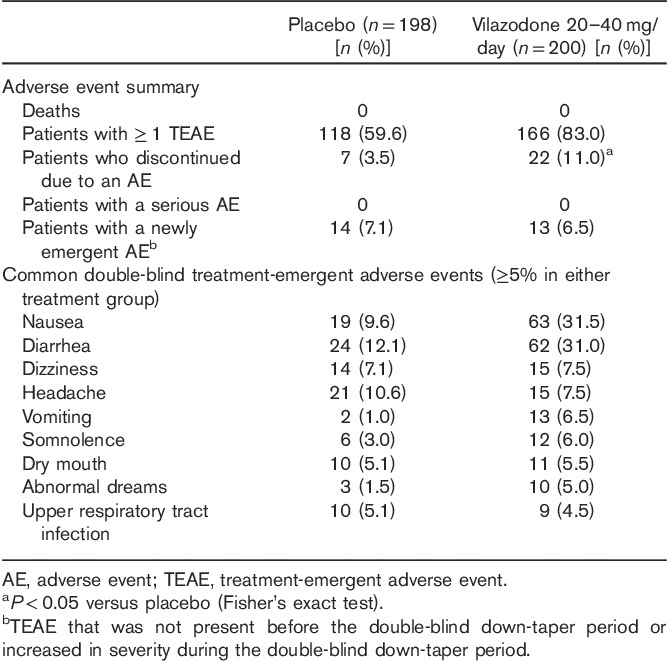
An overall summary of AEs is presented in Table 4. More vilazodone-treated patients (83%) than placebo-treated patients (60%) reported treatment-emergent AEs (TEAEs), and the incidence of discontinuation due to AEs was significantly higher for vilazodone-treated patients than for placebo-treated patients (P<0.05). Diarrhea, nausea, vomiting, somnolence, and abnormal dreams were the TEAEs that were reported in at least 5% of patients in the vilazodone-treatment group and at a rate at least twice that of the placebo-treatment group. The majority of TEAEs were considered mild or moderate in intensity among patients in the placebo-treatment (57%) and vilazodone-treatment (76%) groups. TEAEs were considered to be related to double-blind treatment in 44% of placebo-treated patients and 72% of vilazodone-treated patients. The most common TEAEs reported in the vilazodone group were nausea and diarrhea, 97% of which were considered by the investigator to be mild or moderate. Of patients treated with vilazodone, six (3%) discontinued because of nausea and two (1%) discontinued because of diarrhea; no placebo-treated patients discontinued because of these TEAEs. There were no deaths or serious AEs reported during this study.
Table 4.
Adverse events (safety population)
Clinical laboratory, vital sign, ECG evaluations
Overall, the mean changes in clinical laboratory values and vital signs were low and similar between treatment groups. There were no notable mean changes from baseline to the end of double-blind treatment in liver enzyme/function parameters in either the placebo-treatment group or the vilazodone-treatment group. No patient met the criteria for Hy’s law [alanine aminotransferase or aspartate aminotransferase elevation ≥3× upper limit of normal (ULN), total bilirubin elevation >2× ULN, and alkaline phosphatase <2× ULN], and fewer than 5% of patients in each treatment group shifted from normal baseline values to high values at the end of treatment for alanine aminotransferase, aspartate aminotransferase, and total bilirubin.
There was a higher mean increase in serum creatinine in the vilazodone-treatment group (0.055 mg/dl) compared with the placebo-treatment group (0.0113 mg/dl), but no clinically meaningful difference was observed in the incidence of patients who shifted from normal baseline values to high values at the end of treatment (placebo=5%, vilazodone=8%). A higher percentage of vilazodone-treated compared with placebo-treated patients shifted from normal baseline values to high values at the end of treatment for total cholesterol (18 vs. 11%), glucose (10 vs. 4%), and triglycerides (19 vs. 12%).
Orthostatic hypotension (reduction in systolic blood pressure of ≥20 mmHg or reduction in diastolic blood pressure of ≥10 mmHg while changing from the supine position to the standing position) was low in the placebo-treatment (8%) and vilazodone-treatment (5%) groups. At the end of treatment, there were no notable changes in the mean body weight in either treatment group (placebo=0.21 kg, vilazodone=0.13 kg). No patient had a QTc Bazett or QTc Fridericia interval increase of greater than 500 ms.
Suicidality and suicide-related adverse events
During double-blind treatment, C-SSRS-rated suicidal ideation was reported more often among patients in the placebo group (8%) compared with the vilazodone group (6%); there were no reports of suicidal behavior in either group. One TEAE of suicidal ideation was reported on day 23 of double-blind treatment by a placebo patient; the event was considered to be moderate in severity and unrelated to treatment, and did not result in study discontinuation.
Sexual functioning
Small mean increases in CSFQ scores from baseline to the end of double-blind treatment, which indicate improvement on this scale, were observed in the placebo-treatment and vilazodone-treatment groups (1.6 each group). Small and similar mean increases were also observed in the placebo and vilazodone groups among subgroups of male (1.2 and 1.9, respectively) and female (1.8 and 1.4, respectively) patients. TEAEs associated with sexual function were reported by four vilazodone-treated patients [libido decreased and anorgasmia (one patient), disturbance in sexual arousal, libido increased, ejaculation disorder (one patient each)] and two placebo-treated patients [libido decreased, orgasm abnormal (one patient each)]. All sexual function-related TEAEs were considered mild or moderate in intensity and related to treatment. No patient discontinued treatment nor had a dose reduction in response to a sexual function-related TEAE.
Discussion
This study met its primary efficacy endpoint by demonstrating significantly greater improvement in the mean change from baseline to week 8 in HAMA total score for 20–40 mg/day vilazodone compared with placebo in adult patients with GAD. Improvement in anxiety symptoms in vilazodone-treated patients versus placebo-treated patients was supported by a statistically significant difference in the mean change from baseline in the HAMA psychic anxiety subscale score. Demonstrating statistical separation from placebo on the somatic anxiety subscale may have been more difficult because of a less severe somatic symptom burden, as indicated by lower mean baseline scores on the somatic anxiety subscale (placebo=10.3, vilazodone=11.0) than on the psychic anxiety subscale (placebo=14.5, vilazodone=14.9).
The effect size for vilazodone versus placebo for the mean change in HAMA total score from baseline to week 8 was 0.22. In two additional vilazodone studies that have been conducted in adult patients with GAD, the HAMA effect sizes were 0.18 for the 20 mg/day dose and 0.25 for the 40 mg/day dose in a fixed-dose study (Gommoll et al., 2015), and 0.31 in a flexible-dose study (20–40 mg/day; Sheehan et al., 2015). It is interesting to note that a 2007 analysis found HAMA effect sizes for the treatment of GAD of 0.39 for all drugs versus placebo, 0.50 for pregabalin, 0.42 for the SNRI venlafaxine, 0.38 for benzodiazepines (i.e. alprazolam, diazepam, and lorazepam), 0.36 for SSRIs (i.e. paroxetine, sertraline, fluvoxamine, and escitalopram), and 0.17 for buspirone (Hidalgo et al., 2007). However, caution should be exercised when comparing effect sizes of this nature as they are derived from post-hoc analyses of randomized clinical trials that may differ in design and may be powered for other objectives.
The difference in the change in SDS total score from baseline to week 8 was not statistically significant for vilazodone versus placebo; a statistically significant difference was observed in favor of vilazodone for the SDS work/school item. As improvement in functional impairment may lag behind symptomatic improvement in psychiatric disorders (Hirschfeld et al., 2002), an 8-week study of vilazodone in GAD treatment may not have been long enough for the full treatment effects on functional impairment to be apparent. In addition, delineating improvement in functional impairment using the SDS and its individual domains may be affected by the number of patients included in the analyses.
The mean change in SDS total score was analyzed using a modified ITT population that included only patients with assessments on all three individual SDS domains. Patients may be more likely to participate in activities related to social and family life compared with work or school during the course of a clinical trial, which may leave fewer patients with analysis values for the SDS total score and the work/school item than for the social life and family life items. This was the case in our trial, in which assessments were evaluable for 194 placebo patients and 195 vilazodone patients on the social life and family life items, and for 152 placebo patients and 155 vilazodone patients on the work/school item. Although greater mean score reductions were seen for vilazodone compared with placebo on the SDS total score and on each of the SDS items, the difference between groups was only statistically significant for the SDS work/school item. This finding is interesting as it is more challenging to show statistical separation from placebo and a treatment effect in a smaller sample size.
Minimal clinically important differences (MCIDs) on outcome measures that evaluate the mean change from baseline are often used to indicate the smallest change that a patient may perceive as beneficial. In clinical trials using the SDS as an outcome, it has been suggested that active treatments will generally separate from placebo when the mean change from baseline reaches an MCID of ∼4 points for total score and 1–2 points for an item score (Sheehan and Sheehan, 2008). The validity of these suggested SDS benchmarks was not obvious in our trial, in which the MCID in the mean score change from baseline was reached in the vilazodone group on each SDS component (total score=−8.53; work/school=−2.94; social life=−3.03; family life=−2.74) but the difference was only statistically significant versus placebo on the SDS work/school item. High mean changes from baseline were also observed in the placebo group, which may have made it difficult to detect an efficacy signal for improvement in functional impairment.
For additional efficacy parameters not previously mentioned, the difference between vilazodone and placebo was only statistically significant for the mean change from baseline in the HAMD17 total score. The significant between-group difference in favor of vilazodone on HAMD17 may be of interest as this study was conducted in a nondepressed or mildly depressed patient population, as indicated by baseline HAMD17 scores of ∼13 in both treatment groups (Zimmerman et al., 2013). Although the results from HAMD17 may suggest improvement in depressive symptoms, patients with significant depressive symptoms were excluded from this study and these results should be interpreted accordingly.
Support for the efficacy of vilazodone in the treatment of GAD is demonstrated by its collective clinical trial evidence. Vilazodone has produced statistically significant improvement relative to placebo on the primary efficacy outcome in three of three clinical studies in adult patients with GAD. In addition to the current study, the difference in the mean change from baseline to week 8 in HAMA total score was statistically significant versus placebo in a fixed-dose study of 20 or 40 mg/day vilazodone (Gommoll et al., 2015) and in another flexible-dose study of 20 or 40 mg/day vilazodone (Sheehan et al., 2015). In the fixed-dose study, significant improvement versus placebo was additionally observed for both doses of vilazodone on the HAMA tension item, the CGI-I score at week 8, and the CGI-I rate of response, and for the 40 mg/day dose on the HAMA psychic anxiety subscale, the HAMA somatic anxiety subscale, and the HAMA rate of response. In the flexible-dose study, which had methodology and design that were identical to the current study, statistically significant differences in favor of 20–40 mg/day vilazodone versus placebo were observed on all prospective outcome measures including change from baseline in SDS total score (secondary efficacy parameter), SDS individual item domains, HAMA subscales, and HAMA items. As a considerable amount of variance in effect sizes is generally found in GAD studies, these consistently positive results for vilazodone are noteworthy.
In addition to consistent efficacy, the safety and tolerability of GAD treatment is important to consider because of the chronic nature of the disorder and the recommendation that treatment be continued for at least 1 year to maximize the potential for remission (Allgulander, 2012). In this GAD trial, the safety profile of vilazodone was consistent with that observed in patients with MDD; no new safety concerns were identified. However, more vilazodone patients than placebo patients reported TEAEs and discontinued from the study as a result. The most frequently reported TEAEs for vilazodone were diarrhea and nausea; the vast majority of events were considered mild or moderate and resulted in few discontinuations. No serious AEs were reported. In general, the mean changes in clinical laboratory values and vital signs, and the shifts from normal baseline values to high values at the end of treatment were low and similar between treatment groups; exceptions included triglyceride, glucose, and total cholesterol levels, which had higher incidences of shifts from normal to high values in vilazodone-treated compared with placebo-treated patients.
Since GAD has been associated with suicidality (Revicki et al., 2012, Sareen et al., 2005), the inclusion of a prospective measure of suicidal ideation and behavior in this study was meaningful. Results showed that C-SSRS-rated suicidal ideation was more common in placebo-treated patients than in vilazodone-treated patients and no suicidal behavior was reported during the study. In addition, the only reported suicidal ideation TEAE was reported by a placebo patient. These findings are important as the aggregate benefit of GAD treatment should ideally minimize the humanistic as well as the symptomatic burden of the disorder.
Sexual dysfunction, the most common complication of SSRI treatment in patients with depression (Baldwin, 2004), is also considered to be one of the most unacceptable side effects (Hu et al., 2004). In our study, systematic assessment through the CSFQ suggested that overall sexual function did not worsen during treatment with vilazodone. In addition, only four vilazodone-treated patients reported TEAEs related to sexual function; all these events were considered mild or moderate, and none were associated with treatment discontinuation or dose reduction. As sexual dysfunction is a known effect of SSRI treatment (Kennedy and Rizvi, 2009), it is relevant that treatment with vilazodone and placebo produced similar effects on sexual function according to results from the CSFQ, and few sexual function TEAEs occurred in vilazodone-treated patients.
Limitations of this study include its short duration and lack of an active comparator. No patients with significant depressive symptoms or MDD were enrolled; as such, these findings may not be generalizable to GAD patients with a broader symptom profile or comorbid MDD. Strengths of the study include the prospective inclusion of the CSFQ, which allowed for systematic assessment of sexual functioning during treatment.
Conclusion
This positive treatment trial in GAD demonstrated statistically significant improvement in favor of flexible-dose 20–40 mg/day vilazodone relative to placebo on the primary efficacy measure, HAMA total score change from baseline to week 8. Additional benefits in treating anxiety symptoms and functional impairment associated with GAD were suggested by statistically significant differences for vilazodone compared with placebo on some additional efficacy measures (i.e. HAMA psychic anxiety subscale, the SDS work/school item, HAMD17 total score). Vilazodone was safe and generally well tolerated in patients with GAD; gastrointestinal side effects, similar to those seen in studies of vilazodone in patients with MDD, were common but predominantly considered mild or moderate in intensity. The effect of treatment on sexual function was similar for vilazodone and placebo. Although not specifically approved for the treatment of GAD, in combination with the results of two previously reported positive trials, these results show that vilazodone was effective in treating adults with this common and highly impairing disorder.
Acknowledgements
Writing assistance and editorial support for the preparation of this manuscript were provided by Carol Brown, MS, of Prescott Medical Communications Group, Chicago, Illinois, a contractor of Forest Research Institute, an affiliate of Actavis Inc.
This study was supported by funding from Forest Laboratories, LLC, an affiliate of Actavis Inc. (Jersey City, New Jersey). Forest Laboratories, LLC was involved in the study design, collection (through contracted clinical investigator sites), analysis and interpretation of data, and the decision to present these results.
Conflicts of interest
Angelo Sambunaris, MD, has received consultant and speaking fees from Actavis Inc. and Takeda Pharmaceuticals. He has also received research support from Alkermes, Forest Pharmaceuticals, an affiliate of Actavis Inc., Otsuka, Pfizer, Sunovion, Shire, and Takeda. He was a principal investigator in both vilazodone phase III studies.
Carl Gommoll, Xiongwen Tang, and Suresh Durgam acknowledge a potential conflict of interest as employees of Forest Research Institute, an affiliate of Actavis Inc.; Giovanna Forero, Maju Mathews, and Rene Nunez acknowledge a potential conflict of interest as employees of Forest Research Institute, an affiliate of Actavis Inc., at the time of the study.
References
- Allgulander C. (2012). Generalized anxiety disorder: a review of recent findings. J Exp Clin Med 4:88–91. [Google Scholar]
- Angst J, Gamma A, Baldwin DS, Ajdacic-Gross V, Rössler W. (2009). The generalized anxiety spectrum: prevalence, onset, course and outcome. Eur Arch Psychiatry Clin Neurosci 259:37–45. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders – text revision, Fourth Edition Washington, DC: American Psychiatric Association. [Google Scholar]
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition Arlington, VA: American Psychiatric Association. [Google Scholar]
- Baldwin DS. (2004). Sexual dysfunction associated with antidepressant drugs. Expert Opin Drug Saf 3:457–470. [DOI] [PubMed] [Google Scholar]
- Baldwin DS, Anderson IM, Nutt DJ, Allgulander C, Bandelow B, den Boer JA, et al. (2014). Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol 28:403–439. [DOI] [PubMed] [Google Scholar]
- Bandelow B, Sher L, Bunevicius R, Hollander E, Kasper S, Zohar J, Möller HJ, WFSBP Task Force on Mental Disorders in Primary Care; WFSBP Task Force on Anxiety Disorders, OCD and PTSD (2012). Guidelines for the pharmacological treatment of anxiety disorders, obsessive–compulsive disorder and posttraumatic stress disorder in primary care. Int J Psychiatry Clin Pract 16:77–84. [DOI] [PubMed] [Google Scholar]
- Buoli M, Caldiroli A, Caletti E, Paoli RA, Altamura AC. (2013). New approaches to the pharmacological management of generalized anxiety disorder. Expert Opin Pharmacother 14:175–184. [DOI] [PubMed] [Google Scholar]
- Clayton AH, McGarvey EL, Clavet GJ, Piazza L. (1997). Comparison of sexual functioning in clinical and nonclinical populations using the Changes in Sexual Functioning Questionnaire (CSFQ). Psychopharmacol Bull 33:747–753. [PubMed] [Google Scholar]
- Croft HA, Pomara N, Gommoll C, Chen D, Nunez R, Mathews M. (2014). Efficacy and safety of vilazodone in major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 75:e1291–e1298. [DOI] [PubMed] [Google Scholar]
- Gommoll C, Durgam S, Mathews M, Forero G, Nunez R, Tang X, Thase ME. (2015). A double-blind, randomized, placebo-controlled, fixed-dose phase III study of vilazodone in patients with generalized anxiety disorder. Depress Anxiety 32:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy W. (1976). The clinician global severity and impression scales ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: National Institute of Mental Health; 218–222. [Google Scholar]
- Hamilton M. (1959). The assessment of anxiety states by rating. Br J Med Psychol 32:50–55. [DOI] [PubMed] [Google Scholar]
- Hamilton M. (1960). A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo RB, Tupler LA, Davidson JR. (2007). An effect-size analysis of pharmacologic treatments for generalized anxiety disorder. J Psychopharmacol 21:864–872. [DOI] [PubMed] [Google Scholar]
- Hirschfeld RM, Dunner DL, Keitner G, Klein DN, Koran LM, Kornstein SG, et al. (2002). Does psychosocial functioning improve independent of depressive symptoms? A comparison of nefazodone, psychotherapy, and their combination. Biol Psychiatry 51:123–133. [DOI] [PubMed] [Google Scholar]
- Hu XH, Bull SA, Hunkeler EM, Ming E, Lee JY, Fireman B, Markson LE. (2004). Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry 65:959–965. [DOI] [PubMed] [Google Scholar]
- Kennedy SH, Rizvi S. (2009). Sexual dysfunction, depression, and the impact of antidepressants. J Clin Psychopharmacol 29:157–164. [DOI] [PubMed] [Google Scholar]
- Kenward MG, Roger JH. (1997). Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53:983–997. [PubMed] [Google Scholar]
- Kenward MG, Molenberghs G, Thijs H. (2003). Pattern-mixture models with proper time dependence. Biometrika 90:53–71. [Google Scholar]
- Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen H -U. (2012). Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res 21:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Cutler AJ, Kajdasz DK, Gallipoli S, Athanasiou M, Robinson DS, et al. (2011). A randomized, double-blind, placebo-controlled, 8-week study of vilazodone, a serotonergic agent for the treatment of major depressive disorder. J Clin Psychiatry 72:441–447. [DOI] [PubMed] [Google Scholar]
- Laughren TP, Gobburu J, Temple RJ, Unger EF, Bhattaram A, Dinh PV, et al. (2011). Vilazodone: clinical basis for the US Food and Drug Administration's approval of a new antidepressant. J Clin Psychiatry 72:1166–1173. [DOI] [PubMed] [Google Scholar]
- Liebowitz M, Croft HA, Kajdasz DK, Whalen H, Gallipoli S, Athanasiou M, et al. (2011). The safety and tolerability profile of vilazodone, a novel antidepressant for the treatment of major depressive disorder. Psychopharmacol Bull 44:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews M, Gommoll C, Chen D, Nunez R, Khan A. (2015). Efficacy and safety of vilazodone 20 and 40 mg in major depressive disorder: a randomized, double-blind, placebo-controlled trial. Int Clin Psychopharmacol 30:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. (2011). The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revicki DA, Travers K, Wyrwich KW, Svedsäter H, Locklear J, Mattera MS, et al. (2012). Humanistic and economic burden of generalized anxiety disorder in North America and Europe. J Affect Disord 140:103–112. [DOI] [PubMed] [Google Scholar]
- Rickels K, Athanasiou M, Robinson DS, Gibertini M, Whalen H, Reed CR. (2009). Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 70:326–333. [DOI] [PubMed] [Google Scholar]
- Robinson DS, Kajdasz DK, Gallipoli S, Whalen H, Wamil A, Reed CR. (2011). A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol 31:643–646. [DOI] [PubMed] [Google Scholar]
- Rodriguez BF, Weisberg RB, Pagano ME, Bruce SE, Spencer MA, Culpepper L, Keller MB. (2006). Characteristics and predictors of full and partial recovery from generalized anxiety disorder in primary care patients. J Nerv Ment Dis 194:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen J, Cox BJ, Afifi TO, de Graaf R, Asmundson GJ, ten Have M, Stein MB. (2005). Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry 62:1249–1257. [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Harnett-Sheehan K, Raj BA. (1996). The measurement of disability. Int Clin Psychopharmacol 11 (Suppl 3):89–95. [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Durgam S, Gommoll C, Forero G, Nunez R, Tang X, et al. (2015). A double-blind, randomized, placebo-controlled, flexible-dose study of vilazodone in patients with generalized anxiety disorder. Poster presented at the 168th Annual Meeting of the American Psychiatric Association; 16–20 May 2015; Toronto, Canada.
- Sheehan KH, Sheehan DV. (2008). Assessing treatment effects in clinical trials with the discan metric of the Sheehan Disability Scale. Int Clin Psychopharmacol 23:70–83. [DOI] [PubMed] [Google Scholar]
- Thase ME, Chen D, Edwards J, Ruth A. (2014). Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol 29:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilazodone (2015). Viibryd [package insert]. St. Louis, MO: Forest Pharmaceuticals Inc. [Google Scholar]
- World Health Organization (2001). World Health Report January 2001 Mental health: new understanding, new hope. Geneva, Switzerland: World Health Organization. [Google Scholar]
- Wittchen HU. (2002). Generalized anxiety disorder: prevalence, burden, and cost to society. Depress Anxiety 16:162–171. [DOI] [PubMed] [Google Scholar]
- Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jönsson B, et al. (2011). The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21:655–679. [DOI] [PubMed] [Google Scholar]
- Zimmerman M, Martinez JH, Young D, Chelminski I, Dalrymple K. (2013). Severity classification on the Hamilton Depression Rating Scale. J Affect Disord 150:384–388. [DOI] [PubMed] [Google Scholar]