

Abstract
The present study aimed to investigate the role of the mechanistic target of rapamycin complex 1 (mTORC1) in the regulation of myofibrillar (MyoPS) and mitochondrial (MitoPS) protein synthesis following endurance exercise. Forty-two female C57BL/6 mice performed 1 h of treadmill running (18 m min−1; 5° grade), 1 h after i.p. administration of rapamycin (1.5 mg · kg−1) or vehicle. To quantify skeletal muscle protein fractional synthesis rates, a flooding dose (50 mg · kg−1) of l-[ring-13C6]phenylalanine was administered via i.p. injection. Blood and gastrocnemius muscle were collected in non-exercised control mice, as well as at 0.5, 3 and 6 h after completing exercise (n = 4 per time point). Skeletal muscle MyoPS and MitoPS were determined by measuring isotope incorporation in their respective protein pools. Activation of the mTORC1-signalling cascade was measured via direct kinase activity assay and immunoblotting, whereas genes related to mitochondrial biogenesis were measured via a quantitative RT-PCR. MyoPS increased rapidly in the vehicle group post-exercise and remained elevated for 6 h, whereas this response was transiently blunted (30 min post-exercise) by rapamycin. By contrast, MitoPS was unaffected by rapamycin, and was increased over the entire post-exercise recovery period in both groups (P < 0.05). Despite rapid increases in both MyoPS and MitoPS, mTORC1 activation was suppressed in both groups post-exercise for the entire 6 h recovery period. Peroxisome proliferator activated receptor-γ coactivator-1α, pyruvate dehydrogenase kinase 4 and mitochondrial transcription factor A mRNA increased post-exercise (P < 0.05) and this response was augmented by rapamycin (P < 0.05). Collectively, these data suggest that endurance exercise stimulates MyoPS and MitoPS in skeletal muscle independently of mTORC1 activation.
Key points
Previous studies have shown that endurance exercise increases myofibrillar (MyoPS) and mitochondrial (MitoPS) protein synthesis in skeletal muscle.
The mechanistic target of rapamycin (mTOR) is considered to be a key intracellular nutrient-sensing protein complex, which activates MyoPS in response to anabolic stimuli.
Little is known regarding the regulation of MyoPS and MitoPS in response to endurance exercise.
In the present study, we show that MyoPS and MitoPS increase in skeletal muscle following endurance exercise, despite suppression of mTORC1 during the post-exercise recovery period.
Our data suggests that mTORC1 independent processes regulate both MyoPS and MitoPS following acute endurance exercise.
Introduction
Endurance exercise results in rapid transcriptional and translational remodelling of skeletal muscle, leading to increased mitochondrial biogenesis and, ultimately, enhanced oxidative capacity (Yan et al. 2011). Although the ability of endurance exercise to drive mitochondrial biogenesis has been known for the past 50 years (Holloszy, 1967), the mechanism enabling endurance exercise to drive translational processes involved in skeletal muscle adaptation remains poorly understood (Miller & Hamilton, 2012).
The mechanistic target of rapamycin complex 1 (mTORC1) is a highly conserved serine/threonine kinase protein complex, that is a central regulator of cellular growth in skeletal muscle (Laplante & Sabatini, 2012). The importance of mTORC1 for load- and feeding-induced increases in skeletal muscle protein synthesis has been demonstrated for rodents (Bodine et al. 2001; Goodman et al. 2011) and humans (Drummond et al. 2009; Dickinson et al. 2013); however, the role of mTORC1 in the protein synthetic response to endurance exercise has not been investigated directly. Endurance exercise in rodents increases the phosphorylation of proteins within the mTORC1 signalling cascade that are known to be associated with enhanced translational capacity in skeletal muscle (Edgett et al. 2013). In humans, endurance exercise leads to increases in mTORC1-related signalling and associated increases in both mixed (Harber et al. 2010; Beelen et al. 2011; Hulston et al. 2011) and fractional protein synthesis rates (Breen et al. 2011; Di Donato et al. 2014); however, a causal link between the two has not been measured directly. Collectively, this body of research would suggest an important role for mTORC1 in the adaptive response to endurance exercise (Moore & Stellingwerff, 2012; Moore et al. 2014; Rowlands et al. 2014).
In the present study, we used the mTORC1 inhibitor rapamycin to directly determine the contribution of mTORC1 to the fractional protein synthetic response with respect to a single bout of endurance exercise in mice. We hypothesized that inhibiting mTORC1 would blunt both the myofibrillar (MyoPS) and mitochondrial (MitoPS) responses following endurance exercise during a 6 h post-exercise recovery period.
Methods
Ethical approval
All procedures were approved by the University of California Davis Institutional Animal Care and Use Committee and performed under protocol number 17458. All investigators taking part in the animal experimentation clearly understood the ethical principles under which The Journal of Physiology operates in compliance with the animal ethics checklist (Grundy, 2015).
Exercise protocol
Forty-two female C57BL/6 mice were purchased from Taconic (San Diego, CA, USA) at 12 weeks of age. Following acclimation, mice were fasted for 3 h prior to performing a single bout of uphill treadmill running (18 m min−1 for 1 h at a 5° gradient) to produce a motor endurance exercise stimulus (Roberts, 2002). Following exercise, mice were returned to their cages until muscle collection at 0.5, 3 and 6 h post-exercise (n = 4 per timepoint). Mice had access to water but remained fasted throughout the study. One hour prior to exercise, mice were given an i.p. injection of either rapamycin/PBS or DMSO/PBS (200 μl). Thirty minutes prior to muscle collection, mice received a 50 mg · kg−1 flooding dose of L-[ring-13C6]phenylalanine (Cambridge Isotope Laboratory, MA, USA) via i.p. injection. Gastrocnemius muscles were collected under anaesthesia, with blood collected from the descending aorta.
Rapamycin treatments
Rapamycin (LC Laboratories, Woburn, MA, USA) was dissolved in DMSO to generate a 5 μg · μl−1 stock solution. Each mouse received 1.5 mg · kg−1 rapamycin dissolved in 200 μl of PBS. For the vehicle condition, mice were injected with an equivalent amount of DMSO dissolved in 200 μl of PBS.
Western blot procedure
Phosphorylation-specific immunoblotting was carried out as described previously (Philp et al. 2011). Membranes were incubated overnight with the primary antibodies: phospho-p70S6 kinase (S6K1)Thr389 (#9206), total S6K1 (#9202), phopsho-ribosomal protein S6 (S6)Ser235/236 (#4858), phospho-S6Ser240/244 (#5364), total S6 (#2217), phospho-eIF4E-binding protein 1 (4E-BP1)Thr37/46 (#9459), total 4E-BP1 (#9452), phospho-extracellular signal-regulated protein kinases 1 and 2 (ERK1/2)Thr202/Tyr204 (#4370), total ERK1/2 (#4695), phospho-eukaryotic translation elongation factor 2 (eEF2)Thr56 (#2331), total eEF2 (#2332) and total acetyl-CoA carboxylase (ACC) (#3676) (all obtained from Cell Signalling Technology; New England Biolabs Ltd, Hitchin, UK). Phospho-AMP activated protein kinase (AMPK)Thr172 (07-681SP), total AMPK α1/α2 (07-350SP) and phospho-ACCSer79 (04-1009) were all obtained from Merck Millipore (Watford, UK). The regulated in development and DNA damage responses 1 (REDD1)rabbit polyclonal (#10638-1-AP) antibody was obtained from Proteintech Ltd (Manchester, UK). Immobilon western chemiluminescent HRP susbstrate (Merck Millipore) was used to quantify protein content after IgG binding, and visualized on a G:BOX Chemi XT4 imager using GeneSys capture software (Syngene UK, Cambridge, UK).
S6K1 and AMPK activity assays. In total, 50 mg of gastrocnemius muscle was used for the measurement of S6K1 and AMPKα1/α2 activity as described previously (McGlory et al. 2014).
Quantitative real-time RT-PCR
RNA was extracted from 25 mg of gastrocnemius muscle using the phenol/chloroform method. First-strand cDNA was synthesized on a PCR Mastercycler (Eppendorf, Westbury, NY, USA) from 1 μg of RNA using the reverse transcription system (Promega, Southampton, UK) in accordance with the manufacturer’s instructions. Real-time PCR was performed using an Eppendorf Light Cycler PCR machine, SYBR green PCR plus reagents (Sigma-Aldrich, Poole, UK) and custom designed primers. GAPDH was used as a housekeeping gene control in all of the analysis and the absolute Ct for GAPDH was unchanged by the various interventions. The primer sequences for GAPDH, peroxisome proliferator activated receptor-γ coactivator-1α (PGC-1α), pyruvate dehydrogenase kinase 4 (PDK4) and mitochondrial transcription factor A (TFAM) have been reported previously (Philp et al. 2011).
Plasma and muscle protein subfraction enrichment
Plasma [ring-13C6] phenylalanine enrichments were determined as described previously (Glover et al. 2008) and used as the precursor pool enrichment in the calculation of the muscle protein fractional synthetic rate. Mean ± SEM plasma free phenylalanine enrichment at 30 min post-flood was 43.7 ± 3.1 % t/T (tracer:tracee).
A ∼100 mg piece of wet muscle from the gastrocnemius was homogenized using a glass Dounce homogenizer in ice-cold homogenization buffer (10 μl mg−1; 0.067 msucrose, 0.05 mTris/HCl, 0.05 mKCl, 0.01 mEDTA) with protease and phosphatase inhibitor cocktail tablets (cOmplete Mini, PhosSTOP, Roche Applied Science, Mannheim, Germany). The homogenate was transferred to an Eppendorf tube and centrifuged at 700 g for 15 min at 4°C to pellet myofibrillar proteins. The supernatant was transferred to another Eppendorf tube and centrifuged at 12,000 g for 20 min at 4°C to pellet mitochondria. Amino acids were obtained from the mitochondrial pellet as described previously (Burd et al. 2012). Briefly, the pellet was washed twice with ice-cold homogenization buffer, once with ethanol and then dried under vacuum. Proteins were hydrolysed by adding 6 mHCl and heating at 110°C for 18 h. The free amino acids from the mitochondrial and myofibrillar enriched fractions were purified using cation-exchange chromatography (Dowex 50WX8-200 resin; Sigma-Aldrich, St Louis, MO, USA) and converted to their N-acetyl-n-propyl ester derivatives for gas chromatography combustion isotope ratio mass spectrometry (GC: #6890, Hewlett Packard, Palo Alto, CA, USA; IRMS: Delta Plus XP, Thermo Finnigan, Waltham, MA, USA). An additional cohort of five unlabelled mice was used for the determination of natural isotopic abundance in muscle protein subfractions. Fractional protein synthesis was calculated as:
where Em is the 13C6 isotopic enrichment in muscle protein subfractions minus background basal enrichment, Ep is the mean enrichment in the precursor pool minus background enrichment and t is tracer incorporation in hours (0.5 h). Data are expressed as % · day−1. Background basal enrichment of the gastrocnemius from non-infused wild-type mice (n = 5) was accounted for in the calculation of Em. Background basal enrichment of plasma from non-infused wild-type mice (n = 5) was accounted for in the calculation of Ep.
Statistical analysis
A two-way analysis of variance ANOVA (Prism; GraphPad Software Inc., San Diego, CA, USA) with a Tukey’s posthoc test was used to determine differences in fractional protein synthesis rates, mRNA expression or kinase activity/phosphorylation across time and between groups. A paired Student’s t test was used to determine differences in MyoPS and MitoPS area under the curve. Values are reported as the mean ± SEM, normalized to vehicle-basal levels. P < 0.05 was considered statistically significant.
Results
Fractional synthesis rates increase following acute endurance exercise
Endurance exercise resulted in a rapid increase in MyoPS, increasing by ∼2-fold, 0.5 h post-exercise and remaining elevated at 3 and 6 h after exercise (P < 0.05) (Fig.1A and C). Endurance exercise also resulted in a ∼2–4-fold increase in MitoPS following exercise (Fig.1B and C).
Figure 1.
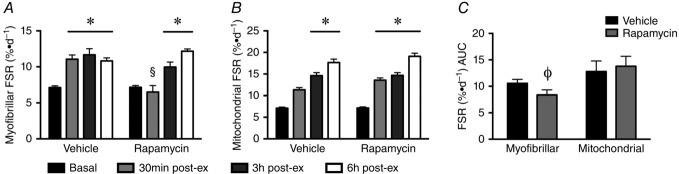
Differential effect of rapamycin on MyoPS and MitoPS following endurance exercise
A, endurance exercise rapidly increased MyoPS in the vehicle group, with this response being sustained during the 6 h recovery period. By contrast, rapamycin blocked MyoPS 0.5 h post-exercise. B, MitoPS was significantly increased post-exercise in both the vehicle and rapamycin groups. C, area under the curve analysis demonstrated that rapamycin suppressed MyoPS without affecting MitoPS. All data are expressed as the mean ± SEM, normalized to vehicle-basal. *Significantly different from vehicle-basal. §Significantly different from vehicle 30 min post-exercise. ΦSignificantly different from vehicle (P < 0.05; n = 4 per group).
Rapamycin delays MyoPS without altering MitoPS
Treating mice with rapamycin 1 h before endurance exercise prevented the initial increase in MyoPS at 0.5 h. However, protein synthesis increased normally (∼2-fold), 3 and 6 h post-exercise (Fig.1A). By contrast to the MyoPS response, rapamycin had no effect on MitoPS, which increased in an identical manner in the presence or absence of rapamycin (Fig.1B). When expressed as area under the curve, the MyoPS response displayed a reduction after rapamycin treatment, whereas MitoPS was unaffected (Fig.1C).
mTORC1 activity following acute endurance exercise
In vehicle-treated mice, endurance exercise had no effect on mTORSer2448 phosphorylation at any time point during the 6 h post-exercise recovery period (Fig.2A). By contrast, S6K1Thr389 phosphorylation was reduced ∼3–5-fold during the post-exercise recovery period (Fig.2B) and S6K1 activity was reduced 5-fold, 0.5 h post-exercise, before returning to basal levels 6 h post-exercise (Fig.2C). Phosphorylation of 4E-BP1Thr37/46 followed a similar trend to S6K1Thr389, being reduced ∼2–3-fold during the 6 h recovery period (Fig.2D).Phosphorylation of S6Ser235/236was reduced 0.5 and 3 h post-exercise (Fig.2E), whereas S6Ser240/244 phosphorylation was reduced at all time points, with the greatest nadir 3 h post-exercise (Fig.2F).
Figure 2.
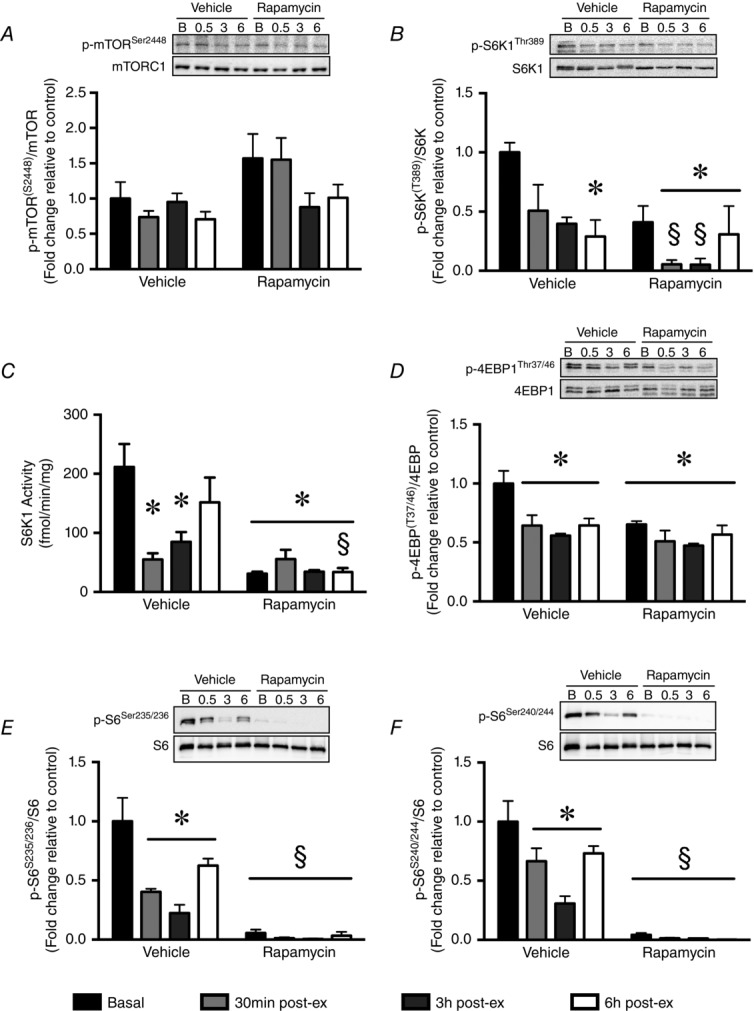
Endurance exercise and rapamycin treatment reduce mTORC1 signalling in skeletal muscle
A, mTORSer2448 was unchanged at any timepoint during the 6 h recovery period. B, S6K1Thr389 phosphorylation was reduced in the vehicle and rapamycin groups post-exercise. C, endurance exercise reduced S6K1 kinase activity in the vehicle group and rapamycin blocked S6K1 activity throughout. D, 4E-BP1Thr37/46 phosphorylation was significantly reduced during the 6 h recovery period in the vehicle group, whereas rapamycin blocked phosphorylation independent of exercise. E, S6Ser235/236 phosphorylation was reduced at 0.5 and 3 h post-exercise in the vehicle group, and completely blunted in the rapamycin group. F, S6Ser240/244 phosphorylation was reduced at 3 h post-exercise in the vehicle group, and completely blunted in the rapamycin group. All data expressed as the mean ± SEM, normalized to vehicle-basal. *Significantly different from vehicle-basal. §Significantly different from vehicle group at the relevant timepoint (P < 0.05; n = 4 per group).
Rapamycin treatment suppresses mTORC1 activity following acute endurance exercise
Neither rapamycin, nor endurance exercise affected mTORSer2448 phosphorylation at any time point (Fig.2A). S6K1Thr389 phosphorylation was significantly reduced compared to vehicle treatment at baseline, as well as 0.5 and 3 h post-exercise (Fig.2B). Basal S6K1 activity was reduced 5-fold compared to vehicle control, and remained significantly blunted up to 6 h post-exercise (Fig.2C). 4E-BP1Thr37/46 phosphorylation was reduced ∼2–3-fold at every time point during the post-exercise period (Fig.2D). S6Ser235/236 and S6Ser240/244 were both reduced ∼10-fold post-exercise throughout the 6 h recovery period (Fig.2E and F).
Rapamycin amplifies PGC-1α mRNA induction following acute endurance exercise
Acute endurance exercise significantly increased PGC-1α gene expression in both the vehicle and rapamycin groups; however, the magnitude of response was 2–3-fold higher in the rapamycin group at each timepoint during the recovery period (Fig.3A). To test the functional significance of this induction, we measured PDK4 and TFAM mRNA expression, both known transcriptional targets of PGC-1α. PDK4 mRNA expression increased post-exercise (∼4-fold) in both groups, with this response being 2–4-fold higher in the rapamycin group (Fig.3B). TFAM followed a similar trend, increasing ∼2-fold and ∼2.5-fold, 6 h post-exercise in the vehicle and rapamycin groups, respectively (Fig.3C).
Figure 3.
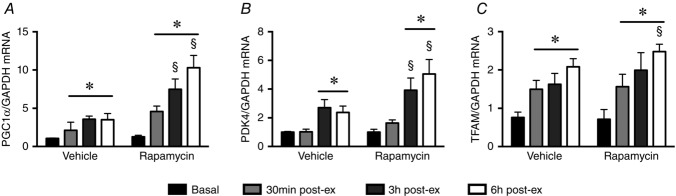
PGC-1α gene expression and associated signalling is increased post-exercise following rapamycin administration
A, endurance exercise increased PGC-1α gene expression in both the vehicle and rapamycin groups post-exercise. Rapamycin amplified the post-exercise response, with PGC-1α gene expression being significantly higher at each timepoint post-exercise compared to vehicle. B, PDK4 gene expression followed a similar pattern, increasing at 3 and 6 h post-exercise in the vehicle group, with this response being enhanced by rapamycin treatment, being significantly higher at 3 and 6 h post-exercise in the rapamycin group. C, TFAM followed a similar trend, increasing 6 h post-exercise in the vehicle group, and throughout the recovery period in the rapamycin group. All data expressed as the mean ± SEM, normalized to vehicle-basal. *Significantly different from vehicle-basal. §Significantly different from vehicle group at the relevant timepoint (P < 0.05; n = 4 per group).
AMPK, eEF2 and ERK1/2 are unchanged following acute endurance exercise
Endurance exercise had no effect on AMPK induction when assessed either via AMPKα1/α2 kinase activity, AMPKThr172 phosphorylation or ACCSer79 phosphorylation (Fig.4A–D). In addition, endurance exercise had no effect on ERK1/2Thr202/Tyr204 (Fig.5A) or eEF2Thr56 phosphorylation (Fig.5B).
Figure 4.
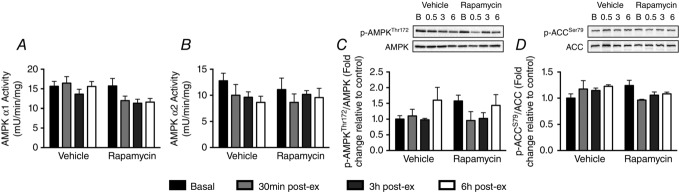
Endurance exercise has no effect on AMPK activity or downstream signalling
AMPK α1 (A) and AMPK α2 (B) activity were unchanged at any timepoint during the 6 h recovery period. In parallel, AMPKThr172(C) and ACCSer79 (D) phosphorylation were unchanged at any timepoint during the 6 h recovery period (n = 4 per group).
Figure 5.
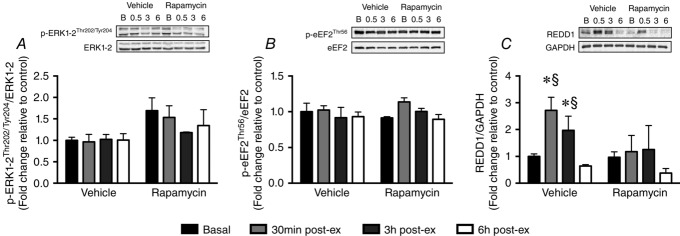
REDD1 but not eEF2 or ERK1/2 is rapidly induced following endurance exercise
ERK1/2Thr202/Tyr204(A) and eEF2Thr56 (B) phosphorylation were unchanged at any timepoint during the 6 h recovery period. By contrast, REDD1 induction (C) occurred rapidly post-exercise in the vehicle group, increasing 3-fold, 0.5 h post-exercise, and remaining elevated 3 h post-exercise, before returning to baseline 6 h post-exercise. REDD1 induction did not occur in the rapamycin group. *Significantly different from vehicle-basal. §Significantly different from rapamycin group at the relevant timepoint (P < 0.05; n = 4 per group).
REDD1 is rapidly induced following acute endurance exercise
REDD1 induction occurred rapidly post-exercise in the vehicle group, increasing 3-fold, 0.5 h post-exercise, and remaining elevated 3 h post-exercise, before returning to baseline 6 h post-exercise (Fig.5C). By contrast, REDD1 induction did not occur in the rapamycin group (Fig.5C).
Discussion
Endurance exercise training results in increased skeletal muscle oxidative capacity and fatigue resistance (Yan et al., 2011). At the molecular level, transcriptional responses appear pivotal in initiating this adaptation (Yan et al., 2011); however, less is known regarding the control of translational responses following endurance exercise (Miller & Hamilton, 2012). The mTORC1 pathway has been reported to undergo activation in response to endurance exercise in rodents (Edgett et al. 2013), inferring a role for mTORC1 in the adaptation to endurance exercise (Moore & Stellingwerff, 2012; Moore et al. 2014; Rowlands et al. 2014). To directly test the role of mTORC1 in endurance exercise adaptation, we examined mTORC1-related signalling, MyoPS and MitoPS during a 6 h post-exercise recovery period in vehicle- and rapamycin-treated mice. Our results demonstrate that acute endurance exercise leads to a rapid and sustained increase in both MyoPS and MitoPS in the hours after exercise. Unexpectedly, the increase in MyoPS and MitoPS occurred despite physiological repression of mTORC1 throughout the post-exercise recovery period and rapamycin only delayed MyoPS. These data suggest that MyoPS and MitoPS are activated independently of mTORC1 following acute endurance exercise.
The absence of increased mTORC1 activity post-exercise, despite an increase in fractional synthesis rates, was unexpected and contradicts previous studies in the literature. For example, high intensity endurance exercise increases the phosphorylation of mTORSer2448(Di Donato et al. 2014) and activity outputs of mTORC1 (Mascher et al. 2011) in conjunction with increased rates of post-exercise mixed muscle or fractional protein synthesis in humans (Mascher et al. 2011; Di Donato et al. 2014). Similarly, in rat skeletal muscle, 2 h of treadmill running resulted in 40% and 265% increases in the phosphorylation of mTORSer2448 and S6K1Thr389, respectively (Edgett et al. 2013). The repression of mTORC1 signalling in the present study, compared to the activation reported in these studies, is probably a result of differences in exercise intensity and exercise modality, as opposed to the effect of endurance exercise per se. For example, steady-state exercise>60% Wmax(Mascher et al. 2007; Di Donato et al. 2014), high-intensity interval exercise (Coffey et al. 2011) and exercise to exhaustion (Edgett et al. 2013) all lead to increases in mTORC1 signalling. By contrast, exercise at a lower intensity (30% Wmax for 1 h) also significantly increased MyoPS, although this occurred without an increase in mTORSer2448 phosphorylation (Di Donato et al. 2014). Taken together, it appears that mTORC1 activity is blunted during and in recovery from (1) low intensity, long duration (<75% Wmax/>60 min) endurance exercise or (2) exercise at a lower percentage of absolute power that does not go to failure. By contrast, at exercise intensities>75% Wmax or when the exercise is taken to failure, mTORC1 activity is increased. The precise mechanisms behind the differential activation of mTORC1 are currently unknown but could potentially be a consequence of greater fibre recruitment and loading during intensive exercise to failure.
At present, how MitoPS is regulated in skeletal muscle, as well as how exercise modulates this response, remains largely unknown (Hallberg & Larsson, 2014). Mitochondria contain their own mitochondrial DNA (mtDNA), encoding 13 genes that are involved in oxidative phosphorylation (Scarpulla, 2008). However, the proteins required for mitochondrial gene expression, proliferation and translation are all nuclear encoded; thus, intricate co-ordination between mitochondrial and nuclear gene pools is required for complete MitoPS. Translation of mtDNA-encoded mRNA is dependent on 24 mtDNA-encoded genes and at least 100 nuclear-encoded genes that regulate ribosomal proteins (Hallberg & Larsson, 2014). Our observation that rapamycin treatment has no effect on MitoPS, even in sedentary mice, would suggest that mitochondrial genes expressed following endurance exercise are translated via a process not requiring mTORC1, potentially within a mitochondrial specific ribosome pool (Pechmann et al. 2013).
In addition to the regulation of protein synthesis, mTORC1 has been linked to mitochondrial respiration and gene regulation via the transcriptional coactivator PGC-1α (Nemoto et al. 2005; Cunningham et al. 2007). For example, rapamycin treatment lowers mitochondrial membrane potential, oxygen consumption and ATP synthetic capacity in E6-1 Jurkat T cells (Schieke et al. 2006). Furthermore, mTORC1 was present in purified mitochondrial fractions and the suppression of mTORC1 activity via gene silencing of mTORC1 components leads to alterations in ATP generation (Schieke et al. 2006). Relevant to skeletal muscle, acute rapamycin treatment causes modest reductions in mitochondrial gene expression in C2C12 myotubes (Cunningham et al. 2007), corroborating findings in E6-1 Jurkat T cells (Schieke et al. 2006). To determine how mTORC1 altered mitochondrial gene expression, Cunningham et al.(2007) used expression profiling in tuberous sclerosis complex 2 (TSC2)−/−mouse embryo fibroblast cells following rapamycin treatment combined with motfiADE analysis to identify cis elements associated with differential expression in response to mTORC1 inhibition. The transcription factor yin-yang 1 (YY1) was identified, which could be coactivated by PGC-1α in the presence of a functional mTORC1 complex (Cunningham et al. 2007). Reducing mTORC1 activity by disruption of the mTOR/raptor interaction blocked the PGC-1α–YY1 interaction and led to decreased mitochondrial gene expression and oxygen consumption (Cunningham et al. 2007).
To test whether mTORC1 activity is required for exercise-induced increases in PGC-1α in vivo, we determined the transcriptional response of PGC-1α and its targets, TFAM and PDK4, during the 6 h recovery period. Rapamycin did not block PGC-1α transcriptional response post-exercise. Indeed, by contrast to the model proposed by Cunningham et al.(2007), PGC-1α gene expression was enhanced to a greater extent after rapamycin treatment at each post-exercise timepoint. A similar pattern was observed for PDK4 and, to a lesser degree, TFAM. Taken together, these data indicate that mTORC1 activity is not a pre-requisite for exercise-induced activation of PGC-1α or increases in post-exercise PGC-1α function. Our data are supported by a recent study reporting that rapamycin treatment during chronic contraction of C2C12 myotubes did not affect increases in mitochondrial protein content or activity following chronic contractile activity, nor the contraction-mediated increase in TFAM or PGC-1α (Carter & Hood, 2012). Thus, skeletal muscle contraction appears to override a regulatory effect that mTORC1 might have on basal mitochondrial function. Furthermore, it is clear that the role of mTORC1 in the regulation of skeletal muscle mitochondrial mass is very different from that of transformed muscle or tumour cells (Cunningham et al. 2007; Carter & Hood, 2012), which display intrinsically high rates of protein synthesis and mTORC1 activity (Schieke et al. 2006; Cunningham et al. 2007; Morita et al. 2013).
Based on the mTORC1 suppression that we observed in both the vehicle and rapamycin groups, we examined whether activation of AMPK, eEF2 or REDD1, which are proteins reported to inhibit mTORC1 (Brugarolas et al. 2004; Rose et al. 2009a; Lantier et al. 2010), might account for mTORC1 repression post-exercise. Endurance exercise had no effect on AMPK induction when assessed either via AMPK α1/α2 kinase activity, AMPKThr172 phosphorylation or ACCSer79 phosphorylation. In addition, endurance exercise had no effect on eEF2Thr56 phosphorylation. The lack of AMPK/eEF2 induction is probably a result of the moderate intensity of our prescribed exercise and the fact that our first muscle collection was 30 min post-exercise (to allow for the tracer flooding dose). The combined effect of these two factors is such that any AMPK/eEF2 activation during exercise may be rapidly lost during the 30 min recovery period. This interpretation is consistent with previously published research in wild-type and AMPK knockout mice showing that AMPK activation is evident immediately post-exercise but rapidly returns to baseline during the post-exercise recovery period (Jorgensen et al. 2005). Similarly, eEF2 activation following endurance exercise has been reported to be intensity-dependent and transient in nature (Rose et al. 2005; Rose et al. 2009a; Rose et al. 2009b). Thus, we suggest that, during moderate intensity exercise, any interference on mTORC1-related signalling by AMPK and/or eEF2 would occur during exercise and not as part of the post-exercise recovery period.
Given the lack of AMPK/eEF2 induction, we focused on REDD1, in accordance with recent reports that REDD1 activity is increased in skeletal muscle following endurance exercise (Murakami et al. 2011; Hayasaka et al. 2014) and correlates with reduced activity of the mTORC1 pathway (Murakami et al. 2011; Hayasaka et al. 2014). In agreement, REDD1 induction occurred rapidly post-exercise in the vehicle group, suggesting that REDD1 may be involved in the repressive effects of endurance exercise on mTORC1. The absence of a REDD1 response in the rapamycin group could potentially be explained by a recent study suggesting that mTORC1 activity is required to stabilize the REDD1 protein (Tan & Hagen, 2013). Therefore, the ablation of mTORC1 activity through rapamycin administration may have blunted the REDD1 post-exercise response. Finally, we examined the phosphorylation of ERK1/2Thr202/Tyr204, given the previous observation that endurance exercise can attenuate ERK1/2-mTORC1 signalling (Williamson et al. 2006). However, in a similar response to AMPK and eEF2, we did not observe any change in ERK1/2Thr202/Tyr204 phosphorylation at any of the time-points measured. As such, these data suggest that ERK1/2 activity is not a prerequisite for endurance exercise-mediated increases in MyoPS and MitoPS.
In summary, we report that skeletal muscle MitoPS and MyoPS are increased following acute endurance exercise despite prolonged suppression of mTORC1 activity in skeletal muscle. These data therefore indicate that, in contrast to resistance exercise adaptive responses requiring mTORC1 activity, endurance exercise stimulates fractional protein synthesis by an as yet unidentified molecular process.
Glossary
- ACC
acetyl-CoA carboxylase
- AMPK
AMP activated protein kinase
- 4E-BP1
eIF4E-binding protein 1
- eEF2
eukaryotic translation elongation factor 2
- ERK1/2
extracellular signal-regulated protein kinases 1 and 2
- MitoPS
mitochondrial protein synthesis
- mtDNA
mitochondrial DNA
- mTORC1
mechanistic target of rapamycin complex 1
- MyoPS
myofibrillar protein synthesis
- PDK4
pyruvate dehydrogenase kinase 4
- PGC-1α
peroxisome proliferator activated receptor-γ coactivator-1α
- REDD1
regulated in development and DNA damage responses 1
- S6
ribosomal protein S6
- S6K1
p70S6 kinase
- TFAM
mitochondrial transcription factor A
- TSC2
tuberous sclerosis complex 2
- YY1
ying-yang 1
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
AP, SS and KB conceived and designed the experiments. AP, SS and KB performed the sample collection. AP, JPS, DLH, LB, EL, SJ and SMP performed the data analysis. AP wrote the manuscript and all authors revised and critically evaluated the manuscript for important intellectual content prior to submission. All authors approved the final version of the manuscript submitted for publication.
Funding
This publication was supported in part by a BBSRC New Investigator AwardBB/L023547/1 to AP; National Institutes of Health (NIH) grants R01 AG043120, R24 HD050837, P30 AR061303 and P30 AR058878 to SS; a Sir Henry Wellcome Post-doctoral Fellowship to JPS; an ACSM Research Endowment and Society for Endocrinology Young Investigator Award to DLH; National Science and Engineering Research Council of Canada grant (RGPIN227870-05) to SMP; and a NIH grant R01 AG045375 to KB.
References
- Beelen M, Zorenc A, Pennings B, Senden JM, Kuipers H. van Loon LJ. Impact of protein coingestion on muscle protein synthesis during continuous endurance type exercise. Am J PhysiolEndocrinol Metab. 2011;300:E945–E954. doi: 10.1152/ajpendo.00446.2010. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ. Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- Breen L, Philp A, Witard OC, Jackman SR, Selby A, Smith K, Baar K. Tipton KD. The influence of carbohydrate-protein co-ingestion following endurance exercise on myofibrillar and mitochondrial protein synthesis. J Physiol. 2011;589:4011–4025. doi: 10.1113/jphysiol.2011.211888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW. Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd NA, Andrews RJ, West DW, Little JP, Cochran AJ, Hector AJ, Cashaback JG, Gibala MJ, Potvin JR, Baker SK. Phillips SM. Muscle time under tension during resistance exercise stimulates differential muscle protein sub-fractional synthetic responses in men. J Physiol. 2012;590:351–362. doi: 10.1113/jphysiol.2011.221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter HN. Hood DA. Contractile activity-induced mitochondrial biogenesis and mTORC1. Am J PhysiolCell Physiol. 2012;303:C540–C547. doi: 10.1152/ajpcell.00156.2012. [DOI] [PubMed] [Google Scholar]
- Coffey VG, Moore DR, Burd NA, Rerecich T, Stellingwerff T, Garnham AP, Phillips SM. Hawley JA. Nutrient provision increases signalling and protein synthesis in human skeletal muscle after repeated sprints. Eur J Appl Physiol. 2011;111:1473–1483. doi: 10.1007/s00421-010-1768-0. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK. Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Di Donato DM, West DW, Churchward-Venne TA, Breen L, Baker SK. Phillips SM. Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am J PhysiolEndocrinol Metab. 2014;306:E1025–E1032. doi: 10.1152/ajpendo.00487.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JM, Drummond MJ, Fry CS, Gundermann DM, Walker DK, Timmerman KL, Volpi E. Rasmussen BB. Rapamycin does not affect post-absorptive protein metabolism in human skeletal muscle. Metabolism. 2013;62:144–151. doi: 10.1016/j.metabol.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond MJ, Fry CS, Glynn EL, Dreyer HC, Dhanani S, Timmerman KL, Volpi E. Rasmussen BB. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol. 2009;587:1535–1546. doi: 10.1113/jphysiol.2008.163816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgett BA, Fortner ML, Bonen A. Gurd BJ. Mammalian target of rapamycin pathway is up-regulated by both acute endurance exercise and chronic muscle contraction in rat skeletal muscle. Appl Physiol Nutr Metab. 2013;38:862–869. doi: 10.1139/apnm-2012-0405. [DOI] [PubMed] [Google Scholar]
- Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K. Rennie MJ. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol. 2008;586:6049–6061. doi: 10.1113/jphysiol.2008.160333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Frey JW, Mabrey DM, Jacobs BL, Lincoln HC, You JS. Hornberger TA. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J Physiol. 2011;589:5485–5501. doi: 10.1113/jphysiol.2011.218255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol. 2015;593:2547–2549. doi: 10.1113/JP270818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg BM. Larsson NG. Making proteins in the powerhouse. Cell Metab. 2014;20:226–240. doi: 10.1016/j.cmet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Harber MP, Konopka AR, Jemiolo B, Trappe SW, Trappe TA. Reidy PT. Muscle protein synthesis and gene expression during recovery from aerobic exercise in the fasted and fed states. Am J PhysiolRegul Integr Comp Physiol. 2010;299:R1254–R1262. doi: 10.1152/ajpregu.00348.2010. [DOI] [PubMed] [Google Scholar]
- Hayasaka M, Tsunekawa H, Yoshinaga M. Murakami T. Endurance exercise induces REDD1 expression and transiently decreases mTORC1 signaling in rat skeletal muscle. Physiol Rep. 2014;2:e12254. doi: 10.14814/phy2.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- Hulston CJ, Wolsk E, Grondahl TS, Yfanti C. Vanhall G. Protein intake does not increase vastus lateralis muscle protein synthesis during cycling. Med Sci Sports Exerc. 2011;43:1635–1642. doi: 10.1249/MSS.0b013e31821661ab. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA. Pilegaard H. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Lantier L, Mounier R, Leclerc J, Pende M, Foretz M. Viollet B. Coordinated maintenance of muscle cell size control by AMP-activated protein kinase. FASEB J. 2010;24:3555–3561. doi: 10.1096/fj.10-155994. [DOI] [PubMed] [Google Scholar]
- Laplante M. Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher H, Andersson H, Nilsson PA, Ekblom B. Blomstrand E. Changes in signalling pathways regulating protein synthesis in human muscle in the recovery period after endurance exercise. Acta Physiol (Oxf) 2007;191:67–75. doi: 10.1111/j.1748-1716.2007.01712.x. [DOI] [PubMed] [Google Scholar]
- Mascher H, Ekblom B, Rooyackers O. Blomstrand E. Enhanced rates of muscle protein synthesis and elevated mTOR signalling following endurance exercise in human subjects. Acta Physiol (Oxf) 2011;202:175–184. doi: 10.1111/j.1748-1716.2011.02274.x. [DOI] [PubMed] [Google Scholar]
- McGlory C, White A, Treins C, Drust B, Close GL, Maclaren DP, Campbell IT, Philp A, Schenk S, Morton JP. Hamilton DL. Application of the [gamma-32P] ATP kinase assay to study anabolic signaling in human skeletal muscle. J Appl Physiol. 2014;116:504–513. doi: 10.1152/japplphysiol.01072.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF. Hamilton KL. A perspective on the determination of mitochondrial biogenesis. Am J PhysiolEndocrinol Metab. 2012;302:E496–E499. doi: 10.1152/ajpendo.00578.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DR, Camera DM, Areta JL. Hawley JA. Beyond muscle hypertrophy: why dietary protein is important for endurance athletes. Appl Physiol Nutr Metab. 2014;39:987–997. doi: 10.1139/apnm-2013-0591. [DOI] [PubMed] [Google Scholar]
- Moore DR. Stellingwerff T. Protein ingestion after endurance exercise: the ‘evolving’ needs of the mitochondria? J Physiol. 2012;590:1785–1786. doi: 10.1113/jphysiol.2011.224188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I. Sonenberg N. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Murakami T, Hasegawa K. Yoshinaga M. Rapid induction of REDD1 expression by endurance exercise in rat skeletal muscle. Biochem Biophys Res Comm. 2011;405:615–619. doi: 10.1016/j.bbrc.2011.01.078. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM. Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Pechmann S, Willmund F. Frydman J. The ribosome as a hub for protein quality control. Mol Cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K. Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TJ. The integrated function of muscles and tendons during locomotion. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:1087–1099. doi: 10.1016/s1095-6433(02)00244-1. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Alsted TJ, Jensen TE, Kobbero JB, Maarbjerg SJ, Jensen J. Richter EA. A Ca(2+)-calmodulin-eEF2K-eEF2 signalling cascade, but not AMPK, contributes to the suppression of skeletal muscle protein synthesis during contractions. J Physiol. 2009;587:1547–1563. doi: 10.1113/jphysiol.2008.167528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Bisiani B, Vistisen B, Kiens B. Richter EA. Skeletal muscle eEF2 and 4EBP1 phosphorylation during endurance exercise is dependent on intensity and muscle fiber type. Am J PhysiolRegul Integr Comp Physiol. 2009;296:R326–R333. doi: 10.1152/ajpregu.90806.2008. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Broholm C, Kiillerich K, Finn SG, Proud CG, Rider MH, Richter EA. Kiens B. Exercise rapidly increases eukaryotic elongation factor 2 phosphorylation in skeletal muscle of men. J Physiol. 2005;569:223–228. doi: 10.1113/jphysiol.2005.097154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands DS, Nelson AR, Phillips SM, Faulkner JA, Clarke J, Burd NA, Moore D. Stellingwerff T. Protein-leucine fed dose effects on muscle protein synthesis after endurance exercise. Med Sci Sports Exerc. 2014;47:547–555. doi: 10.1249/MSS.0000000000000447. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP, Jr, Aponte AM, Shen RF, Balaban RS. Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- Tan CY. Hagen T. mTORC1 dependent regulation of REDD1 protein stability. PloS ONE. 2013;8:e63970. doi: 10.1371/journal.pone.0063970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DL, Kubica N, Kimball SR. Jefferson LS. Exercise-induced alterations in extracellular signal-regulated kinase 1/2 and mammalian target of rapamycin (mTOR) signalling to regulatory mechanisms of mRNA translation in mouse muscle. J Physiol. 2006;573:497–510. doi: 10.1113/jphysiol.2005.103481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Okutsu M, Akhtar YN. Lira VA. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J Appl Physiol. 2011;110:264–274. doi: 10.1152/japplphysiol.00993.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]