

Abstract
Pulmonary hypertension (PH) is a disorder of lung vasculature characterized by arterial narrowing. Phosphatase-and-tensin homolog on chromosome 10 (PTEN), associated in the progression of multiple cancers, is implicated in arterial remodeling. However, the involvement of PTEN in PH remains unclear. The objective of the present study was to determine the role of PTEN in pulmonary vascular remodeling using established models of PH. The study used rat models of PH, induced by monocrotaline (MCT) administration (60 mg/kg) or continuous hypoxic exposure (10% oxygen) for 3 weeks. Pulmonary artery smooth muscle cells (SMCs) were used for in vitro confirmation. Development of PH was verified by hemodynamic, morphological and histopathology analyses. PTEN and key downstream proteins in pulmonary and cardiac tissues were analyzed by western blotting and RT-PCR. PTEN was significantly decreased (MCT, 53%; Hypoxia, 40%), pAkt was significantly increased (MCT, 42%; Hypoxia, 55%) in tissues of rats with PH. Similar results were observed in SMCs exposed to hypoxia (1% oxygen) for 48 h. Ubiquitination assay showed that PTEN degradation occurs via proteasomal degradation pathway. Western blotting demonstrated a significant downregulation of cell-cycle regulatory proteins p53 and p27, and upregulation of cyclin-D1 in the lungs of both models. The results showed that PTEN-mediated modulation of PI3K pathway was independent of the focal adhesion kinase and fatty acid synthase. The study, for the first time, established that PTEN plays a key role in the progression of pulmonary hypertension. The findings may have potential for the treatment of pulmonary hypertension using PTEN as a target.
Keywords: PTEN, Pulmonary hypertension, Monocrotaline, Hypoxia, Proteasomal degradation
Introduction
Pulmonary hypertension (PH) is a disorder of the lung vasculature that manifests itself when the mean pulmonary arterial pressure increases to one-fourth of the systemic blood pressure. PH results from constriction, or stiffening, of the pulmonary arteries that supply blood to the lungs. PH promotes vascular remodeling, leading to the development of neointimal formation and vascular occlusion [1]. Consequently, it becomes more difficult for the heart to pump blood forward through the lungs. This stress on the heart leads to enlargement of the right heart. In the clinical setting, PH may occur as a result of heart failure, pulmonary parenchymal or vascular disease, thromboembolism, or a combination of these factors [2]. Irreversible PH, with a fixed pulmonary vascular resistance of more than 4 Wood units, has long been considered a contraindication to transplantation in heart failure patients [3]. However, the reversibility of PH in patients is now well recognized, and it is believed that if reversibility could be achieved acutely with pharmacologic intervention, there would be a significant decrease in mortality and morbidity due to PH [4].There is a significant gap in the understanding of the mechanisms involved with the development and progression of PH, and if identified, could provide key therapeutic targets [5–7].
Phosphatase-and-tensin homolog on chromosome 10 (PTEN) is a multifunctional lipid phosphatase that was initially identified as a tumor suppressor gene [5, 7, 8]. Active PTEN protein serves as a modulator of several cellular functions, including cell survival, migration, proliferation, and apoptosis [9–11]. PTEN has recently been found to play an important role in the cardiovascular and pulmonary systems [12, 13]. Activation of PTEN regulates cardiomyocyte hypertrophy and survival, and also regulates pulmonary smooth muscle cell (SMC) proliferation and cell survival [12, 13]. The inactivation of PTEN in the setting of vascular injury leads to constitutive Akt activation, which is an early and critical event involved in neointimal formation and the activation of pulmonary SMCs [14]. Targeted deletion of PTEN in SMCs results in vascular remodeling and PH [6]. In contrast, overexpression and stabilization of PTEN in vitro and in vivo have been shown to inhibit vascular SMC proliferation and survival leading to neointimal hyperplasia and PH [5, 6, 15]. PTEN activation regulates both cell proliferation and survival genes that control the progression of cardiovascular and pulmonary diseases by targeting the Akt pathway [12, 13]. PTEN inhibits the phosphatidyl-inositol-3-kinase (PI3K) and downstream functions, including activation of Akt, cell survival, and cell proliferation in tumor cells carrying mutant or deletion-type PTEN [16]. However, the role of PTEN in PH, particularly with respect to pulmonary arterial SMC proliferation, survival-signaling pathways, and the mechanisms involved remain unclear. It is possible that PTEN expression is directly downregulated by factors promoting the development of PH, including hypoxia [7, 17], or could be due to other factors associated with the remodeling process [18, 19].
In the present study, we hypothesized that PTEN is involved in the development of PH, irrespective of the etiology of the process. Accordingly, the aim of this study was to determine the involvement of PTEN in the progression of PH. The studies were conducted using established in vivo rat models of PH induced either by monocrotaline (MCT) administration [20] or long-term hypoxic exposure [7, 17]. Pulmonary arterial smooth muscle cells (SMC) exposed to hypoxic conditions were used for in vitro experiments. The results showed that PTEN expression levels were significantly compromised in both in vivo models. In vitro studies using SMCs showed the loss of PTEN occurring through the proteasomal degradation pathway.
Materials and Methods
Reagents
Dimethyl sulfoxide (DMSO) and antibody directed against α-smooth muscle actin (α-SMA) were obtained from Sigma-Aldrich (St. Louis, MO). Polyvinylidene fluoride (PVDF) membrane and molecular-weight markers were obtained from Bio-Rad (Hercules, CA). Antibodies directed against Akt, pAkt (Ser473), PTEN, and pPTEN (Ser380 and Thr381/382) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies directed against cyclin-D1, p53, p21, and p27 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Enhanced chemiluminescence reagents were obtained from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK).
Monocrotaline (MCT) Model
All animal experiments were performed according to the guidelines approved by the IACUC of the Ohio State University. Rats were administrated a single, subcutaneous (sc) dose of MCT (60 mg/kg BW). The rats were euthanized by intra-peritoneal (i.p) injection of pentobarbital 2 weeks after MCT exposure. An age-matched control group with vehicle (saline)-only treatment was used for comparison. The lungs and hearts from both groups were collected and homogenized for protein expression analysis.
Chronic Hypoxia (CH) Model
Rats were exposed to 10% oxygen continuously for 3 weeks using a custom-built hypoxia chamber (BioSpherix-A chamber equipped with ProOx oxygen level controller; BioSpherix, Lacona, NY). The addition of nitrogen to ambient air was used to modify the oxygen level. The rats were euthanized, as described in the previous model at 3 weeks after continuous exposure. An age-matched control group of animals exposed to normoxia (21% oxygen) for 3 weeks was used for comparison. Analysis of protein expression was performed in the homogenized lungs in both groups.
Human Pulmonary Artery Smooth Muscle Cells
The human pulmonary artery smooth muscle cells (SMC) used in this study were obtained at passage 3 (Lonza CC-2581) and maintained in culture. The cells were briefly thawed according to the vendor’s protocol and maintained in a humidified incubator at 37°C/5% CO2. The SmGM-2 “bullet kit” (Lonza CC-3182) contains one 500 ml SMC basal medium, and the following growth factor supplements: hEGF 0.5 ml; Insulin 0.5 ml; hFGF-B 1 ml; FBS 10 ml; GA-1000 0.5 ml; 10 U/ml penicillin; and 1 mg/ml streptomycin. Cells were trypsinized and passaged at 95% confluence. Studies were performed on cells at passages 4–6 and 60–90% confluence. The cells were exposed to 1% hypoxia for 48 h, and cells of the same passage were exposed to 20% oxygen (normoxia) for the same time periods that were used for controls. Cell counting, protein analysis, and ubiquitination assays were performed using these cells. Cell counting was performed using a Nucleo-Counter (New Brunswick Scientific).
Histopathology and Immunohistochemistry
Lungs were inflated with 4% paraformaldehyde at 20-cm water pressure and fixed in 10% neutral buffered formalin for 48 h at room temperature. Paraffin-embedded tissues were cut into 5-μm thick sections and stained with hematoxylin and eosin (H&E) by standard methods. Immunohistochemistry (IHC) using PTEN antibody (catalog no. 9559, 1:100 dilution; Cell Signaling Technology) was performed on the tissue sections with 3,3′-diaminobenzidine peroxidase substrate (SK-4100, Vector Labs) and counterstained with hematoxylin following the manufacturer’s protocol. Representative photomicrographs were taken using an inverted fluorescence microscope (Nikon TE 2000, Japan).
Reverse-Transcription PCR
Total RNA was isolated from the lungs of hypoxia-exposed rats and controls, using the RNeasy system according to the manufacturer’s instructions and analyzed using a previously reported procedure [21].
Western Blotting
Western blotting was used to determine protein expression in the cardiac and pulmonary tissues. The specimens were homogenized with nondenaturing lysis buffer (10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 0.3 mM phenylmethylsulfonyl fluoride, 0.2 mM sodium orthovanadate, 0.5% NP40, 1 lg/ml aprotinin, and leupetin). The lysates were centrifuged at 10,000×g for 20 min at 4°C, and the supernatant was separated from the solid material. The protein concentration in the lysates was determined using a Pierce detergent-compatible protein assay kit. The protein lysate was denatured in 2× sample buffer and subjected to SDS-PAGE on a 10 or 12% tris–glycine gel. The separated proteins were transferred to a PVDF membrane, and the membrane was blocked with 5% nonfat milk powder (w/v) in TBST (10 mM Tris, 100 mM NaCl, 0.1% Tween 20) for 1 h at room temperature or overnight at 4°C. The membranes were incubated with primary antibodies directed against or known to cross-react with samples of rat origin. Actin was used as the loading control, and was detected by the corresponding primary antibody. The bound antibodies were detected using appropriate horseradish peroxidase (HRP)-labeled secondary antibodies using an enhanced chemiluminescence-detection system (ECL Advanced kit; Amersham Biosciences). Protein expression was quantified using Image Gauge version 3.45.
Ubiquitination Assay
SMCs were cultured under hypoxic conditions for 48 h and subsequently treated with 25 lM MG-132 (Calbiochem) for 2 h. Cells were then collected and lysed in lysis buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA (pH 8.0), 1 mM EGTA (pH 8.0), 0.1 mM sodium fluoride, 0.1 mM sodium orthovanadate, 1 mM dithiothreitol, 2 μg/ml aprotinin, and 2 μg/ml leuptin). Cell lysates were centrifuged at 10,000×g for 20 min at 4°C, and the supernatant was separated. A 500-μg sample of the total protein was used for immunoprecipitation. PTEN, pPTEN, or ubiquitin antibody was incubated with cell lysates for 12 h, followed by addition of 20 μl of recombinant agarose G-protein for 2 h. The matrices were washed four times with the same lysis buffer. After being boiled for 8 min in the presence of 2-mercaptoethanol, samples containing cell lysate protein were separated on a 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE gel) and then transferred onto equilibrated PVDF membranes. After blocking with 5% nonfat milk powder (w/v), the membranes were incubated with the primary antibodies described above. The bound antibodies were detected with horseradish peroxidase (HRP)-labeled donkey anti-rabbit IgG using an enhanced chemiluminescence-detection system (ECL Advanced kit; Amersham Biosciences).
Magnetic Resonance Imaging (MRI)
MRI was performed using a 9.4 T horizontal-bore small animal imaging system (Bruker Biospin) with cardiac gating. The rats were anesthetized using an isofluorane mixture (1.5–2% in carbogen gas), monitored for cardiac and respiratory function, and kept warm throughout the imaging period. LV end-systolic, LV end-diastolic, RV end-systolic, and RV end-diastolic volumes were calculated to determine cardiac function and dimensions. The image data were analyzed using ImageJ software (NIH).
Hemodynamic (pressure) Measurements
Hemodynamic measurements were measured in rats kept under isofluorane (2%) anesthesia. A Millar catheter (SPR-1000) was advanced through the LV directly. The RV systolic pressure (RVSP), LV systolic pressure (LVSP), pulmonary arterial pressure (PAP), LV end-diastolic pressure (LVEDP) were measured and analyzed using a PowerLab data-acquisition system (model ML866; Colorado Springs, CO).
Statistical Analysis
Data were expressed as mean ± SEM. Comparisons between groups were performed using the Student’s t test or ANOVA as appropriate. The significance level was set at P < 0.05.
Results
PTEN Expression in Monocrotaline-Induced Pulmonary Hypertension
Western blotting of lung and heart tissues obtained from rats 2 weeks after MCT administration showed a significant decrease in PTEN and phosphorylated-PTEN (pPTEN) levels, and a significant increase in phosphorylated-Akt (pAkt-Ser473) when compared to control tissues in the lung and RV, but not in the LV (Fig. 1a, b). In both the lung and RV tissues, the pPTEN/PTEN ratios were significantly lower in the MCT-administered animals when compared with control, suggesting a decrease in PTEN activation in PH (Fig. 1c). The results indicated that MCT-induced PH was associated with a substantial loss of PTEN activity with a concomitant increase of Akt activation in the lung and RV tissues, but not in the LV tissue.
Fig. 1.

PTEN expression in MCT-induced pulmonary hypertension. Western-blot analysis was performed on lung and heart tissues obtained 2 weeks after administration of a single dose of MCT (60 mg/kg). a Representative blots of pPTEN, PTEN, and Akt in the control (C) and MCT-induced PH (MCT) tissues. b Quantitative results of the blots of pPTEN, PTEN and pAkt. Data are shown as mean ± SEM (n = 8). *P < 0.05 versus respective control. The results show a significant decrease in pPTEN and PTEN expressions along with an increase in pAkt in both the lung and RV, but not in the LV. c Ratios of pPTEN to PTEN in the lung, and ventricles. *P < 0.05 versus respective control. The ratios show a significant decrease in pPTEN levels in the lung and RV, but an increase in the LV
Cell-Cycle Regulatory Proteins in Monocrotaline-Induced Pulmonary Hypertension
Western blot analysis of cell-cycle regulatory proteins in the lung and heart tissues obtained from rats 2 weeks after MCT administration showed a significant decrease in p53 in all tissues when compared with control (Fig. 2). However, p21 and p27 were decreased in the lung and RV, but not LV of MCT group. Cyclin-D1, which promotes progression of the cell-cycle, was significantly upregulated in all the three tissues of the MCT-administered rats. The results suggested that MCT-administration upregulated cell-cycle progression in lung and RV tissues.
Fig. 2.

Cell-cycle regulatory proteins in MCT-induced pulmonary hypertension. Western-blot analysis was performed on lung and heart tissues obtained 2 weeks after administration of a single dose of MCT (60 mg/kg). a Representative blots show the downregulation of p53, p21, and p27, and upregulation of cyclin-D1 expression. b Quantitative results of the blots of p53 and cyclin-D1 in the lung, RV, and LV. Data represent mean ± SEM (n = 8). *P < 0.05 versus respective control (C). The results show a significant decrease of p53 and increase of cyclin-D1 in the lung, RV and LV
Changes in Cardio-Pulmonary Functions upon Chronic Exposure to Hypoxia
Changes in cardiac function upon exposure to hypoxia were measured using MRI. Representative short-axis images of the heart, obtained 3 weeks after exposure to hypoxia, are shown in Fig. 3a. Analysis of the MRI data showed an increasing trend, although not significant, in both end-systolic and end-diastolic volumes of the ventricles (Fig. 3b). These results suggested a tendency toward cardiac dysfunction possibly due to development of PH upon chronic exposure of the animals to hypoxia. Ventricular and pulmonary arterial pressures were measured to confirm the development of PH in rats exposed to 3 weeks of continuous hypoxia. There was a significant increase in RV systolic pressure and mean pulmonary arterial pressure when compared with normoxic controls (Fig. 4). However, there was no significant change in the LVSP upon hypoxic exposure. The results suggested the development of PH upon chronic exposure of the animals to hypoxia.
Fig. 3.

Changes in cardiac function upon chronic exposure to hypoxia. Cardiac MRI of rat hearts was performed before exposure to hypoxia and also at 3 weeks post-hypoxia exposure. a Representative short-axis MRI images of the heart at baseline and after 3 weeks of hypoxia exposure. The images show significant RV hypertrophy in the end-diastolic image on hypoxia exposure. There is also an increase in LV wall thickness on hypoxia exposure in the endsystolic image. b End-diastolic (EDV) and end-systolic (ESV) volumes of the LV and RV. The results (mean ± SEM; n = 4/group) show an increasing trend in ventricular EDV and ESV in both ventricles on exposure to hypoxia
Fig. 4.
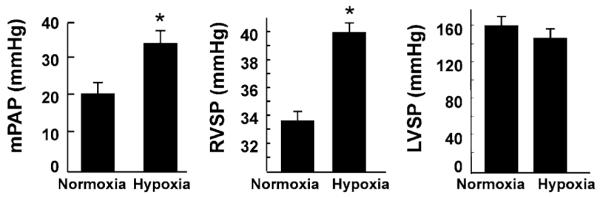
Changes in cardiac and pulmonary arterial pressure upon chronic exposure to hypoxia. Hemodynamic measurements were performed in rats exposed to hypoxia for 3 weeks. The results show a significant elevation in mean pulmonary arterial pressure (PAP) and RV systolic pressure (RVSP) in hypoxia-exposed rats when compared with the normoxic controls. *P < 0.05 versus normoxic conrols (mean ± SEM; n = 4). There was no significant change in the LV systolic pressure (LVSP)
Histopathology of Lung Tissue upon Chronic Exposure to Hypoxia
Histopathology and immunohistochemical analyses of the lung sections obtained from age-matched rats after 3 weeks of hypoxia showed a widespread medial hypertrophy of arteries in comparison with the normoxic controls (Fig. 5). The peripheral arteries showed hypertrophy and hyperplasia of medial smooth muscle and deposition of extracellular matrix in the perivascular space. There was also marked muscularization of the peripheral small arteries. Expression of PTEN was decreased in the expanded smooth muscle compartment in the vascular wall in the lungs of hypoxia-exposed rats. The results clearly indicated a loss of PTEN expression in the proliferating smooth muscle cells in the lung vasculature.
Fig. 5.

Histopathological and Immunohistochemical analysis of lung vascular SMC hyperplasia in hypoxia-exposed rats. Lung sections obtained from age-matched rats exposed to 3 weeks of hypoxia show marked muscularization of peripheral small arteries. The arteries show vascular smooth muscle hyperplasia in the tunica media. Immunohistochemical analysis also reveals a loss of PTEN expression, confined to the proliferating smooth muscle cells of the tunica media
PTEN and Cell-Cycle Regulatory Protein Expression in Hypoxia-Induced Pulmonary Hypertension
The deregulation of PTEN was further studied in the hypoxia model of PH. RT-PCR analysis in the lungs of hypoxia-exposed rats showed a decrease in the expression of PTEN at the mRNA level (Fig. 6a). Western-blot analysis of PTEN and related proteins showed a significant decrease in the levels of PTEN and pPTEN (Fig. 6b). Cyclin-D1 expression was significantly upregulated, while p53 was downregulated in the hypoxia-exposed animals. The results suggested that chronic hypoxia administration downregulated PTEN expression and upregulated cell-cycle regulatory protein cyclin-D1 in the lung.
Fig. 6.
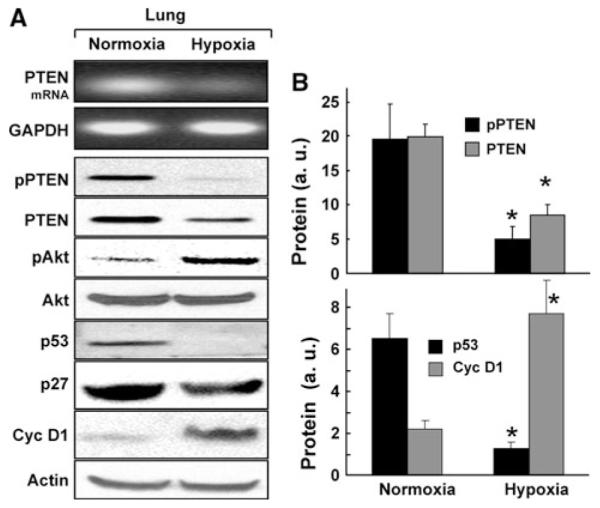
Expression of PTEN and related downstream proteins in the hypoxia model of PH. Rats were continuously exposed to 10% oxygen (hypoxia) for 3 weeks to induce PH. Reverse Transcription-PCR and western-blot analysis were performed on lung tissues. a Representative RT-PCR and western blot images of PTEN and related proteins. The RT-PCR image shows a decrease in PTEN expression at the mRNA level. Western-blot images show decreases in PTEN, pPTEN, and p53, and increase in pAkt and cyclin-D1 protein levels. b Quantitative results of the blots. Data represent mean ± SEM (n = 8). *P < 0.05 versus respective normoxic control. Significant decreases in both PTEN and pPTEN was observed when compared with the normoxic controls. The results also show a significant downregulation of p53 and upregulation of cyclin-D1 in the PH tissues
Hypoxia Induces SMC Proliferation by Modulating PTEN
We next determined the effect of hypoxia on PTEN expression using SMCs exposed to 1% oxygen in vitro for 24 or 48 h. Hypoxia exposure for 24 h had no effect on cell proliferation (data not shown). However, following 48-h exposure, there was a significant increase in cell proliferation when compared with normoxic-cultured cells (Fig. 7a). Western-blot analysis of the 24-h hypoxia-exposed cells showed no changes in the expression of PTEN, as well as in its upstream regulators (FAS and FAK) and downstream proteins (Akt and p21). However, 48-h exposure showed changes in PTEN, Akt, and p21 while there were no changes in the expressions of FAS and FAK. PTEN was downregulated, with a corresponding increase in Akt and decrease in p21 (Fig. 7b). It is known that the intracellular amount of rapid-turnover proteins, including PTEN, is tightly regulated by the ubiquitin-dependent proteolytic pathway [22]. To determine whether hypoxia can induce proteasomal degradation of PTEN, we performed ubiquitination assays on SMCs cultured underhypoxic conditions. Incubation with MG-132, an inhibitor of proteasomal degradation, caused an accumulation of polyubiquitin-tagged proteins on pPTEN and PTEN (Fig. 7c), suggesting that PTEN is degraded via a proteasome-mediated mechanism under hypoxic conditions.
Fig. 7.

Hypoxia induces pulmonary arterial SMC proliferation by activating pAkt and downregulating PTEN expressions. Human pulmonary artery smooth muscle cells (SMCs) were incubated at 1% oxygen for 48 h. a Quantitative analysis of SMC proliferation at 48 h. *P < 0.05 versus respective normoxic control. There was a significant increase in proliferation in the cells cultured without FBS in comparison with the respective normoxic controls. b Western-blot analysis of the downstream regulators of the PI3K pathway. Results show that, at 24 h, there are no changes in the levels of FAS, FAK, pAkt, pPTEN, PTEN and p21. PTEN, pPTEN, and p21 are decreased in the SMC exposed to hypoxia for 48 h. There is also a corresponding increase in pAkt with no changes in FAS and FAK levels. c Hypoxia promotes the proteasomal degradation of PTEN expression in SMC. A predominant ubiquitination of PTEN and pPTEN is seen in the SMC cells exposed to hypoxia via proteasomal inhibition using MG-132 (20 μmol/l). The results show that PTEN is downregulated in the setting of hypoxia exposure via the Akt-dependent pathway
Discussion
The results of the present study showed, for the first time, that PTEN is substantially dysregulated in the progression of PH. Both the MCT-administered and hypoxia-exposed in vivo models of PH showed a significant decrease in the expression of active PTEN in the lung and RV, but not in the LV. The decrease in PTEN was accompanied by an increase in pAkt and alteration in the cell-cycle promoting proteins in both models. The results of the in vivo models of PH were corroborated by in vitro studies using vascular smooth muscle cells exposed to hypoxia. Taken together, our findings confirm that PTEN plays a role in vascular remodeling in experimentally induced pulmonary hypertension.
In the MCT model of PH, we observed a decrease in PTEN with a concomitant increase in pAkt in the RV and lung tissues, suggesting upregulation of the PI3K pathway. However, PTEN and Akt were not affected in the LV tissue, suggesting that the MCT had no effect on the expression of these proteins in LV. Akt is a downstream target of the PI3K pathway and plays a central role in cell growth and survival [16, 23, 24]. The underlying mechanisms through which Akt is involved in cell proliferation may be via the inactivation of p53, p21, and p27, which are the inhibitors of cell-cycle progression as well as the accumulation of cyclin-D1 [21, 25, 26]. Activation of Akt exerts anti-apoptotic effects through phosphorylation of substrates that directly regulate the apoptotic machinery such as Bad [8] and caspase-9 [8, 24], or phosphorylation of substrates that indirectly inhibit apoptosis, such as inhibitors of NF-κB kinases [27, 28] and MDM2 [8]. Our results show upregulation of the PI3K pathway by PTEN, leading to Akt-dependent phosphorylation and inactivation of p53, p21, and p27.
The chronic hypoxia model of PH produced similar results as the MCT model with respect to PTEN-regulated Akt activation. Cyclin-dependent kinase inhibitor p27, a target of Akt, has been proposed as a downstream mediator through which PTEN may negatively regulate cell-cycle progression [9, 29]. The lipid–phosphatase activity of PTEN regulates Akt phosphorylation and p27 expression levels, whereas its protein–phosphatase activity regulates phosphorylation of mitogen-activated protein kinase (MAPK) and cyclin-D1 expression [11]. The activation of Akt/protein kinase B (PKB) is regulated in a complex manner via phosphorylation of Akt on Thr308 and Ser473 by protein-dependent kinase-1 (PDK1) and integrin-linked kinase (ILK), respectively [13, 21, 25]. Suppression of PTEN, such as that which occurs in PH, will constitutively activate the Akt/PKB pathway [30]. There is also activation of cyclin-D1, a controller of cell-cycle progression in both the models of PH used. This may signify that the vasculature has undergone hyperplasia, which is validated by the histopathological data [31, 32]. The effect of hypoxia on RV occurs in the initial stages of PH, leading to RV dilation which usually occurs terminally and is reflective of clinical scenarios. LV hypertrophy occurs in hypoxia due to decreased oxygen content in the coronary circulation and is a global phenomenon. We, however, did not see any LV hypertrophy in our hypoxia-exposed animals; it may possibly be too early to see the manifestation of this disorder in the LV [33, 34].
We observed that both PTEN and pPTEN were suppressed in SMCs exposed to hypoxia for 48 h, with a corresponding increase in pAkt and a decrease in p21 expression. p21 is a key regulator of cell-cycle progression when cells are exposed to oxidative stress, and this plays an important role in SMC proliferation [26, 29]. One of the downstream targets of PI3K is the serine/threonine kinase Akt, which mediates cell growth through stabilization of cyclin-D1 and downregulation of cyclin-dependent kinase (CDK) inhibitors p27 and p21 [21, 29]. We observed that a reduction in PTEN expression promotes SMC proliferation through activation of Akt, which requires phosphorylation. This, in turn, phosphorylates cellular-signaling molecules relevant for cell proliferation, including p27.
PTEN is an unstable protein and is known to undergo constant proteasome-mediated degradation in cells. The decrease in PTEN levels in the hypoxia-exposed pulmonary vasculature may be due to its accelerated degradation as has been observed in our laboratory and others [22, 35]. Our results using ubiquitination assays on hypoxia-exposed SMCs suggest that, the decrease in PTEN was caused by polyubiquitin-mediated proteasomal degradation. However, this observation does not rule out the possibility of inhibition of PTEN and its expression at the mRNA level by hypoxia exposure, as has been shown in a number of cancer cells [5, 22]. Overall, the results suggest that stabilization of PTEN by inhibition of its proteasomal degradation or increase of its expression at the mRNA level maybe a potential therapeutic opportunity to restore PTEN levels and inhibit the progression of PH.
Fatty acid synthase (FAS) is an essential factor for diverse cellular functions including energy storage, membrane formation, signal transduction, and protein acylation [11]. FAS is regulated by the tumor suppressor gene PTEN in vitro [11, 36]. This regulation is thought to be mediated by a PI3K/Akt-dependent pathway, since overexpression of Akt enhanced the expression of the FAS gene, and the treatment of cells with a PI3K inhibitor reduced the FAS expression [11]. FAK is a key molecule implicated in integrin-signaling pathways, and is a target and effector of PTEN action [36]. PTEN interacts with FAK, directly dephosphorylating the active form of the molecule [37]. We determined that the PTEN-mediated regulation of the PI3K pathway, in our in vitro hypoxic exposure experiments, were independent of the FAS- and FAK-signaling pathways.
Pulmonary arterial remodeling in PH occurs due to noninvasive injury. This is in contrast to the vascular injury observed in coronary and carotid arteries, which is usually due to invasive injury (placement of stents) or atherosclerotic plaque rupture. Early elevation in pulmonary pressure is known to be mediated by hypoxia-induced pulmonary vasoconstriction which eventually (under chronically elevated pressures) leads to the development of PH. The development of irreversible PH may result in vascular remodeling by proteasomal degradation-mediated downregulation PTEN levels.
In the present study, we observe that PTEN is suppressed in both models of PH. Thus, compounds capable of promoting or stabilizing PTEN expression could have an inhibitory effect on vascular remodeling [10, 15, 20, 22]. Thus, inhibition of SMC proliferation by PTEN activation may prove to be an attractive therapeutic strategy [17, 30,38–40]. In our previous study, we have tested the effect of HO-3867, a novel synthetic compound, on SMC proliferation through upregulation of PTEN [10, 22]. Future studies will need to evaluate the use of such PTEN-stabilizing compounds to inhibit the vascular remodeling associated with PH.
As mentioned in the introduction, the primary objective of this study was to determine the involvement of PTEN in PH, and hence the study used two well-established animal models of PH. Since both the monocrotaline and hypoxia models of PH have been well established, the present study did not attempt to fully characterize the model. In regard to translational aspects of our findings, we also recognize species variation in the pathogenesis of PH in rodents when compared to that in humans [35, 41]. Also, our studies do not exclude the potential involvement of other mediators in the models of PH used [20, 38, 39, 42–44]. Recent evidence that PTEN is deregulated in cardiac diseases with similar pathologies provides some assurance that the models used mimic alterations in PTEN accurately [13, 45].
Our findings clearly indicate that PTEN plays a significant role in vascular remodeling involved in PH. While the mechanism of PTEN dysregulation in vascular remodeling has been studied in other vascular tissues, it has not been investigated in the pulmonary vasculature under the conditions of chronically elevated pulmonary pressures that may occur secondary to hypoxia-induced pulmonary vasoconstriction. Nevertheless, the results of the present study indicate that it may be useful to exploit PTEN as a potential therapeutic target for the management PH or any other pathology in which vascular SMC-induced remodeling plays a role.
Acknowledgments
This study was supported by NIH grant HL095066. The authors thank Alex Dayton and Lakshmi Kuppusamy for providing cells and their help with some measurements. The text embodied in this manuscript was presented at the American Heart Association Annual Scientific Meeting, Nov 13-17, 2010, Chicago, USA, and the abstract is published in Circulation 122: A20181 (2010).
Abbreviations
- LAD
Left anterior descending coronary artery
- LVSP
Left ventricular systolic pressure
- MAP
Pulmonary arterial pressure
- MAP
Mean aortic pressure
- MCT
Monocrotaline
- MRI
Magnetic resonance imaging
- PH
Pulmonary hypertension
- PTEN
Phosphatase and tensin homolog deleted on chromosome 10
- RVSP
Right ventricular systolic pressure
- SMC
Smooth muscle cell
Footnotes
Conflict of interest All authors have no relevant disclosures.
Contributor Information
Yazhini Ravi, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA; Department of Surgery, Davis Heart and Lung Research Institute, The Ohio State University, N 816 Doan Hall, West 10th Avenue, Columbus, OH 43210, USA.
Karuppaiyah Selvendiran, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
Sarath Meduru, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
Lucas Citro, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
Shan Naidu, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
Mahmood Khan, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
Brian K. Rivera, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA
Chittoor B. Sai-Sudhakar, Department of Surgery, Davis Heart and Lung Research Institute, The Ohio State University, N 816 Doan Hall, West 10th Avenue, Columbus, OH 43210, USA
Periannan Kuppusamy, Department of Internal Medicine, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH 43210, USA.
References
- 1.Rabinovitch M. Cellular and molecular pathobiology of pulmonary hypertension conference summary. Chest. 2005;128:642S–646S. doi: 10.1378/chest.128.6_suppl.642S. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal R, Gomberg-Maitland M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. American Heart Journal. 2011;162:201–213. doi: 10.1016/j.ahj.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics–2010 update: A report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 4.Sehgal PB, Mukhopadhyay S. Pulmonary arterial hypertension: A disease of tethers, SNAREs and SNAPs? American Journal of Physiology—Heart and Circulation Physiology. 2007;293:H77–H85. doi: 10.1152/ajpheart.01386.2006. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Kontos CD. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:745–751. doi: 10.1161/01.atv.0000016358.05294.8d. [DOI] [PubMed] [Google Scholar]
- 6.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, et al. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1alpha. Circulation Research. 2008;102:1036–1045. doi: 10.1161/CIRCRESAHA.107.169896. [DOI] [PubMed] [Google Scholar]
- 7.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, et al. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. American Journal of Respiratory Cell and Molecular Biology. 2010;42:482–490. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu J, Tamura M, Yamada KM. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. Journal of Cell Biology. 1998;143:1375–1383. doi: 10.1083/jcb.143.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon SK, Kim HM, Kim CH. PTEN induces G1 cell cycle arrest and inhibits MMP-9 expression via the regulation of NF-kappaB and AP-1 in vascular smooth muscle cells. Archives of Biochemistry and Biophysics. 2004;421:267–276. doi: 10.1016/j.abb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Tamguney T, Stokoe D. New insights into PTEN. Journal of Cell Science. 2007;120:4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- 11.Van de Sande T, De Schrijver E, Heyns W, Verhoeven G, Swinnen JV. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Research. 2002;62:642–646. [PubMed] [Google Scholar]
- 12.Oudit GY, Penninger JM. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovascular Research. 2009;82:250–260. doi: 10.1093/cvr/cvp014. [DOI] [PubMed] [Google Scholar]
- 13.Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. Journal of Molecular and Cellular Cardiology. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, Vienney N, et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circulation Research. 2011;109:543–553. doi: 10.1161/CIRCRESAHA.111.241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J, Kontos CD. PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. Journal of Biological Chemistry. 2002;277:10760–10766. doi: 10.1074/jbc.M110219200. [DOI] [PubMed] [Google Scholar]
- 16.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 17.Voelkel NF, Tuder RM. Hypoxia-induced pulmonary vascular remodeling: A model for what human disease? Journal of Clinical Investigation. 2000;106:733–738. doi: 10.1172/JCI11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: Efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation. 2008;117:2626–2636. doi: 10.1161/CIRCULATIONAHA.107.737031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng N, Hoover DB, Paolocci N. Forever young? Nerve growth factor, sympathetic fibers, and right ventricle pressure overload. Circulation Research. 2007;100:1670–1672. doi: 10.1161/CIRCRESAHA.107.155861. [DOI] [PubMed] [Google Scholar]
- 20.Itoh T, Nagaya N, Fujii T, Iwase T, Nakanishi N, Hamada K, et al. A combination of oral sildenafil and beraprost ameliorates pulmonary hypertension in rats. American Journal of Respiratory and Critical Care Medicine. 2004;169:34–38. doi: 10.1164/rccm.200303-346OC. [DOI] [PubMed] [Google Scholar]
- 21.Selvendiran K, Kuppusamy ML, Bratasz A, Tong L, Rivera BK, Rink C, et al. Inhibition of vascular smooth-muscle cell proliferation and arterial restenosis by HO-3867, a novel synthetic curcuminoid, through up-regulation of PTEN expression. Journal of Pharmacology and Experimental Therapeutics. 2009;329:959–966. doi: 10.1124/jpet.108.150367. [DOI] [PubMed] [Google Scholar]
- 22.Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, et al. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. Journal of Biological Chemistry. 2007;282:28609–28618. doi: 10.1074/jbc.M703796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ, et al. Phosphorylation of HDM2 by Akt. Oncogene. 2002;21:1955–1962. doi: 10.1038/sj.onc.1205276. [DOI] [PubMed] [Google Scholar]
- 24.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 25.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno S, Bogaard HJ, Voelkel NF, Umeda Y, Kadowaki M, Ameshima S, et al. Hypoxia regulates human lung fibroblast proliferation via p53-dependent and -independent pathways. Respiration Research. 2009;10:17. doi: 10.1186/1465-9921-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 28.Robbins IM. Advancing therapy for pulmonary arterial hypertension: Can animal models help? American Journal of Respiratory and Critical Care Medicine. 2004;169:5–6. doi: 10.1164/rccm.2310015. [DOI] [PubMed] [Google Scholar]
- 29.Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, et al. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. American Journal of Physiology Lung Cellular and Molecular Physiology. 2011;300:L753–L761. doi: 10.1152/ajplung.00286.2010. [DOI] [PubMed] [Google Scholar]
- 30.Penumathsa SV, Maulik N. Resveratrol: A promising agent in promoting cardioprotection against coronary heart disease. Canadian Journal of Physiology and Pharmacology. 2009;87:275–286. doi: 10.1139/Y09-013. [DOI] [PubMed] [Google Scholar]
- 31.Chung JH, Ostrowski MC, Romigh T, Minaguchi T, Waite KA, Eng C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Human Molecular Genetics. 2006;15:2553–2559. doi: 10.1093/hmg/ddl177. [DOI] [PubMed] [Google Scholar]
- 32.Weng LP, Brown JL, Eng C. PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Human Molecular Genetics. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- 33.Chen L, Zhang J, Gan TX, Chen-Izu Y, Hasday JD, Karmazyn M, et al. Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. Journal of Applied Physiology. 2008;104:218–223. doi: 10.1152/japplphysiol.00301.2007. [DOI] [PubMed] [Google Scholar]
- 34.Kimura H, Kasahara Y, Kurosu K, Sugito K, Takiguchi Y, Terai M, et al. Alleviation of monocrotaline-induced pulmonary hypertension by antibodies to monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Laboratory Investigation. 1998;78:571–581. [PubMed] [Google Scholar]
- 35.Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Current Opinion in Cell Biology. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- 36.Heath D. The rat is a poor animal model for the study of human pulmonary hypertension. Cardioscience. 1992;3:1–6. [PubMed] [Google Scholar]
- 37.Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. Journal of Biological Chemistry. 1999;274:20693–20703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 38.Huang J, Niu XL, Pippen AM, Annex BH, Kontos CD. Adenovirus-mediated intraarterial delivery of PTEN inhibits neointimal hyperplasia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:354–358. doi: 10.1161/01.ATV.0000151619.54108.a5. [DOI] [PubMed] [Google Scholar]
- 39.Osman N, Ballinger ML, Dadlani HM, Getachew R, Burch ML, Little PJ. p38 MAP kinase mediated proteoglycan synthesis as a target for the prevention of atherosclerosis. Cardiovasc Hematol Disord Drug Targets. 2008;8:287–292. doi: 10.2174/187152908786786205. [DOI] [PubMed] [Google Scholar]
- 40.Vidavalur R, Swarnakar S, Thirunavukkarasu M, Samuel SM, Maulik N. Ex vivo and in vivo approaches to study mechanisms of cardioprotection targeting ischemia/reperfusion (i/r) injury: Useful techniques for cardiovascular drug discovery. Current Drug Discovery Technologies. 2008;5:269–278. doi: 10.2174/157016308786733555. [DOI] [PubMed] [Google Scholar]
- 41.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez OC, Lai EW, Vissapragada S, Cromelin C, Avetian M, Salinas P, et al. A reduction in Pten tumor suppressor activity promotes ErbB-2-induced mouse prostate adenocarcinoma formation through the activation of signaling cascades downstream of PDK1. American Journal of Pathology. 2009;174:2051–2060. doi: 10.2353/ajpath.2009.080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basu P, Sen U, Tyagi N, Tyagi SC. Blood flow interplays with elastin: collagen and MMP: TIMP ratios to maintain healthy vascular structure and function. Journal of Vascular Health and Risk Management. 2010;6:215–228. doi: 10.2147/vhrm.s9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perez J, Torres RA, Rocic P, Cismowski MJ, Weber DS, Darley-Usmar VM, et al. PYK2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. American Journal of Physiology—Cell Physiology. 2011;301:C242–C251. doi: 10.1152/ajpcell.00315.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: Moving beneath the cell surface. Nature Reviews Drug Discovery. 2007;6:617–635. doi: 10.1038/nrd2193. [DOI] [PubMed] [Google Scholar]