

Abstract
With the increase in the ageing population, neurodegenerative disease is devastating to families and poses a huge burden on society. The brain and spinal cord are extraordinarily complex: they consist of a highly organized network of neuronal and support cells that communicate in a highly specialized manner. One approach to tackling problems of such complexity is to address the scientific questions in simpler, yet analogous, systems. The fruit fly, Drosophila melanogaster, has been proven tremendously valuable as a model organism, enabling many major discoveries in neuroscientific disease research. The plethora of genetic tools available in Drosophila allows for exquisite targeted manipulation of the genome. Due to its relatively short lifespan, complex questions of brain function can be addressed more rapidly than in other model organisms, such as the mouse. Here we discuss features of the fly as a model for human neurodegenerative disease. There are many distinct fly models for a range of neurodegenerative diseases; we focus on select studies from models of polyglutamine disease and amyotrophic lateral sclerosis that illustrate the type and range of insights that can be gleaned. In discussion of these models, we underscore strengths of the fly in providing understanding into mechanisms and pathways, as a foundation for translational and therapeutic research.
Keywords: FlyBook, Drosophila, ALS, FTD, genetic pathways, polyglutamine disease, protein toxicity
THE World Health Organization, the World Bank, and the Harvard School of Public Health describe neurological disease as one of the greatest burdens to public health, and they expect this burden to advance to a globally unmanageable problem (Aarli et al. 2006). Multiple scientific approaches are required to understand disease etiology, progression, and management, to aid understanding of onset and risk factors, as well as treatment and intervention design. With >600 neurological disorders listed by the National Institute of Neurological Disorders and Stroke—categories include neurodevelopmental, stroke, traumatic injury, cancer, and neurodegenerative—the disease range is complex. An approach toward disease insight is to model disease mechanisms and identify disease-modifying pathways in less complex, yet analogous, organisms. The fruit fly Drosophila melanogaster has yielded great advances into the underpinnings of many neurological and neurodegenerative diseases, not only providing understanding of biological pathways impaired in disease, but also the foundation for strategies for intervention approaches in mammalian systems.
The fly was introduced into biological research >100 years ago and quickly became a valuable tool that has been paramount to our understanding of genes, chromosomes, and the inheritance of genetic information (St Johnston 2002; Bellen et al. 2010). Ionizing and chemical mutational techniques were developed that allowed researchers to probe gene function by analyzing the biological impact of mutations. Given the relatively rapid lifespan of the fly, coupled with its propensity to produce large numbers of progeny, large-scale mutagenic screens identified many critical genes in a vast range of biological processes, including embryogenesis, neural development, and complex behaviors; many of these genes were later found to be conserved in sequence and function in mammals. One of the early genetic screens was designed to identify mutants with abnormal behavior, including the so-named drop dead (drd) mutant with a shortened lifespan and a degenerate brain (Benzer 1971). The discovery that the drd mutant underwent brain deterioration suggested that the fly could model not only puzzling behaviors like phototaxis and circadian cycles, but also human late-onset degenerative disorders. Later studies led to the isolation of additional fly brain mutants with features reminiscent of human brain disease pathology: spongecake and eggroll develop membrane vacuoles and multilamellar structures reminiscent of the pathological structures that form in Creutzfeldt-Jakob disease and Tay Sachs disease (Min and Benzer 1997). These genes have yet to be defined; however, the gene encoding swiss cheese (sws), a mutant with an age-dependent loss in motor activity and brain degeneration shows homology to the human gene encoding neuropathy target esterase PNPLA6 (Kretzschmar et al. 1997; Lush et al. 1998). Mutations in PNPLA6 have since been identified as causal for motor neuron disease and the neurodegenerative disorders Boucher-Neuhauser and Gordon Holmes syndromes (Rainier et al. 2008; Deik et al. 2014; Synofzik et al. 2014). These and other studies (for reviews, see Lessing and Bonini 2009; Zu et al. 2011) underscore the extraordinary contributions that the fly can make toward a fundamental understanding of gene function in brain maintenance through forward genetic screens.
Although powerful, the use of classical genetic screens to provide insight into human neurodegenerative disease can be slow because of several limitations. For example, mutations may not be evident in the precise fly homolog of the human disease gene; rather, the fly highlights the pathway or process that is reflected by the human disease (Reiter and Bier 2002). Classical fly mutant screens also typically isolate alleles that are loss of function, whereas in human disease, mutations can have complex presentations, including loss of function, gain of function, new functions, or a combination of these.
The sequencing of the fly and human genomes revealed remarkable gene and pathway conservation between the fly and human (Rubin et al. 2000), with an estimated two-thirds of known human disease-causing genes being present in the fly (Rubin et al. 2000; Reiter et al. 2001). With the genome information, a different approach to disease could be introduced whereby the fly can be “given” the human disease: by expressing the disease form of the human gene in the animal, the animals can be analyzed in detail to assess the biological effects and pathways involved in the disease process (Figure 1). Among ways to do this is the popular approach of the GAL4-UAS binary system (Brand and Perrimon 1993). GAL4 is a yeast transcription factor that can be used to control both the spatial and temporal expression of target genes, to direct gene activity at a specific time (for example, the adult), and a specific cell type (for example, neurons). Alternatively, the human disease gene can be directly expressed in a nonessential tissue such as the eye, given the frequently dominant nature of such human disease mutations and their lethality. Initial fly models of this type recreated human polyglutamine (polyQ) disease in the animal by expressing normal and mutant versions of the genes associated with spinocerebellar ataxia type 3 (SCA3) (Warrick et al. 1998) and Huntington’s disease (Jackson et al. 1998). The explosion in the application of the fly as a model for such human neurodegenerative diseases since that introduction is aided by the vast array of available genetic tools that allow for the up- and downregulation of gene expression and ability to perform genome-wide unbiased screens (Perrimon et al. 2010; Venken and Bellen 2014): modifiers can be rapidly identified to define genes and biological pathways that impact disease susceptibility, onset, and progression.
Figure 1.

Steps for generating and characterizing a Drosophila model for a human neurodegenerative disease. Here, we outline what we consider to be basic steps in the generation and characterization of a Drosophila neurodegenerative disease model. These steps generally apply to any human disease. Genes that are known to be involved in disease, based on genetic studies or pathological examination of human neural tissue usually serve as a starting point. Drosophila orthologs are identified to enable loss-of-function and gain-of-function gene manipulation. Alternatively, or in addition, the human gene or the genetic lesion associated with the disease (e.g., hexanucleotide repeat expansion as found in ALS/FTD) can be expressed in the fly. Multifarious strategies for genetic manipulation exist in the fly, allowing tissue-specific and age-specific expression of transgenes for both up- and downregulation of gene expression. Examples for loss-of-function studies include hypomorphic or null alleles, deficiency lines in which a stretch of DNA that includes all or part of the gene of interest or a region of the genome covering many genes has been deleted, and shRNA and dsRNA transgenes to knockdown gene expression. For upregulation, a commonly used strategy is expression of a transgene under the control of the upstream activating sequence (UAS). In this bipartite system, the GAL4 transcriptional activator is placed under a cell-type-specific promoter, such as gmr-GAL4 for expression in the fly eye, enabling spatial and/or temporal control of transgene expression. Genes can also be directly driven by a promoter in a tissue of interest, and techniques can be used to create the human mutation within the fly ortholog under endogenous regulatory control. A plethora of GAL4 lines driven by defined DNA fragments for distinct cellular patterns in the developing and adult brain are available for very select expression patterns (Jenett et al. 2012; Li et al. 2014). Upon establishment of a model, the effects of the disease gene are examined and compared to known manifestations and features of the human disease, and those observed in other models. Detailed functional studies of the fly model can reveal new insight into the disease process. Functional characterization and genome-wide screens in the fly can allow additional insight into disease pathways and mechanisms. Genes and processes identified through these studies can then be examined in human samples or mammalian models to aid discovery of novel disease-associated genes, risk factors, and pathological hallmarks. Fly models can also be used for compound screening.
A combination of several key factors makes the fly a uniquely powerful animal model for neuroscience research. First, Drosophila has a short life cycle of ∼10 days to the adult and is easily and relatively inexpensively maintained in the laboratory in large numbers. Since the days of Thomas Hunt Morgan who used Drosophila to discover the role of chromosomes in hereditary, and of Hermann J. Muller who found that irradiation causes an increase in the mutation rate in Drosophila (Morgan et al. 1915), tools to manipulate and control Drosophila genetics have been developed, offering researchers a comprehensive toolbox. One example are so-called balancer chromosomes that allow homozygous lethal mutations to be maintained in heterozygous “balanced” fly stocks and are typically marked with visible dominant markers.
Second, gene manipulation in Drosophila can be fast and relatively simple. Over the years, the fly community has developed a staggering number of mutant and transgenic fly lines. Drosophila stocks cannot be frozen, and all fly lines are maintained alive and are commonly shared. The Bloomington Drosophila Stock Center at Indiana University (Cook et al. 2010) maintains a comprehensive collection of mutant, RNAi, misexpression, as well as other stocks. During 2014, this stock center maintained >53,000 Drosophila stocks and shipped >230,000 fly cultures. Additional stock centers, such as the Harvard Transgenic RNAi Project (TRiP) (Ni et al. 2011) and the Vienna Drosophila Research Center (VDRC) (Dietzl et al. 2007), have generated genome-wide RNAi collections. This availability of tools to exquisitely manipulate expression of nearly every gene dramatically facilitates research capabilities. Precise spatiotemporal control over gene expression is easily attained in Drosophila, using large collections of cell-type-specific drivers, as well as compound- or temperature-activated driver lines. The versatile genetic toolbox of Drosophila surpasses that of any other multicellular organism and is constantly being expanded, allowing more elaborate manipulation of the Drosophila genome. FlyBase (www.flybase.org)—the website of Drosophila genes and genomes—curates published phenotypes, gene expression, genetic and physical interactions, and many other datasets concerning Drosophila genetics (St Pierre et al. 2014), allowing researchers access to high-quality and comprehensive information.
Third, for the study of the nervous system, the fly also offers unique advantages. While less complicated than the mammalian brain, the fly has a complex central nervous system with neurons and glia, is protected by a blood–brain barrier, and shares striking organizational similarities with the vertebrate brain. An example of such similarities, which were even observed by the great neuroscientist Ramon Y. Cajal, are the neuronal circuits of vision: as noted by Sanes and Zipursky (2010), Cajal turned to the fly with hopes of finding a simple circuitry to allow easier tracing of neuronal connectivity. Instead, he found a complicated system rivaling that of vertebrates. Functional, developmental, and molecular similarities between these systems further testify that basic principles of neural circuitry are conserved from flies to humans.
Fourth, the smaller size of the Drosophila genome (1.2 × 108 base pairs) compared to the human genome (3.3 × 109 base pairs) and the smaller number of genes (∼14,000 vs. ∼20,000–25,000 protein-coding genes) have an important practical implication: many genes in humans are part of gene families composed of paralogues with redundant and/or overlapping functions. In Drosophila, the reduced genome complexity allows easier interpretation of loss-of-function studies. Taken together, these advantages exemplify the reasons for which Drosophila has made paramount contributions to biological research in general and to neuroscience research in particular.
The approach of recreating a simplified version of human neurological disease by expressing (or knocking down) the human mutant gene in the fly has now been used to model features of many neurological and neurodegenerative disorders beyond polyQ disease. These include, but are not limited to, Parkinson’s disease (Online Mendelian Inheritance of Man, OMIM no. 168600), Alzheimer’s disease (OMIM no. 104300), fragile X syndrome (OMIM no. 300624), fragile X tremor/ataxia syndrome (FXTAS; OMIM no. 300623), and additional spinocerebellar ataxias (Feany and Bender 2000; Fernandez-Funez et al. 2000; Wittmann et al. 2001; Dockendorff et al. 2002; Morales et al. 2002; Jin et al. 2003; Mutsuddi et al. 2004). When combined with findings from human patient pathology, human population genetics and other biological understanding, such fly models provide mechanistic insight into the molecular pathways of disease that in turn may be of importance for future translational applications.
Given the current vast breadth of the field of fly models of neurodegenerative disease, here we focus on two disease classes for illustrative purposes: polyQ diseases and amyotrophic lateral sclerosis (ALS; OMIM no. 105400)/frontotemporal dementia (FTD; OMIM no. 600274) spectrum. These studies were selected to highlight how a disease model can be established in the fly, and how studies in Drosophila can be combined with basic research in human genetics and patient pathological findings to provide novel understanding. These examples also include emphasis on both protein-based disease toxicity models and contrasting RNA-pathway based models. These highlight two of the biologically diverse pathways known to contribute to major human neurodegenerative diseases (although also reveal that pathways may be more similar than initially suspected).
Our goal in this review is to illustrate the template and work flow of such studies using this extraordinary model system, by highlighting the methodology, as well as approaches that lead to surprising discoveries. We have selected studies that illustrate key points, as the foundation for developing and using fly models of disease. Because the study of neurodegenerative disease in Drosophila is a vigorous and broad field, many outstanding reviews are available that highlight additional disorders and aspects of study (see Shulman et al. 2003; Bier 2005; Bilen and Bonini 2005; Marsh and Thompson 2006; Lessing and Bonini 2009; Lu and Vogel 2009; Bakhoum and Jackson 2011; Jaiswal et al. 2012).
We include a figure illustrating the strategy plan, with the various steps that can be involved in development and characterization of a human disease model (Figure 1) and highlight biological assays that are frequently used (Figure 2). In addition, for readers’ reference, we include a table of the range of neurodegenerative diseases modeled in the fly, beyond polyQ and ALS, with an initial citation that presents the model (Table 1). Models include those for Alzheimer’s disease, Parkinson’s disease, motor neuron disease, fragile X tremor/ataxia syndrome, myotonic dystrophy, as well as a range of polyQ diseases, ALS, and frontotemporal dementia. Thus, this area of research is broad and vigorous, with many diverse and important models. As noted, here we focus on select studies, often launching from work in our laboratory, to illustrate how the fly can be used and how studies in the fly can be instrumental hand in hand with other studies to provide key insight into mechanism.
Figure 2.

Examples of robust assays to assess neural degeneration and dysfunction in Drosophila. We illustrate assays that can be used in disease models for a neurodegenerative effect. We note that similar approaches, but geared toward the features of the disease of interest, can be used in the characterization of fly models for other classes of human disease. (A) Using an eye-specific GAL4 driver, both the disease gene and candidate modifiers can be expressed in the eye. As the eye is a nonvital tissue, the effects of highly toxic genes can be assessed in adult flies without concerns of lethality. For large-scale screens, the external eye (top panel) offers a rapid readout. Compared with the normal eye, the degenerative eye can show disruption of ommatidial structure, reduced size, and loss of pigmentation. Internal sections (bottom panel) allow examination of the retinal tissue. The degenerative eye shows collapse of the retina (arrows). (B) The pseudopupil assay allows a quantitative measure of degeneration in adult flies by assessing the structure of the photoreceptor cells by counting the number of intact rhabdomeres (the specialized organelles of the photoreceptor cells). Each ommatidium is composed of eight photoreceptor cells. Using back illumination of the head, seven rhadomeres of each ommaditium can be visualized (left panel) and their structural deterioration over time due to degeneration can be monitored (right panel). (C) Labeling of specific neuronal cell types, either by using selective antibodies (such as antityrosine hydroxylase to label dopaminergic neurons) or by expression of markers using the GAL4-UAS system can allow one to assess the integrity of neurons in the brain [reprinted from Auluck et al. (2002)]. (D) Expression of disease genes in motor neurons, followed by immunostaining for presynaptic (red, anti-HRP) and postsynaptic (green, anti-Dlg1) neuromuscular junction (NMJ) markers can reveal NMJ abnormalities such as reduced bouton numbers and increase in “ghost boutons” that lack opposing postsynaptic structure (Kim et al. 2013b). Images are courtesy of N. C. Kim and J. P. Taylor (St. Judes Medical Center). (E) Lifespan analysis of flies expressing disease-related genes. Transgene expression can be limited to the nervous system, to specific neuronal cell types, or to the adult stage to bypass developmental effects by using the range of driver lines available. (F) The negative geotaxis climbing reflex of Drosophila can be used to examine motor deficits with age. Flies are tapped to the bottom of an empty vial and the number of flies that can climb above a certain height is recorded. (G) Brain degeneration in Drosophila is often accompanied by formation of vacuoles as shown here upon expression of a CAG-repeat RNA [reprinted from Li et al. (2008)]. Image of Drosophila in center, is courtesy of wiki commons.
Table 1. A listing of various fly models for human neurodegenerative disease.
Listed are a range of fly models for various human neurodegenerative diseases. These models highlight the approaches and understanding achieved by the variety of disease models, and the range of diseases and disease genes that have been expressed in Drosophila to provide insight into pathways involved, hand-in-hand with analysis in mammalian or patient tissue systems. Although there are many human disease genes, and many different models (we apologize that not all could be referenced here), this table can serve as a launching point for examples of the generation and characterization of disease models in Drosophila for readers.
Modeling Protein-Based Toxicity in Drosophila with PolyQ Disease
Establishing a model
The human polyQ diseases encompass a set of diseases whereby a CAG-trinucleotide repeat expansion occurs within the open reading frame of the respective gene. These diseases include Huntington’s disease (HD; OMIM no. 143100), spinal and bulbar muscular atrophy (SBMA; OMIM no. 313200), dentatorubral-pallidoluysian atrophy (DRPLA; OMIM no. 125370), and spinocerebellar ataxias types 1 (OMIM no. 164400), 2 (OMIM no. 183090), 3 (OMIM no. 109150), 6 (OMIM no. 183086), 7 (OMIM no. 164500), and 17 (OMIM no. 607136) (Gusella and MacDonald 2000; Orr and Zoghbi 2007). In the ataxias, regions of the brain that control motor coordination and balance degenerate, and patients often become permanently disabled. Spinocerebellar ataxia type 3 (SCA3) is the most common dominantly inherited ataxia (Ruano et al. 2014). The mutation in SCA3 is an expansion of the CAG-trinucleotide repeat in the coding region of the ATAXIN 3 (ATXN3) gene and results in an expanded polyQ tract within the protein (Kawaguchi et al. 1994). The pathological hallmark of polyQ disease is the presence of insoluble ubiquitinated protein inclusions in the brain that contain the expanded polyQ protein, as well as proteins such as molecular chaperones (which normally facilitate protein folding) and components of protein degradation pathways (Paulson et al. 1997; Cummings et al. 1998; Gusella and MacDonald 2000; Orr and Zoghbi 2007).
The generation of transgenic flies (flies that stably carry exogenous DNA elements) has been instrumental to the generation of the vast array of tools available to Drosophila researchers (Venken and Bellen 2012). To provide in vivo models for polyQ disease, transgenic flies were generated for SCA3 and HD, such that the proteins encoded by the ATXN3 gene and HD gene HUNTINGTIN (HTT), respectively, with normal or expanded mutant polyQ repeat lengths, were stably inserted into the fly genome (Jackson et al. 1998; Warrick et al. 1998). The SCA3 and HD transgenes were expressed in specific cell types of the fly to determine the cellular consequences of the pathogenic polyQ protein. The fly eye is composed of thousands of neuronal and nonneuronal cells, and the glass multiple reporter (gmr) promoter and gmr-GAL4 driver are active in the photoreceptor neurons and other cell types of the eye throughout development (Ellis et al. 1993). To test whether the pathogenic SCA3 and HD transgenes resulted in degeneration, they were expressed in the eye and disease-associated toxicity was measured through analysis of the integrity of the adult retina.
Expression of an Ataxin-3 expanded polyQ protein (SCA3trQ78) in the eye by gmr-GAL4 conferred toxicity that was remarkably reminiscent of human polyQ disease: whereas a polyQ repeat within the normal range (SCA3trQ27) had no effect, expression of the disease form with the long expansion conferred late-onset progressive degeneration (Warrick et al. 1998) (Figure 3). Similarly, expression of a fragment of the HD protein (called exon1) with a short polyQ repeat (Q2) had no effect, whereas expression of HD exon1 with disease-associated polyQ expansions (Q75 and Q120) induced late-onset, progressive retinal deterioration (Jackson et al. 1998). In addition to gross degeneration of the eye, these studies showed that the fly could recapitulate detailed molecular and neural pathological aspects of human polyQ disease. For example, in polyQ disease, there is a general correlation of disease severity with the length of the CAG-repeat expansion; this could be seen with the fly HD model. In humans, a neuropathological hallmark of polyQ disease is the accumulation of proteinaceous neuronal inclusions that contain the pathological polyQ protein, a feature shared not only by mouse models, but also by the fly (Jackson et al. 1998; Warrick et al. 1998). These initial studies of HD and SCA3 revealed the striking similarities between the fly with mouse models and human disease (Davies et al. 1997; DiFiglia et al. 1997; Paulson et al. 1997; Sapp et al. 1997) and launched the fly as a robust and remarkable model for approaching human degenerative disease.
Figure 3.

Expression of pathogenic human polyQ protein in Drosophila causes severe eye degeneration that is suppressed by Hsp70. Eyes (A–C) and retinal sections (D–F) of flies expressing expanded polyQ protein alone or with human Hsp70, using the gmr-GAL4 driver to express UAS-transgenes. (A and D) Control eye structure. These are flies expressing human hsp70 gene and the eye structure is nearly normal. (B and E) Flies expressing the expanded polyglutamine protein SCA3tr-Q78. These flies have severely degenerate eyes that lack pigment and show severe loss of structure. (C and F) Flies expressing both SCA3tr-Q78 and Hsp70. Hsp70 expression mitigates the degenerative effects of the pathogenic polyQ protein to restore the external eye completely and the internal eye back toward normal [reprinted from Warrick et al. (1999)].
Modifier pathways: molecular chaperones in polyQ toxicity and beyond
These initial polyQ models illustrated the generation of fly models that recapitulate pathological features of human disease. Subsequent studies include the generation of a wide array of fly disease models (Table 1) and also genetic modifier screens that paved the way for the discovery of novel pathways involved in the disease process. We delineate these approaches below for polyQ disease, highlighting the overall steps in Figure 1 and Figure 2. Such an approach can be applied not only to neurodegenerative and neurological disease, but to other disorders as well.
Studies in mammalian cell culture systems showed that polyQ inclusions could be cleared by the upregulation of molecular chaperones (Cummings et al. 1998; Chai et al. 1999; Stenoien et al. 1999)—but could these pathways have an effect in vivo in the organism, and could they modulate neurotoxicity? To address this, the molecular chaperone Hsp70 was analyzed in the SCA3 fly model (Warrick et al. 1999). The nuclear inclusions that form in the fly by the SCA3trQ78 protein were found to colocalize with Hsp70, signifying that the polyQ inclusions in the fly have similar pathology to polyQ inclusions that form in human brain tissue (Paulson et al. 1997; Cummings et al. 1998). Importantly, the upregulation of chaperones in the SCA3trQ78 fly model powerfully protected against the ability of the protein to induce toxicity: the retinal degeneration induced by the pathogenic polyQ protein was dramatically prevented, restoring an eye to nearly normal structure (Warrick et al. 1999) (Figure 3). Interestingly, these studies also revealed that a compromise of chaperone activity can accelerate and enhance toxicity of the polyQ protein. This finding of dosage sensitivity of pathogenic polyQ protein to chaperone activity indicates that molecular chaperone function is a critical player in the presentation of toxicity in vivo: not only does upregulation modify the toxicity of the protein, but chaperone function normally contributes to the disease process.
The above studies examined the role of a specific player in the polyQ disease process: molecular chaperones. They illustrate how molecular genetic approaches can assess whether up- and downregulation of target genes of special interest can modulate the toxicity of a disease protein and thereby contribute to disease progression. These and other studies have revealed a number of effects of various molecular chaperone proteins, chaperone helper proteins, and protein quality control pathways on disease pathogeneisis in fly models (Warrick et al. 1999; Chan et al. 2000; Kazemi-Esfarjani and Benzer 2000; Vos et al. 2010; Kuo et al. 2013). These studies have also revealed links between the ubiquitin–proteasome pathway and autophagy pathways, as these processes dynamically interact in vivo to affect protein degradation, normally and in disease (Pandey et al. 2007). Studies on the role of moleular chaperones in neurodegenerative disease have revealed that genes of the heat shock response pathway, as well as protein degradation pathways, which help to handle misfolded proteins in times of stress, are a plausible approach for mitigating disease protein toxicity. Intriguingly, compromise of just chaperone activity alone was also noted to cause effects reminiscent of degeneration (Elefant and Palter 1999; Bonini 2002), suggesting that too little chaperone activity—on its own in the absence of a specific disease protein—is deleterious.
The yeast heat shock protein 104 (Hsp104) is a highly potent disaggregase that resolubilizes protein aggregates (Shorter 2008). Despite being absent from the metazoan genome, the disaggregase activity of Hsp104 is effective in vivo at mitigating the toxicity of the HD protein in the mouse and the toxicity of proteins associated with Parkinson’s disease in the rat (Vacher et al. 2005; Lo Bianco et al. 2008). The ability of Hsp104 to mitigate toxicity of a variety of different disease proteins highlights the possibilty of a common therapeutic strategy to human neurodegeneration. The studies of Hsp104 in the mouse and rat involved coexpression of the disease protein with the Hsp104 disaggregase. But the question arises as to how effective would such a modifier be at mitigating protein toxicity after disease onset? The fly is an amenable system to address such a question. A drug-inducible adaptation of the GAL4 binary system called GAL4 GeneSwitch (GS) activates the GAL4 transcription factor in the presence of the drug mifepristone (RU486) (Osterwalder et al. 2001; Roman and Davis 2002). Cushman-Nick et al. (2013), used a gmrGS-GAL4 driver to show that Hsp104 was able to not only reduce Ataxin-3 aggregation when expressed prior to inclusion formation, but also to suppress toxicity of the pathogenic polyQ protein when expressed after the onset of degeneration (Cushman-Nick et al. 2013). Unexpectedly, whereas Hsp104 mitigated toxicity of the truncated Ataxin-3 protein, Hsp104 enhanced the toxic effect of full-length pathogenic Ataxin-3 protein, highlighting the importance of protein context in which the polyQ repeat is embedded (discussed below), even for interactions with cellular pathways such as molecular chaperones.
The introduction of the fly as a model for neurodegenerative disease rapidly led to additional models; among these was a Parkinson’s disease model mimicked by the expression of α-synuclein (Feany and Bender 2000), a protein associated pathologically and by mutation with Parkinson’s disease (OMIM no. 168600) (Kruger et al. 1998; Spillantini et al. 1998; Singleton et al. 2003; Zarranz et al. 2004; Lesage et al. 2013; Proukakis et al. 2013). Here we illustrate how findings from study of polyQ disease in the fly led to insight into Parkinson’s disease. Parkinson’s disease is the most common movement disorder and is characterized by resting tremor, bradykinesia (slowed movement), impaired balance and muscular rigidity. Pathologically, the disease involves the degeneration of dopaminergic neurons in the substantia nigra and is accompanied by widespread intracellular protein accumulations of α-synuclein called Lewy bodies and Lewy neurites (Lees et al. 2009; Goedert et al. 2013; Irwin et al. 2013). Causal association of α-synuclein is underscored by mutations in, and increased copy number of, the ALPHA-SYNUCLEIN (SNCA) gene in patients with Parkinson’s disease (Kruger et al. 1998; Singleton et al. 2003; Zarranz et al. 2004; Lesage et al. 2013; Proukakis et al. 2013). The neurodegenerative capacity of α-synuclein was assessed in the nervous system of the fly using both global neuronal expression and expression directed selectively to dopaminergic neurons (Feany and Bender 2000). Expression of α-synuclein in flies confers age-dependent motor difficulties and a compromise of dopaminergic neurons, observed by a loss of tyrosine hydroxylase staining (Feany and Bender 2000; Trinh et al. 2008). α-Synuclein accumulations, reminiscent of Lewy bodies, occur in a small percentage of fly neurons and, similar to human Parkinson’s, these accumulations are not restricted to the dopaminergic neurons (Feany and Bender 2000; Goedert et al. 2013). Although it is not clear that the neurons are gone in all models of α-synuclein expression (Auluck et al. 2005; Trinh et al. 2008), the neurons appear compromised for tyrosine hydroxylase immunostaining, allowing study of the pathological processes and pathways involved.
A common feature of many neurodegenerative diseases, including Parkinson’s disease (Irizarry et al. 1998; Spillantini et al. 1998), is the ubiquitination of the protein aggregates. With the finding of chaperone suppression of degeneration conferred by toxic polyQ proteins, one could ask whether similar pathways may protect against the toxicity of other human disease proteins. Auluck et al. (2002) hypothesized that because the Lewy body and Lewy neurites are ubiquitinated, molecular chaperone pathways may be involved. To test this, Hsp70 was coexpressed with α-synuclein in the fly; indeed, this led to mitigation of the effects of α-synuclein on tyrosine hydroxylase activity of the dopaminergic neurons (Auluck et al. 2002). In addition, α-synuclein aggregates in the fly brain were found to be ubiquitinated and immunostained for Hsp70. This provided the impetus to determine whether immunostaining for molecular chaperones was also a feature of human disease associated with Lewy pathology. Pathological analysis of human postmortem Parkinson’s disease brain confirmed that Lewy bodies and Lewy neurites are similarly associated with molecular chaperones (Auluck et al. 2002; Klucken et al. 2006; Uryu et al. 2006; Durrenberger et al. 2009). Taken together, these studies suggest that several pathways involved in the cellular response to protein misfolding and aggregation appear conserved in their role in degenerative disease between human and flies. Such studies raised the prospect that chemical compounds that boost the stress response may be of benefit. A naturally occurring antimicrobial ansamycin, geldanamycin, promotes the heat shock response pathway (Onuoha et al. 2007). Flies expressing α-synuclein were fed geldanamycin, which led to protection of the dopaminergic neurons compared to vehicle-fed animals (Auluck and Bonini 2002). Geldanamycin has since been shown to reduce toxicity of α-synuclein in several other model systems and in additional disease models (McLean et al. 2004; Shen et al. 2005; Waza et al. 2005, 2006; Batulan et al. 2006; Thomas et al. 2006; Liu et al. 2009). Derivatives of the ansamycin family are being generated to identify inhibitors with reduced inherent toxicity for future therapeutics (Kitson et al. 2013).
Taken together, these studies illustrate how a modulator pathway can be identified in the fly system, how its normal role in the disease process can be revealed, and then how the pathway can be targeted with compounds for the hope of therapeutic intervention. The specific approach of protein quality control pathways originally defined in these studies is the subject of much additional research, focusing on the involvement of heat shock response proteins in brain diseases, as well as other processes such as the biology of ageing (see Powers et al. 2009 for a review). In addition, many more exciting studies have been performed on the toxicity pathways of α-synuclein in flies for important new insight, including the role of protein modifications of α-synuclein (Takahashi et al. 2003; Chen et al. 2009) and interactions between α-synuclein with proteins of other diseases, such as Alzheimer’s and HD (Roy and Jackson 2014; Pocas et al. 2015).
Modifier pathways: histone acetyltransferase activity in HD toxicity
Pathogenic polyQ inclusions can form in nuclei and disrupt nuclear protein–protein interactions, such as those involved in transcriptional regulation (Huang et al. 1998; Steffan et al. 2000; Nucifora et al. 2001). Remarkable insight was revealed when studies to assess mechanisms by which the HD protein causes degeneration were pursued. CREB-binding protein (CBP) and CBP-associated factor (p300) are two ubiquitously expressed histone acetyltransferases that acetylate lysine residues within histone tails, facilitating the recruitment of transcriptional regulators (Valor et al. 2013). Intrigingly, a specific interaction between CBP and the HD protein was found, such that CBP becomes sequestered in polyQ accumulations in HD, leading to the loss of CBP activity and repression of CBP/p300-associated gene expression (Steffan et al. 2000; Nucifora et al. 2001). Added CBP suppresses polyQ-associated degeneration, indicating that CBP is functionally important for polyQ-associated toxicity (McCampbell et al. 2000). Additionally, in HD mammalian cell models, histone acetylation is decreased (Steffan et al. 2001).
These studies raised the possibility that restoring histone acetylation to normal levels could be a promising strategy. To pursue this approach, histone deacetylase (HDAC) inhibitors, sodium butyrate and SAHA, which increase the global levels of acetylated histones, were tested for ability to prevent polyQ-associated neurodegeneration by feeding flies these compounds and assessing the effect on neurodegeneration. This showed that HDAC inhibitors had the ability to mitigate the degenerative disease by protecting against retinal deterioration, providing the first evidence of HDAC inhibitors as a potential treatment option (Figure 4) (Steffan et al. 2001). This direction has been pursued with many additional studies to define the specific HDACs involved, as well as pioneering combinations of agents that may be appropriate for mitigation of disease pathogenesis (Agrawal et al. 2005; Kazantsev and Thompson 2008; Pallos et al. 2008). Misregulation of histone acetylation to impact gene expression has been confirmed in a number of additional polyQ models, as well as several other diseases of the central nervous system, including motor neuron disease (reviewed in Kazantsev and Thompson 2008; Valor et al. 2013; Valor and Guiretti 2014). The use of HDAC inhibitors has been well advanced in the field of cancer, with many tested in clinical trials, and three approved for cancer therapy (reviewed in Mottamal et al. 2015). Phase II clinical trials for the use of HDAC inhibitors for the treatment of the motor neuron disease, spinal muscular atrophy (SMA), yielded initial encouraging results, although these were not confirmed or extended by later studies (Mercuri et al. 2004, 2007; Swoboda et al. 2010; Kissel et al. 2011, 2014). More research on the specific targets of acetylation, the specific enzymes involved, and the development of more directed compounds are likely required. Nevertheless, these studies highlight how the fly can be employed to provide data to accelerate research on potential therapeutic pathways and approaches.
Figure 4.

Neurodegeneration induced by the Htt exon1 protein, assayed using the pseudopupil in the fly eye, is suppressed by treating the flies with HDAC inhibitors. (A) The retinal structure of the eye rhabdomeres seen with the pseudopupil assay shows degeneration from the normal number of seven, with expression of a Q48 htt exon1 transgene. (B) Feeding the flies HDAC inhibitors SAHA (2 μM) or butyrate (100 mM) mitigates the degeneration seen at 6 days. (C) Photographs of the pseudopupil from Q48-expressing flies with and without added HDAC inhibitors [reprinted by permission from Macmillan Publishers: Nature (Steffan et al. 2001), copyright 2001)].
Genome-wide screens to define modifier players
The cellular and molecular pathways involved in polyQ-mediated neurodegeneration are diverse and include autophagy, cell death pathways, the ubiquitin proteasome pathway, as well as transcriptional regulation (Fernandez-Funez et al. 2000; McCampbell et al. 2000; Chan et al. 2002; Ravikumar et al. 2004; Sang et al. 2005). Large-scale and candidate-based genetic-modifier screens can be used to identify novel pathways involved in the disease process. The availability of the vast array of fly reagents for both upregulation and loss of function of the genes (see Introduction) make the fly highly conducive for such screens. One type of upregulation screen involves crossing the disease-bearing animals to fly lines that contain a transposon insertion that can direct the expression of the downstream gene in the presence of the GAL4 transcription factor, a so-called EP screen (Rorth 1996). Downregulation screens can also be performed, one popular approach being to use the GAL4-UAS system to direct reagents to reduce gene expression, such as double-stranded RNA (dsRNA) or short hairpin RNA (shRNA) (Dietzl et al. 2007; Ni et al. 2011). The approaches of knocking down and upregulating genes yield different, but complementary, information. If the knockdown of a gene affects the degenerative phenotype, then that gene is expected to normally function in this process. Thus, this tells you about the normal biology involved in pathogenesis, as well as pathways to target for interference. A modifier found through upregulation, on the other hand, suggests that added activity can impact the process, although the modifier may or may not normally contribute to pathogenesis. Modifier screens are distinct from classical forward genetic screens: for example, it is often difficult to discern whether the modifier pathway is acting directly in the pathway of disease toxicity or if the modifier pathway functions in parallel. Additional cell biological studies (such as the observations noted that polyQ protein accumulations normally immunostain for molecular chaperone activity or that the HD protein interacts directly with CBP/p300) can help discriminate these possibilities. We note that such understanding can be helpful for basic biological insight, but regardless of direct or indirect involvement, the uncovered pathway could still be a potential therapeutic target.
Large-scale and candidate-based genetic-modifier screens have been extremely successful in revealing pathways novel to polyQ disease protein toxicity (Fernandez-Funez et al. 2000; Kazemi-Esfarjani and Benzer 2000; Bilen and Bonini 2007; Nedelsky et al. 2010). Examples include a large-scale EP screen for polyQ modifiers that revealed new roles for microRNAs (miRNAs) in maintaining the integrity of the nervous system (Bilen et al. 2006; Liu et al. 2012). Studies on the function of the Atrophin gene, and of other miRNAs, have led to intriguing roles for additional such pathways to modulate neural function and integrity that may extend to mammals (Karres et al. 2007; Verma et al. 2015). Other types of insight from large-scale modifier screens include new toxic components of the gene mutation. For example, the toxicity of the CAG-trinucleotide mutation was believed to be conferred solely by the polyQ protein domain within the disease gene. However, a modifier screen using the SCA3 fly revealed that the RNA binding protein Muscleblind could modulate polyQ toxicity (Li et al. 2008). Muscleblind is an important modifier of myotonic dystrophy, in which a noncoding CTG repeat expansion in the 3′ UTR of the DMPK gene (myotonic dystrophy protein kinase) leads to disease through RNA toxicity mechanisms that include sequestration of key players such as Muscleblind (Miller et al. 2000; Mankodi et al. 2001; Jiang et al. 2004). Similar to CTG repeat RNAs, CAG repeat RNA will form unusual hairpin structures (Sobczak et al. 2003) that can bind and sequester RNA binding proteins; notably, Muscleblind binds CAG repeat RNA as well as CUG repeat RNA (Ho et al. 2005). Toxicity of the CAG repeat RNA transcript was tested by disrupting the hairpin structure of the CAG repeat: CAA codon interruptions of the CAG-trinucleotide repeat prevent the RNA from forming a hairpin structure, while maintaining the protein coding sequence for polyQ. Expression of such a CAA/G repeat in the fly brain led to the same polyQ protein expressed at the same level, yet the biological outcome was a twofold delayed onset of degeneration. These data indicate that toxicity associated with a CAG-trinucleotide expansion arises from both the polyQ protein and aspects of the CAG-repeat RNA (Li et al. 2008). Additional intriguing mechanisms of RNA toxicity have more recently been revealed, including that such hairpin repeats can be translated into proteins despite lacking conventional protein coding indicators (Zu et al. 2011; discussed below).
Small-scale genetic modifer screens that target specific biological processes have also been fruitful in uncovering disease-associated pathways. In human brain tissue and mouse models of HD, SCA1, and SCA3, abnormalities in dendritic structure are a prominent pathology (Graveland et al. 1985; Clark et al. 1997; Guidetti et al. 2001; Cemal et al. 2002). The dendritic arborization neurons in the Drosophila nervous system are well suited for the examination of disease-associated neuritic abnormalities (Grueber et al. 2002). Expression of mutant Ataxin-3 and Ataxin-1 in the dendritic arborization neurons results in morphological defects that precede neuronal cell loss (Lee et al. 2011). Careful examination of the fly revealed that the abnormalities arose from the preferential distruption of F-actin cytoskeletal structures. The defects were partially suppressed by the upregulation of F-actin polymerization through the Rac-PAK signaling pathway (Lee et al. 2011). These findings provide an excellent launching ground for future studies into the disease process and possible therapeutic strategies targeted to the cytoskeleton in more complex model systems.
Integrative approaches that combine the fly model system with other model systems are being used to rapidly ascertain disease-associated pathways that are likely to be affected in the human. In a mouse model for SCA1, the phosphorylation of Ataxin-1 at serine position 776 (S776) leads to enhanced toxicity (Emamian et al. 2003). An integrative approach to elucidate potential kinases involved in Ataxin-1 toxicity combined a screen for kinases that could decrease the protein levels of Ataxin-1 in human cells in culture and a fly modifier screen of Drosophila kinases that could modify Ataxin-1 toxicity (Park et al. 2013). The overlap between these two screens identified the RAS-MAPK-MSK1 signaling pathway as a regulator of Ataxin-1 levels and toxicity, and, stunningly, this was via the phosphorylation of Ataxin-1 at S776 by the MSK1 kinase (Park et al. 2013). Further work in a mouse model expressing Ataxin-1 with an expanded polyQ repeat confirmed that reduction of MSK1 and MSK2 kinases mitigates Purkinje cell loss in the cerebellum (Park et al. 2013). These studies highlight the power of the fly at recapitulating very precise disease features, such as specific protein phosphorylation sites, and how the fly can be applied in combination with other approaches and model systems to define key players in the disease process.
How normal protein function and protein context relates to disease phenotype
A polyQ expansion mutation is causitive for distinct human neurodegenerative diseases (Gusella and MacDonald 2000; Orr and Zoghbi 2007); yet, despite the same molecular mutation and common neuropathological hallmarks of ubiquitinated polyQ inclusions, the polyQ disorders can be clinically distinct. The protein context of the polyQ mutation likely defines the observed differences, with cell-specific factors that selectively interact with the specific disease protein, and the expanded polyQ version, influencing the disease state. The fly has been powerfully utilized to investigate the differences in toxicity produced by a “raw” polyQ tract and a polyQ tract within the context of a disease-associated protein, as well as the effect of the mutant polyQ tract in the distinct disease proteins.
Intriguing initial studies of a Drosophila polyQ-containing protein, Disheveled, addressed whether the polyQ mutation is sufficient for toxicity or whether the toxicity of a polyQ mutation is the result of an altered function of the protein in which the polyQ resides (Marsh et al. 2000). These data showed that expansion of the polyQ repeat from Q27 to Q108 in Dishevelled compromised the normal function, although not to null activity. Removal of the polyQ repeat had very mild loss-of-function effects. Importantly, these studies also showed a striking mitigation of the toxicity of an exceedingly long, highly toxic, “raw” polyQ tract upon its insertion into the Dishevelled protein (Marsh et al. 2000). This highlights the critical importance and impact of protein context to the toxicity conferred by a polyQ expansion. This has since been pursued in a number of ways in other systems, including how modulation of flanking sequences by post-translational protein modification as noted in the section above with Ataxin-1, among other studies, profoundly impacts polyQ toxicity (Emamian et al. 2003; Aiken et al. 2009).
Mounting evidence suggests that in several of the polyQ syndromes, including SCA3, the disease protein undergoes cleavage to produce toxic polyQ fragments (Wellington et al. 1998; Berke et al. 2004; Haacke et al. 2006; Evers et al. 2014). To establish in vivo evidence for how polyQ protein cleavage modulates toxicity of the Ataxin-3 protein, a combination of insect cell culture analysis with in vivo fly models was used to reveal that Ataxin-3 with an expanded polyQ undergoes a caspase-dependent cleavage, which can be prevented by mutation of putative caspase sites (Berke et al. 2004; Jung et al. 2009; Liman et al. 2014). Interestingly polyQ-associated toxicity is alleviated in transgenic fly lines expressing uncleavable Ataxin-3 with a polyQ expansion (Jung et al. 2009), lending support to the notion that the generation of polyQ fragments promotes disease severity.
Studies on the Ataxin-3 protein also revealed that the normal protein has activity to mitigate the toxicity of the mutant protein, leading to reduced levels of the pathogenic form (Warrick et al. 2005). Detailed molecular domain studies identified the ubiquitin protease domain as critical: the Ataxin-3 protein with an expanded polyQ repeat retains this protective activity, suggesting that in disease, cleavage of Ataxin-3 to remove the protective ubiquitin protease domain would enhance pathogenesis (Warrick et al. 2005). Further molecular analysis in mammalian cells identified the Ubiquitin Binding Site 2 (UbS2) of Ataxin-3 to negatively regulate the protein’s turnover, via interaction with Rad23A and Rad23B (Blount et al. 2014). Interestingly, decreasing the levels of Rad23 reduces SCA3 disease-associated toxicity. These studies hint at complex interactions and regulation of the proteasome with normal and mutant Ataxin-3, which may play a critical role in disease. These studies also highlight how detailed anaysis in mammalian model systems can be rapidly translated to the fly to gain functional in vivo insight.
To date there is minimal evidence to support that protein cleavage occurs in SCA1 (Klement et al. 1998). This suggests that pathogenesis in the context of Ataxin-1 may occur by other mechanisms. Particularly intriguing results emerged with studies of the normal and disease Ataxin-1 protein using Drosophila. These studies showed that expression of the normal Ataxin-1 protein with a normal length polyQ repeat was capable of producing deleterious effects in the fly that appear similar to Ataxin-1 protein with an expanded polyQ repeat (Figure 5) (Fernandez-Funez et al. 2000). This finding was confirmed in the mouse, where upregulation of the normal protein led to features reminiscent of disease (Fernandez-Funez et al. 2000). These data suggest that the polyQ repeat, at least in this case, may lead to increased levels of the protein, contributing to or causing neurodegeneration. Such findings underscore the importance of regulation of the expression and accumulation level of these proteins. Many other important studies have been performed on the Ataxin-1 protein, integrating fly with mammalian studies to gain important functional insight (Tsuda et al. 2005; Lam et al. 2006; Tong et al. 2011).
Figure 5.

The effect of the Ataxin-1 protein with a normal and expanded polyQ domain in the fly eye. (A–C) Scanning electron microscopic images of the eyes of flies that are control (A), expressing a normal Ataxin-1 protein (B), or an Ataxin-1 protein with a pathogenic expanded polyQ repeat (C). Insets show higher magnification view of the ommatidia. (D–F) Sections through the retina, show that the interior of the eye is affected. Degeneration is very severe with Ataxin-1 bearing an expanded polyQ and mild with the normal protein. Detailed studies as highlighted in the text, have revealed much insight into the critical role of protein context in polyQ pathogenesis in studies of the Ataxin-1 protein [reprinted by permission from Macmillan Publishers: Nature, (Fernandez-Funez et al. 2000) copyright 2000)].
Additional findings in Drosophila have been highly revealing for the normal function of these disease-associated proteins. For example, Drosophila that have the homolog of the Huntington’s disease gene deleted develop normally, but show age-related neurodegenerative phenotypes (Zhang et al. 2009) and reveal a regulatory role for the HD protein in autophagy (Ochaba et al. 2014; Rui et al. 2015). Studies on the full-length rather than the truncated version of the HD protein bearing a pathogenic expansion in the fly have shown that there are early effects to enhance neurotransmission and that these impacts on neurotransmission may be central to subsequent degenerative effects (Romero et al. 2008).
Finally, a stunning example of the critical importance of protein context was illustrated with Drosophila models for the Androgen receptor, whose pathogenic repeat expansions are associated with SBMA. In this situation, expression of the androgen receptor protein containing a pathogenic expanded repeat completely failed to confer any deleterious effect on the animal—despite robust expression of a presumably highly toxic protein bearing an expanded polyQ domain—unless the animals were fed an androgen receptor ligand that translocated the protein into the nucleus (Takeyama et al. 2002). These studies highlight not only the fundamental—and astounding—impact that protein context can have, but also indicated a remarkable feature of localization of the protein within the cell for pathogenesis—in this case in the nucleus. In the fly, both agonists and antagonists translocated the protein and thus allowed the protein to convey toxicity. Subsequent studies revealed that nuclear translocation and native functions of the protein were both required to confer toxicity, indicating that not only nuclear translocation, but also DNA binding of the protein is required (Nedelsky et al. 2010). Ligand-dependent toxicity was supported by antiandrogen treatments in transgenic mouse models for the disease, which mitigated degeneration; there are now data to support clinical endeavors, with mixed outcomes, for the testing of antiandrogen agents as treatments for SBMA (Katsuno et al. 2002, 2003; Chevalier-Larsen et al. 2004; Fischbeck and Bryan 2009).
Taken together, these studies highlight some of the striking findings regarding how normal function of the disease proteins can be uncovered using the fly system. We have attempted to illustrate some of the dramatic ways in which the context/molecular domains of the disease proteins have been shown to influence toxicity. Whereas early findings suggested a pathogenic polyQ expansion was the signature of disease, as in many fields, detailed studies have revealed a more complex story with protein context and disease protein function playing critical roles, with fly studies contributing importantly toward this understanding.
Modeling RNA-Based Mechanisms of Toxicity of ALS/FTD in the Fly
Modeling amyotrophic lateral sclerosis in the fly
ALS (OMIM no. 105400) is the most prevelant motor neuron disease, with a prevalence of ∼3.9 cases per 100,000 individuals (Mehta et al. 2014). A devastating adult-onset disease, it impacts motor neurons in the motor cortex and spinal cord, leading to paralysis and death. The first gene mutations associated with ALS were identified in SOD1, a metabolic gene encoding for copper/zinc ion binding superoxide dismutase (Rosen et al. 1993). In 2006, insight into ALS changed dramatically with the discovery that TDP-43 (transactive response (TAR) DNA binding protein of 43 kDa) is mislocalized to the cytoplasm in disease, where it forms phosphorylated and ubiquitinated accumulations in nearly all cases of ALS, as well as about half of a related disorder, FTD (OMIM no. 600274) (Neumann et al. 2006). Rare mutations associated with disease have been identified in TDP-43, with a preponderance within the C-terminal low complexity prion-like domain (Lagier-Tourenne and Cleveland 2009; Mackenzie et al. 2010). Since this discovery, enhanced genomic technologies have led to an explosion in the identification of genes associated with ALS (Ling et al. 2013; Renton et al. 2014). Similar to TDP-43, many of these genes encode proteins involved in RNA processing (Table 1), including proteins with similar domains, such as FUS (Andersson et al. 2008; Kwiatkowski et al. 2009; Vance et al. 2009; Bentmann et al. 2012). The association of several genes involved in RNA processing with ALS suggests that RNA biogenesis and regulation is involved in the pathology of the disease.
Here we focus on TDP-43 and a recently identified mutation that is the most common mutation associated with ALS and FTD in European ethnic populations—a GGGGCC-hexanucleotide repeat expansion within the C9orf72 gene (DeJesus-Hernandez et al. 2011; Renton et al. 2011; Majounie et al. 2012). Again, we focus on use of the fly to reveal new insight and highlight how these studies have led to a convergence of mechanism, and gene overlap, with polyQ protein-based diseases. Our goal is to illustrate with select examples the power of the application of Drosophila to the study of disease and its mechanisms. As highlighted previously, many models of many diseases, including other genetic contributors to ALS, have been generated that provide exceptional insight (Table 1).
Modeling TDP-43 toxicity in the fly and the discovery of novel ATXN2 mutations
Several TDP-43 fly models have been generated and characterized for TDP-43-associated pathways. In summary, expression of human TDP-43 in the fly causes gross pathologies similar to those observed in ALS patients: neuronal degeneration, motor neuron deficits, and shortened lifespan (Figure 6) (Elden et al. 2010; Hanson et al. 2010; Li et al. 2010; Ritson et al. 2010; Voigt et al. 2010; Estes et al. 2011; Kim et al. 2014). At a more detailed level, the TDP-43-expressing flies also mimic molecular pathologies such as mislocalization of the protein to the cytoplasm and disease-associated phosphorylation (Hanson et al. 2010; Li et al. 2010; Estes et al. 2011; Choksi et al. 2014). Mutant forms of TDP-43, that lack the ability to bind RNA confer little toxicity in the fly compared to the normal protein (Voigt et al. 2010), indicating that it is not aggregation, but the disruption to RNA pathways that is important to TDP-43 toxicity.
Figure 6.

TDP-43 toxicity in Drosophila leads to eye degeneration and loss of climbing ability. (A) Expression of human TDP-43 in Drosophila causes degeneration, seen on the external eye and internal structure. Expression is moderate (M) or strong (S). Notice that with moderate expression, despite a mild effect on the external eye, the internal retina is severely disrupted. (B) TDP-43 causes climbing defects when expressed in motor neurons with the D42-GAL4 driver. A mutant form of TDP-43, TDP-43.Q331K, causes a more severe loss of motility than the normal protein at the same level of expression [reprinted from Elden et al. (2010)].
As highlighted with polyQ disease models, the fly has become a well-embraced model to address complex questions of disease-associated cytotoxicity. Coupling these screens with in-depth analysis of human genetics, as well as pathological examination of patient tissue, can reveal new pathways relevant to the human condition. We use the following examples to illustrate how studies in the fly, along with other model organisms, can lead to defining new associations of mutations with human disease.
An integration of genetic modifier screens in yeast and fly identified ATXN2, the spinocerebellar ataxia type 2 (SCA2) gene, as a modifier of TDP-43: downregulation of Ataxin-2 mitigated TDP-43 toxicity, and upregulation of Ataxin-2 enhanced TDP-43 toxicity (Figure 7) (Elden et al. 2010). Subsequent analyses in human ALS spinal cord revealed abnormal accumulation of Ataxin-2, suggesting that ATXN2 could be central to TDP-43-associated disease (Elden et al. 2010). In SCA2, an expanded CAG-trinucleotide repeat leads to a polyQ expansion of Q34 or longer in the Ataxin-2 protein (Imbert et al. 1996; Pulst et al. 1996; Sanpei et al. 1996; Lastres-Becker et al. 2008). However, the interaction between TDP-43 and ATXN2 raised the possibility of ATXN2 repeat expansions in ALS: indeed, sequence analysis revealed a significant association of CAG-repeat expansions of intermediate length—higher than normal (∼Q22–23) but below the threshold for SCA2 (∼Q27–33) associated with patients with ALS (Figure 7) (Elden et al. 2010). This finding has been confirmed in many different patient populations (Chen et al. 2011a; Ross et al. 2011; Conforti et al. 2012; Gellera et al. 2012; Gijselinck et al. 2012; Gispert et al. 2012; Liu et al. 2013; van den Heuvel et al. 2014). Importantly, SCA2 and ALS have been reported in the same family where both the SCA2 and ALS cases had ATXN2 repeat expansions, implicating ATXN2 as a bona fide ALS disease gene; the findings suggest screening for such ATXN2 mutations in family pedigrees that present with both ALS and SCA2 (Tazen et al. 2013). The fly studies showed that repeat expansions of only 10 CAG repeats, into the ALS disease range, are sufficient to enhance TDP-43 interactions with ATXN2 (Kim et al. 2014). More recent human patient studies suggest that a longer repeat in Ataxin-2 of >Q31 is associated with shorter survival in ALS (Chio et al. 2015). Moreover, the fly studies indicate that Ataxin-2 could be an important target in ALS, as normal Ataxin-2 function promotes TDP-43 neural toxicity. These studies highlight the tremendous impact that relatively rapid genetic analyses in simple model organisms can have on human genetic understanding.
Figure 7.

Studies in fly and yeast led to testing for Ataxin-2 polyQ expansions in ALS human disease. (A) The fly Ataxin-2 homolog modulates the toxicity of TDP-43. Flies expressing TDP-43 or Drosophila Ataxin-2 (dAtx2) alone (dAtx2EP; latter not shown here) have a mild effect on retinal structure, but TDP-43 toxicity is markedly more severe upon added upregulation of dAtx2 (dAtx2EP314) and markedly reduced by heterozygous reduction of dAtx2 (null allele dAtx2X1). (B) The ATXN2 gene is a polyQ disease gene, with a repeat region normally of 22–23Q units. Expansions of >Q34 are associated with SCA2. Fly and yeast data revealed a striking interaction between TDP-43 and Ataxin-2 suggesting that expansions longer than normal, but less than that associated with SCA2, may be found in ALS. (C) The distribution of Ataxin-2 polyQ repeat lengths in ALS and controls. In Elden et al. (2010), polyQ lengths ≥Q27 were found to be significantly associated with ALS. Figure panels are adapted from Elden et al. (2010).
The Ataxin-2 protein bears two evolutionarily conserved protein domains: an RNA binding motif (LSM domain), and a PolyA binding protein (PABP)-interacting motif 2 (PAM2 motif) (Satterfield and Pallanck 2006). A direct link of a physical association of Ataxin-2 with RNA came from studies in the fly that found that the fly Ataxin-2 (dAtx2) protein physically interacts with pABp and the polyribosome via the PAM2 motif (Satterfield and Pallanck 2006). In the fly, deletion of the PAM2 motif in ATXN2 eliminates the enhancement of TDP-43 toxicity, indicating that the interaction between Ataxin-2 and TDP-43 is via PABP; indeed downregulation of PABP in the fly mitigates TDP-43 toxicity (Kim et al. 2014).
Upon stress, TDP-43 has been reported to localize to cytoplasmic stress granules—cytoplasmic accumulations containing translationally repressed RNA, abortive initiation complexes, and messenger RNA (mRNA) binding proteins such as Ataxin-2 and PABP (Anderson and Kedersha 2006; Colombrita et al. 2009; Freibaum et al. 2010; Cohen et al. 2011; Bentmann et al. 2012). Given the association of these proteins, among others in ALS such as FUS, stress granule formation may play a role in disease. PABPC1 also pathologically mislocalizes in ALS motorneurons (Figure 8) (Dormann et al. 2010; Bentmann et al. 2013; Kim et al. 2014; McGurk et al. 2014). Intriguingly, therapeutic modulation of the stress granule pathway is a possible avenue for therapeutic research. A nucleating factor for stress granule formation is the phosphorylation of elongation initiation factor 2α (eIF2α) by stress-activated kinases such as protein kinase R (PKR) and PKR like-ER-kinase (PERK) (Anderson and Kedersha 2006). A small molecule inhibitor PERKi (GSK2606414) was developed by GlaxoSmithKline that specifically inhibits PERK function (Axten et al. 2012). Strikingly, feeding flies this inhibitor mitigates TDP-43 toxicity, such that animals retain locomotor ability over time (Kim et al. 2014). Extending this to mammalian cortical neurons showed that treatment by the PERKi inhibitor mitigates risk of death of primary rat cortical neurons expressing TDP-43 (Kim et al. 2014). The approach of targeting stress granule formation is a potentially rich area for investigation as stress granule modulation pathways have been implicated in mouse models of Alzheimer’s (Ma et al. 2013) and prion disease (Moreno et al. 2012, 2013); in the latter case, treatment by the PERKi strikingly mitigates degeneration (Moreno et al. 2013). The association of these processes with several different disease models, and perhaps normal memory (Sidrauski et al. 2013, 2015; Sekine et al. 2015), suggests that inhibition of translation via phosphorylation of eIF2α coupled to stress granule formation, may be a unifying pathway common to a number of disease and other situations.
Figure 8.
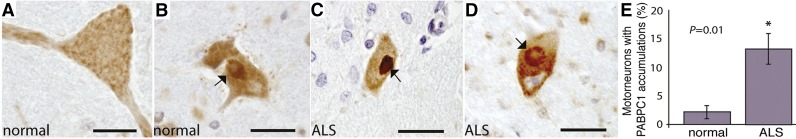
Fly studies highlighting interactions with PABP led to testing for PABPC1 mislocalization in motor neurons of ALS patient tissue. Fly data suggested that PABP may be involved in ALS; here human patient tissue was examined to assess that possibility. (A–D) Immunostaining for PABPC1 (cytoplasmic PABP) of spinal cord motor neurons in control and ALS patients. PABPC1 is present and localized throughout the cytoplasm in normal motor neurons, but shows striking accumulation in motor neurons of patients with ALS. Bars, 30 µm. (E) Quantitation of the accumulations [reprinted from Kim et al. (2014)].
In ALS, the accumulation of TDP-43 in the cytoplasm coincides with a clearance of TDP-43 from the nucleus (Neumann et al. 2006), raising the possibility that neurodegeneration could result from either a loss-of-function or gain-of-function mechanism, or contributions of both mechanisms. Whereas the aforementioned studies highlighted toxic mechanisms upon expression of TDP-43, studies in the fly on the endogenous TDP-43 homolog, TBPH, indicate that normal TBPH function is critical for the development and maintenance of the nervous system. Flies lacking TBPH have motor defects and reduced lifespan—features markedly similar to humans with ALS (Feiguin et al. 2009; Lu et al. 2009; Lin et al. 2011; Diaper et al. 2013). These data from the fly support the idea that disease-associated toxicity of TDP-43 could involve both loss-of-function and gain-of-function mechanisms (reduced nuclear function and accumulation in the cytoplasm, respectively). Interestingly, gene expression analysis using the fly has uncovered similar transcriptional alterations when TDP-43 is upregulated or deleted (Vanden Broeck et al. 2013), suggesting that nuclear clearing and cytoplasmic accumulation could potentially reinforce similar patterns of pathological gene expression.
A GGGGCC repeat expansion associated with ALS/FTD
As noted, FTD, the second most common presenile dementia, shares clinical and pathological overlap with ALS (Gunnarsson et al. 1991; Lomen-Hoerth et al. 2002; Mackenzie and Feldman 2005; Neumann et al. 2006; Murphy et al. 2007; Geser et al. 2009). In 2011, the most common mutation in European ethnic-based populations associated with autosomal dominant FTD and ALS was defined as a hexanucleotide repeat expansion of GGGGCC in the first intron of the C9orf72 gene (DeJesus-Hernandez et al. 2011; Renton et al. 2011; Gijselinck et al. 2012; Majounie et al. 2012). Although variable in length, the repeat is typically 3 or 12 units in controls, with expansions of ∼30 to far greater (>1000 units) in disease. The GGGGCC repeat expansion leads to an accumulation of RNA containing the repeat in nuclear foci, translation of dipeptide repeat proteins through repeat associated non-ATG translation, and may also be associated with reduction of C9orf72 gene expression. These data underscore that both loss-of-function of the gene, as well as gain-of-function mechanisms associated with the accumulation of the expanded repeat RNA may underlie toxicity associated with the GGGGCC expansion mutation (DeJesus-Hernandez et al. 2011; Renton et al. 2011). Studies of the role of orthologs of the C9orf72 protein will be important (Levine et al. 2013); however, here we focus on the underlying biology of the RNA gain-of-function mechanisms, describing two examples by which the fly was used to tease out contributing processes.
TDP-43 pathological inclusions characterize cases harboring GGGGCC expansions in the C9orf72 gene (reviewed in Mackenzie et al. 2014). However, pathology specific to C9orf72 carriers include RNA foci hybridizing to both the sense GGGGCC repeat RNA, as well as the antisense GGCCCC repeat RNA, in spinal cord and multiple brain regions, implicating an RNA gain-of-function mechanism (DeJesus-Hernandez et al. 2011; Simon-Sanchez et al. 2012; Donnelly et al. 2013; Gendron et al. 2013; Lagier-Tourenne et al. 2013; Lee et al. 2013; Mizielinska et al. 2013). An additional pathology found in GGGGCC repeat carriers is the cerebellar, hippocampal, and cortical accumulation of TDP-43 negative inclusions that are positive for proteins in the ubiquitin–proteasome pathway (Mackenzie et al. 2014). The underlying mechanism is interesting and suggests protein translation from the accumulated repeat RNA in the absence of canonically considered protein translation sequences. This finding stems from studies of the disease-causing mutation of spinocerebellar ataxia type 8 (SCA8) (OMIM no. 608768), which revealed that this CAG-trinucleotide repeat RNA underwent repeat associated non-ATG (RAN) translation. That is, the accumulated expanded repeat RNA could undergo protein translation, and in all three reading frames, to produce polyQ, polyserine (polyS), and polyalanine (polyA), with polyQ and polyA accumulations detectable in patient brain tissue (Zu et al. 2011). The RNA lacks an AUG start codon, but nevertheless, repeat-containing RNAs were shown able to produce protein when expressed in cultured cells (Zu et al. 2011). As polyQ protein can be toxic, evidence was provided to suggest that these various generated proteins, polyQ and the others, could be toxic to cells. The ability to generate proteins from such accumulated disease RNAs was shown for a number of different expanded repeat RNAs, including CAG repeat RNAs. This raised a question of what mechanisms of RNA toxicity are conferred by the GGGGCC repeat expansion in patients.
Translation of the open reading frames of the sense and the antisense expanded repeat RNA associated with the C9orf72 mutation is predicted to give rise to five different polypeptides [referred to as dipeptide repeat proteins (DPRs)]: glycine-alanine (GA), glycine-arginine (GR), and glycine-proline (GP) from the sense strand and proline-alanine (PA) and proline-arginine (PR) from the antisense strand. Upon raising antibodies specific to these DPRs, various DPRs have been detected in the TDP-43-negative accumulations in the cerebellum, hippocampus, and neocortex of C9orf72 patient tissue (Ash et al. 2013; Gendron et al. 2013; Mackenzie et al. 2013; Mann et al. 2013; Mori et al. 2013a,b; Zu et al. 2013). Cell culture studies indicate that GR and PR peptides can be toxic to cultured neurons (May et al. 2014; Wen et al. 2014; Zhang et al. 2014; Yamakawa et al. 2015). Although it is still unclear if DPRs are toxic in disease in vivo, these pathological and cell culture findings indicate that the expanded GGGGCC repeat RNA could confer toxicity either because the RNA on its own may be toxic and also because the RNA produces RAN-translated DPRs at least some of which may be toxic. Fly models for GGGGCC-associated toxicity have provided important mechanistic insight into this question.
Insight Into GGGGCC-associated toxicity from Drosophila
Expression in Drosophila of a repeat of 30 GGGGCC units confers a disrupted eye morphology, and, when expressed in motor neurons, an age-dependent decline in motor function (Xu et al. 2013). Studies in vitro on the repeat RNA identified Pur-α as an RNA binding protein that could bind the expanded GGGGCC repeat RNA. To investigate the significance of this interaction, directed genetic tests revealed that downregulation of Pur-α mitigated GGGGCC toxicity (Xu et al. 2013). Immunohistochemical analysis showed that Pur-α was sequestered into the GGGGCC RNA foci in the fly eye. The relevance of these findings to disease is underscored by studies of human postmortem brain tissue, where Pur-α colocalized with GGGGCC RNA foci. These studies support the idea that RNA toxicity of the accumulated GGGGCC RNA may play a role in disease by sequestering RNA binding proteins. In this manner, the RNA toxicity may be akin to that of Muscleblind and other RNA binding protein interactions with an expanded CTG repeat, where the RNA that accumulates in disease binds and sequesters key proteins, thereby disrupting their function (Ranum and Cooper 2006; Wheeler and Thornton 2007).
A second hypothesis is that the RNA is indeed driving the expression of polypeptides that are toxic. This was examined using Drosophila models for a range of features of the GGGGCC RNA. Fly models were generated that expressed a series of constructs that probed both the potential toxic role of a GGGGCC RNA that lacked the ability to be translated, as well as the toxicity of the predicted protein products produced by such an RNA. First, pathogenic length repeats of GGGGCC were created bearing stop codon interruptions within the sequence, rendering the RNA incapable of being translated into DPRs. The phenotypes of these animals were compared to flies expressing pure, uninterrupted repeats of GGGGCC RNA that could be translated into protein (Mizielinska et al. 2014). Remarkably, while both pure and stop-codon interrupted repeats could form RNA foci reminescent of human disease, the interrupted GGGGCC repeats of up to 288 unit repeats did not confer degenerative eye effects, whereas pure repeats of 33 or 103 conferred toxicity (Mizielinska et al. 2014). These data indicate that a pure repeat capable of being translated into protein is toxic. Flies were then generated that could express DPR proteins, although not by a pure GGGGCC repeat RNA, but by an RNA of alternate codons such that the RNA sequence was not a GGGGCC repeat (along similar lines to earlier studies on the pure vs. CAA interrupted CAG repeat). Comparison of these flies expressing various DPRs predicted to be encoded by the GGGGCC repeat and its antisense showed that the GR and PR peptides have toxicity (lengths of 36 and 100), while PA and GA peptides in lengths up to 100 DPR units do not (Figure 9). Thus, peptides of GR and PR can confer marked toxicity in vivo in the nervous system.
Figure 9.

Drosophila was used to parse possible mechanisms of toxicity by the hexanucleotide GGGGCC repeat expansion in the C9orf72 gene. Expansion of a GGGGCC repeat in the first intron of the C9orf72 gene is a major genetic cause of ALS and FTD. The underlying mechanisms may include RNA-mediated toxicity due to sequestration of RNA-binding proteins, and also dipeptide repeat (DPR) protein-mediated toxicity due to repeat associated non-ATG (RAN) translation. To assess potential contributions of these distinct mechanisms, Mizielinska et al. (2014) generated transgenic flies expressing either “RNA-only” GGGGCC repeats, which contained interrupting stop codons to prevent protein translation, or “protein only” transgenes of the predicted translated DPRs. Expression of these transgenes in the fly eye indicates that while the RNA-only repeats and the glycine/alanine (GA) or proline/alanine (PA) fail to confer toxicity, glycine/arginine (GR) and proline/arginine (PR) show robust toxicity. Fly lines are from the Bloomington Stock Center. Images were generated and kindly shared by Lindsey Goodman (Bonini laboratory).
The strategy of using Drosophila to study mechanisms of toxicity that could be conferred by the GGGGCC mutation has provided important insight into potential pathogenic pathways that may function in vivo in patients bearing the C9orf72 repeat expansion mutation. Additional studies of fly models for this mutation promise to be highly revealing for biological pathways involved, as well as to dissect the mechanisms of this presumed atypical protein translation. Taken together, these studies of ALS disease genes exemplify how the fly can be used to discover new associations of mutations in human disease, as well as mechanisms at play, utilizing an in vivo system that is highly amenable to genetic manipulations.
Concluding Remarks
The fly is an exceptional research tool for providing insight into many biological processes; here we have emphasized how it has been used for directed studies of neurodegenerative disease. As a well-established model, it mirrors—with remarkable similarities—mammalian neurodegenerative disease pathologies. We selected examples that emphasize the power of the fly in this endeavor, and have also highlighted examples of genetic modifier screens that uncover the pathways perturbed (see Figure 1 and Figure 2). We have also tried to give examples of how the fly system has been integrated with mammalian studies, human genetics, and patient samples, for a new breadth of understanding.
With improvements in human genomic sequencing technologies, Drosophila can provide functional support in additional ways for human molecular genetic findings. One recent example integrated disease-associated mutations discovered by exome sequencing and linkage analysis in heterogeneous nuclear ribonucleoproteins (hnRNPs) A2B1 and A1 in families with multisystem proteinopathy and ALS, with fly data for functional effects (Kim et al. 2013a). These proteins have a low complexity prion-like domain like TDP-43 and FUS, and the mutations identified accelerate aggregation of the protein. The fly was instrumental in showing the pathogenicity of the disease-associated mutations and the association of toxicity with the prion domain.
Genome-wide association studies (GWAS) are important to reveal modifiers that may affect disease risk in humans; however, a challenge with GWAS is that the association domains are broad and include many loci, making it difficult to identify the actual gene implicated in the disease process. Beyond that is how the gene associated with disease-risk is involved in disease outcome—Does it mitigate or enhance? One approach to defining the important factors is to test the impact of the genes within a GWAS region in a disease model: whereas the human data identifies genomic regions of interest, coupling that with fly genetics to examine functional interactions can narrow down gene targets to reveal those with the potential to have an impact on the disease process (Shulman et al. 2014). Another example is that the prion-like spread of disease proteins within the brain has now been shown for the HD protein in flies, promising advances into mechanism and biological impact (Goedert et al. 2010; Pearce et al. 2015). These are some of the ways fly models have been extended. The continued commitment of Drosophila scientists to generate new, exciting applications and methods, coupled with new discoveries into disease physiology, assures that the fly will continue as a vital biological complement for studies of human disease.
Such ever-evolving approaches to manipulate the fly genome, including more precise ways in which to induce mutations with the CRISPR/cas9 system to generate humanized mutations in situ (Housden et al. 2014), and the continuous addition of new tools to manipulate genes (Riabinina et al. 2015) assures that the fly will continue as a formidable and critical complementary system to reveal important biology. These tools can be coupled with more sophisticated techniques to appreciate the biology and workings of the neural circuits of brain function. These approaches promise to lead to a deeper molecular understanding of basic biological processes, as well as how these processes are impacted in disease, thus unveiling the secrets of brain function, its response to ageing, and the dysfunctional state. Research using Drosophila will continue to provide essential groundwork necessary for the development of therapeutics needed to mitigate these devastating diseases of the brain.
Acknowledgments
We thank our colleagues Leslie Thompson, Juan Botas, Nam Kim, J. Paul Taylor, and Lindsey Goodman for generously sharing images. The Bonini laboratory receives funding from the National Institutes of Health (F32-NS084667 to A.B. and R01-NS0736690, R01-NS078283, and R21-NS088370 to N.M.B.) and Target ALS (L.M. and N.M.B.). We apologize to any colleagues whose work could not be covered due to space and focus limitations.
Note added in proof: Two genetic screens of GGGGCC repeat expressing Drosophila, an unbiased large-scale screen and a candidate-based screen, identified components of the nuclear pore complex and related genes that impact nuclear transport as modifiers of GGGGCC-expanded repeat toxicity (Freibaum et al. 2015; Zhang et al. 2015). These studies converge on the finding that expression of an expanded GGGGCC repeat RNA disrupts nucleocytoplasmic transport. These findings were extended to mammalian cells and neurons derived from human patient tissue. A third screening approach, in the model organism yeast, showed that similar genes modify toxicity of the PR peptide that can be generated from the GGGGCC hexanucleotide repeat (Jovicic et al. 2015).
Footnotes
Communicating editor: J. Truman
These authors contributed equally to this work.
Literature Cited
- Aarli J. A., Dua T., Janca A., Muscetta A., 2006. Neurological Disorders. Public Health Challenges, World Health Organization, Geneva. [Google Scholar]
- Agrawal N., Pallos J., Slepko N., Apostol B. L., Bodai L., et al. , 2005. Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc. Natl. Acad. Sci. USA 102: 3777–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C. T., Steffan J. S., Guerrero C. M., Khashwji H., Lukacsovich T., et al. , 2009. Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284: 29427–29436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambegaokar S. S., Jackson G. R., 2010. Interaction between eye pigment genes and tau-induced neurodegeneration in Drosophila melanogaster. Genetics 186: 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambegaokar S. S., Jackson G. R., 2011. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 20: 4947–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P., Kedersha N., 2006. RNA granules. J. Cell Biol. 172: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M. K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., et al. , 2008. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P. E., Bieniek K. F., Gendron T. F., Caulfield T., Lin W. L., et al. , 2013. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck P. K., Bonini N. M., 2002. Pharmacological prevention of Parkinson disease in Drosophila. Nat. Med. 8: 1185–1186. [DOI] [PubMed] [Google Scholar]
- Auluck P. K., Chan H. Y., Trojanowski J. Q., Lee V. M., Bonini N. M., 2002. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295: 865–868. [DOI] [PubMed] [Google Scholar]
- Auluck P. K., Meulener M. C., Bonini N. M., 2005. Mechanisms of suppression of {alpha}-synuclein neurotoxicity by geldanamycin in Drosophila. J. Biol. Chem. 280: 2873–2878. [DOI] [PubMed] [Google Scholar]
- Axten J. M., Medina J. R., Feng Y., Shu A., Romeril S. P., et al. , 2012. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 55: 7193–7207. [DOI] [PubMed] [Google Scholar]
- Bakhoum M. F., Jackson G. R., 2011. Demise of the flies: why Drosophila models still matter. Prog. Mol. Biol. Transl. Sci. 100: 483–498. [DOI] [PubMed] [Google Scholar]
- Batulan Z., Taylor D. M., Aarons R. J., Minotti S., Doroudchi M. M., et al. , 2006. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 24: 213–225. [DOI] [PubMed] [Google Scholar]
- Bellen H. J., Tong C., Tsuda H., 2010. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat. Rev. Neurosci. 11: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D., et al. , 2012. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287: 23079–23094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann E., Haass C., Dormann D., 2013. Stress granules in neurodegeneration: lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 280: 4348–4370. [DOI] [PubMed] [Google Scholar]
- Benzer S., 1971. From the gene to behavior. JAMA 218: 1015–1022. [PubMed] [Google Scholar]
- Berke S. J., Schmied F. A., Brunt E. R., Ellerby L. M., Paulson H. L., 2004. Caspase-mediated proteolysis of the polyglutamine disease protein ataxin-3. J. Neurochem. 89: 908–918. [DOI] [PubMed] [Google Scholar]
- Bier E., 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 6: 9–23. [DOI] [PubMed] [Google Scholar]
- Bilen J., Bonini N. M., 2005. Drosophila as a model for human neurodegenerative disease. Annu. Rev. Genet. 39: 153–171. [DOI] [PubMed] [Google Scholar]
- Bilen J., Bonini N. M., 2007. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 3: 1950–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J., Liu N., Burnett B. G., Pittman R. N., Bonini N. M., 2006. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell 24: 157–163. [DOI] [PubMed] [Google Scholar]
- Blount J. R., Tsou W. L., Ristic G., Burr A. A., Ouyang M., et al. , 2014. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 5: 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini N. M., 2002. Chaperoning brain degeneration. Proc. Natl. Acad. Sci. USA 99(Suppl 4): 16407–16411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N., 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- Cao W., Song H. J., Gangi T., Kelkar A., Antani I., et al. , 2008. Identification of novel genes that modify phenotypes induced by Alzheimer’s beta-amyloid overexpression in Drosophila. Genetics 178: 1457–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemal C. K., Carroll C. J., Lawrence L., Lowrie M. B., Ruddle P., et al. , 2002. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet. 11: 1075–1094. [DOI] [PubMed] [Google Scholar]
- Chai A., Withers J., Koh Y. H., Parry K., Bao H., et al. , 2008. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 17: 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y., Koppenhafer S. L., Bonini N. M., Paulson H. L., 1999. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci. 19: 10338–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R., Vepuri V., Mhatre S. D., Paddock B. E., Miller S., et al. , 2011. Characterization of a Drosophila Alzheimer’s disease model: pharmacological rescue of cognitive defects. PLoS One 6: e20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. Y., Warrick J. M., Gray-Board G. L., Paulson H. L., Bonini N. M., 2000. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 9: 2811–2820. [DOI] [PubMed] [Google Scholar]
- Chan H. Y., Warrick J. M., Andriola I., Merry D., Bonini N. M., 2002. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum. Mol. Genet. 11: 2895–2904. [DOI] [PubMed] [Google Scholar]
- Chan Y. B., Miguel-Aliaga I., Franks C., Thomas N., Trulzsch B., et al. , 2003. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum. Mol. Genet. 12: 1367–1376. [DOI] [PubMed] [Google Scholar]
- Chen H. J., Anagnostou G., Chai A., Withers J., Morris A., et al. , 2010. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 285: 40266–40281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Periquet M., Wang X., Negro A., McLean P. J., et al. , 2009. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 119: 3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., R. Huang, Y. Yang, K. Chen, W. Song et al., 2011a Ataxin-2 intermediate-length polyglutamine: a possible risk factor for Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 32: 1925.e1–5. [DOI] [PubMed]
- Chen Y., Yang M., Deng J., Chen X., Ye Y., et al. , 2011b Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E. S., O’Brien C. J., Wang H., Jenkins S. C., Holder L., et al. , 2004. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J. Neurosci. 24: 4778–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A., Calvo A., Moglia C., Canosa A., Brunetti M., et al. , 2015. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 84: 251–258. [DOI] [PubMed] [Google Scholar]
- Choksi D. K., Roy B., Chatterjee S., Yusuff T., Bakhoum M. F., et al. , 2014. TDP-43 Phosphorylation by casein kinase Iepsilon promotes oligomerization and enhances toxicity in vivo. Hum. Mol. Genet. 23: 1025–1035. [DOI] [PubMed] [Google Scholar]
- Clark H. B., Burright E. N., Yunis W. S., Larson S., Wilcox C., et al. , 1997. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 17: 7385–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., et al. , 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166. [DOI] [PubMed] [Google Scholar]
- Cohen T. J., Lee V. M., Trojanowski J. Q., 2011. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 17: 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., et al. , 2009. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Conforti F. L., Spataro R., Sproviero W., Mazzei R., Cavalcanti F., et al. , 2012. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 79: 2315–2320. [DOI] [PubMed] [Google Scholar]
- Cook K. R., Parks A. L., Jacobus L. M., Kaufman T. C., Matthews K. A., 2010. New research resources at the Bloomington Drosophila Stock Center. Fly (Austin) 4: 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J., Hart M. P., Shorter J., DeJesus-Hernandez M., Erion R., et al. , 2011. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 108: 20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J., Hart M. P., Erion R., King O. D., Diaz Z., et al. , 2012. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 21: 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings C. J., Mancini M. A., Antalffy B., DeFranco D. B., Orr H. T., et al. , 1998. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19: 148–154. [DOI] [PubMed] [Google Scholar]
- Cushman-Nick M., Bonini N. M., Shorter J., 2013. Hsp104 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet. 9: e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., et al. , 1997. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537–548. [DOI] [PubMed] [Google Scholar]
- de Haro M., Al-Ramahi I., De Gouyon B., Ukani L., Rosa A., et al. , 2006. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 15: 2138–2145. [DOI] [PubMed] [Google Scholar]
- Deik A., Johannes B., Rucker J. C., Sanchez E., Brodie S. E., et al. , 2014. Compound heterozygous PNPLA6 mutations cause Boucher-Neuhauser syndrome with late-onset ataxia. J. Neurol. 261: 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., et al. , 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper D. C., Adachi Y., Sutcliffe B., Humphrey D. M., Elliott C. J., et al. , 2013. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet. 22: 1539–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K. C., Barinova Y., et al. , 2007. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448: 151–156. [DOI] [PubMed] [Google Scholar]
- DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., et al. , 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277: 1990–1993. [DOI] [PubMed] [Google Scholar]
- Dockendorff T. C., Su H. S., McBride S. M., Yang Z., Choi C. H., et al. , 2002. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34: 973–984. [DOI] [PubMed] [Google Scholar]
- Donnelly C. J., Zhang P. W., Pham J. T., Haeusler A. R., Mistry N. A., et al. , 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., et al. , 2010. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29: 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrenberger P. F., Filiou M. D., Moran L. B., Michael G. J., Novoselov S., et al. , 2009. DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J. Neurosci. Res. 87: 238–245. [DOI] [PubMed] [Google Scholar]
- Elden A. C., Kim H. J., Hart M. P., Chen-Plotkin A. S., Johnson B. S., et al. , 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elefant F., Palter K. B., 1999. Tissue-specific expression of dominant negative mutant Drosophila HSC70 causes developmental defects and lethality. Mol. Biol. Cell 10: 2101–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A. J., Parkes T. L., Kirby K., St George-Hyslop P., Boulianne G. L., et al. , 1999. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic. Biol. Med. 26: 1332–1338. [DOI] [PubMed] [Google Scholar]
- Ellis M. C., O’Neill E. M., Rubin G. M., 1993. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development 119: 855–865. [DOI] [PubMed] [Google Scholar]
- Emamian E. S., Kaytor M. D., Duvick L. A., Zu T., Tousey S. K., et al. , 2003. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375–387. [DOI] [PubMed] [Google Scholar]
- Estes P. S., Boehringer A., Zwick R., Tang J. E., Grigsby B., et al. , 2011. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20: 2308–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers M. M., Toonen L. J., van Roon-Mom W. M., 2014. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: current insights and emerging therapeutic strategies. Mol. Neurobiol. 49: 1513–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany M. B., Bender W. W., 2000. A Drosophila model of Parkinson’s disease. Nature 404: 394–398. [DOI] [PubMed] [Google Scholar]
- Feiguin F., Godena V. K., Romano G., D’Ambrogio A., Klima R., et al. , 2009. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583: 1586–1592. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P., Nino-Rosales M. L., de Gouyon B., She W. C., Luchak J. M., et al. , 2000. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 408: 101–106. [DOI] [PubMed] [Google Scholar]
- Finelli A., Kelkar A., Song H. J., Yang H., Konsolaki M., 2004. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol. Cell. Neurosci. 26: 365–375. [DOI] [PubMed] [Google Scholar]
- Fischbeck K. H., Bryan W. W., 2009. Anti-androgen treatment for spinal and bulbar muscular atrophy. Ann. Neurol. 65: 119–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossgreen A., Bruckner B., Czech C., Masters C. L., Beyreuther K., et al. , 1998. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc. Natl. Acad. Sci. USA 95: 13703–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum B. D., Chitta R. K., High A. A., Taylor J. P., 2010. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9: 1104–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum B. D., Lu Y., Lopez-Gonzalez R., Kim N. C., Almeida S., et al. , 2015. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin B. A., Dolph M. J., Deleault N. R., Geoghegan J. C., Khurana V., et al. , 2006. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Straussler-Scheinker syndrome. J. Neurosci. 26: 12408–12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellera, C., N. Ticozzi, V. Pensato, L. Nanetti, A. Castucci et al., 2012 ATAXIN2 CAG-repeat length in Italian patients with amyotrophic lateral sclerosis: Risk factor or variant phenotype? Implication for genetic testing and counseling. Neurobiol. Aging 33: 1847.e15–21. [DOI] [PubMed]
- Gendron T. F., Bieniek K. F., Zhang Y. J., Jansen-West K., Ash P. E., et al. , 2013. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126: 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geser F., Martinez-Lage M., Robinson J., Uryu K., Neumann M., et al. , 2009. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch. Neurol. 66: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., et al. , 2012. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11: 54–65. [DOI] [PubMed] [Google Scholar]
- Gispert S., Kurz A., Waibel S., Bauer P., Liepelt I., et al. , 2012. The modulation of amyotrophic lateral sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol. Dis. 45: 356–361. [DOI] [PubMed] [Google Scholar]
- Goedert M., Clavaguera F., Tolnay M., 2010. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33: 317–325. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Del Tredici K., Braak H., 2013. 100 years of Lewy pathology. Nat. Rev. Neurol. 9: 13–24. [DOI] [PubMed] [Google Scholar]
- Graveland G. A., Williams R. S., DiFiglia M., 1985. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227: 770–773. [DOI] [PubMed] [Google Scholar]
- Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B., et al. , 2003. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 100: 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve I., Kretzschmar D., Tschape J. A., Beyn A., Brellinger C., et al. , 2004. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 24: 3899–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber W. B., Jan L. Y., Jan Y. N., 2002. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 129: 2867–2878. [DOI] [PubMed] [Google Scholar]
- Guidetti P., Charles V., Chen E. Y., Reddy P. H., Kordower J. H., et al. , 2001. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp. Neurol. 169: 340–350. [DOI] [PubMed] [Google Scholar]
- Gunnarsson L. G., Dahlbom K., Strandman E., 1991. Motor neuron disease and dementia reported among 13 members of a single family. Acta Neurol. Scand. 84: 429–433. [DOI] [PubMed] [Google Scholar]
- Gusella J. F., MacDonald M. E., 2000. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1: 109–115. [DOI] [PubMed] [Google Scholar]
- Haacke A., Broadley S. A., Boteva R., Tzvetkov N., Hartl F. U., et al. , 2006. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 15: 555–568. [DOI] [PubMed] [Google Scholar]
- Hanson K. A., Kim S. H., Wassarman D. A., Tibbetts R. S., 2010. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J. Biol. Chem. 285: 11068–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. H., Savkur R. S., Poulos M. G., Mancini M. A., Swanson M. S., et al. , 2005. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J. Cell Sci. 118: 2923–2933. [DOI] [PubMed] [Google Scholar]
- Housden B. E., Lin S., Perrimon N., 2014. Cas9-based genome editing in Drosophila. Methods Enzymol. 546: 415–439. [DOI] [PubMed] [Google Scholar]
- Houseley J. M., Wang Z., Brock G. J., Soloway J., Artero R., et al. , 2005. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum. Mol. Genet. 14: 873–883. [DOI] [PubMed] [Google Scholar]
- Hsu T. C., Wang C. K., Yang C. Y., Lee L. C., Hsieh-Li H. M., et al. , 2014. Deactivation of TBP contributes to SCA17 pathogenesis. Hum. Mol. Genet. 23: 6878–6893. [DOI] [PubMed] [Google Scholar]
- Huang C. C., Faber P. W., Persichetti F., Mittal V., Vonsattel J. P., et al. , 1998. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 24: 217–233. [DOI] [PubMed] [Google Scholar]
- Iijima K., Liu H. P., Chiang A. S., Hearn S. A., Konsolaki M., et al. , 2004. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 101: 6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert G., Saudou F., Yvert G., Devys D., Trottier Y., et al. , 1996. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 14: 285–291. [DOI] [PubMed] [Google Scholar]
- Irizarry M. C., Growdon W., Gomez-Isla T., Newell K., George J. M., et al. , 1998. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol. 57: 334–337. [DOI] [PubMed] [Google Scholar]
- Irwin D. J., Lee V. M., Trojanowski J. Q., 2013. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 14: 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson G. R., Salecker I., Dong X., Yao X., Arnheim N., et al. , 1998. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21: 633–642. [DOI] [PubMed] [Google Scholar]
- Jackson G. R., Wiedau-Pazos M., Sang T. K., Wagle N., Brown C. A., et al. , 2002. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34: 509–519. [DOI] [PubMed] [Google Scholar]
- Jackson S. M., Whitworth A. J., Greene J. C., Libby R. T., Baccam S. L., et al. , 2005. A SCA7 CAG/CTG repeat expansion is stable in Drosophila melanogaster despite modulation of genomic context and gene dosage. Gene 347: 35–41. [DOI] [PubMed] [Google Scholar]
- Jaiswal M., Sandoval H., Zhang K., Bayat V., Bellen H. J., 2012. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu. Rev. Genet. 46: 371–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenett A., Rubin G. M., Ngo T. T., Shepherd D., Murphy C., et al. , 2012. A GAL4-driver line resource for Drosophila neurobiology. Cell Reports 2: 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Mankodi A., Swanson M. S., Moxley R. T., Thornton C. A., 2004. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13: 3079–3088. [DOI] [PubMed] [Google Scholar]
- Jin P., Zarnescu D. C., Zhang F., Pearson C. E., Lucchesi J. C., et al. , 2003. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 39: 739–747. [DOI] [PubMed] [Google Scholar]
- Jovicic A., Mertens J., Boeynaems S., Bogaert E., Chai N., et al. , 2015. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 18: 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., Xu K., Lessing D., Bonini N. M., 2009. Preventing Ataxin-3 protein cleavage mitigates degeneration in a Drosophila model of SCA3. Hum. Mol. Genet. 18: 4843–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karres J. S., Hilgers V., Carrera I., Treisman J., Cohen S. M., 2007. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131: 136–145. [DOI] [PubMed] [Google Scholar]
- Katsuno M., Adachi H., Kume A., Li M., Nakagomi Y., et al. , 2002. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35: 843–854. [DOI] [PubMed] [Google Scholar]
- Katsuno M., Adachi H., Doyu M., Minamiyama M., Sang C., et al. , 2003. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med. 9: 768–773. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., et al. , 1994. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8: 221–228. [DOI] [PubMed] [Google Scholar]
- Kazantsev A. G., Thompson L. M., 2008. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7: 854–868. [DOI] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P., Benzer S., 2000. Genetic suppression of polyglutamine toxicity in Drosophila. Science 287: 1837–1840. [DOI] [PubMed] [Google Scholar]
- Kim H. J., Kim N. C., Wang Y. D., Scarborough E. A., Moore J., et al. , 2013a Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495: 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Raphael A. R., LaDow E. S., McGurk L., Weber R. A., et al. , 2014. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 46: 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N. C., Tresse E., Kolaitis R. M., Molliex A., Thomas R. E., et al. , 2013b VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissel J. T., Scott C. B., Reyna S. P., Crawford T. O., Simard L. R., et al. , 2011. SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One 6: e21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissel J. T., Elsheikh B., King W. M., Freimer M., Scott C. B., et al. , 2014. SMA valiant trial: a prospective, double-blind, placebo-controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle Nerve 49: 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson R. R., Chang C. H., Xiong R., Williams H. E., Davis A. L., et al. , 2013. Synthesis of 19-substituted geldanamycins with altered conformations and their binding to heat shock protein Hsp90. Nat. Chem. 5: 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement I. A., Skinner P. J., Kaytor M. D., Yi H., Hersch S. M., et al. , 1998. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95: 41–53. [DOI] [PubMed] [Google Scholar]
- Klucken J., Ingelsson M., Shin Y., Irizarry M. C., Hedley-Whyte E. T., et al. , 2006. Clinical and biochemical correlates of insoluble alpha-synuclein in dementia with Lewy bodies. Acta Neuropathol. 111: 101–108. [DOI] [PubMed] [Google Scholar]
- Kretzschmar D., Hasan G., Sharma S., Heisenberg M., Benzer S., 1997. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J. Neurosci. 17: 7425–7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R., Kuhn W., Muller T., Woitalla D., Graeber M., et al. , 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18: 106–108. [DOI] [PubMed] [Google Scholar]
- Kuo Y., Ren S., Lao U., Edgar B. A., Wang T., 2013. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 4: e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T. J., Jr, Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., et al. , 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Cleveland D. W., 2009. Rethinking ALS: the FUS about TDP-43. Cell 136: 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Baughn M., Rigo F., Sun S., Liu P., et al. , 2013. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 110: E4530–E4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Y. C., Bowman A. B., Jafar-Nejad P., Lim J., Richman R., et al. , 2006. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127: 1335–1347. [DOI] [PubMed] [Google Scholar]
- Lanson N. A., Jr, Maltare A., King H., Smith R., Kim J. H., et al. , 2011. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 20: 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastres-Becker I., Rub U., Auburger G., 2008. Spinocerebellar ataxia 2 (SCA2). Cerebellum 7: 115–124. [DOI] [PubMed] [Google Scholar]
- Latouche M., Lasbleiz C., Martin E., Monnier V., Debeir T., et al. , 2007. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J. Neurosci. 27: 2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. B., Bagley J. A., Lee H. Y., Jan L. Y., Jan Y. N., 2011. Pathogenic polyglutamine proteins cause dendrite defects associated with specific actin cytoskeletal alterations in Drosophila. Proc. Natl. Acad. Sci. USA 108: 16795–16800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. B., Chen H. J., Peres J. N., Gomez-Deza J., Attig J., et al. , 2013. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Reports 5: 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees A. J., Hardy J., Revesz T., 2009. Parkinson’s disease. Lancet 373: 2055–2066. [DOI] [PubMed] [Google Scholar]
- Lesage S., Anheim M., Letournel F., Bousset L., Honore A., et al. , 2013. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73: 459–471. [DOI] [PubMed] [Google Scholar]
- Lessing D., Bonini N. M., 2009. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat. Rev. Genet. 10: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine T. P., Daniels R. D., Gatta A. T., Wong L. H., Hayes M. J., 2013. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29: 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. H., Kroll J. R., Lennox S. M., Ogundeyi O., Jeter J., et al. , 2014. A GAL4 driver resource for developmental and behavioral studies on the larval CNS of Drosophila. Cell Reports 8: 897–908. [DOI] [PubMed] [Google Scholar]
- Li L. B., Xu K., Bonini N. M., 2007. Suppression of polyglutamine toxicity by the yeast Sup35 prion domain in Drosophila. J. Biol. Chem. 282: 37694–37701. [DOI] [PubMed] [Google Scholar]
- Li L. B., Yu Z., Teng X., Bonini N. M., 2008. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453: 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ray P., Rao E. J., Shi C., Guo W., et al. , 2010. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 107: 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman J., Deeg S., Voigt A., Vossfeldt H., Dohm C. P., et al. , 2014. CDK5 protects from caspase-induced Ataxin-3 cleavage and neurodegeneration. J. Neurochem. 129: 1013–1023. [DOI] [PubMed] [Google Scholar]
- Lin M. J., Cheng C. W., Shen C. K., 2011. Neuronal function and dysfunction of Drosophila dTDP. PLoS One 6: e20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S. C., Polymenidou M., Cleveland D. W., 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zhang J. P., Shi M., Quinn T., Bradner J., et al. , 2009. Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J. Neurosci. 29: 1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Landreh M., Cao K., Abe M., Hendriks G. J., et al. , 2012. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482: 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., M. Lu, L. Tang, N. Zhang, D. Chui et al., 2013 ATXN2 CAG repeat expansions increase the risk for Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 34: 2236.e5–8. [DOI] [PubMed]
- Liu Z., Wang X., Yu Y., Li X., Wang T., et al. , 2008. A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. USA 105: 2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C., Shorter J., Regulier E., Lashuel H., Iwatsubo T., et al. , 2008. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Invest. 118: 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C., Anderson T., Miller B., 2002. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59: 1077–1079. [DOI] [PubMed] [Google Scholar]
- Lu B., Vogel H., 2009. Drosophila models of neurodegenerative diseases. Annu. Rev. Pathol. 4: 315–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Ferris J., Gao F. B., 2009. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lush M. J., Li Y., Read D. J., Willis A. C., Glynn P., 1998. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem. J. 332(Pt 1): 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T., Trinh M. A., Wexler A. J., Bourbon C., Gatti E., et al. , 2013. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16: 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R., Feldman H. H., 2005. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J. Neuropathol. Exp. Neurol. 64: 730–739. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Rademakers R., Neumann M., 2010. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9: 995–1007. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Arzberger T., Kremmer E., Troost D., Lorenzl S., et al. , 2013. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126: 859–879. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Frick P., Neumann M., 2014. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 127: 347–357. [DOI] [PubMed] [Google Scholar]
- Majounie E., Renton A. E., Mok K., Dopper E. G., Waite A., et al. , 2012. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A., Urbinati C. R., Yuan Q. P., Moxley R. T., Sansone V., et al. , 2001. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10: 2165–2170. [DOI] [PubMed] [Google Scholar]
- Mann D. M., Rollinson S., Robinson A., Bennion Callister J., Thompson J. C., et al. , 2013. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. L., Thompson L. M., 2006. Drosophila in the study of neurodegenerative disease. Neuron 52: 169–178. [DOI] [PubMed] [Google Scholar]
- Marsh J. L., Walker H., Theisen H., Zhu Y. Z., Fielder T., et al. , 2000. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum. Mol. Genet. 9: 13–25. [DOI] [PubMed] [Google Scholar]
- May S., Hornburg D., Schludi M. H., Arzberger T., Rentzsch K., et al. , 2014. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128: 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCampbell A., Taylor J. P., Taye A. A., Robitschek J., Li M., et al. , 2000. CREB-binding protein sequestration by expanded polyglutamine. Hum. Mol. Genet. 9: 2197–2202. [DOI] [PubMed] [Google Scholar]
- McGurk L., Lee V. M., Trojanowksi J. Q., Van Deerlin V. M., Lee E. B., et al. , 2014. Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 73: 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean P. J., Klucken J., Shin Y., Hyman B. T., 2004. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 321: 665–669. [DOI] [PubMed] [Google Scholar]
- Mehta P., Antao V., Kaye W., Sanchez M., Williamson D., et al. , 2014. Prevalence of amyotrophic lateral sclerosis: United States, 2010–2011. MMWR Surveill. Summ. 63(Suppl 7): 1–14. [PubMed] [Google Scholar]
- Mercuri E., Bertini E., Messina S., Pelliccioni M., D’Amico A., et al. , 2004. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul. Disord. 14: 130–135. [DOI] [PubMed] [Google Scholar]
- Mercuri E., Bertini E., Messina S., Solari A., D’Amico A., et al. , 2007. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 68: 51–55. [DOI] [PubMed] [Google Scholar]
- Merdes G., Soba P., Loewer A., Bilic M. V., Beyreuther K., et al. , 2004. Interference of human and Drosophila APP and APP-like proteins with PNS development in Drosophila. EMBO J. 23: 4082–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre S. D., Michelson S. J., Gomes J., Tabb L. P., Saunders A. J., et al. , 2014. Development and characterization of an aged onset model of Alzheimer’s disease in Drosophila melanogaster. Exp. Neurol. 261: 772–781. [DOI] [PubMed] [Google Scholar]
- Miguel, L., T. Avequin, M. Delarue, S. Feuillette, T. Frebourg et al., 2012 Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging 33: 1008.e1–15.
- Miller J. W., Urbinati C. R., Teng-Umnuay P., Stenberg M. G., Byrne B. J., et al. , 2000. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19: 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K. T., Benzer S., 1997. Spongecake and eggroll: two hereditary diseases in Drosophila resemble patterns of human brain degeneration. Curr. Biol. 7: 885–888. [DOI] [PubMed] [Google Scholar]
- Mizielinska S., Lashley T., Norona F. E., Clayton E. L., Ridler C. E., et al. , 2013. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126: 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S., Gronke S., Niccoli T., Ridler C. E., Clayton E. L., et al. , 2014. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345: 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J., Hiesinger P. R., Schroeder A. J., Kume K., Verstreken P., et al. , 2002. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34: 961–972. [DOI] [PubMed] [Google Scholar]
- Moreno J. A., Radford H., Peretti D., Steinert J. R., Verity N., et al. , 2012. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485: 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno J. A., Halliday M., Molloy C., Radford H., Verity N., et al. , 2013. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5: 206ra138. [DOI] [PubMed] [Google Scholar]
- Morgan T. H., Sturtevant A. H., Muller H. J., Bridges C. B., 1915. The Mechanism of Mendelian Heredity, Henry Holt, New York. [Google Scholar]
- Mori K., Arzberger T., Grasser F. A., Gijselinck I., May S., et al. , 2013a Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126: 881–893. [DOI] [PubMed] [Google Scholar]
- Mori K., Weng S. M., Arzberger T., May S., Rentzsch K., et al. , 2013b The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339: 1335–1338. [DOI] [PubMed] [Google Scholar]
- Mottamal M., Zheng S., Huang T. L., Wang G., 2015. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20: 3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad A., Flores I., Zhang H., Yu R., Staniszewski A., et al. , 2008. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA 105: 7327–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. M., Henry R. G., Langmore S., Kramer J. H., Miller B. L., et al. , 2007. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch. Neurol. 64: 530–534. [DOI] [PubMed] [Google Scholar]
- Mutsuddi M., Marshall C. M., Benzow K. A., Koob M. D., Rebay I., 2004. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr. Biol. 14: 302–308. [DOI] [PubMed] [Google Scholar]
- Nedelsky N. B., Pennuto M., Smith R. B., Palazzolo I., Moore J., et al. , 2010. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 67: 936–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., et al. , 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. [DOI] [PubMed] [Google Scholar]
- Ni J. Q., Zhou R., Czech B., Liu L. P., Holderbaum L., et al. , 2011. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8: 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y., Yalgin C., Akimoto S., Doumanis J., Sasajima R., et al. , 2010. Selection of behaviors and segmental coordination during larval locomotion is disrupted by nuclear polyglutamine inclusions in a new Drosophila Huntington’s disease-like model. J. Neurogenet. 24: 194–206. [DOI] [PubMed] [Google Scholar]
- Nisoli I., Chauvin J. P., Napoletano F., Calamita P., Zanin V., et al. , 2010. Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ. 17: 1577–1587. [DOI] [PubMed] [Google Scholar]
- Nucifora F. C., Jr, Sasaki M., Peters M. F., Huang H., Cooper J. K., et al. , 2001. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423–2428. [DOI] [PubMed] [Google Scholar]
- Ochaba J., Lukacsovich T., Csikos G., Zheng S., Margulis J., et al. , 2014. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 111: 16889–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuoha S. C., Mukund S. R., Coulstock E. T., Sengerova B., Shaw J., et al. , 2007. Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J. Mol. Biol. 372: 287–297. [DOI] [PubMed] [Google Scholar]
- Orr H. T., Zoghbi H. Y., 2007. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30: 575–621. [DOI] [PubMed] [Google Scholar]
- Osterwalder T., Yoon K. S., White B. H., Keshishian H., 2001. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. USA 98: 12596–12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallos J., Bodai L., Lukacsovich T., Purcell J. M., Steffan J. S., et al. , 2008. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 17: 3767–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., et al. , 2007. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447: 859–863. [DOI] [PubMed] [Google Scholar]
- Park J., Lee S. B., Lee S., Kim Y., Song S., et al. , 2006. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161. [DOI] [PubMed] [Google Scholar]
- Park J., Al-Ramahi I., Tan Q., Mollema N., Diaz-Garcia J. R., et al. , 2013. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature 498: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson H. L., Perez M. K., Trottier Y., Trojanowski J. Q., Subramony S. H., et al. , 1997. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19: 333–344. [DOI] [PubMed] [Google Scholar]
- Pearce M. M., Spartz E. J., Hong W., Luo L., Kopito R. R., 2015. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 6: 6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrimon N., Ni J. Q., Perkins L., 2010. In vivo RNAi: today and tomorrow. Cold Spring Harb. Perspect. Biol. 2: a003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocas G. M., Branco-Santos J., Herrera F., Outeiro T. F., Domingos P. M., 2015. alpha-Synuclein modifies mutant huntingtin aggregation and neurotoxicity in Drosophila. Hum. Mol. Genet. 24: 1898–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E., 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78: 959–991. [DOI] [PubMed] [Google Scholar]
- Proukakis C., Dudzik C. G., Brier T., MacKay D. S., Cooper J. M., et al. , 2013. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80: 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst S. M., Nechiporuk A., Nechiporuk T., Gispert S., Chen X. N., et al. , 1996. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14: 269–276. [DOI] [PubMed] [Google Scholar]
- Rainier S., Bui M., Mark E., Thomas D., Tokarz D., et al. , 2008. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 82: 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra T. K., Gonsalvez G. B., Walker M. P., Shpargel K. B., Salz H. K., et al. , 2007. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J. Cell Biol. 176: 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum L. P., Cooper T. A., 2006. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 29: 259–277. [DOI] [PubMed] [Google Scholar]
- Ratnaparkhi A., Lawless G. M., Schweizer F. E., Golshani P., Jackson G. R., 2008. A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS One 3: e2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., et al. , 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36: 585–595. [DOI] [PubMed] [Google Scholar]
- Reiter L. T., Bier E., 2002. Using Drosophila melanogaster to uncover human disease gene function and potential drug target proteins. Expert Opin. Ther. Targets 6: 387–399. [DOI] [PubMed] [Google Scholar]
- Reiter L. T., Potocki L., Chien S., Gribskov M., Bier E., 2001. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11: 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., et al. , 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Chio A., Traynor B. J., 2014. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riabinina O., Luginbuhl D., Marr E., Liu S., Wu M. N., et al. , 2015. Improved and expanded Q-system reagents for genetic manipulations. Nat. Methods 12: 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritson G. P., Custer S. K., Freibaum B. D., Guinto J. B., Geffel D., et al. , 2010. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30: 7729–7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman G., Davis R. L., 2002. Conditional expression of UAS-transgenes in the adult eye with a new gene-switch vector system. Genesis 34: 127–131. [DOI] [PubMed] [Google Scholar]
- Romero E., Cha G. H., Verstreken P., Ly C. V., Hughes R. E., et al. , 2008. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorth P., 1996. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc. Natl. Acad. Sci. USA 93: 12418–12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., et al. , 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62. [DOI] [PubMed] [Google Scholar]
- Ross O. A., Rutherford N. J., Baker M., Soto-Ortolaza A. I., Carrasquillo M. M., et al. , 2011. Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B., Jackson G. R., 2014. Interactions between Tau and alpha-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 23: 3008–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano L., Melo C., Silva M. C., Coutinho P., 2014. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42: 174–183. [DOI] [PubMed] [Google Scholar]
- Rubin G. M., Yandell M. D., Wortman J. R., Gabor Miklos G. L., Nelson C. R., et al. , 2000. Comparative genomics of the eukaryotes. Science 287: 2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y. N., Xu Z., Patel B., Chen Z., Chen D., et al. , 2015. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17: 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes J. R., Zipursky S. L., 2010. Design principles of insect and vertebrate visual systems. Neuron 66: 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang T. K., Li C., Liu W., Rodriguez A., Abrams J. M., et al. , 2005. Inactivation of Drosophila Apaf-1 related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum. Mol. Genet. 14: 357–372. [DOI] [PubMed] [Google Scholar]
- Sanpei K., Takano H., Igarashi S., Sato T., Oyake M., et al. , 1996. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 14: 277–284. [DOI] [PubMed] [Google Scholar]
- Sapp E., Schwarz C., Chase K., Bhide P. G., Young A. B., et al. , 1997. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann. Neurol. 42: 604–612. [DOI] [PubMed] [Google Scholar]
- Satterfield T. F., Pallanck L. J., 2006. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 15: 2523–2532. [DOI] [PubMed] [Google Scholar]
- Satterfield T. F., Jackson S. M., Pallanck L. J., 2002. A Drosophila homolog of the polyglutamine disease gene SCA2 is a dosage-sensitive regulator of actin filament formation. Genetics 162: 1687–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidner G. A., Ye Y., Faraday M. M., Alvord W. G., Fortini M. E., 2006. Modeling clinically heterogeneous presenilin mutations with transgenic Drosophila. Curr. Biol. 16: 1026–1033. [DOI] [PubMed] [Google Scholar]
- Sekine Y., Zyryanova A., Crespillo-Casado A., Fischer P. M., Harding H. P., et al. , 2015. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348: 1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. Y., He J. C., Wang Y., Huang Q. Y., Chen J. F., 2005. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J. Biol. Chem. 280: 39962–39969. [DOI] [PubMed] [Google Scholar]
- Shorter J., 2008. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals 16: 63–74. [DOI] [PubMed] [Google Scholar]
- Shulman J. M., Shulman L. M., Weiner W. J., Feany M. B., 2003. From fruit fly to bedside: translating lessons from Drosophila models of neurodegenerative disease. Curr. Opin. Neurol. 16: 443–449. [DOI] [PubMed] [Google Scholar]
- Shulman J. M., Imboywa S., Giagtzoglou N., Powers M. P., Hu Y., et al. , 2014. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet. 23: 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., Acosta-Alvear D., Khoutorsky A., Vedantham P., Hearn B. R., et al. , 2013. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2: e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C., Tsai J. C., Kampmann M., Hearn B. R., Vedantham P., et al. , 2015. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife 4: e07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J., Dopper E. G., Cohn-Hokke P. E., Hukema R. K., Nicolaou N., et al. , 2012. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135: 723–735. [DOI] [PubMed] [Google Scholar]
- Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., et al. , 2003. alpha-synuclein locus triplication causes Parkinson’s disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Sobczak K., de Mezer M., Michlewski G., Krol J., Krzyzosiak W. J., 2003. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 31: 5469–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M., 1998. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 95: 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D., 2002. The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 3: 176–188. [DOI] [PubMed] [Google Scholar]
- St Pierre S. E., Ponting L., Stefancsik R., McQuilton P., and FlyBase Consortium, 2014. FlyBase 102: advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42: D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan J. S., Kazantsev A., Spasic-Boskovic O., Greenwald M., Zhu Y. Z., et al. , 2000. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 97: 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan J. S., Bodai L., Pallos J., Poelman M., McCampbell A., et al. , 2001. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413: 739–743. [DOI] [PubMed] [Google Scholar]
- Stempfle D., Kanwar R., Loewer A., Fortini M. E., Merdes G., 2010. In vivo reconstitution of gamma-secretase in Drosophila results in substrate specificity. Mol. Cell. Biol. 30: 3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien D. L., Cummings C. J., Adams H. P., Mancini M. G., Patel K., et al. , 1999. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 8: 731–741. [DOI] [PubMed] [Google Scholar]
- Stokin G. B., Almenar-Queralt A., Gunawardena S., Rodrigues E. M., Falzone T., et al. , 2008. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum. Mol. Genet. 17: 3474–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda K. J., Scott C. B., Crawford T. O., Simard L. R., Reyna S. P., et al. , 2010. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 5: e12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M., Gonzalez M. A., Lourenco C. M., Coutelier M., Haack T. B., et al. , 2014. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 137: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Kanuka H., Fujiwara H., Koyama A., Hasegawa M., et al. , 2003. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 336: 155–158. [DOI] [PubMed] [Google Scholar]
- Takeyama K., Ito S., Yamamoto A., Tanimoto H., Furutani T., et al. , 2002. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 35: 855–864. [DOI] [PubMed] [Google Scholar]
- Tazen S., Figueroa K., Kwan J. Y., Goldman J., Hunt A., et al. , 2013. Amyotrophic lateral sclerosis and spinocerebellar ataxia type 2 in a family with full CAG repeat expansions of ATXN2. JAMA Neurol. 70: 1302–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray A. M., Muhammad F., Zhang C., Denyer M., Spiropoulos J., et al. , 2012. Prion-induced toxicity in PrP transgenic Drosophila. Exp. Mol. Pathol. 92: 194–201. [DOI] [PubMed] [Google Scholar]
- Thomas M., Harrell J. M., Morishima Y., Peng H. M., Pratt W. B., et al. , 2006. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum. Mol. Genet. 15: 1876–1883. [DOI] [PubMed] [Google Scholar]
- Tong X., Gui H., Jin F., Heck B. W., Lin P., et al. , 2011. Ataxin-1 and Brother of ataxin-1 are components of the Notch signalling pathway. EMBO Rep. 12: 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh K., Moore K., Wes P. D., Muchowski P. J., Dey J., et al. , 2008. Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson’s disease. J. Neurosci. 28: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H., Jafar-Nejad H., Patel A. J., Sun Y., Chen H. K., et al. , 2005. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 122: 633–644. [DOI] [PubMed] [Google Scholar]
- Uryu K., Richter-Landsberg C., Welch W., Sun E., Goldbaum O., et al. , 2006. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am. J. Pathol. 168: 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher C., Garcia-Oroz L., Rubinsztein D. C., 2005. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 14: 3425–3433. [DOI] [PubMed] [Google Scholar]
- Valor L. M., Guiretti D., 2014. What’s wrong with epigenetics in Huntington’s disease? Neuropharmacology 80: 103–114. [DOI] [PubMed] [Google Scholar]
- Valor L. M., Viosca J., Lopez-Atalaya J. P., Barco A., 2013. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 19: 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel D. M., Harschnitz O., van den Berg L. H., Pasterkamp R. J., 2014. Taking a risk: A therapeutic focus on ataxin-2 in amyotrophic lateral sclerosis? Trends Mol. Med. 20: 25–35. [DOI] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobagyi T., De Vos K. J., Nishimura A. L., et al. , 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323: 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck L., Naval-Sanchez M., Adachi Y., Diaper D., Dourlen P., et al. , 2013. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Reports 3: 160–172. [DOI] [PubMed] [Google Scholar]
- Venderova K., Kabbach G., Abdel-Messih E., Zhang Y., Parks R. J., et al. , 2009. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet. 18: 4390–4404. [DOI] [PubMed] [Google Scholar]
- Venken K. J., Bellen H. J., 2012. Genome-wide manipulations of Drosophila melanogaster with transposons, Flp recombinase, and PhiC31 integrase. Methods Mol. Biol. 859: 203–228. [DOI] [PubMed] [Google Scholar]
- Venken K. J., Bellen H. J., 2014. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods 68: 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P., Augustine G. J., Ammar M. R., Tashiro A., Cohen S. M., 2015. A neuroprotective role for microRNA miR-1000 mediated by limiting glutamate excitotoxicity. Nat. Neurosci. 18: 379–385. [DOI] [PubMed] [Google Scholar]
- Voigt A., Herholz D., Fiesel F. C., Kaur K., Muller D., et al. , 2010. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One 5: e12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M. J., Zijlstra M. P., Kanon B., van Waarde-Verhagen M. A., Brunt E. R., et al. , 2010. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 19: 4677–4693. [DOI] [PubMed] [Google Scholar]
- Warrick J. M., Chan H. Y., Gray-Board G. L., Chai Y., Paulson H. L., et al. , 1999. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 23: 425–428. [DOI] [PubMed] [Google Scholar]
- Warrick J. M., Morabito L. M., Bilen J., Gordesky-Gold B., Faust L. Z., et al. , 2005. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 18: 37–48. [DOI] [PubMed] [Google Scholar]
- Warrick J. M., Paulson H. L., Gray-Board G. L., Bui Q. T., Fischbeck K. H., et al. , 1998. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93: 939–949. [DOI] [PubMed] [Google Scholar]
- Watson M. R., Lagow R. D., Xu K., Zhang B., Bonini N. M., 2008. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 283: 24972–24981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waza M., Adachi H., Katsuno M., Minamiyama M., Sang C., et al. , 2005. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 11: 1088–1095. [DOI] [PubMed] [Google Scholar]
- Waza M., Adachi H., Katsuno M., Minamiyama M., Tanaka F., et al. , 2006. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med (Berl) 84: 635–646. [DOI] [PubMed] [Google Scholar]
- Wellington C. L., Ellerby L. M., Hackam A. S., Margolis R. L., Trifiro M. A., et al. , 1998. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 273: 9158–9167. [DOI] [PubMed] [Google Scholar]
- Wen X., Tan W., Westergard T., Krishnamurthy K., Markandaiah S. S., et al. , 2014. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84: 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M., Thornton C. A., 2007. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 20: 572–576. [DOI] [PubMed] [Google Scholar]
- Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., et al. , 2001. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711–714. [DOI] [PubMed] [Google Scholar]
- Xia R., Liu Y., Yang L., Gal J., Zhu H., et al. , 2012. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener. 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Poidevin M., Li X., Li Y., Shu L., et al. , 2013. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 110: 7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa M., Ito D., Honda T., Kubo K., Noda M., et al. , 2015. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 24: 1630–1645. [DOI] [PubMed] [Google Scholar]
- Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., et al. , 2006. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 103: 10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Fortini M. E., 1999. Apoptotic activities of wild-type and Alzheimer’s disease-related mutant presenilins in Drosophila melanogaster. J. Cell Biol. 146: 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Goodman L. D., Shieh S. Y., Min M., Teng X., et al. , 2015. A fly model for the CCUG-repeat expansion of myotonic dystrophy type 2 reveals a novel interaction with MBNL1. Hum. Mol. Genet. 24: 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz J. J., Alegre J., Gomez-Esteban J. C., Lezcano E., Ros R., et al. , 2004. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55: 164–173. [DOI] [PubMed] [Google Scholar]
- Zhang S., Feany M. B., Saraswati S., Littleton J. T., Perrimon N., 2009. Inactivation of Drosophila Huntingtin affects long-term adult functioning and the pathogenesis of a Huntington’s disease model. Dis. Model. Mech. 2: 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. J., Jansen-West K., Xu Y. F., Gendron T. F., Bieniek K. F., et al. , 2014. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 128: 505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Donnelly C. J., Haeusler A. R., Grima J. C., Machamer J. B., et al. , 2015. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T., Gibbens B., Doty N. S., Gomes-Pereira M., Huguet A., et al. , 2011. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 108: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T., Liu Y., Banez-Coronel M., Reid T., Pletnikova O., et al. , 2013. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 110: E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]