

Abstract
NF-κB signaling plays a crucial role in regulating proliferation and differentiation in the epidermis. Alterations in the NF-κB pathway can lead to skin pathologies with a significant burden to human health such as psoriasis and cutaneous squamous cell carcinoma (cSCC). Caspase recruitment domain (CARD)-containing scaffold proteins are key regulators of NF-κB signaling by providing a link between membrane receptors and NF-κB transcriptional subunits. Mutations in the CARD family member, CARD14, have been identified in patients with the inflammatory skin diseases psoriasis and pityriasis rubra pilaris. Here, we describe that the gene coding for another CARD scaffold protein, CARD11, is mutated in more than 38% of 111 cSCCs, and show that novel variants outside of the coiled-coil domain lead to constitutively activated NF-κB signaling. CARD11 protein expression was detectable in normal skin and increased in all cSCCs tested. CARD11 mRNA levels were comparable with CARD14 in normal skin and CARD11 mRNA was increased in cSCC. In addition, we identified CARD11 mutations in peritumoral and sun-exposed skin, suggesting that CARD11-mediated alterations in NF-κB signaling may be an early event in the development of cSCC.
Skin cancers are the most frequent human malignancy and present an increasing health burden with a steep increase in cutaneous squamous cell carcinoma (cSCC) occurring in the aging population.1 Although metastasis is relatively uncommon, advanced, often inoperable, locoregional disease is frequent with cSCC and such patients would benefit from systemic or directly delivered (intralesional) chemotherapy. Nonsurgical therapies also would be desirable for high-risk patients with a considerable cSCC burden caused by immunosuppression, for which repeated surgical procedures bring significant morbidity.2 For those cSCCs that do metastasize, 5-year survival rates are documented at less than 30%,3 and as many as one in three skin cancer–related deaths reported to cancer registries are caused by cSCC (Informational Services Division, NHS National Services, Scotland; http://www.isdscotland.org/Health-Topics/Cancer/Cancer-Statistics/Skin/#non-melanoma, last accessed April 17, 2015).
Recent exome sequencing has shown that cSCC harbors a mutation burden greater than any other known cancer with metastatic potential,4–6 only exceeded by basal cell carcinoma of the skin.7 These and other studies in human cSCC have identified key driver mutations in NOTCH receptors, TP53, CDKN2A, and RAS, whereas numerous murine models and functional analyses have implicated disruption of major signaling pathways such as epidermal growth factor receptor, mitogen-activated protein kinase, and transforming growth factor β.8 In particular, NF-κB signaling has been shown to both promote9,10 and inhibit SCC in a variety of model systems.11,12
The NF-κB family consists of five transcription factors, RelA (P65), RelB, c-Rel, NF-κB1 (P100/P50), and NF-κB2 (P105/P52), which form a variety of homodimers and heterodimers that regulate the expression of genes associated with a variety of processes, such as inflammatory and stress responses. NF-κB can be activated via at least two different pathways: the so-called canonical and noncanonical pathways.13 Canonical signaling begins with receptor-ligand stimulation of a kinase cascade that phosphorylates the inhibitor of NF-κB proteins via the inhibitor of NF-κβ kinase (IKK) complex. The IKK complex consists of two catalytic subunits, IKKα and IKKβ, and the regulatory subunit, NF-κB–essential modulator (also known as IKKγ).14,15 Once phosphorylated, the inhibitor of NF-κB proteins is targeted for degradation, which releases NF-κB subunits (predominantly P65 and P50) sequestered in the cytoplasm. The released subunits translocate to the nucleus and bind to DNA recognition motifs of target genes, as a dimer, to regulate transcription. In contrast, the noncanonical pathway is triggered by different stimuli and activation is mediated by an alternative IKK complex (ie, essentially independent of the NF-κB–essential modulator), and leads to transcriptional regulation via the RelB and P52 subunits.13,16
In normal skin, NF-κB has been shown to be a critical regulator of proliferation and differentiation and is key to overall tissue homeostasis, which is essential for the protective role of this self-renewing organ.17 Targeted ablation of multiple components of this pathway in mice, coupled with in vitro cellular experimentation, has driven much of our understanding of how NF-κB regulates epidermal homeostasis.18 Considerable data point to a growth inhibitory/prodifferentiation role for NF-κB in epidermal keratinocytes.19,20 However, more recent murine data have indicated that the primary role of NF-κB in epidermal keratinocytes is to maintain immune homeostasis. A disruption of this status quo results in changes in proliferation and apoptosis as a result of immunologic cross-talk.21,22 A likely explanation for this controversy may lie in the complexity of the pathway, the different models used, or cross-talk with other major signaling axes such as TP53 and mitogen-activated protein kinase.23 One emerging aspect, which largely was ignored until recently, is the involvement of scaffold proteins that provide a link between membrane receptors and the IKK complex. A direct association between human skin disease and a scaffold protein recently was established with the identification of CARD14 mutations in the common skin disease psoriasis24,25 and the much rarer condition pityriasis rubra pilaris.26 Both skin conditions share significant clinical overlap, and molecular characterization has shown common mechanisms driven by mutations in CARD14 that activate NF-κB–centric programs of gene expression.24–26
Caspase recruitment domain (CARD)-containing proteins are conserved across many species and are characterized by the presence of different functional domains shared between family members.27 Structurally, CARD10, CARD11, and CARD14 (also known as CARMA3, CARMA1, and CARMA2) contain a CARD and a coiled-coil domain located at their amino-terminus, followed by a PDZ domain, then a SRC homology 3 (SH3) domain and a guanylate kinase-like (GUK) domain, which together constitute the membrane-associated GUK region at the carboxy-terminus. After phosphorylation, CARD proteins undergo conformational change that enables association with the downstream signaling components B-cell lymphoma protein 10 (BCL10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1). Formation of the complex CARD/BCL10/MALT1 is believed to be an essential step for triggering the subsequent events that lead to activation of NF-κB.27 CARD11 is essential for antigen receptor–mediated NF-κB activation in lymphocytes,28 and mutations in the CARD and coiled-coil domains are prevalent in diffuse large B-cell lymphoma (DLBCL), leading to hyperactive NF-κB signaling and increased cell survival.29–32
In this study, we present data supporting a tumorigenic role for CARD11 in human epidermis.
Materials and Methods
Sequencing
Sample collection and sequencing was performed as described previously.6 Primers were designed and validated by Fluidigm Corporation (San Francisco, CA) per the recommended guidelines for Roche Titanium sequencing (Roche, Mannheim, Germany). Each primer included a sample-specific Fluidigm 454 barcode primer and adapter sequences. Primer sequences are detailed in Table 1. Reactions contained 50 ng genomic DNA, 1 μmol/L forward and reverse tagged amplification primers, 400 nmol/L forward and reverse barcode primers, 1× Access Array Loading Reagent, 1× FastStart High Fidelity Reaction Buffer, 4.5 mmol/L MgCl2, 5% dimethyl sulfoxide, 0.05 U FastStart High Fidelity Enzyme Blend, and 200 μmol/L PCR-grade nucleotide mix (Roche). Thermal cycling was performed on the Fluidigm FC1 Cycler per the manufacturer's guidelines. The resultant libraries were harvested and collected on a microtiter plate and were normalized and pooled before purification using the Agencourt AMPure XP system (Beckman Coulter, High Wycombe, UK) per the manufacturer's protocol. Library components were amplified clonally using the GSJunior emPCR Lib-A Kit (Roche) by inputting one molecule of library DNA per capture bead. Pyrosequencing was performed using the GS Junior system (Roche). Mapping and variant calling were performed as described.6 cSCC mutations were compared with those reported previously for this tumor type and DLBCL using the online catalog of somatic mutations in cancer (http://cancer.sanger.ac.uk/cosmic, last accessed April 17, 2015).
Table 1.
CARD11 Primer Sequences
| Assay_ID | Assay_name | Forward | Reverse |
|---|---|---|---|
| AAA0043704 | CARD11_1 | 5′-CAGGGTGCCTGCCTCATAG-3′ | 5′-TATAGGGAGAAGCAAGGCAGGG-3′ |
| AAA0029061 | CARD11_2 | 5′-TTTACAAAAGGGGTGCATGAGGT-3′ | 5′-GAGTGTCCAGACTCATGTGGG-3′ |
| AAA0051943 | CARD11_3 | 5′-CGGGGAGGTGAAAACAACTCTTC-3′ | 5′-TTTAAACATGGCTGGAAGGGGAT-3′ |
| AAA0029077 | CARD11_4 | 5′-GGAAGTCGTCCTGGTTCATGG-3′ | 5′-AGGTGACCTCAGACACTGGAT-3′ |
| AAA0029066 | CARD11_5 | 5′-CCCTCACAAGGACCGATGTTC-3′ | 5′-CTGCTGGGAGTCTCCTGCTTTTA-3′ |
| AAA0029078 | CARD11_6 | 5′-TATGAAAGTGGGATTTGCTTCCC-3′ | 5′-CTCCAGTTCTCACCAGCATCATT-3′ |
| AAA0029067 | CARD11_7 | 5′-GGATTGCATTTGTTACTGCCTCT-3′ | 5′-ACACAGATACAAGGAGCCACATC-3′ |
| AAA0029069 | CARD11_8 | 5′-CTATAAGCCCCTCTGCTGATGTT-3′ | 5′-CCCCATACTGATAGACAGCTGGA-3′ |
| AAA0051944 | CARD11_9 | 5′-TCCCTGTGTGCTCTCCCA-3′ | 5′-CTCGCTCCCGCAGCTC-3′ |
| AAA0051940 | CARD11_10 | 5′-GTCTGACCACCTTCCAGCTC-3′ | 5′-GTCCCAGTAGGGCTTTTCTCTTT-3′ |
| AAA0029054 | CARD11_11 | 5′-AGGAGAACAAAGCCCACATGAAT-3′ | 5′-GAACCTCAGAGACCCATCTCCT-3′ |
| AAA0029062 | CARD11_12 | 5′-AAAGAACAAGCTCACAGGGCATC-3′ | 5′-CTCTCAGAAGCAAGGCCACTC-3′ |
| AAA0029071 | CARD11_13 | 5′-GTAAGGAGGCTTGGCATGGT-3′ | 5′-GTCCTCCCTGTGAGCAGATG-3′ |
| AAA0051942 | CARD11_14 | 5′-GTGGTGGGCATGAACTGGG-3′ | 5′-GAGCCCTCAAGGGAGAGAAGAG-3′ |
| AAA0029083 | CARD11_15 | 5′-CAGGATTGTTCGTTACAGTGGGA-3′ | 5′-TGGGAGGAGAGCTTGTCTATACC-3′ |
| AAA0029081 | CARD11_16 | 5′-AGGATTTCCAGCTTACAGGGTTG-3′ | 5′-CTAACGCCAGCTTTTTCTCTGTC-3′ |
| AAA0029072 | CARD11_17 | 5′-CTCAGTGGGCACCAGGG-3′ | 5′-GACGAGAGCCACAGACGATG-3′ |
| AAA0029075 | CARD11_18 | 5′-GTTGCACTGGACAAAACACTCTG-3′ | 5′-CAACAGTCAGATAGTCGGTTCCC-3′ |
| AAA0029068 | CARD11_19 | 5′-ATCTAGTCCCTGGAAAGGAGTGT-3′ | 5′-TCCGTATCTGTTGCTGGTTCTTC-3′ |
| AAA0029076 | CARD11_20 | 5′-CAGGTTCATCGTTTCCCCCAA-3′ | 5′-AAGAAGGAGAGAGGGGCATAACT-3′ |
| AAA0029057 | CARD11_21 | 5′-GGTGCATAGTGGACCTAGAACTC-3′ | 5′-AGGATGAGAAGAAGCAGATGACG-3′ |
| AAA0029065 | CARD11_22 | 5′-CTTGGGCACCCCTCACC-3′ | 5′-CTTTTTCCACTTCCCAGTCCTCA-3′ |
| AAA0051941 | CARD11_23 | 5′-TGAACACCCACTTCTCTTGGTTC-3′ | 5′-TCACAGAGTGATCTGGTCTTTCC-3′ |
| AAA0029063 | CARD11_24 | 5′-CGGACCCCAGTTTAAAAATCCCT-3′ | 5′-GCTGTTCCAGTGAGACTGCTTTAT-3′ |
| AAA0029058 | CARD11_25 | 5′-GTGGAGGGCTGCAGTATGTTATG-3′ | 5′-ACTTCCTGGCCTTTTCTTCCAAA-3′ |
P65 Activation Assays
NF-κB activity was determined using the TransAM NF-κB transactivation assay (Active Motif, Rixensart, Belgium). The initial assessment of CARD11 F130V, R818E, P833L, and R1016K using individual assays for P50, P52, P65, RELB, and c-REL identified only P50 and P65 increased activation. All subsequent assays were performed using the P65 TransAM assay. Briefly, human embryonic kidney (HEK) 293 cells (3 × 106 cells/6-cm dish) were transfected with DDK-tagged wild-type or mutant CARD11 plasmid DNA using Lipofectamine 2000 (Invitrogen, Paisley, UK). Cells were harvested at 42 hours after transfection and both cytoplasmic and nuclear fractions were isolated using a Nuclear Extract Kit (Active Motif). Protein concentration was determined by the Bradford assay and CARD11 expression was analyzed by immunoblotting with an anti-DDK antibody (TA50011; Origene, Rockville, MD). Wild-type CARD11 nuclear extract (10 μg) was loaded per well of an enzyme-linked immunosorbent assay plate. To account for variation in protein expression between different constructs, loading amounts were normalized against wild-type CARD11. The assay was performed according to the manufacturer's instructions and absorbance readings were taken using a Versamax plate reader (Molecular Devices, Sunnyvale, CA).
Recombinant CARD11 Immunofluorescence
HEK 293 cells were seeded on coverslips at 2 × 105 cells/well in 24-well plates and transfected 24 hours later as described earlier. Forty-two hours after transfection the cells were washed in phosphate-buffered saline (PBS) and fixed for 20 minutes in 4% paraformaldehyde in PBS at room temperature. Cells were washed three times with PBS before incubating with 50 mmol/L NH4CL/PBS and after a further wash in PBS were permeabilized with 0.2% Triton X-100/PBS (Sigma-Aldrich, St. Louis, MO) for 4 minutes. After a PBS wash cells were blocked (10% fetal calf serum, 0.5% bovine serum albumin, and 0.2% Tween in PBS) for 40 minutes and then incubated with anti-DDK antibody at 1:1000 dilution in blocking buffer for 1 hour at room temperature. After washing in PBS, Alexa 594–conjugated secondary antibody (Dako, Tokyo, Japan) was added in blocking buffer for 30 minutes, followed by washes in PBS, then distilled water, and coverslips were mounted on slides using Prolong Gold Antifade plus DAPI (Invitrogen) as a counterstain. Slides were viewed on a Zeiss Axiovision fluorescent microscope (Zeiss, Cambridge, UK).
Immunohistochemistry
Immunohistochemical staining was performed on formalin-fixed, paraffin-embedded cSCC tissue specimens and normal skin specimens. Tissue sections were cut at thicknesses of 3 μm (cSCC) and 5 μm (normal skin) onto 3-aminopropyltrioxysilane–coated slides and dried overnight in a 45°C oven, followed by de-waxing and dehydration in xylene and industrial methylated spirits. Antigen retrieval was performed by incubating sections in TRIS-EDTA-citrate antigen unmasking solution (5 g EDTA, 2.5 g Tris base, and 3.2 g sodium citrate in 1 L of distilled water, pH adjusted to 8.1 with sodium hydroxide) for 35 minutes in a microwave set to full power. Endogenous peroxidase activity was blocked at ambient temperature in 3% hydrogen peroxide in distilled water for 15 minutes (cSCC) or 45 minutes (normal skin). A polyclonal antibody against recombinant human CARD11 (ab113409; Abcam, Cambridge, UK) was applied as the primary antibody for 40 minutes at ambient temperature (cSCC) at a dilution of 1:100. Incubation with secondary biotinylated antibody followed by streptavidin-biotin-peroxidase complex was performed using the Vector Elite ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions with diaminobenzidine as the chromogen and hematoxylin as counterstain. Human cerebellum was used as the positive control tissue. Negative control reactions were performed on duplicate tissue sections by omitting the primary antibody incubation step.
Immunofluorescence
Immunofluorescence staining was performed on formalin-fixed, paraffin-embedded cSCC tissue and normal skin specimens that were processed as for immunohistochemistry (described earlier), with omission of the peroxidase blocking step. The CARD11 antibody (1:200 final dilution) was mixed with a monoclonal antibody raised against the active subunit of P65 (MAB3026, clone 12H11; EMD Millipore, Billerica, MA) (1:50 final dilution) and applied as the primary antibody for 1 hour at ambient temperature. A mix of Alexa Fluor secondary antibodies (Alexa Fluor 488 goat anti-mouse, 1:250 final dilution, and Alexa Fluor 594 goat anti-rabbit, 1:800 final dilution) and DAPI (1×) (Life Technologies, Grand Island, NY) were applied for 1 hour at ambient room temperature. Sections were mounted and documented using the EVOS FL AutoCell Imaging System (Life Technologies).
Results
Missense Mutations in CARD11 Are Frequent Events in cSCC
Our initial studies aimed to identify mutated genes that segregated with poorly differentiated cSCC histologic grade and more aggressive biological behavior. To do this we interrogated our previous data set describing whole-exome sequencing of 10 well-differentiated and 10 poorly differentiated human cSCC tumors.6 As described in this study, unsupervised hierarchical clustering of 20 tumors failed to separate cSCCs based on histologic grade and mutation profile. This was consistent with a recent study of poorly differentiated cSCCs that failed to identify a clear tumor suppressor or driver in high-grade tumors.5 However, from our exome data set we identified nine genes that were mutated in >60% of poorly differentiated and <20% of well-differentiated cSCCs (Supplemental Figure S1). Of these, CARD11 was the only gene to be mutated preferentially in poorly differentiated tumors (8 of 10) and not in well-differentiated tumors (Supplemental Figure S1 and Supplemental Table S1). To validate this initial finding in our discovery cohort we used targeted re-sequencing with Fluidigm PCR amplification and 454 pyrosequencing in a cohort of 91 sporadic human cSCC samples as previously described.6CARD11 mutation did not stratify with histologic grade in this cohort, but the overall mutation frequency was high (37%) and similar when compared with the discovery cohort (40%), thus identifying CARD11 as heavily mutated in human cSCC (38% of 111 cSCC; Supplemental Tables S1 and S2).
Missense Mutations in CARD11 Can Be Detected in Normal Skin
Our previous sequencing analysis identified mutations in the driver genes HRAS, NOTCH1, and NOTCH2 present in normal, peritumoral, and sun-exposed skin from patients with cSCC.6 Nine of 10 of these normal skin samples had sufficient CARD11 sequencing coverage to determine whether CARD11 mutation also could be detected. In four of these nine samples, CARD11 mutations readily were detected in four or more sequencing reads, albeit at an allelic frequency lower than the detection threshold of 10% for tumors (average, 9.3% allelic frequency in skin compared with 19.2% in cSCC). No equivalent mutations were identified in germline DNA isolated from peripheral blood.
CARD11 Mutations External to the Coiled-Coil Domain Mediate Aberrant NF-κB Signaling
We next compared the spectrum of CARD11 mutations in cSCC with those reported for other tumors in the catalog of somatic mutations in cancer.33CARD11 mutations (n = 88) were distributed more evenly in cSCC compared with those reported in DLBCL, in which mutations in the CARD and coiled-coil domains of CARD11 lead to hyperactive NF-κB signaling and increased cell survival29–31 (Figure 1). Given the even distribution of mutations in cSCC we speculated that CARD11 variants external to these domains also may lead to dysregulated NF-κB activity. To test this hypothesis we generated a series of DDK-tagged CARD11 constructs comprising mutations against all described protein domains and assessed their effect on NF-κB activity in vitro after transfection into HEK 293 cells. Exogenous expression of wild-type CARD11 and a single DLBCL-specific mutation (F130V29) confirmed activation of NF-κB signaling using enzyme-linked immunosorbent assay–based detection of active NF-κB subunits. Specifically, P65 and P50 activity were stimulated compared with the empty vector control with no increase in c-Rel, RelB, or P52, consistent with activation of the canonical NF-κB pathway (data not shown). Subsequently, we identified five cSCC-specific mutants that, when expressed in HEK 293 cells, induced a significant increase in P65 activity compared with wild-type CARD11 and at a comparable level with F130V. Surprisingly, we also identified two variants whose expression resulted in complete attenuation of the CARD11-induced NF-κB response (Figure 2, A and B). Together, our data indicate that mutations in multiple domains of CARD11 result in aberrant functionality with subsequent consequences for NF-κB signaling.
Figure 1.

Cutaneous squamous cell carcinoma (cSCC) caspase recruitment domain (CARD)11 mutations (n = 88) distribute evenly compared with diffuse large B-cell lymphoma (DLBCL) (n = 85). CARD11 mutations (colored vertical lines) are distributed evenly throughout all protein domains in cSCC compared with clustering within the coiled-coil and CARD domains in DLBCL. Mutations are indicated by vertical bars and were compiled from this study (66 cSCC) and the catalog of somatic mutations in cancer (22 cSCC and 85 DLBCL) (http://cancer.sanger.ac.uk/cosmic; last accessed April 15, 2015). Green lines represent missense; red lines represent nonsense; and yellow lines represent an insertion/deletion. Vertical bars above the CARD11 colored boxes indicate two or more mutations present at the same amino acid position. Orange bars above the cSCC CARD11 cartoon indicate two regions of low sequence coverage in cSCC (2 to 10 reads).
Figure 2.

Multidomain cutaneous squamous cell carcinoma (cSCC) mutations in CARD11 result in dysregulated NF-κB activity. A: Individual mutations were transfected into HEK 293 cells and P65/NF-κB activity was analyzed using a TransAM enzyme-linked immunosorbent assay 42 hours after transfection. The F130V is a diffuse large B-cell lymphoma–specific CARD11 mutation to increase P65 activation (data not shown). All other mutations are cSCC specific. The initial assessment of CARD11 F130V, R818E, P833L, and R1016K, using individual assays for P50, P52, P65, RELB, and c-REL, identified only P50 and P65 increased activation. All subsequent assays were performed using the P65 TransAM assay. B: The mean of P65 activity relative to wild-type CARD11 for each individual mutation from three to four separate experiments. Red bars indicate CARD11 variants with increased activity compared with CARD11 wild-type, whereas black bars indicate no significant difference. Samples with only background activity comparable with the empty vector control are indicated by yellow bars. C: DDK immunostaining shows the assembly of CARD11 into cytoplasmic aggregates. HEK 293 cells were seeded on coverslips and transfected 24 hours later as described. Forty-two hours after transfection the cells were incubated with an anti-DDK antibody followed by Alexa 594–conjugated secondary antibody (Dako). Arrows indicate representative foci that are reduced in wild-type CARD11-transfected cells. D: A minimum of 200 cells from two separate experiments were scored for the presence of cytoplasmic aggregates and the graph shows the aggregate-positive fraction for each indicated CARD11 transfection. E: Co-localization of DDK-CARD11 and MALT1 in cytoplasmic aggregates (arrows). Immunofluorescence was performed using anti-DDK antibody and anti-MALT1 antibody. ∗∗P < 0.01, ∗∗∗P < 0.001, two-sample t-test comparison with wild-type CARD11.
Mutant CARD11 Assembles in Cytoplasmic Aggregates Associated with Constitutive NF-κB Activity
Previous studies have shown that expression of activating CARD11 coiled-coil domain mutants results in multimeric aggregation into cytoplasmic complexes, or signalosomes, in contrast to wild-type CARD11, which can be found diffusely throughout the cytoplasm.31,32 This spontaneous complex formation was correlated previously with constitutive, receptor-independent, NF-κB activation. Here, we analyzed the subcellular distribution of DDK-CARD11 proteins by immunofluorescence after transfection into HEK 293 cells. Consistent with lymphoma-derived coiled-coil domain mutants, the F130V DLBCL mutant was found extensively in large fluorescent foci throughout the cytoplasm, which were reduced significantly in wild-type CARD11 samples (Figure 2, C and D). Similar results were seen with cSCC-specific mutant CARD11, including those outside of the coiled-coil domain, whereas foci were reduced in cells expressing mutant constructs not associated with altered NF-κB activity (Figure 2D). Interestingly, cytoplasmic aggregates also were extensive after expression of the R47C and L1015F CARD and GUK domain mutants despite the inactivation of downstream NF-κB activity (Figure 2, B and D). Therefore, to confirm that these aggregates represented bona fide signalosomes, we co-stained for the CARD11 signaling partner MALT1. This showed a clear co-localization of CARD11 and MALT1 within cytoplasmic aggregates, showing the enhanced formation of signaling complexes on expression of those CARD11 mutants that impacted P65 activity (Figure 2E).
CARD11 Expression Is Detected in all cSCC Where it Is Up-Regulated and Localized to Specific Regions within Tumors
Little is known about the role of CARD11 outside the hematopoietic system and descriptions in epithelial tissue and cancers are rare. The related protein CARD14 recently was identified as being expressed in the basal layer of normal skin, which subsequently is up-regulated and altered in distribution in involved skin from patients with psoriasis and pityriasis rubra pilaris.25,26 Therefore, to ascertain whether CARD11 was present in human skin and cSCC we analyzed expression in vivo using immunohistochemistry and immunofluorescence. A CARD11-specific antibody identified expression in cSCC, with strong positive foci throughout all samples (n = 38) (Figure 3). Quantitation scoring signal intensity as absent (0), low (1), medium (2), or high (3), showed a slight but significant increase in CARD11 expression in tumors harboring CARD11 mutations (n = 9) compared with tumors in which CARD11 mutations were not detected (n = 9) (Figure 3A). CARD11 expression in cSCC was not uniform, rather the strong positive staining was greatest in areas with increased keratinocyte differentiation and in some larger tumors the expression was very focal (Figure 3, E and F), indicating the possibility of a selective signaling switch in response to pressures such as reduced vascularization. Variation in staining intensity and positive foci within individual cells was reminiscent of the signalosome aggregation observed in transfected 293 cells (Figures 2C and 3, B–I, and Supplemental Figure S2). Immunofluorescence using breast and foreskin epidermis (n = 5) showed that CARD11 expression was detected throughout normal epidermis at substantially lower levels compared with cSCC (Figure 4A).
Figure 3.

Caspase recruitment domain (CARD)11 is expressed in cutaneous squamous cell carcinoma (cSCC) and is increased in tumors harboring the CARD11 mutation. A: Quantitation of immunohistochemistry in 38 separate cSCC samples using ab113409 antibody (Abcam) raised against recombinant human CARD11 using a scale of 0 (absent) to 3 (high expression). The CARD11 mutation status was determined in 18 of the 38 samples: nine were mutated and nine were not mutated. The mutation status was unknown in the remaining 20 samples. B–I: cSCC (n = 38) showed foci of high CARD11 expression often associated with central regions of larger tumors (E and F). E and F as well as H and I, respectively, show high and low magnification of the same field. Human cerebellum was used as the positive control tissue for CARD11 (not shown). Negative control reactions were performed on duplicate tissue sections by omitting the primary antibody incubation step (not shown). ∗P < 0.05, two-sample t-test comparison; all other pairwise comparisons were not significant.
Figure 4.

Caspase recruitment domain (CARD)11 is expressed in human skin and increased levels in cutaneous squamous cell carcinoma (cSCC) are associated with nuclear P65. A: Immunofluorescence using a polyclonal antibody raised against recombinant human CARD11 detects low levels of expression in normal human skin compared with cSCC tumors. Control indicates the omission of the primary antibody in normal human skin, DAPI highlights nuclei. B: Raw signal intensities from bead-array experiments performed previously34 were searched for CARD11 and CARD14 expression in vitro (left graph) and in vivo (right graph). Each graph (1 × CARD11, 3 × CARD14) represents a separate probe on the array. In each case expression of CARD11, but not CARD14, was increased in cSCC (n = 5 in vitro, n = 9 in vivo) compared with NHK or normal skin (n = 5). C: Dual immunofluorescence with polyclonal anti-CARD11 and monoclonal anti-active P65 antibodies detects nuclear P65 in cSCC with high CARD11 expression. Scale bars: 100 um (A and C). ∗∗P < 0.01, two-sample t-test comparison. NHK, normal human keratinocytes (n = 4).
CARD11 mRNA Expression Is Increased in cSCC Compared with Normal Skin
Consistent with our immunofluorescence data, analysis of our previous microarray data34 showed that CARD11 mRNA expression was increased in primary cultured cSCC keratinocytes compared with normal primary keratinocytes and significantly increased in cSCC tissue compared with normal skin (P = 0.0067). CARD14 expression was comparable with CARD11 in vitro and in vivo but was not altered in cSCC (Figure 4B).
Nuclear P65 Is Associated with High CARD11 Expression in cSCC
To correlate CARD11 expression with NF-κB activation in vivo, we co-incubated sections from cSCC (n = 8) with antibodies raised against CARD11 and nuclear P65. Specific nuclear P65 expression was evident only in regions of tumors with high levels of CARD11 expression (Figure 4C), indicating that in tumors with high levels of CARD11, NF-κB activation is evident.
Discussion
The findings described here point to a role for CARD11 in human cSCC development. Consistent with a role for NF-κB signaling in promoting cSCC,35,36 CARD11 cSCC variants increased NF-κB activation in the absence of exogenous cytokine stimulation (Figure 2). The mechanism for NF-κB activation by CARD11 variants in the CARD and coiled-coil domain has been described.29,31 For those cSCC variants within the C-terminal, membrane-associated GUK region that lead to enhanced NF-κB activity, a likely mechanism is a negative regulatory function in keeping with the importance of the SH3 and GUK domains for CARD11-mediated NF-κB signaling.37 SH3 and GUK domains likely regulate activity by controlling subcellular localization and crucial protein–protein interactions.38,39 Furthermore, enhanced activation has been reported upon loss of MAGUK domains40 raising the possibility that cSCC-specific variants may increase activity by abrogating this inhibitory action. Exactly how the mutations we identify here affect CARD11 function and impact NF-κB signaling in the epidermis currently is unclear, but because downstream activation is comparable with oncogenic coiled-coil domain mutants (also identified in cSCC), it suggests that CARD11-mediated NF-κB signaling is regulated at multiple levels in human cSCC.
The parallel between CARD11 mutation in cSCC and CARD14 mutation in psoriasis is intriguing given the similarities, and the differences, in these two diseases. Each show increased proliferation, inflammation, and delayed differentiation, however, cSCC is malignant and psoriasis benign. We and others have exploited these properties to identify gene expression changes specific to cSCC,34,41 and it is tempting to speculate that at least some of the differences between these two diseases may be mediated through differential CARD signal transduction. It is important to note that CARD scaffold proteins can signal through pathways other than NF-κB, such as Jun N-terminal kinase.42,43 The identification of cSCC mutations in the CARD and GUK domains that abolish CARD11-mediated NF-κB activation yet maintain signalosome formation may be indicative of differential signaling. Similar variants in CARD14, which reduced NF-κB activation in 293 cells in vitro, also were identified in psoriasis patients,24 therefore further work will be needed to determine whether CARD11 and CARD14 do indeed mediate differential signaling in keratinocytes. Another possibility is that cSCC variants that abolish NF-κB activation reflect the published context-dependent role of NF-κB signaling in keratinocytes10,11,44 and, given the correct context, reduction of NF-κB signaling through CARD11 mutation may be tumor promoting. Those CARD11 variants that abolish NF-κB activation may result from disruption of essential protein–protein interactions such as association with BCL1040,43 and PDK1,39 both of which are crucial for pathway activation.
Although sharing a high degree of homology, CARD10, CARD11, and CARD14 are reported to have different tissue expression patterns, suggesting that they mediate different signaling pathways through differential expression. An analysis of gene expression data sets indicated significant overlap; CARD10 is expressed in a broad range of tissues, and CARD11, although expressed most highly in the hematopoietic system, is co-expressed with CARD14 in mucosal tissue such as tonsil and tongue papilla.28 Skin was not included in this analysis, and it now is clear from our work and that of others that both CARD14 and CARD11 are expressed in the epidermis25 (Figure 4A). The functional significance of CARD11 expression in the skin is shown by severe dermatitis, erythematous eruptions, and hyperkeratosis, which develop in unmodulated mice that carry an inactivating point mutation in CARD11.45 Thus, in mice, CARD14 expression in the skin is unable to compensate for CARD11 inactivation.
Our analysis of nuclear P65 in a small subset of tumor samples shows that in regions of cSCC with high CARD11 expression, NF-κB activation is apparent (Figure 4C). This was observed regardless of CARD11 mutation status, suggesting that CARD11 expression, rather than mutation, dictates NF-κB activation. However, semiquantitative analysis of CARD11 expression levels also identified a small, but significant, increase in CARD11 in mutated tumors compared with tumors in which no CARD11 mutations were detected (Figure 3A), suggesting that CARD11 mutation in cSCC increases NF-κB activation, pointing toward a protumorigenic role. In support of a driver role for CARD11 we note that a recent study using targeted sequencing of 29 cSCC lymph node metastasis also highlighted mutations in CARD11.46 Such a role, however, is not supported by the even distribution of mutations observed in cSCC when compared with DLBCL (Figure 1), and future functional experiments are necessary to determine whether the CARD11 mutation truly is contributing to cSCC development or is a consequence of a high mutation rate in this tumor type. The observation of high levels of CARD11 expression in specific regions of the tumor does raise the possibility that spatiotemporal regulation of NF-κB occurs through the localized expression of CARD11, mutant or wild-type. Indeed, in psoriasis and pityriasis rubra pilaris, mutant CARD14 expression increases and relocates, presumably in response to disease pathology. Further work to determine whether gain-of-function or loss-of-function CARD11 mutants provide a context-dependent selective advantage for tumor cells in human cSCC pathogenesis is ongoing. Therapies targeting the signaling complex, such as MALT1 inhibitors,46,47 may well provide an opportunity for targeting progressive disease.
In conclusion, we present data that identify high-frequency somatic mutations in CARD11 as a feature of human cSCC and show that novel mutations in regions of CARD11 external to the coiled-coil domain result in aberrant NF-κB signaling with potential consequences for disease pathogenesis. We show that CARD11 is expressed throughout normal skin and is up-regulated significantly in cSCC, in which high levels of CARD11 correlate with nuclear P65 and therefore NF-κB activation. Together, our data increase our understanding of human cSCC and highlight potentially novel targets for therapeutic development.
Footnotes
Supported by Cancer Research UK programme grant A13044 (I.M.L., C.A.H., C.M.P., A.P.S., K.J.P., S.A.W.), British Skin Foundation grant 4054 (C.P., A.P.S.), European Research Council advanced investigator award no 250170 (I.M.L., A.P.S.), and strategic grant 098439/Z/12/2 from the Wellcome Trust (I.M.L.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.05.018.
Supplemental Data
Supplemental Figure S1.
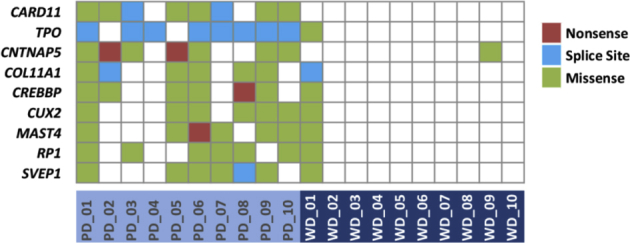
Whole-exome sequencing identifies candidate mutated genes associated with histologic grade in human cutaneous squamous cell carcinoma. Genes mutated in more than 60% of poorly differentiated (PD) tumors and less than 20% in well-differentiated (WD) tumors are shown with mutation type in individual tumor indicated by color coding.
Supplemental Figure S2.
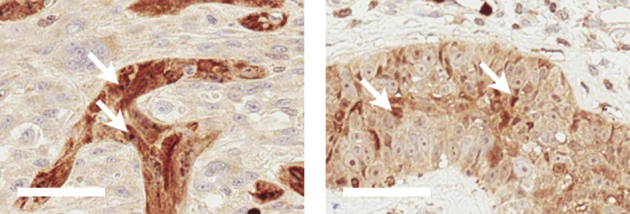
By using an antibody raised against CARD11, expression is detected throughout cutaneous squamous cell carcinoma where regions of strong immune-reactivity also show positive foci within individual tumor cells (arrows).
References
- 1.Karia P.S., Han J., Schmults C.D. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. 2013;68:957–966. doi: 10.1016/j.jaad.2012.11.037. [DOI] [PubMed] [Google Scholar]
- 2.Harwood C.A., Mesher D., McGregor J.M., Mitchell L., Leedham-Green M., Raftery M., Cerio R., Leigh I.M., Sasieni P., Proby C.M. A surveillance model for skin cancer in organ transplant recipients: a 22-year prospective study in an ethnically diverse population. Am J Transplant. 2013;13:119–129. doi: 10.1111/j.1600-6143.2012.04292.x. [DOI] [PubMed] [Google Scholar]
- 3.Cherpelis B.S., Marcusen C., Lang P.G. Prognostic factors for metastasis in squamous cell carcinoma of the skin. Dermatol Surg. 2002;28:268–273. doi: 10.1046/j.1524-4725.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- 4.Durinck S., Ho C., Wang N.J., Liao W., Jakkula L.R., Collisson E.A., Pons J., Chan S.W., Lam E.T., Chu C., Park K., Hong S.W., Hur J.S., Huh N., Neuhaus I.M., Yu S.S., Grekin R.C., Mauro T.M., Cleaver J.E., Kwok P.Y., LeBoit P.E., Getz G., Cibulskis K., Aster J.C., Huang H., Purdom E., Li J., Bolund L., Arron S.T., Gray J.W., Spellman P.T., Cho R.J. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011;1:137–143. doi: 10.1158/2159-8290.CD-11-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickering C.R., Zhou J.H., Lee J.J., Drummond J.A., Peng S.A., Saade R.E., Tsai K.Y., Curry J., Tetzlaff M.T., Lai S.Y., Yu J., Muzny D.M., Doddapaneni H., Shinbrot E., Covington K.R., Zhang J., Seth S., Caulin C., Clayman G.L., El-Naggar A.K., Gibbs R.A., Weber R.S., Myers J.N., Wheeler D.A., Frederick M.J. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582–6592. doi: 10.1158/1078-0432.CCR-14-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.South A.P., Purdie K.J., Watt S.A., Haldenby S., den Breems N.Y., Dimon M., Arron S.T., Kluk M.J., Aster J.C., McHugh A., Xue D.J., Dayal J.H., Robinson K.S., Rizvi S.M., Proby C.M., Harwood C.A., Leigh I.M. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014;134:2630–2638. doi: 10.1038/jid.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jayaraman S.S., Rayhan D.J., Hazany S., Kolodney M.S. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol. 2014;134:213–220. doi: 10.1038/jid.2013.276. [DOI] [PubMed] [Google Scholar]
- 8.Huang P.Y., Balmain A. Modeling cutaneous squamous carcinoma development in the mouse. Cold Spring Harb Perspect Med. 2014;4:a013623. doi: 10.1101/cshperspect.a013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duffey D.C., Chen Z., Dong G., Ondrey F.G., Wolf J.S., Brown K., Siebenlist U., Van Waes C. Expression of a dominant-negative mutant inhibitor-kappaBalpha of nuclear factor-kappaB in human head and neck squamous cell carcinoma inhibits survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res. 1999;59:3468–3474. [PubMed] [Google Scholar]
- 10.Loercher A., Lee T.L., Ricker J.L., Howard A., Geoghegen J., Chen Z., Sunwoo J.B., Sitcheran R., Chuang E.Y., Mitchell J.B., Baldwin A.S., Jr., Van Waes C. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64:6511–6523. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- 11.Dajee M., Lazarov M., Zhang J.Y., Cai T., Green C.L., Russell A.J., Marinkovich M.P., Tao S., Lin Q., Kubo Y., Khavari P.A. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 12.van Hogerlinden M., Rozell B.L., Ahrlund-Richter L., Toftgard R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59:3299–3303. [PubMed] [Google Scholar]
- 13.Hayden M.S., Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zandi E., Rothwarf D.M., Delhase M., Hayakawa M., Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 15.Rothwarf D.M., Zandi E., Natoli G., Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 16.Hacker H., Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 17.Pasparakis M. Role of NF-kappaB in epithelial biology. Immunol Rev. 2012;246:346–358. doi: 10.1111/j.1600-065X.2012.01109.x. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman C.K., Fuchs E. It's got you covered. NF-kappaB in the epidermis. J Cell Biol. 2000;149:999–1004. doi: 10.1083/jcb.149.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seitz C.S., Lin Q., Deng H., Khavari P.A. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci U S A. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beg A.A., Sha W.C., Bronson R.T., Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 21.Pasparakis M., Courtois G., Hafner M., Schmidt-Supprian M., Nenci A., Toksoy A., Krampert M., Goebeler M., Gillitzer R., Israel A., Krieg T., Rajewsky K., Haase I. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–866. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- 22.Omori E., Matsumoto K., Sanjo H., Sato S., Akira S., Smart R.C., Ninomiya-Tsuji J. TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. J Biol Chem. 2006;281:19610–19617. doi: 10.1074/jbc.M603384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oeckinghaus A., Hayden M.S., Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 24.Jordan C.T., Cao L., Roberson E.D., Duan S., Helms C.A., Nair R.P., Duffin K.C., Stuart P.E., Goldgar D., Hayashi G., Olfson E.H., Feng B.J., Pullinger C.R., Kane J.P., Wise C.A., Goldbach-Mansky R., Lowes M.A., Peddle L., Chandran V., Liao W., Rahman P., Krueger G.G., Gladman D., Elder J.T., Menter A., Bowcock A.M. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan C.T., Cao L., Roberson E.D., Pierson K.C., Yang C.F., Joyce C.E., Ryan C., Duan S., Helms C.A., Liu Y., Chen Y., McBride A.A., Hwu W.L., Wu J.Y., Chen Y.T., Menter A., Goldbach-Mansky R., Lowes M.A., Bowcock A.M. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012;90:784–795. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs-Telem D., Sarig O., van Steensel M.A., Isakov O., Israeli S., Nousbeck J., Richard K., Winnepenninckx V., Vernooij M., Shomron N., Uitto J., Fleckman P., Richard G., Sprecher E. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. 2012;91:163–170. doi: 10.1016/j.ajhg.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang C., Lin X. Regulation of NF-kappaB by the CARD proteins. Immunol Rev. 2012;246:141–153. doi: 10.1111/j.1600-065X.2012.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blonska M., Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011;21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong G., Chanudet E., Zeng N., Appert A., Chen Y.W., Au W.Y., Hamoudi R.A., Watkins A.J., Ye H., Liu H., Gao Z., Chuang S.S., Srivastava G., Du M.Q. A20, ABIN-1/2, and CARD11 mutations and their prognostic value in gastrointestinal diffuse large B-cell lymphoma. Clin Cancer Res. 2011;17:1440–1451. doi: 10.1158/1078-0432.CCR-10-1859. [DOI] [PubMed] [Google Scholar]
- 30.Chan W., Schaffer T.B., Pomerantz J.L. A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol Cell Biol. 2013;33:429–443. doi: 10.1128/MCB.00850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenz G., Davis R.E., Ngo V.N., Lam L., George T.C., Wright G.W., Dave S.S., Zhao H., Xu W., Rosenwald A., Ott G., Muller-Hermelink H.K., Gascoyne R.D., Connors J.M., Rimsza L.M., Campo E., Jaffe E.S., Delabie J., Smeland E.B., Fisher R.I., Chan W.C., Staudt L.M. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 32.Snow A.L., Xiao W., Stinson J.R., Lu W., Chaigne-Delalande B., Zheng L., Pittaluga S., Matthews H.F., Schmitz R., Jhavar S., Kuchen S., Kardava L., Wang W., Lamborn I.T., Jing H., Raffeld M., Moir S., Fleisher T.A., Staudt L.M., Su H.C., Lenardo M.J. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med. 2012;209:2247–2261. doi: 10.1084/jem.20120831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forbes S.A., Bhamra G., Bamford S., Dawson E., Kok C., Clements J., Menzies A., Teague J.W., Futreal P.A., Stratton M.R. The catalogue of somatic mutations in cancer (COSMIC) Curr Protoc Hum Genet. 2008;Chapter 10 doi: 10.1002/0471142905.hg1011s57. Unit 10 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watt S.A., Pourreyron C., Purdie K., Hogan C., Cole C.L., Foster N., Pratt N., Bourdon J.C., Appleyard V., Murray K., Thompson A.M., Mao X., Mein C., Bruckner-Tuderman L., Evans A., McGrath J.A., Proby C.M., Foerster J., Leigh I.M., South A.P. Integrative mRNA profiling comparing cultured primary cells with clinical samples reveals PLK1 and C20orf20 as therapeutic targets in cutaneous squamous cell carcinoma. Oncogene. 2011;30:4666–4677. doi: 10.1038/onc.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim C., Pasparakis M. Epidermal p65/NF-kappaB signalling is essential for skin carcinogenesis. EMBO Mol Med. 2014;6:970–983. doi: 10.15252/emmm.201303541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poligone B., Hayden M.S., Chen L., Pentland A.P., Jimi E., Ghosh S. A role for NF-kappaB activity in skin hyperplasia and the development of keratoacanthomata in mice. PLoS One. 2013;8:e71887. doi: 10.1371/journal.pone.0071887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCully R.R., Pomerantz J.L. The protein kinase C-responsive inhibitory domain of CARD11 functions in NF-kappaB activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol Cell Biol. 2008;28:5668–5686. doi: 10.1128/MCB.00418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D., Matsumoto R., You Y., Che T., Lin X.Y., Gaffen S.L., Lin X. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol Cell Biol. 2004;24:164–171. doi: 10.1128/MCB.24.1.164-171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee K.Y., D'Acquisto F., Hayden M.S., Shim J.H., Ghosh S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science. 2005;308:114–118. doi: 10.1126/science.1107107. [DOI] [PubMed] [Google Scholar]
- 40.Bertin J., Wang L., Guo Y., Jacobson M.D., Poyet J.L., Srinivasula S.M., Merriam S., DiStefano P.S., Alnemri E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 41.Haider A.S., Peters S.B., Kaporis H., Cardinale I., Fei J., Ott J., Blumenberg M., Bowcock A.M., Krueger J.G., Carucci J.A. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J Invest Dermatol. 2006;126:869–881. doi: 10.1038/sj.jid.5700157. [DOI] [PubMed] [Google Scholar]
- 42.Scudiero I., Vito P., Stilo R. The three CARMA sisters: so different, so similar: a portrait of the three CARMA proteins and their involvement in human disorders. J Cell Physiol. 2014;229:990–997. doi: 10.1002/jcp.24543. [DOI] [PubMed] [Google Scholar]
- 43.Gaide O., Favier B., Legler D.F., Bonnet D., Brissoni B., Valitutti S., Bron C., Tschopp J., Thome M. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:836–843. doi: 10.1038/ni830. [DOI] [PubMed] [Google Scholar]
- 44.Ren Q., Kari C., Quadros M.R., Burd R., McCue P., Dicker A.P., Rodeck U. Malignant transformation of immortalized HaCaT keratinocytes through deregulated nuclear factor kappaB signaling. Cancer Res. 2006;66:5209–5215. doi: 10.1158/0008-5472.CAN-05-4158. [DOI] [PubMed] [Google Scholar]
- 45.Jun J.E., Wilson L.E., Vinuesa C.G., Lesage S., Blery M., Miosge L.A., Cook M.C., Kucharska E.M., Hara H., Penninger J.M., Domashenz H., Hong N.A., Glynne R.J., Nelms K.A., Goodnow C.C. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18:751–762. doi: 10.1016/s1074-7613(03)00141-9. [DOI] [PubMed] [Google Scholar]
- 46.Li Y.Y., Hanna G.J., Laga A.C., Haddad R.I., Lorch J.H., Hammerman P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res. 2015;21:1447–1456. doi: 10.1158/1078-0432.CCR-14-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young R.M., Staudt L.M. A new “brew” of MALT1 inhibitors. Cancer Cell. 2012;22:706–707. doi: 10.1016/j.ccr.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.