

Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) is a cancer that is characterized by its high morbidity and mortality rates. While tobacco use and alcohol consumption are two major contributing factors for HNSCC carcinogenesis, how the combination of tobacco and alcohol increases HNSCC risk is not understood.
Methods
We combined the 4-nitroquinoline-1-oxide (4-NQO) oral carcinogenesis and Meadows-Cook alcohol mouse models to elucidate the molecular events and to identify novel biomarkers associated with oral cancer development.
Results
By genome-wide RNA-seq of tongue samples (three mice per group) we identified changes in transcripts that mediate alcohol metabolism and oxidative stress (Aldh2, Aldh1a3, Adh1, Adh7, and Cyp2a5) in mice treated with 4-NQO followed by ethanol (4-NQO/EtOH) as compared to the vehicle control/untreated samples (V.C./Untr.). We measured major, global increases in specific histone acetylation and methylation epigenetic marks (H3K27ac, H3K9/14ac, H3K27me3, and H3K9me3) in the oral cavities of V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH treatment groups compared to the V.C./Untr. group. We detected changes in histone epigenetic marks near regulatory regions of genes involved in ethanol metabolism by chromatin immunoprecipitation (ChIP). For instance, the Aldh2 promoter showed increased H3K27me3 marks, and Aldh2 mRNA levels were reduced by 10-fold in 4NQO/EtOH vs. V.C./Untr. tongue samples. 4-NQO/EtOH treatment also caused increases in markers of oxidative stress, including 4-HNE, MCT4/Slc16a3, and TOM20, as measured by immunohistochemistry.
Conclusions
We delineate a mechanism by which 4-NQO and ethanol can regulate gene expression during the development of HNSCC, and suggest that histone epigenetic marks and oxidative stress markers could be novel biomarkers and targets for the prevention of HNSCC.
Keywords: head and neck cancer, epigenetics, oxidative stress, tobacco, alcohol
Introduction
Cancers of the head and neck include cancers of the pharynx, larynx, esophagus, and oral cavity, and are typically characterized by their aggressiveness and high risk of death (Hashibe et al., 2007b, Rothenberg and Ellisen, 2012). In the U.S. alone, it is estimated that over 40,000 will be diagnosed in 2014, with 8,400 of these cancers resulting in death (Siegel et al., 2014). Tobacco and alcohol consumption are the leading risk factors for oral cancer, and alcohol consumption together with tobacco use synergize to greatly increase cancer risk (Hashibe et al., 2007a, Seitz and Stickel, 2007, Bauman et al., 2012, Rothenberg and Ellisen, 2012). While the risk of developing head and neck squamous cell carcinoma (HNSCC) compared to never users is 7 times higher in only smokers and 3 times higher in only drinkers, the risk of developing HNSCC is 38 times higher in chronic users of both tobacco and alcohol (Morita et al., 2010, Seitz and Stickel, 2007). However, the molecular mechanisms by which ethanol and tobacco together promote HNSCC are not completely understood.
Carcinogens found within cigarette smoke, such as nitrosamines and polycyclic aromatic hydrocarbons (PAHs), can promote HNSCC through gene mutations and chromosomal aberrations resulting from the formation of DNA and protein adducts (Seitz and Stickel, 2007, Chen et al., 2011). While alcohol contributes to these effects, other potential mechanisms by which alcohol may promote tumorigenesis include inhibition of metabolic pathways involved in cancer prevention; the carcinogenic effects of its metabolite acetaldehyde (AcH); and the induction of aberrant epigenetic changes (Seitz and Stickel, 2007).
The disruption of cellular respiration through mitochondrial dysfunction is a major factor for many types of cancers, and epidemiological studies have found that human HNSCC patients experience increases in oxidative mitochondrial metabolism and oxidative stress. Mitochondrial dysfunction causes the electron transport chain to generate free radicals and reactive oxidative species (ROS) as by-products, which leads to DNA and protein damage (Choudhari et al., 2014). Both tobacco and alcohol use can alter the oxidant/antioxidant balance in the oral mucosa (Seitz and Stickel, 2007, Choudhari et al., 2014), implicating both tobacco and alcohol in ROS production during the development of HNSCC.
Ethanol metabolism is a multi-step process in which ethanol is oxidized to the carcinogen AcH by the alcohol dehydrogenase (ADH) family of enzymes (ADH1, ADH3, ADH4), or cytochrome P450 2E1 (CYP2E1) (Fig. 2(a)) (Black and Vasiliou, 2009). AcH is then converted to acetate by the aldehyde dehydrogenase (ALDH) family of enzymes (ALDH1, ALDH2, ALDH3) (Black and Vasiliou, 2009). Genetic mutations that lead to increased ADH1A, ADH1B, and/or CYP2E1 activity, or decreased ALDH2 enzyme activity increase cancer susceptibility, since these different mutations result in increased levels of AcH after alcohol consumption (Seitz and Stickel, 2006, Black and Vasiliou, 2009). AcH can exert damaging effects by inducing point mutations, sister chromatid exchanges, chromosomal aberrations, and the formation of protein and DNA adducts (Seitz and Stickel, 2007).
Fig. 2.
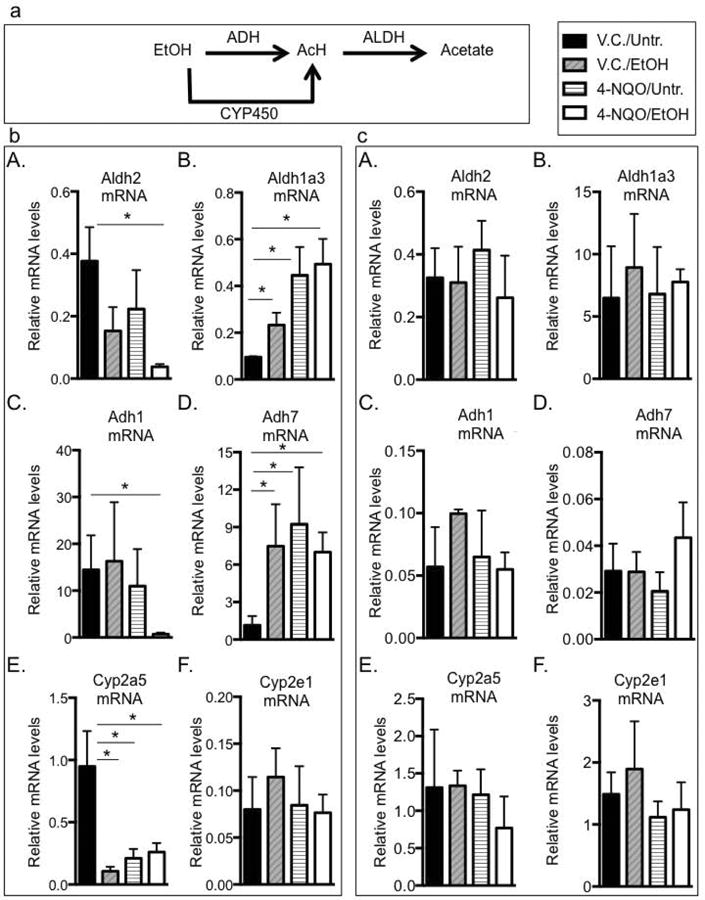
Ethanol and 4-NQO change transcript levels of genes associated with ethanol metabolism. (a) Schematic representation of ethanol metabolism. Ethanol (EtOH) is metabolized to acetaldehyde (AcH) primarily by the alcohol dehydrogenase (ADH) family of enzymes. Cytochrome P450 (Cyp P450) enzymes can also metabolize ethanol to AcH. AcH is metabolized to acetate by aldehyde dehydrogenases (ALDH). ((b) A-F) Validation of RNA-seq analysis, where reverse transcribed RNA isolated from tongues was subjected to qRT-PCR analysis to measure mRNA levels compared to Gapdh (control) mRNA levels, n=8. ((c) A-F) Reverse transcribed RNAs isolated from kidneys were subjected to qRT-PCR analysis to determine mRNA levels compared to Gapdh (control) mRNA levels. (A) Aldh2 (Gene ID 11669), (B) Aldh1a3 (Gene ID 56847), (C) Adh1 (Gene ID 11522), (D) Adh7 (Gene ID 11529), (E) Cyp2a5 (Gene ID 13087), (F) Cyp2e1 (Gene ID 13106). One-way ANOVA followed by Tukey's post hoc analysis, n=3, *p<0.05 between all treatment groups and the V.C./Untr. samples.
Another mechanism by which ethanol may promote HNSCC is through induction of epigenetic changes. Histone tails are subjected to a multitude of post-translational modifications that can mark a gene promoter as being in an active or inactive state (Shukla et al., 2008). Acetylation at histone 3 lysine residues 9, 14, and 27, (H3K9/14ac or H3K27ac) and mono-methylation at histone 3 lysine 4 (H3K4me1) are typically associated with increased gene expression (Shukla et al., 2008, Urvalek et al., 2014). Conversely, tri-methylation at lysine 27 or 9 of histone 3 (H3K27me3 and H3K9me3) is associated with epigenetic silencing (Shukla et al., 2008, Urvalek and Gudas, 2014). Changes in the epigenome can predict the prognosis of different cancers (Chervona and Costa, 2012), and alcohol induces epigenetic changes in breast, lung, and colon cancers (Seitz and Becker, 2007, Seitz and Stickel, 2007, Chervona and Costa, 2012). However, few studies have examined the role of alcohol-induced epigenetic changes in oral cancers, nor have histone modifications been examined as potential biomarkers in HNSCC.
Head and neck cancers are typically diagnosed at later stages, at least in part explaining the poor survival rate of oral cancer patients (approximately 50%) (Hashibe et al., 2007a). Despite improved treatments, the overall survival rate has not significantly improved over recent decades (Bauman et al., 2012). As a result, it is important to identify preventative measures and early detection techniques in order to prevent oral cancer related deaths. Our laboratory has developed a mouse model that combines the 4-NQO oral carcinogenesis and the Meadows-Cook alcohol models to delineate the mechanisms by which ethanol promotes oral carcinogenesis (Osei-Sarfo et al., 2013, Tang et al., 2004). This model is powerful since it mimics many aspects of human behavior in which tobacco and alcohol are used together (Squier and Kremer, 2001). Here we show that ethanol together with 4-NQO cause changes in gene expression that can alter ethanol metabolism and induce oxidative stress. We show that one mechanism by which ethanol induces changes in gene expression is by regulating epigenetic histone marks near the transcriptional start sites of these genes, which can lead to oxidative stress.
Materials and Methods
RNA-seq Analysis of the mRNA Transcriptome
RNA-seq was performed as previously described (Tang et al., 2014) and in Supplemental Materials. n=3 for each treatment group. The Gene Expression Omnibus accession number is GSE62125. Gene Ontology (GO) analysis was performed using the QFAB website (http://www.qfab.org/next-generation-sequencing).
Animals and Treatments
Animals used and treatments were performed as previously described (Osei-Sarfo et al., 2013). Briefly, eight-week female C57BL/6 wild type mice (Jackson Laboratory, Bar Harbor, ME) were randomized and divided into four groups, 15 mice per group. Control mice were treated with propylene glycol (vehicle control, V.C.) in their drinking water for 10 weeks, followed by a 15-week period with normal water (V.C./Untreated). Two groups were chronically treated with 100 μg/ml 4-NQO (Cat# N8141; Sigma, St Louis, MO) in their drinking water for 10 weeks. After this period these mice were treated with normal water (4-NQO/Untr.), or 20% ethanol (EtOH) (4-NQO/EtOH) for 15 weeks. A fourth group was treated with 20% EtOH for 15 weeks (V.C./EtOH). Mice were chronically exposed to ethanol, and blood alcohol levels were measured throughout the study (Supplemental Fig. 4). The care and use of animals in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of WCMC.
Immunohistochemistry (IHC)
Experiments were performed as previously described (Osei-Sarfo et al., 2013, Tang et al., 2014). Sections were incubated with primary antibodies overnight at 4 °C (1:200 TOM20 (sc-11415, Santa Cruz, Santa Cruz, CA), 1:200 4-HNE (Ab48506, Abcam, Cambridge, MA), 1:100 MCT4 (sc-50329, Santa Cruz), 1:400 H3K27ac (Ab4729, Abcam), 1:1,000 H3K9/14ac (06-599, Millipore, Billerica, MA), 1:400 H3K27me3 (07-449, Millipore), 1:1,000 H3K9me3 (07-473, Millipore), 1:1,000 H3K4me1 (Ab8895, Abcam)). The antibody signal was visualized by peroxidase reaction using 3,3′-diaminobenzidine. Methyl green was used as a nuclear counterstain. At least three mice were used for each treatment group, and for each sample at least four non-contiguous regions were photographed and analyzed. Image J (http://imagej.nih.gov) software was used to measure positively stained tongue epithelial area compared to total epithelial area. Ratios were calculated and converted to percent positive area.
mRNA Analysis
qPCR was performed as previously described (Urvalek and Gudas, 2014). Primers were designed for specific genes (Table 1). Samples were normalized to a GAPDH internal control.
Table 1. Primer sequences used for qPCR and predicted product size. (bp, base pair).
| Target | Forward (5′-3′) | Reverse (3′-5′) | Band Size (bp) |
|---|---|---|---|
| Aldh2 | TTCAACGCTGCAGTCGCCCTCC | TTCCTGCTGACGGCGTCGTGCC | 269 |
| Aldh1a3 | AAGACTCGTCAGACCCAGG | ATG ACG ATC TTT CCT ACA GCC | 326 |
| Adh1 | GACATAGAAGTCGCACCCCC | AACCGAAACACAAGCCCTGA | 219 |
| Adh7 | CCTATGACCCAATGCTGCTCT | CCCAGGTCTCTGGAACTCAAA | 237 |
| Cyp2e1 | TTCCGAGGATATGTCATCCCCAAGG | TTCTCCAACACACACGCGCTTTCC | 189 |
| Cyp2a5 | TAGCTCAATTGTCTTCGGGGAC | AAG TCC TCC AGG CCC TGC | 195 |
| Aldh1a3 TSS | TCACTCCCGCCGAAGTTTTG | ATTCCCAAGGGTCGGACATC | 151 |
| Aldh2 TSS | ATGAATTCTCTTCGCCGCCA | GCTCCCAAAGCCAAGTCAGA | 133 |
| Adh1 TSS | TACACACCCCATGGACTGGA | ACGTGATCTGGCCCTTTACG | 275 |
| Adh7 TSS | AGCTTGCACGTAGAAGGCAT | TGAGGCACGGTTCACTCTTC | 342 |
| Cyp2e1 TSS | ATTTGACAACGTGAAATGGGATCC | TACTGATCTGACAAGGATGAGGC | 394 |
| Cyp2a5 TSS | TAGCTCAATTGTCTTCGGGGAC | AAGTCCTCCAGGCCCTGC | 195 |
Chromatin Immunoprecipitation
25 mg of snap frozen mouse tongue tissue was minced and incubated in PBS + protease inhibitors with 37% formaldehyde for 20 min at room temperature. Cross-linking was quenched using 0.5 M glycine for 5 min. Samples were centrifuged at 1,500 rpm for 5 min, washed with PBS, and replaced with 1 ml PBS + protease inhibitors. Tissues were homogenized to a single cell solution using a Dounce homogenizer. Lysates were centrifuged, supernatants were removed, and pellets were resuspended in 1 mL RIPA buffer supplemented with DTT. Samples were incubated on ice for 10 min, followed by incubation with 7.5 μl micrococcal nuclease for 20 min at 37° C with frequent mixing. Reactions were neutralized using 0.5 M EDTA, and after centrifugation the pellets were resuspended in 100 μl RIPA buffer + protease inhibitors. Lysates were sonicated 5 times for 15 seconds each. Immunoprecipitation and qPCR was performed as previously described (Urvalek and Gudas, 2014).
Statistical Analysis
Statistical analysis was performed on at least three samples from each treatment group (n=3 or >3) using the Graph Pad Prism 6.0 software. ANOVA followed by Tukey's post hoc analysis was used when comparing three or more groups. Student's t-test was used to determine statistically significant changes between two independent populations, where p<0.05 was considered statistically significant. Graphs represent the means, where error bars depict ± S.E.
Results
RNA-seq analysis reveals major changes in transcript levels after 4-NQO and ethanol exposure
All oral tissue is covered by a stratified epithelium that shows a similar pattern of differentiation (Squier and Kremer, 2001). Tongue tissue also has this epithelial structure, and therefore is representative of most oral tissue. 4-NQO can induce mouse oral cavity cancers that are similar to human HNSCC by causing DNA adduct formation, increased reactive oxidative species, and changes in expression of genes associated with human tumorigenesis (Rubin et al., 1995, Serewko et al., 2002, Tang et al., 2004). To determine how the combination of 4-NQO and ethanol promotes oral carcinogenesis we performed RNA-seq analysis on the tongues of control mice (V.C./Untr.), mice exposed to ethanol (V.C./EtOH), to 4-NQO (4-NQO/Untr.), or to 4-NQO followed by ethanol (4-NQO/EtOH).
We observed that ethanol, 4-NQO, and 4-NQO/EtOH caused major changes (defined as at least a 2-fold change) in transcript levels when compared to the V.C./Untr. mice (Fig. 1A&B). Ethanol alone induced increases in transcript levels of 312 genes and decreases in 183 transcripts, while 4-NQO caused increases in 1087 mRNAs and decreases in 720 mRNAs (Fig. 1C&D). Interestingly, we discovered that 4-NQO followed by ethanol caused the greatest number of changes in transcripts, with 1,666 transcripts increased and 1,939 transcripts decreased compared to the V.C./Untr. group (Fig. 1C&D). We also found 626 transcripts that increased, and 1,158 transcripts that decreased in the 4-NQO/EtOH compared to 4-NQO/Untr. group (Fig. 1E&F).
Fig. 1.
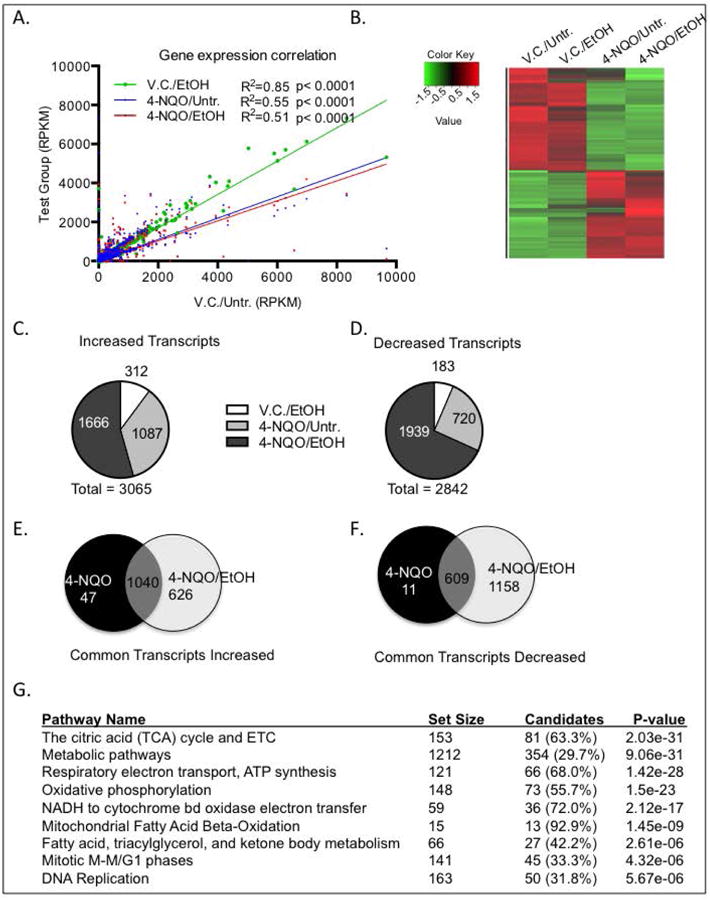
Ethanol and 4-NQO alter transcript levels in the tongues on a genome-wide scale. RNA was isolated from the tongues of the four different treatment groups and subjected to RNA-seq. (A) Gene expression correlation of total number of transcript levels (RPKM, reads per kilobase of transcript per million reads mapped) with at least a 2-fold change in in treatment groups (y-axis) compared to V.C./Untr. group (x-axis). Pearson correlation where p<0.05, R2=goodness of fit. (B) Heat map depicting genome-wide changes in transcript levels among all groups. Color scale is log2, red depicting increases and green depicting decreases. p<0.05 with at least a 2-fold change. (C & D) Pie charts depicting the numbers of transcripts increased (C), or (D) decreased compared to the V.C./Untr. group. (E & F) Venn diagrams depicting increased (E) or decreased (F) transcripts that are in common or unique to either 4-NQO/Untr. or 4-NQO/EtOH compared to the V.C./Untr. group. (G) Gene ontology analysis showing the top ten signaling pathways with the lowest p-values in the 4-NQO/EtOH treatment group that are changed compared to the V.C./Untr. group.
Using gene ontology (GO) analysis we showed that many genes in the major signaling pathways associated with cellular respiration were changed in the 4-NQO/EtOH group compared to V.C./Untr. (Fig. 1G). This is significant since ROS production mainly occurs during mitochondrial electron transport (Choudhari et al., 2014). Many of these genes involved in cellular respiration are also associated with alcohol and carcinogen metabolism (Fig. 2(a) and Supplementary Fig. 1). For example, RNA-seq revealed a 1.6-fold decrease in Aldh2 (Gene ID 11669) transcripts (the gene that is primarily responsible for metabolizing AcH to acetate) in 4-NQO/EtOH-treated mice compared to V.C./Untr. Other transcripts that were changed in the 4-NQO/EtOH treatment group compared to V.C./Untr. and that play a role in both cellular respiration and ethanol and/or carcinogen metabolism include Adh1 (gene ID 11522), Adh7 (gene ID 11529), Aldh1a3 (gene ID 56847), Cyp2e1 (gene ID 13106), and Cyp2a5 (gene ID 13087). A summary of the major functions of these genes is shown in Table 2.
Table 2. Enzymes examined that are associated with ethanol metabolism and their function.
| Gene | Function | Refs |
|---|---|---|
| Aldh2 | Metabolizes AcH to acetate | Seitz & Becker, 2007; Black et al, 2009; Singh et al., 2013 |
| Aldh1a3 | Metabolizes retinaldehyde to retinoic acid | Seitz & Becker, 2007; Black et al, 2009; Singh et al., 2013 |
| Adh1 | Metabolizes EtOH to AcH | Seitz & Becker, 2007; Jelski et al, 2006 |
| Adh7 | Metabolizes EtOH to AcH | Seitz & Becker, 2007; Jelski et al, 2006 |
| Cyp2e1 | Metabolizes xenobiotics (EtOH to AcH) | Seitz & Becker, 2007; Lu et al., 2011 |
| Cyp2a5 | Metabolizes drugs and toxins (i.e. nicotine) (Cyp2a6 is human orthologue) | Seitz & Becker, 2007; Lu et al, 2011; Xu et al., 2002 |
Transcript levels of genes involved in ethanol metabolism and ROS production are changed after 4-NQO and ethanol exposure
To validate our RNA-seq results we performed qPCR on a larger number of samples. We found that changes in mRNAs by qPCR were consistent with our RNA-seq samples. We observed changes in transcript levels in the 4-NQO/EtOH treatment group compared to V.C./Untr. (Fig. 2(b)A-E), with the exception of the Cyp2e1 transcript, which did not show statistically significant changes (Fig. 2(b)F). By qPCR we detected a major, 10-fold decrease in Aldh2 transcripts in the 4-NQO/EtOH vs. the V.C./Untr. group (Fig. 2(b)A). To determine if the results we observed are specific to the oral cavity, we performed qPCR on the kidneys of the same samples and showed that treatment with 4-NQO and ethanol had no effect on transcript levels of these genes in the kidney (Fig. 2(c)A-F).
4-NQO and ethanol induce epigenetic changes associated with changes in gene expression
Histone tails are subjected to many different posttranslational modifications that can alter gene expression (Hon et al., 2009, Urvalek et al., 2014), and aberrant epigenetic modifications are found in both early and late stages of carcinogenesis (Chen et al., 2011). In hepatocytes ethanol can affect these epigenetic changes, potentially by regulating the availability of substrates for epigenetic modifications, including acetyl-CoA (for acetylation) and S-andenosyl-methionine (for methylation) (Shukla et al., 2008, Seitz and Stickel, 2007, Seitz and Becker, 2007). Whether ethanol and/or 4-NQO can alter epigenetic histone modifications in the oral cavity is not well studied. Using immunohistochemistry (IHC) we determined that the levels of the transcriptional permissive H3K27ac mark increased 1.4 ± 0.2 fold in the V.C./EtOH and 2.5 ± 0.4 fold in the 4-NQO/EtOH group, but we detected no changes in the levels of the H3K27ac mark in the 4-NQO/Untr. compared to the V.C./Untr. group (Fig. 3A) (Hon et al., 2009). Additionally, H3K27ac levels increased 1.5 ± 0.1 fold in the 4-NQO/EtOH compared to V.C./EtOH group, and 1.8 ± 0.1 fold in the 4-NQO/EtOH compared to the 4-NQO/Untr. group (Fig. 3A). These results indicate that 4-NQO followed by ethanol has additive effects on the deposition of the H3K27ac histone mark.
Fig. 3.
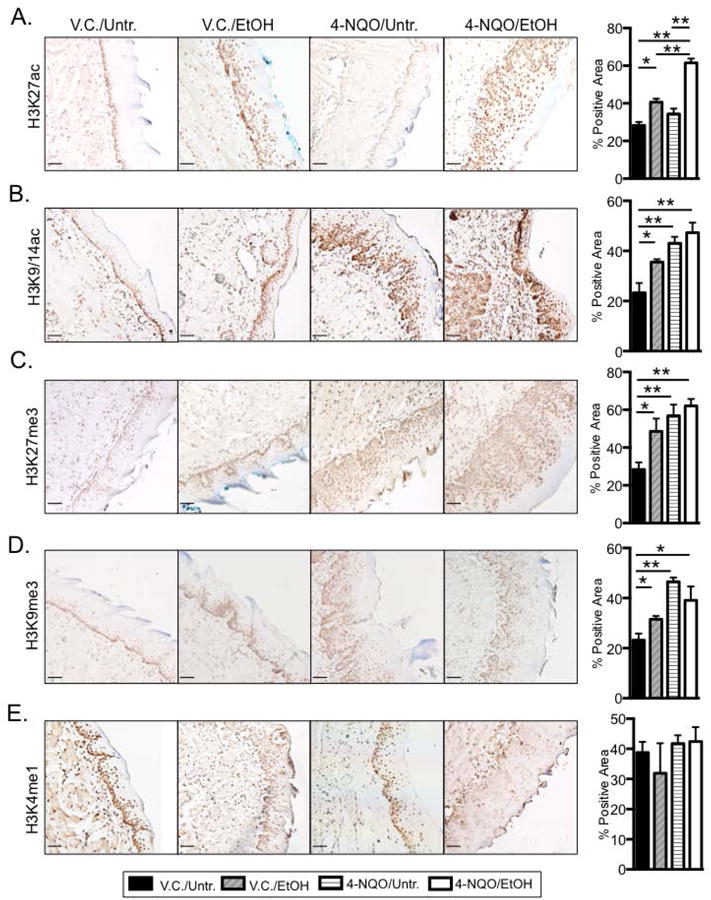
Ethanol and 4-NQO induce global changes in histone acetylation and methylation in the tongue epithelia. Immunohistochemistry (IHC) using antibodies specific for (A) H3K27ac, (B) H3K9/14ac, (C) H3K27me3, (D) H3K9me3, or (E) H3K4me1. Shown are representative images (200× magnification; scale bar, 50 μm) from at least three individual mice from each treatment group, and quantitation of percent area with positive staining for the indicated antibody. Brown color indicates positively stained cells. (Blue trapping of methyl green in keratinized layers). Panels at right indicate quantitation of percent positively stained cells within the entire epithelia layer. One-way ANOVA followed by Tukey's post hoc analysis, n≥3, *p<0.05, **p<0.001.
Similarly, we found that compared to the V.C./Untr. group, the H3K9/14ac transcriptionally permissive mark increased by 1.5 ± 0.3 fold in the V.C./EtOH group, 1.8 ± 0.3 fold in the 4-NQO/Untr. group, and 2.0 ± 0.3 fold in the 4-NQO/EtOH group (Fig. 3B). We interpret these data to indicate that both ethanol and 4-NQO can globally increase levels of the H3K9/14ac mark in the tongue.
We also examined general transcriptional repressive marks, including H3K27me3 and H3K9me3. We observed a 1.6 ± 0.3 fold increase in the levels of H3K27me3 in the V.C./EtOH, a 1.8 ± 0.12 fold in the 4-NQO/Untr., and a 2.0 ± 0.2 fold increase in in the 4-NQO/EtOH compared to the V.C./Untr. Samples (Fig. 3C). Similarly, we found that the H3K9me3 mark increased by 1.4 ± 0.1 fold in the V.C/EtOH, 2.0 fold ± 0.1 in the 4-NQO/Untr., and 1.7 ± 0.1 fold in the 4-NQO/EtOH groups compared to the V.C./Untr. group (Fig. 3D). These data show that ethanol and 4-NQO can also increase levels of the H3K27me3 and H3K9me3 marks.
The H3K4me1 mark is found at poised transcriptional enhancer regions, and interestingly (Hon et al., 2009), we found no significant changes in the H3K4me1 mark in the V.C./EtOH, 4-NQO/Untr., and 4-NQO/EtOH groups compared to the V.C./Untr. group (Fig. 3E). Overall, these findings suggest that both 4-NQO and ethanol can regulate histone acetylation and methylation at specific histone lysine residues, which likely play a role in regulating gene expression in the oral cavity. These specific epigenetic marks could be markers of early carcinogenesis.
To determine whether these changes in histone acetylation and methylation affected the deposition of these marks near the transcriptional start sites (TSSs) of genes that regulate ethanol metabolism and oxidative stress, we next performed chromatin immunoprecipitation (ChIP) on tongue tissues. Near the Aldh2 TSS we measured 2.8 ± 0.1 and 2.6 ± 0.6 fold increases in the levels of the H3K27me3 mark in the 4-NQO/Untr. and 4-NQO/EtOH treated tongue samples, respectively, compared to the V.C./Untr. samples (Fig. 4A). This is consistent with our finding that Aldh2 transcript levels are lower in the 4-NQO/Untr. and 4-NQO/EtOH compared to V.C./Untr. samples (Fig. 2(b)A). In contrast, we did not detect changes in H3K27ac levels near the Aldh2 TSS (Fig. 4A).
Fig. 4.
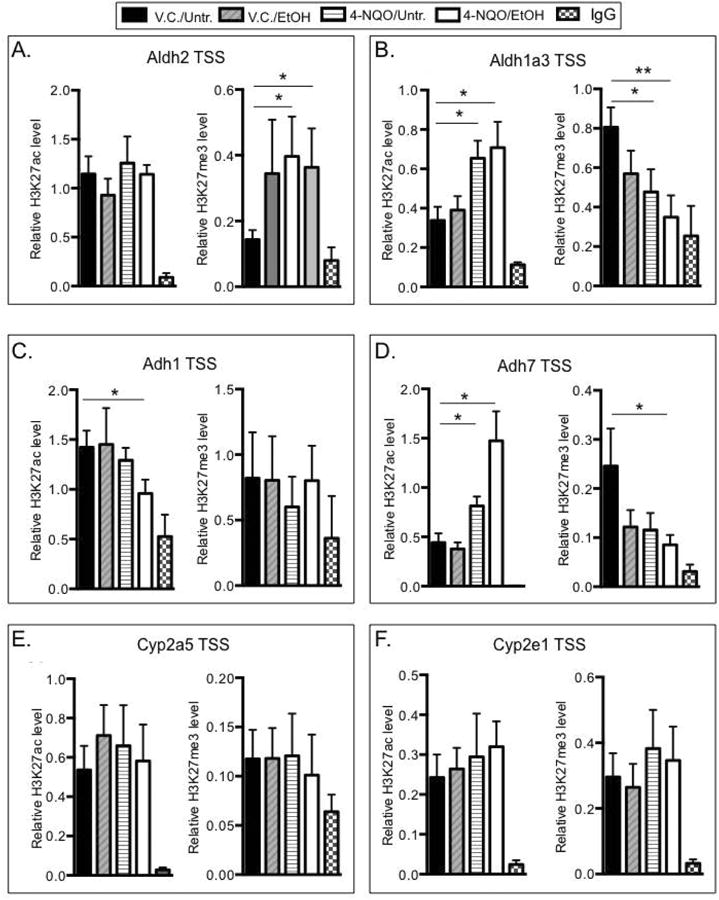
Ethanol and 4-NQO induce changes in histone acetylation and methylation at transcriptional start sites of some genes involved in ethanol metabolism. Mouse tongue tissue was homogenized, and pre-cleared lysates (15 μg of DNA) were immunoprecipitated (IP'ed) using 0.5 μg of antibodies specific for H3K27ac, H3K27me3, or IgG (negative control). Purified DNA was used for qPCR analysis using primers specific for transcriptional start site (TSS) regions (Table 1) of the (A) Aldh2, (B) Aldh1a3, (C) Adh1, (D) Adh7, (E) Cyp2a5, or (F), Cyp2e1 genes. Binding is expressed relative to the pre-IP input DNA. Error bars represent standard errors of independent mouse tongue samples where n≥5. One-way ANOVA followed by Tukey's post hoc analysis, *p<0.05, **p<0001.
Near the Aldh1a3 TSS we detected a 1.9 ± 0.4 fold increase in the H3K27ac mark in the 4-NQO/Untr. and a 2.1 ± 1.1 fold increase in the 4-NQO/EtOH group relative to the V.C./Untr group. Conversely, compared to the V.C./Untr. group we detected 1.7 ± 0.3 and 2.3 ± 0.1 fold decreases in the H3K27me3 mark in the 4-NQO/Untr. and 4-NQO/EtOH groups, respectively (Fig. 4B). This again is consistent with the increased Aldh1a3 transcript levels in these two groups compared to the V.C./Untr. group (Fig. 2(b)B). The Adh1 TSS region exhibited a 1.5 ± 0.2 fold decrease in the level of H3K27ac and no changes in the H3K27me3 levels in the 4-NQO/EtOH vs. the V.C./Untr. samples (Fig. 4C), consistent with the decrease in Adh1 transcripts in the 4-NQO/EtOH compared to the V.C./Untr. samples (Fig. 2(b)C). We measured 1.8 ± 0.6 and 3.3 ± 0.7 fold increases in the H3K27ac mark near the Adh7 TSS in the 4-NQO/Untr. and 4-NQO/EtOH groups, respectively, compared to the V.C./Untr. samples. We also observed a 2.8 ± 0.2 fold decrease in the H3K27me3 near the Adh7 TSS in the 4-NQO/EtOH vs. the V.C./Untr. samples (Fig. 4D). These data agree with the increased Adh7 transcript levels in these samples (Fig. 2(b)D). We did not detect changes in H3K27ac or H3K27me3 levels at the Cyp2a5 TSS (Fig. 4E), despite the decreases in Cyp2a5 transcript levels in the V.C./EtOH, 4-NQO/Untr., and 4-NQO/EtOH compared to the V.C./Untr. group (Fig. 2(b)E). We did not observe changes in H3K27ac or H3K27me3 levels at the Cyp2e1 TSS (Fig. 4F), which agrees with the lack of any change in Cyp2e1 transcript levels (Fig. 2(b)F). Overall, our results show that the deposition of the H3K27ac and H3K27me3 marks near the transcriptional start sites of the genes examined is differentially regulated after ethanol and/or 4-NQO treatment.
4-NQO and ethanol alter mitochondrial function and promote oxidative stress
To determine if 4-NQO and/or ethanol change cellular respiration and induce oxidative stress in the tongue epithelia, we examined markers of both cellular respiration and oxidative stress by immunohistochemistry. Increased expression of the TOM20 (gene ID 67952) protein correlates with increased mitochondrial mass and oxidative phosphorylation, and we measured a 1.7 ± 0.3 fold increase in TOM20 protein levels in the tongue epithelia of the V.C./EtOH compared to V.C./Untr. group (Fig. 5A&D) (Curry et al., 2013, Wurm et al., 2011). We also observed 2.3 ± 0.4 and 3.3 ± 0.3 fold increases in TOM20 in the 4-NQO/Untr. and 4-NQO/EtOH, respectively, compared to the V.C./Untr. group (Fig. 5A&D). Interestingly, compared to the V.C./EtOH and 4-NQO/Untr. groups respectively, we observed a 1.9 ± 0.6 and 1.4 ± 0.2 fold increase in TOM20 in the 4-NQO/EtOH group (Fig. 5A&D). These data suggest that increased oxidative phosphorylation and aerobic respiration occur in the tongue epithelial cells in all three treatment groups (V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH) relative to the V.C./Untr. group, and that 4-NQO/EtOH treatment increases TOM20 expression to a greater extent than ethanol or 4-NQO alone (Fig. 5D).
Fig. 5.
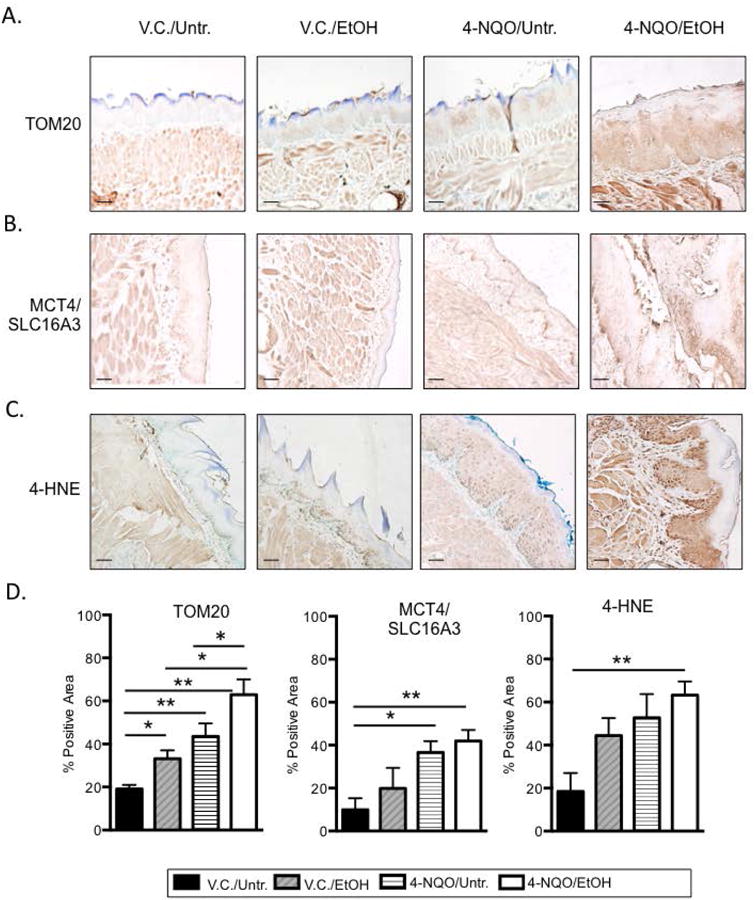
4-NQO and ethanol induce oxidative stress in the tongue epithelia. IHC using antibodies specific for (A) TOM20, (B) MCT4/SLC16A3, or (C) 4-HNE was performed. Shown are representative images (200× magnification; scale bar, 50 μm) from at least three mice from each experimental group with three images per mouse quantitated for (D). Brown represents positively stained cells. (D) Quantitation of percent area with positive protein staining for the indicated antibody. One-way ANOVA followed by Tukey's post hoc analysis, n≥3, *p<0.05, **p<0.001.
MCT4 (Slc16a3) (gene ID 80879) is a marker of oxidative stress and increased glycolysis, and high MCT4/Slc16a3 expression in non-proliferating cells correlates with a poor outcome in head and neck cancer patients (Curry et al., 2013, Ganapathy et al., 2009, Halestrap and Price, 1999). We found 3.2 ± 1.5 and 4.3 ± 1.5 fold increases in MCT4/Slc16a3 protein levels in the 4-NQO/Untr. and 4-NQO/EtOH groups, respectively, compared to the V.C./Untr. group (Fig. 5B&D). 4-Hydroxynonenal (4-HNE) is a second marker of oxidative stress and lipid peroxidation (Singh et al., 2013), and we measured a 3.0 ± 0.2 fold increase in 4-HNE levels in the 4-NQO/EtOH compared to the V.C./Untr. group (Fig. 5C&D). Thus elevations in these markers of oxidative stress could be prognostic markers and preventative treatment targets for those who use both tobacco and alcohol, and are at a high risk for developing HNSCC.
Discussion
Transcript levels of enzymes that metabolize ethanol are altered in the V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH groups
While it is clear that tobacco use together with drinking greatly increase the risk of head and neck cancer (Hashibe et al., 2007b, Seitz and Stickel, 2007), the mechanisms by which this occurs are not understood. We have previously shown that 4-NQO-induced oral carcinogenesis is associated with genome-wide changes in gene expression (Tang et al., 2013, Tang et al., 2004, Tang et al., 2009, Osei-Sarfo et al., 2013, Tang et al., 2014), and here we show that 4-NQO treatment followed by ethanol treatment (4-NQO/EtOH) altered the largest numbers of transcripts as compared to V.C./Untr., V.C./EtOH or 4-NQO/Untr. groups (Fig. 1). Importantly, we found that transcripts of genes involved in ethanol metabolism were changed, suggesting that alterations in ethanol metabolism play a major role in 4-NQO/EtOH-mediated HNSCC carcinogenesis. Furthermore, we found that only 4-NQO followed by ethanol treatment induced epigenetic changes at specific gene regulatory regions (Fig. 4) and increased the oxidative stress marker 4-HNE (Fig. 5C&D).
ALDH2 is the major enzyme that converts AcH to acetate, and decreased ALDH2 expression can lead to increased levels of the AcH carcinogen, increased DNA- and protein-adduct formation, and increased ROS levels (Seitz and Stickel, 2007, Yokoyama and Omori, 2003). Importantly, humans that possess a mutant ALDH2 genotype (ALDH2*2) show decreased ALDH2 activity and are more susceptible to HNSCC, highlighting the importance of ALDH2 activity in reducing alcohol-associated carcinogenesis in the oral cavity (Yokoyama and Omori, 2003). Our qPCR analysis showed that Aldh2 mRNA levels are decreased by 10-fold in the 4-NQO/EtOH group compared to V.C./Untr. (Fig. 2(b)A). Thus, 4-NQO/EtOH associated decreases in Aldh2 mRNA levels may play a major role in oral tumorigenesis. Interestingly, in the liver ethanol caused decreases in the Aldh2 mRNA levels and ALDH2 activity, contributing to liver steatosis (Liu et al., 2012). This result is consistent with our finding that ethanol exposure causes decreases in Aldh2 mRNA levels in the oral cavity.
While in the same family as ALDH2, ALDH1A3 converts retinaldehyde to retinoic acid and shows little capability to metabolize AcH to acetate. In contrast to Aldh2, our qPCR analysis showed a 7-fold increase in Aldh1a3 transcripts in the 4-NQO/EtOH compared to the V.C./Untr. group (Fig. 2(b)B) (Eriksson, 1977). This increase is consistent with the finding that ALDH1A3 is overexpressed in human HNSCC, where it is a marker of poor prognosis (Masood et al., 2013). Importantly, ALDH1A3 is also a common marker of cancer stem cell (CSC) populations in breast cancer (Marcato et al., 2011). We previously found that horizontal expansion of stem/progenitor cell populations takes place in the tongue basal epithelia during carcinogenesis, and these cells are likely tumor initiating cells in HNSCCs (Osei-Sarfo et al., 2013, Tang et al., 2013). Thus, our results suggest that Aldh1a3 could be a marker of HNSCC CSCs.
Ethanol is metabolized to AcH by the ADH family of enzymes (Seitz and Becker, 2007). We observed increased Adh7 transcript levels in V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH tongues compared to V.C./Untr. tongue samples (Fig. 2(b)D), which could increase AcH levels in the oral cavity. Conversely, Adh1 transcripts were decreased after 4-NQO/EtOH treatment (Fig. 2(b)C). It is interesting to note that although both ADH1 and ADH7 catalyze the metabolism of ethanol, we observe opposite changes in gene expression of these enzymes in our samples. However, in the upper gastrointestinal tract ADH7 shows the highest metabolism of ethanol to AcH out of the ADH enzymes (Wei et al., 2010). Additionally, others have found that ADH1 levels are decreased in breast cancers, and that ADH1 is inhibited during chronic alcohol abuse in liver, suggesting that decreased ADH1 expression may have other physiological effects besides ethanol metabolism that contribute to HNSCC (Seitz and Becker, 2007, Jelski et al., 2006). In fact, ADH1 is important for reducing 4-HNE levels (Singh et al., 2013), and we detected increased 4-HNE levels in the 4-NQO/EtOH treatment group relative to the V.C./Untr. group (Fig. 5C&D).
Two additional genes associated with alcohol metabolism are Cyp2a5 and Cyp2e1 (Jelski et al., 2006, Lu et al., 2011). In our model we observed a decrease in Cyp2a5 transcripts (Fig. 2(b)E), and no significant changes in Cyp2e1 transcripts (Fig. 2(b)F) in V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH groups compared to the V.C./Untr. group. CYP2E1 polymorphisms that alter CYP2E1 enzymatic activity have been observed in head and neck cancer patients, suggesting that regulation of Cyp2e1 at the transcript level may not be a major mechanism (Tang et al., 2010). CYP2A5 also metabolizes nicotine, and inactivating polymorphisms in the Cyp2a5 human ortholog Cyp2a6 lead to decreases in nicotine metabolism and increased lung and esophageal cancer risk (Xu et al., 2002).
Ethanol and 4-NQO increase various histone acetylation and methylation marks
We discovered that 4-NQO and ethanol globally alter epigenetic modifications during the carcinogenesis process. We showed that ethanol increases H3K9/14 and H3K27 acetylation (Fig. 3A&B). This is the first report of global increases in histone acetylation during oral carcinogenesis, which could explain the dramatic increases in transcripts in the 4-NQO/EtOH group as compared to the V.C./EtOH and 4-NQO/Untr. groups (Fig. 1).
We also detected increases in global H3K27me3 and H3K9me3 marks in all treatment groups (V.C./EtOH, 4-NQO/Untr. and 4-NQO/EtOH) compared to the V.C./Untr. control (Fig. 3C&D). However, we did not observe changes in H3K4me1 levels in treatment groups, consistent with a previous report that H3K4me1 levels did not change in human oral carcinomas compared to healthy mucosa (Mancuso et al., 2009). Our findings in this oral cancer model are significant, as other studies in lung, prostate, breast, esophageal, and liver cancers have shown that increases in the levels of histone marks, including H3K9ac, H3K27me3, H3K9me3, and H3K4me1, are often correlated with poor prognosis, increased metastasis, and increased recurrence rates (Chervona and Costa, 2012). This further highlights changes in histone marks as potential biomarkers for HNSCC and assessment of cancer risk.
Currently, the mechanisms by which ethanol and 4-NQO can increase both histone acetylation and methylation are not known. One potential mechanism is through regulation of the expression of histone modifying genes. In stem cells we observed an increase in the H3K27ac mark at specific gene promoters when histone deacetylase 1 (HDAC1) was knocked down (Urvalek and Gudas, 2014). By RNA-seq we found that HDAC1 mRNA decreased by 1.5-fold in the 4-NQO/EtOH compared to the V.C./Untr. group (data not shown), so this decrease in HDAC1 could result in greater H3K27ac and H3K9/14ac levels in 4-NQO/EtOH treated tongues. Similarly, the enzyme EZH2, which trimethylates H3K27, is overexpressed in 50% of human HNSCC tumor samples, where it adds the H3K27me3 inhibitory mark at many differentiation associated genes (Gannon et al., 2013, Kidani et al., 2009). We found that EZH2 transcripts are increased by 1.8- and 1.6-fold in tongues from the 4-NQO/Untr. and 4-NQO/EtOH groups, respectively (data not shown). Additionally, by IHC we observed an increase in EZH2 protein expression after 4-NQO treatment of the tongue epithelia (Supplemental Fig. 4). It will be important to determine the enzymatic activities of co-regulators (i.e. HDAC1, EZH2), and whether they interact near the transcriptional start sites and enhancers of genes involved in 4-NQO/EtOH-mediated oral carcinogenesis.
Unlike permanent genetic alterations such as gene mutations, epigenetic alterations are reversible, making cancer-associated epigenetic changes attractive targets for cancer prevention and therapy. Epigenetic changes are believed to precede, and potentially to cause neoplastic changes, and inhibitors of epigenetic modifying enzymes are currently being examined as potential therapies for HNSCC (Issa, 2008). For example, The EZH2 inhibitor DZNep has selective cancer toxicity in HNSCC (Gannon et al., 2013).
Markers of oxidative stress are increased in the tongue after 4-NQO/Untr. and 4-NQO/EtOH treatments
Oxidative stress promotes carcinogenesis by inducing DNA, RNA, lipid, and protein damage, leading to increased cellular proliferation, invasion, metastasis, and evasion of apoptosis (Choudhari et al., 2014). We observed increased TOM20 (Fig. 5A&D), a marker of oxidative phosphorylation, which was one of the signaling pathways we found to be altered in our RNA-seq analysis (Fig. 1D, and Supplementary Fig. 1A) (Wurm et al., 2011). Increased oxidative phosphorylation can lead to increased ROS production, causing lipid peroxidation, which results in 4-HNE protein and DNA adduct formation (Singh et al., 2013). We found increased 4-HNE staining in the 4-NQO/EtOH as compared to the V.C./Untr. group (Fig. 5C&D). Interestingly, 4-HNE can bind to specific HDACs, leading to their inactivation (Doyle and Fitzpatrick, 2010), and 4-HNE interaction with HDACs may be another mechanism by which histone acetylation levels are increased in the 4-NQO/EtOH treatment group in our model.
Others have shown that carcinogens can crosslink chromatin remodeling proteins such as HDAC1 to gene regulatory regions, thereby altering histone modifications at these sites (Schnekenburger et al., 2007). Additionally, many toxicants, including the cigarette smoke carcinogen acrolein, alter various histone mark levels, including H3K9/14ac, H3K27me3, and H3K4me3, by disrupting chromatin assembly and nucleosome occupancy at gene promoters (Chen et al., 2013, Zhou et al., 2009). These findings suggest that DNA and protein adduct formation induced by ethanol and 4-NQO could alter histone modifications globally and at specific gene promoters through similar mechanisms.
MCT4/SLC16A3 is a marker for increased glycolysis (Ganapathy et al., 2009, Halestrap and Price, 1999), and we found increased MCT4/SLC16A3 protein in 4-NQO/Untr. and 4-NQO/EtOH groups compared to the V.C./Untr. group (Fig. 5B&D). Other researchers reported that MCT4/SLC16A3 expression in cancer-associated fibroblasts resulted from oxidative stress, which protected cancer cells from cell death (Curry et al., 2013). These results indicate that MCT4/SLC16A3 inhibitors could be anti-cancer therapies for HNSCC, specifically targeting hypoxic cells.
In summary, we have identified alterations in epigenetic marks and increased oxidative stress as mechanisms by which 4-NQO followed by ethanol can promote oral cavity carcinogenesis. This suggests that specific epigenetic marks and/or markers of oxidative stress could be used as effective biomarkers for oral cavity carcinogenesis. In the future, our model will be useful for testing drugs directed towards epigenetic modifying enzymes or oxidative stress-associated proteins.
Supplementary Material
Acknowledgments
We would like to thank Weixun Wang for technical assistance, the Weill Cornell Genomics Core Facility, and members of the Gudas laboratory for helpful discussions and comments.
Financial Support: This research was supported by the National Institute of Health [R01-AA018332 to L.J.G.]; [F32-AA021045 to A.M.U.]; and for a portion of this work, [T32-CA062948 to A.M.U and K.O.S.)].
References
- Bauman JE, Michel LS, Chung CH. New promising molecular targets in head and neck squamous cell carcinoma. Curr Opin Oncol. 2012;24:235–242. doi: 10.1097/CCO.0b013e3283517920. [DOI] [PubMed] [Google Scholar]
- Black W, Vasiliou V. The aldehyde dehydrogenase gene superfamily resource center. Hum Genomics. 2009;4:136–142. doi: 10.1186/1479-7364-4-2-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Fang L, Li H, Tang MS, Jin C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J Biol Chem. 2013;288:21678–21687. doi: 10.1074/jbc.M113.476630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RJ, Chang LW, Lin P, Wang YJ. Epigenetic effects and molecular mechanisms of tumorigenesis induced by cigarette smoke: an overview. J Oncol. 2011;2011:654931. doi: 10.1155/2011/654931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervona Y, Costa M. Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res. 2012;2:589–597. [PMC free article] [PubMed] [Google Scholar]
- Chikamatsu K, Ishii H, Murata T, Sakakura K, Shino M, Toyoda M, Takahashi K, Masuyama K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013;104:1468–1475. doi: 10.1111/cas.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhari SK, Chaudhary M, Gadbail AR, Sharma A, Tekade S. Oxidative and antioxidative mechanisms in oral cancer and precancer: a review. Oral Oncol. 2014;50:10–18. doi: 10.1016/j.oraloncology.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, Leiby B, Cognetti DM, Sotgia F, Lisanti MP, Martinez-Outschoorn UE. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013;12:1371–1384. doi: 10.4161/cc.24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle K, Fitzpatrick FA. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem. 2010;285:17417–17424. doi: 10.1074/jbc.M109.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson CJ. Acetaldehyde metabolism in vivo during ethanol oxidation. Adv Exp Med Biol. 1977;85A:319–341. doi: 10.1007/978-1-4899-5181-6_21. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121:29–40. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Gannon OM, Merida de Long L, Endo-Munoz L, Hazar-Rethinam M, Saunders NA. Dysregulation of the repressive H3K27 trimethylation mark in head and neck squamous cell carcinoma contributes to dysregulated squamous differentiation. Clin Cancer Res. 2013;19:428–441. doi: 10.1158/1078-0432.CCR-12-2505. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343(Pt 2):281–299. [PMC free article] [PubMed] [Google Scholar]
- Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, Wunsch-Filho V, Franceschi S, Hayes RB, Herrero R, Koifman S, La Vecchia C, Lazarus P, Levi F, Mates D, Matos E, Menezes A, Muscat J, Eluf-Neto J, Olshan AF, Rudnai P, Schwartz SM, Smith E, Sturgis EM, Szeszenia-Dabrowska N, Talamini R, Wei Q, Winn DM, Zaridze D, Zatonski W, Zhang ZF, Berthiller J, Boffetta P. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007a;99:777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, Wünsch-Filho V, Franceschi S, Hayes RB, Herrero R, Koifman S, La Vecchia C, Lazarus P, Levi F, Mates D, Matos E, Menezes A, Muscat J, Eluf-Neto J, Olshan AF, Rudnai P, Schwartz SM, Smith E, Sturgis EM, Szeszenia-Dabrowska N, Talamini R, Wei Q, Winn DM, Zaridze D, Zatonski W, Zhang ZF, Berthiller J, Boffetta P. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007b;99:777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Ren B. Predictive chromatin signatures in the mammalian genome. Hum Mol Genet. 2009;18:R195–201. doi: 10.1093/hmg/ddp409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res (Phila) 2008;1:219–222. doi: 10.1158/1940-6207.CAPR-08-0029. [DOI] [PubMed] [Google Scholar]
- Jelski W, Chrostek L, Szmitkowski M, Markiewicz W. The activity of class I, II, III and IV alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in breast cancer. Clin Exp Med. 2006;6:89–93. doi: 10.1007/s10238-006-0101-z. [DOI] [PubMed] [Google Scholar]
- Kang H, Gillespie TW, Goodman M, Brodie SA, Brandes M, Ribeiro M, Ramalingam SS, Shin DM, Khuri FR, Brandes JC. Long-term use of valproic acid in US veterans is associated with a reduced risk of smoking-related cases of head and neck cancer. Cancer. 2014;120:1394–1400. doi: 10.1002/cncr.28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidani K, Osaki M, Tamura T, Yamaga K, Shomori K, Ryoke K, Ito H. High expression of EZH2 is associated with tumor proliferation and prognosis in human oral squamous cell carcinomas. Oral Oncol. 2009;45:39–46. doi: 10.1016/j.oraloncology.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Liu S, Yeh TH, Singh VP, Shiva S, Krauland L, Li H, Zhang P, Kharbanda K, Ritov V, Monga SP, Scott DK, Eagon PK, Behari J. β-catenin is essential for ethanol metabolism and protection against alcohol-mediated liver steatosis in mice. Hepatology. 2012;55:931–940. doi: 10.1002/hep.24766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhuge J, Wu D, Cederbaum AI. Ethanol induction of CYP2A5: permissive role for CYP2E1. Drug Metab Dispos. 2011;39:330–336. doi: 10.1124/dmd.110.035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Matassa DS, Conte M, Colella G, Rana G, Fucci L, Piscopo M. H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim Pol. 2009;56:405–410. [PubMed] [Google Scholar]
- Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, Helyer L, Pan L, Leidal A, Gujar S, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- Masood R, Hochstim C, Cervenka B, Zu S, Baniwal SK, Patel V, Kobielak A, Sinha UK. A novel orthotopic mouse model of head and neck cancer and lymph node metastasis. Oncogenesis. 2013;2:e68. doi: 10.1038/oncsis.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Kumashiro R, Kubo N, Nakashima Y, Yoshida R, Yoshinaga K, Saeki H, Emi Y, Kakeji Y, Sakaguchi Y, Toh Y, Maehara Y. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: epidemiology, clinical findings, and prevention. Int J Clin Oncol. 2010;15:126–134. doi: 10.1007/s10147-010-0056-7. [DOI] [PubMed] [Google Scholar]
- Osei-Sarfo K, Tang XH, Urvalek AM, Scognamiglio T, Gudas LJ. The molecular features of tongue epithelium treated with the carcinogen 4-nitroquinoline-1-oxide and alcohol as a model for HNSCC. Carcinogenesis. 2013;34:2673–2681. doi: 10.1093/carcin/bgt223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012;122:1951–1957. doi: 10.1172/JCI59889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JS, Qiu L, Etkind P. Amplification of the Int-2 gene in head and neck squamous cell carcinoma. J Laryngol Otol. 1995;109:72–76. doi: 10.1017/s0022215100129305. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M, Talaska G, Puga A. Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol Cell Biol. 2007;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Becker P. Alcohol metabolism and cancer risk. Alcohol Res Health. 2007;30:38–41. 44–37. [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem. 2006;387:349–360. doi: 10.1515/BC.2006.047. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- Serewko MM, Popa C, Dahler AL, Smith L, Strutton GM, Coman W, Dicker AJ, Saunders NA. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res. 2002;62:3759–3765. [PubMed] [Google Scholar]
- Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- Singh S, Brocker C, Koppaka V, Chen Y, Jackson BC, Matsumoto A, Thompson DC, Vasiliou V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med. 2013;56:89–101. doi: 10.1016/j.freeradbiomed.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squier CA, Kremer MJ. Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr. 2001:7–15. doi: 10.1093/oxfordjournals.jncimonographs.a003443. [DOI] [PubMed] [Google Scholar]
- Tang K, Li Y, Zhang Z, Gu Y, Xiong Y, Feng G, He L, Qin S. The PstI/RsaI and DraI polymorphisms of CYP2E1 and head and neck cancer risk: a meta-analysis based on 21 case-control studies. BMC Cancer. 2010;10:575. doi: 10.1186/1471-2407-10-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Albert M, Scognamiglio T, Gudas LJ. A DNA methyltransferase inhibitor and all-trans retinoic acid reduce oral cavity carcinogenesis induced by the carcinogen 4-nitroquinoline 1-oxide. Cancer Prev Res (Phila) 2009;2:1100–1110. doi: 10.1158/1940-6207.CAPR-09-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res. 2004;10:301–313. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- Tang XH, Osei-Sarfo K, Urvalek AM, Zhang T, Scognamiglio T, Gudas LJ. Combination of bexarotene and the retinoid CD1530 reduces murine oral-cavity carcinogenesis induced by the carcinogen 4-nitroquinoline 1-oxide. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1404828111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Scognamiglio T, Gudas LJ. Basal stem cells contribute to squamous cell carcinomas in the oral cavity. Carcinogenesis. 2013;34:1158–1164. doi: 10.1093/carcin/bgt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urvalek A, Laursen KB, Gudas LJ. The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. Subcell Biochem. 2014;70:129–149. doi: 10.1007/978-94-017-9050-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urvalek AM, Gudas LJ. Retinoic Acid and Histone Deacetylases Regulate Epigenetic Changes in Embryonic Stem Cells. J Biol Chem. 2014;289:19519–19530. doi: 10.1074/jbc.M114.556555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Liu Z, Zhao H, Niu J, Wang LE, El-Naggar AK, Sturgis EM, Wei Q. A single nucleotide polymorphism in the alcohol dehydrogenase 7 gene (alanine to glycine substitution at amino acid 92) is associated with the risk of squamous cell carcinoma of the head and neck. Cancer. 2010;116:2984–2992. doi: 10.1002/cncr.25058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm CA, Neumann D, Lauterbach MA, Harke B, Egner A, Hell SW, Jakobs S. Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc Natl Acad Sci U S A. 2011;108:13546–13551. doi: 10.1073/pnas.1107553108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Goodz S, Sellers EM, Tyndale RF. CYP2A6 genetic variation and potential consequences. Adv Drug Deliv Rev. 2002;54:1245–1256. doi: 10.1016/s0169-409x(02)00065-0. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol. 2003;33:111–121. doi: 10.1093/jjco/hyg026. [DOI] [PubMed] [Google Scholar]
- Zhou X, Li Q, Arita A, Sun H, Costa M. Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol Appl Pharmacol. 2009;236:78–84. doi: 10.1016/j.taap.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.