

Abstract
Bone neoplasms, such as osteosarcoma, exhibit a propensity for systemic metastases resulting in adverse clinical outcome. Traditional treatment consisting of aggressive chemotherapy combined with surgical resection, has been the mainstay of these malignances. Therefore, bone-targeted non-toxic therapies are required. We previously conjugated the aminobisphosphonate alendronate (ALN), and the potent anti-angiogenic agent TNP-470 with N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer. HPMA copolymer-ALN-TNP-470 conjugate exhibited improved anti-angiogenic and anti-tumor activity compared with the combination of free ALN and TNP-470 when evaluated in a xenogeneic model of human osteosarcoma. The immune system has major effect on toxicology studies and on tumor progression. Therefore, in this manuscript we examined the safety and efficacy profiles of the conjugate using murine osteosarcoma syngeneic model. Toxicity and efficacy evaluation revealed superior anti-tumor activity and decreased organ-related toxicities of the conjugate compared with the combination of free ALN plus TNP-470. Finally, comparative anti-angiogenic activity and specificity studies, using surrogate biomarkers of circulating endothelial cells (CEC), highlighted the advantage of the conjugate over the free agents. The therapeutic platform described here may have clinical translational relevance for the treatment of bone-related angiogenesis-dependent malignances.
Keywords: Polymer therapeutics, HPMA copolymer, TNP-470, Alendronate, Osteosarcoma
1. Introduction
Osteosarcoma is the most common primary tumor of bone and considered to be the third most common malignancy in children and adolescents. It is a highly vascular and extremely destructive malignancy that most commonly arises in the metaphyseal ends of long bones [1]. Over the past two decades, multimodality treatment consisting of aggressive chemotherapy combined with radical surgical resection, has been the mainstay of osteosarcoma management, with achievable 5-year survival rates of 50–70% in patients who do not have metastatic disease at presentation [2]. Surgery remains the vital modality for treating the primary osteosarcoma tumor, whereas adjuvant chemotherapy (i.e. methotrexate, cisplatin and doxorubicin) plays an essential role in the control of subclinical metastatic disease [3]. Another class of skeletal malignancies is bone metastases originated from soft tissues such as breast and prostate cancers [3]. Tumor cells in the bone microenvironment produce a large number of cytokines that stimulate osteoclastic activity [4]. Increased osteoclastic activity, in turn, leads to production of a variety of lymphokines and growth factors that increase tumor cell proliferation [5]. This vicious circle can be disrupted by inhibition of osteoclast activity. Aminobisphosphonates (NBP) such as alendronate (ALN) are a class of drugs with high affinity to the bone mineral hydroxyapatite (HA), capable of inhibiting osteoclast recruitment and function, and bone resorption. Consequently, the aggressive interaction between osteoclasts and tumor cells is disrupted [6]. Moreover, adjacent to bone resorption activity, NBP have direct anti-tumor and anti-angiogenic effects, therefore can be used for bone metastases treatment [7]. Recently, highly potent NBP (i.e. Zolendronic acid, Pamidronate and ALN) became available for clinical use and represent an important adjunct in the management of bone metastases originating from breast and prostate cancer, multiple myeloma, and other types of malignancies [8,9]. Despite the advantages described above, potential adverse effects associated with NBP therapy limit their use in some patients [10]. These include unwanted side effects such as gastroesophageal irritation, nausea, transient hypocalcemia with secondary hyper-parathyroidism, severe musculoskeletal pain, increased risk of esophageal cancer, osteonecrosis of the jaw, severe suppression of bone turnover and atrial fibrillation [11,12]. A different class of drugs used for the treatment of bone malignancy is angiogenesis inhibitors [13]. The importance of new blood vessels formation in osteosarcoma pathogenesis and progression has been highlighted by previous studies, showing correlation of increased tumor vascularity with metastatic potential, and poorer prognosis in osteosarcoma [14–16]. Anti-angiogenic therapy aims either, to prevent formation of new vessels, sequester angiogenic stimulators, or damage existing vessels [17]. Anti-angiogenic treatment reduces the incidence of drug resistance and does not cause the side effects associated with conventional chemotherapy (i.e. bone-marrow suppression, hair loss, severe vomiting, diarrhea, and weakness) [18,19]. Nevertheless, some angiogenesis inhibitors increase the incidence of thrombotic complications hypertension, intratumoural bleeding and neurological dysfunction [20,21]. There is accumulating data showing that combined treatment of classical chemotherapy with angiogenesis inhibitors can lead to a synergistic and more effective treatment of osteosarcoma and bone metastases [22–24]. Most of these drugs, however, are low Mw agents delivered systemically, and consequently exhibit a non-specific biodistribution, deprived pharmacokinetic profile and severe side effects [25]. Therefore, utilization of combination therapy advantages with an improved tumor-selective drug delivery system is a desirable goal for the treatment of osteosarcoma, bone neoplasms and osteoidic metastatic lesions.
Here we describe the synthesis and characterization of a 30 kDa dual-targeted conjugate comprised of N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer backbone, the potent anti-angiogenic agent TNP-470 and ALN. TNP-470 is a low Mw synthetic analog of fumagillin able to selectively inhibit endothelial growth in vitro [26,27]. In clinical trials, it showed promising results when used in combination with conventional chemotherapy [28]. However, due to dose limiting neurotoxicity, TNP-470 has not made further progress past Phase II [29]. We have recently shown that caplostatin, an HPMA copolymer conjugate of TNP-470, prolonged the circulating life of the drug, increased the accumulation of the drug in angiogenic tissue through the enhanced permeability and retention (EPR) effect and prevented it from crossing the blood brain barrier (BBB), therefore abrogating neurotoxicity [21,30]. Using the reversible addition-fragmentation chain transfer (RAFT) polymerization technique, we conjugated ALN and TNP-470 via a Glycine-Glycine-Proline-Norleucine (Gly-Gly-Pro-Nle) cathepsin K-cleavable linker to HPMA copolymer backbone. RAFT polymerization is a versatile controlled/“living” free radical polymerization technique resulting in predetermined Mw with narrow polydispersity index (PDI) [31]. Polymer conjugation increases the half-life of free low Mw drugs (e.g. ALN and TNP-470), increases their tumor accumulation by the EPR effect and reduces their toxicity [32]. Previous biodistribution and pharmacokinetic studies showed that ALN can facilitate specific and rapid accumulation of HPMA copolymer in bone tissues [33–35]. Therefore, in the conjugate platform presented here, ALN is used both as a targeting moiety (due to its high affinity to HA) and as a therapeutic agent. HPMA copolymer-ALN-TNP-470 conjugate was designed to target the neoplastic sites within the bones and metastatic calcified neoplasms in other organs. The rationale behind the conjugate design is that following accumulation of the conjugate at the target site, the Gly-Gly-Pro-Nle linker will be cleaved by cathepsin K, a cysteine protease overexpressed at resorption sites in bony tumors, and active ALN and TNP-470 will be released [36,37]. The anti-angiogenic and anti-tumor potency of HPMA copolymer-ALN-TNP-470 conjugate were previously evaluated on a xenogeneic model of severe combined immunodeficiency (SCID) mice bearing MG-63-Ras human osteosarcoma. The conjugate had superior anti-angiogenic and anti-tumor activity compared with the combination of free ALN and TNP-470 [38].
An appropriate preclinical tumor model that reflects the clinical settings can provide insight into key processes such as angiogenesis, tumor progression, bone metastases, and therapeutic response. The use of human xenografts in mice still remains the gold standard in developing therapeutics for cancer therapy and evaluating their efficacy [39]. However, xenogeneic models lack human stroma and immune cells, which are important for tumor initiation and progression [40]. Moreover, host–tumor interactions and the immune system are important parameters that might affect the outcome of toxicology and efficacy studies [41]. In relevance to the targeted delivery system described here, both ALN and TNP-470 are capable of inhibiting neo-vascularization and overall tumor development in part through key inflammatory-cell-derived mediators and other important cytokines [30,42]. Therefore, it is essential to validate the safety and efficacy profiles of HPMA copolymer-ALN-TNP-470 conjugate in a syngeneic model that preserve the natural microenvironmental host–tumor interactions. Following these principles, we performed the safety and efficacy studies in a model of Balb/c mice bearing K7M2 murine osteosarcoma in the tibia [43]. Although this orthotropic model provides clinically relevant outcome parameters, the anatomical location of K7M2 tumors in the tibia sets a technical obstacle in terms of monitoring tumor progression. Therefore, we used mCherry-infected K7M2 osteosarcoma cells that allow tumor progression monitoring by non-invasive intravital fluorescence imaging techniques. The ability to induce neovascularization is crucial for the development and progression of osteosarcoma and bone metastases [13]. Thus, in addition to safety and efficacy studies, we assessed the ability of the conjugate to inhibit angiogenesis. Most standard assays for assessing the anti-angiogenic effect of therapeutics use tumor tissue in order to perform immunohistochemistry analysis of angiogenesis markers and microvessel density. This generally requires termination of the experiment and removal of the entire tumor from the animals. An alternative approach is the use of surrogate angiogenic biomarkers such as circulating endothelial cells (CEC) and/or their progenitor subset (CEP) [44,45]. CEC and CEP have been found to correlate with preclinical and clinical efficacy of anti-angiogenic therapy [46–48]. Here we evaluated the anti-angiogenic effect of the conjugate in non-tumor and tumor-bearing Balb/c mice. The use of CEC and CEP enables simple, repeated and relatively non-invasive assessment of tumor angiogenesis in mice. Furthermore, it allowed us to compare the anti-angiogenic activity of the conjugate in non-tumor bearing and tumor-bearing mice using the same procedure. In summary, in the search for improved targeted polymer therapy for bone neoplasms and osteoidic metastatic lesions, we used solid phase peptide synthesis (SPPS) and RAFT chemistry to synthesize dual-targeted HPMA copolymer-ALN-TNP-470 conjugate. Integration of syngeneic orthotropic osteosarcoma model, surrogate biomarkers and functional non-invasive imaging tools allowed us to examine the potential of our conjugate to decrease the non-specific organ toxicities associated with free ALN and TNP-470, and its anti-angiogenic and anti-tumor efficacy.
2. Materials and methods
2.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM) and XTT reagent were purchased from Biological Industries Ltd. (Kibbutz Beit Haemek, Israel). Fetal bovine serum (FBS), Penicillin, Streptomycin, Nystatin and L-glutamine were from Biological Industries, Israel. pEGFPLuc plasmid was from Clontech (Mountain View, CA, USA). Nuclear staining was from Procount, BD Pharmingen, San Jose, CA. 7-amino-actinomycin D (7AAD) was from Chemicon, Billerica, MA. All other chemical reagents, including salts and solvents, were purchased from Sigma–Aldrich. All reactions requiring anhydrous conditions were performed under an Ar or N2 atmosphere. Chemicals and solvents were either A.R. grade or purified by standard techniques.
2.2. Ethics statement
All animal procedures were performed in compliance with Tel Aviv University, Sackler School of Medicine guidelines and protocols approved by the Institutional Animal Care and Use Committee. Mice’s body weight and tumor size were measured three times a week.
2.3. Synthesis of HPMA copolymer-ALN-TNP-470 conjugate
2.3.1. Synthesis of ALN containing monomer (MA-Gly-Gly-Pro-Nle-ALN)
MA-Gly-Gly-Pro-Nle was synthesized by solid phase peptide synthesis (SPPS) and manual Fmoc/tBu strategy using 2-chlorotrityl chloride beads with 80% of loading. MA-Gly-Gly-Pro-Nle-OH (100 mg, 0.24 mmol) and 2-mercaptothiazoline (TT, 33 mg, 0.28 mmol) were dissolved in a mixture of 2 ml 1,4-dioxane and 1 ml tetrahydrofuran (THF), and cooled to 4 °C. DCC (60 mg, 0.29 mmol) in 1 ml of 1,4-dioxane was added dropwise and the reaction mixture stirred overnight at 4 °C. After the reaction, DCU was removed by filtration and the filtrate was added to ALN aqueous solution (70 mg, 4 ml; pH was adjusted to ~7.4 by NaHCO3 solution). The reaction mixture was stirred overnight at room temperature. The solvent was removed on a rotary evaporator, the residue was re-dissolved in water and extracted with ethyl acetate 3 times to remove TT. The product was purified by preparative high performance liquid chromatography (HPLC, buffer A: H2O 0.1% TFA, buffer B: acetonitrile 0.1% TFA; gradient method: buffer B 0–20%/ 20 min; 2.5 ml/min; elution time 9.7 min) with a yield of 83 mg. The MALDI-TOF spectrum negative ion: m/z = 640 (M − H+), 662 (M-mono-Na salt − H+); positive ion: m/z = 642 (M + H+), 664 (M-mono-Na salt + H+), 686 (M-mono-Na salt + Na+).
2.3.2. Synthesis of amine containing monomer (MA-Gly-Gly-Pro-Nle-NH(CH2)2NH2)
MA-Gly-Gly-Pro-Nle-NH(CH2)2NH2 was synthesized by SPPS using 1.5 g of 2-chlorotrityl chloride beads. First, 6 times excess of ethylenediamine in anhydrous THF was applied, followed by Fmoc-amino acids, and capping with MA-Gly-Gly-OH. The final peptide was cleaved by 5% TFA in dichloromethane (DCM) with a yield of 0.85 g, 89%. The purity of product was proved by HPLC (buffer A: H2O 0.1% TFA, buffer B: acetonitrile 0.1% TFA; gradient method: buffer B 2%–60%/30 min; 1 ml/min; single peak, elution time 8.27 min). The MALDI-TOF spectrum (Fmoc derivative) positive ion: m/z = 697 (M + Na+), 713 (M + K+).
2.3.3. Synthesis FITC containing monomer (MA-FITC)
Fluoresceinisothiocyanate (FITC) (1 g, 2.57 mmol) and N-(3-aminopropyl) methacrylamide hydrochloride (0.92 g, 5.14 mmol) were dissolved in 5 ml dimethylformamide (DMF) and cooled to 4 °C, then N,N-diisopropylethylamine (DIPEA) (1.79 ml, 10.3 mmol) in 2 ml of DMF was added dropwise. The reaction mixture was stirred at 4 °C for 2 days. Then, the reaction mixture was poured into 100 ml water (pH ~4–5) and pH adjusted to ~4 by 6 N HCl. The precipitate was filtered off, washed with water, and vacuum dried over P2O5 [49].
2.3.4. Synthesis of polymer precursor containing ALN, NH2, and (optionally) FITC groups (HPMA copolymer-ALN-NH2)
Reversible addition-fragmentation chain transfer (RAFT) polymerization technique was used to synthesize HPMA copolymer-ALN-TNP-470 conjugate. MA-Gly-Gly-Pro-Nle-ALN (293 mg), MA-Gly-Gly-Pro-Nle-NH(CH2)2NH2 (215 mg), HPMA (948 mg), MA-FITC (0 mg or 4 mg), 2,2′-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044 1.6 mg) as initiator and S,S′-bis(α,α′-dimethyl-α″-acetic acid) trithiocarbonate as chain transfer agent (TTC 4.2 mg) were dissolved in 7.5 ml of water. The solution was bubbled with N2(g) for 30 min, sealed in ampoule, and the mixture polymerized at 30 °C for 48 h. Both resulting polymers were purified by dissolving in water and precipitating into an excess of acetone; following each precipitation, the precipitate was washed with acetone. Finally, the polymers were dissolved in 15 ml of water; pH adjusted to 12 with 1 N NaOH, and dialyzed against DI water for 24 h at 4 °C (MWCO 12–14 kDa) to remove ALN monomer. The samples were freeze-dried after dialysis.
2.3.5. Binding of TNP-470 to HPMA copolymer-ALN
HPMA copolymer-ALN-NH2 (150 mg) was dissolved in 6 ml of DMF (if necessary, a small amount of water was added to dissolve the polymer) and cooled to 4 °C. Then, TNP-470 (150 mg) in 1 ml DMF was added. The reaction mixture was stirred at 4 °C in dark for 12 h. Following the reaction, the conjugate was precipitated into acetone and purified by reprecipitation (3 times) from an aqueous solution into an excess of acetone. The precipitate was washed with acetone and the residue was dissolved in water and dialyzed for 1 day at 4 °C (MWCO 12–14 kDa) against DI water. The conjugate was isolated by freeze-drying.
2.4. Determination of ALN content
The formation of chromophoric complex between ALN and Fe3+ ions in perchloric acid solution was used to determine the ALN content by spectrophotometry [50]. Briefly, 0.1 ml conjugate (conc. 2–10 mg/ml) was mixed with 0.1 ml 4 mM FeCl3 and 0.8 ml 0.2 M HClO4 and absorbance at 300 nm was measured against blank. The calibration curve was prepared by using ALN solution at concentration range 0–3 mM.
2.5. Estimation of TNP-470 content
The content of TNP-470 was estimated from the content of NH2 groups in the HPMA copolymer-ALN-NH2 precursor (Scheme 1). It was assumed that the TNP-470 binding was quantitatively equal. The content of NH2 groups was determined by ninhydrin method using an amine containing monomer [N-(3-aminopropyl) methacrylamide] as the calibration sample (modified from Ref. [51]).
Scheme 1.

Synthesis of HPMA copolymer-ALN-TNP-470 conjugate. Table in Scheme 1. Physicochemical Characterization of HPMA copolymer conjugates. a-Determined by Fe3+ complexation method. b-NH2 determined by ninhydrin method; it was assumed that TNP-470 content is similar. c-Determined using extinction coefficient of MA-FITC 78000 M−1 cm−1 in 0.1 M borate. d-Measured by spectroscopy using extinction coefficient for Tyr (295 nm) 2700 M−1 cm−1 in 1 N NaOH (the background of the polymer was subtracted). e-Determined by DLS in PBS (pH 7.4). f-Determined by SEC on AKTA/FPLC system (Pharmacia), using Superose 6 HR 10/30 column, buffer 0.1 M acetate/30% acetonitrile, pH 5.5. g-Was calculated from SEC profiles data.
2.6. Determination of HPMA copolymer-ALN-TNP-470 conjugate molecular weight and polydispersity index (PDI)
Molecular weight and polydispersity index (PDI) were measured using size-exclusion chromatography (SEC) on an ÄKTA/FPLC system (GE Healthcare) equipped with three online detectors: three angle light scattering detector miniDAWN TREOS (wavelength 658 nm), RI detector Optilab-rEX (Wyatt Technology, Santa Barbara, CA), UV detector UPC-900 (AKTA set for 280 nm detection), using GE Healthcare columns: Superose 6 HR10/30 or Superose 12 HR10/30 column; flow rate 0.4 ml/min. The dn/dc value (0.17; PBS) was calculated using ASTRA software assuming 100% recovery of sample.
2.7. Measurement of effective diameter of HPMA copolymer-ALN-TNP-470 conjugate
A dynamic light scattering method for effective diameter of the conjugates was performed on a Brookhaven BI-200SM goniometer and BI-9000 AT digital correlator equipped with a He–Ne laser (λ = 633 nm) at 90° angle room temperature (25 °C). The conjugate was dissolved in double DI water at a concentration of 5 mg/ml and filtered through 0.2 μm PTFE filter before measurement. The effective diameter and PDI were calculated as an average of 6 runs.
2.8. Synthesis and characterization of HPMA copolymer-FITC-Tyr conjugate
HPMA (280 mg), MA-FITC (10 mg, 1 mol%), N-methacryloyltyrosineamide (MA-Tyr-NH2, 10 mg, 2 mol%), 4-cyanopentanoic acid dithiobenzoate (1.4 mg) and VA-044 (0.5 mg) were dissolved in 2.4 ml of MeOH/H2O (2/1), bubbled with N2 for 30 min, sealed and polymerized at 37 °C for 24 h. The conjugate was purified by dissolution-precipitation method in methanol–acetone and dried under reduced pressure at room temperature. The Mw and PDI of HPMA copolymer-FITC-Tyr conjugate were measured using AKTA FPLC system (Pharmacia/GE Healthcare) equipped with miniDAWN TREOS and OptilabEX detectors (Wyatt Technology, Santa Barbara, Ca), using a Superose 6 HR10/30 column with PBS (pH = 7.3) as the mobile phase. The content of FITC and Tyr was measured using Varian Cary 400 Bio UV-visible spectrophotometer and calculated using ε(495) = 80,000 cm−1 (0.1 N borate) for FITC and ε(280) = 1600 cm−1 M−1 for Tyr-NH2. After subtraction of polymer background, the content of FITC and Tyr-NH2 was 0.038 and 0.1 mmol/g respectively.
2.9. Cell culture
K7M2 murine osteosarcoma cells were obtained from the American Type Culture Collection (ATCC). Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 μg/ml Penicillin, 100 U/ml Streptomycin, 12.5 U/ml Nystatin and 2 mM L-glutamine. Cells were grown at 37 °C in 5% CO2.
2.10. Red blood cell (RBC) lysis assay
Red blood cells (RBC) lysis assay was performed as previously described [52]. Briefly, a 2% w/w rat RBC solution was incubated with serial dilutions of free combination of ALN plus TNP-470 or HPMA copolymer-ALN-TNP-470 conjugate for 1 h at 37 °C. Negative controls were PBS and Dextran (Mw ~70000) while positive controls were 1% w/v solution of Triton X100 (100% lysis) and polyethyleneimine (PEI). Following centrifugation, the supernatant was drawn off and its absorbance measured at 550 nm using a microplate reader (Genios, TECAN). The results were expressed as percentage of hemoglobin released relative to the positive control (Triton × 100).
2.11. Peripheral total white blood cells (WBC) count
Balb/c male mice aged 6 weeks were injected subcutaneously (s.c.) with a combination of free ALN and TNP-470 (1:1, 30 mg/kg), HPMA copolymer-ALN-TNP-470 conjugate (30 mg/kg q.o.d. TNP-470-equivalent dose) or saline (n = 5 mice/group). On day 14, a capillary pipette containing anticoagulant (0.1% EDTA) was inserted in the lateral canthus and blood was collected from the retro-orbital sinus. After collection, the pipette was removed and bleeding stopped when the eye returned to a normal position. EDTA anti-coagulated blood samples were used to obtain a complete blood count. Samples were counted no longer than 5 min after blood was drawn from mice. Ten μl of blood samples were mixed with 90 μl of track solution (1% acetic acid in DDW), and cells were counted by a Z1 Coulter® Particle Counter (Beckman Coulter™). Data is expressed as mean ± standard error of the mean (s.e.m.).
2.12. Evaluation of HPMA copolymer-ALN-TNP-470 conjugate neurotoxicity in non-tumor bearing mice
Balb/c male mice aged 6 weeks were injected s.c. with a combination of free ALN and TNP-470 (1:1, 30 mg/kg), HPMA copolymer-ALN-TNP-470 conjugate (30 mg/kg q.o.d. TNP-470-equivalent dose) or saline (n = 5 mice/group). On day 14, motor coordination and balance were measured using standard accelerating Rotarod treadmill as previously described [21]. Briefly, mice were placed on the rotating rod for 30 s to acclimate. The time of each animal remained on the rotating rod was recorded. Data is expressed as mean ± s.e.m.
2.13. Measurement of CEC and CEP by flow cytometry
Balb/c male mice aged 6 weeks were injected s.c. with a combination of free ALN and TNP-470 (1:1, 30 mg/kg), HPMA copolymer-ALN-TNP-470 conjugate (30 mg/kg q.o.d. TNP-470-equivalent dose) or saline (n = 5 mice/group). On day 14, blood was obtained from anaesthetized mice by retro-orbital sinus bleeding. CEP and CEC were quantitated using flow cytometry, as described previously [47]. Briefly, 24 h after drug treatment, blood was collected in tubes containing EDTA to avoid clotting. Monoclonal antibodies were used to detect all populations with the following antigenic phenotypes: for CEP: CD13+/VEGFR2+/CD117+/CD45-/dim; for CEC: CD13+/VEGFR2+/CD45−/dim. Nuclear staining was used in some experiments to exclude platelets or cellular debris. 7-aminoactinomycin D (7AAD) was used to distinguish apoptotic and dead cells from viable cells. After red cell lysis, cell suspensions were analyzed and at least 200,000 cells per sample were acquired Analyses were considered informative when an adequate number of events (i.e. >50, typically 50–150) was collected in the CEP or CEC enumeration gate in untreated control animals. Percentages of stained cells were determined and compared with appropriate negative controls. Positive staining was defined as being greater than non-specific background staining. Flow cytometry studies were performed on Cyan ADP flow cytometer (Beckman Coulter) and analyzed with Summit (Beckman Coulter) software. All monoclonal antibodies were purchased from BD Biosciences and used for flow cytometry analysis in accordance with the manufacturer’s protocols. Data is expressed as mean ± s.e.m.
2.14. Cell viability assay
K7M2 murine osteosarcoma cells were plated at 2500 cells/well in DMEM supplemented with 5% FBS and incubated for 24 h (37 °C; 5% CO2). The medium was then replaced with DMEM supplemented with 10% FBS. Cells were exposed to ALN, TNP-470, and HPMA copolymer-ALN-TNP-470 conjugate or with equivalent concentrations of combinations of free ALN and TNP-470 at serial dilutions. Viable cells were counted by XTT reagent respectively after 72 h of incubation. Data is expressed as mean ± standard deviation (s.d.).
2.15. Biodistribution studies
FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate (30 mg/kg TNP-470-equivalent dose in 100 μl saline/mouse) or FITC-labeled HPMA copolymer (0.2 mol% FITC equivalent dose in 100 μl saline/mouse) were administered to mCherry-K7M2 tumor-bearing Balb/c mice (~20 g, 5 mice/group) via tail vein injection. Eight hours post injection, vascular PBS perfusion was performed by cannulating the left ventricle and draining from the right atrium in an open-chest procedure. Major organs including heart, kidneys, liver, spleen, and bones (spine and ribs) were isolated, processed, and analyzed using CRI Maestro™ imaging system. Auto-fluorescence and undesired background signals were eliminated by spectral analysis and linear unmixing algorithm. FITC-fluorescence unmixed specific signal was quantified as total signal of photons/exposure time (s)/tissue weight (g). Data is expressed as mean ± s.e.m.
2.16. Generation of mCherry-infected K7M2 murine osteosarcoma cell line
mCherry was subcloned from pART7-mCherry (kindly provided by A. Avni from Tel Aviv University), into pQCXIP. Human embryonic kidney 293T (HEK 293T) cells were co-transfected with pQC-mCherry and the compatible packaging plasmids (pMD.G.VSVG and pGagpol.gpt). Forty eight hours following transfection, the pQC-mCherry retroviral particles containing supernatant was collected. K7M2 murine osteosarcoma cells were infected with the retroviral particles media, and 48 h following the infection, mCherry positive cells were selected by puromycin resistance.
2.17. Establishment of K7M2 murine osteosarcoma model
For inoculations of tumor cells into mice tibia, confluent mCherry-labeled K7M2 murine tumor cells were trypsinized, washed 3 times in PBS and then adjusted to a final concentration of 1 ×106 cells/100 μl. Each mouse was injected with 0.5 ×106 cells (in 50 μl PBS) intratibialy (i.t.) using a 26 g needle. mCherry-labeled tumor progression was monitored using CRI Maestro™ non-invasive fluorescence imaging system.
2.18. Evaluation of anti-tumor activity of HPMA copolymer-ALN-TNP-470 conjugate
Mice bearing K7M2 tumors were injected s.c. with a combination of free ALN and TNP-470 (1:1, 30 mg/kg), HPMA copolymer-ALN-TNP-470 conjugate (30 mg/kg q.o.d. TNP-470-equivalent dose) or saline (n = 5 mice/group). Therapy was initiated at a relatively early state in order to imitate early-detected primary osteosarcoma. Tumor progression was monitored by CRI™ Maestro non-invasive intravital imaging system. At termination, tumors were dissected, weighed and analyzed. Data is expressed as mean total signal/exposure time (ms) ± s.e.m.
2.19. Intravital non-invasive imaging of mCherry-labeled K7M2 tumor-bearing mice
CRI Maestro™ non-invasive fluorescence imaging system was used to follow tumor progression of mice bearing mCherry-labeled K7M2 murine osteosarcoma tumors and for biodistribution studies of FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate. Mice were maintained on a non-fluorescent diet (Harlan) for the whole period of the experiment. Mice were anesthetized using ketamine (100 mg/kg) and xylazine (12 mg/kg), treated with a depilatory cream (Veet®) and placed inside the imaging system. Alternatively, selected organs from mice were dissected and placed inside the imaging system. Multispectral image-cubes were acquired through 550–800 nm spectral range in 10 nm steps using excitation (575–605 nm) and emission (645 nm longpass) filter set. Mice autofluorescence and undesired background signals were eliminated by spectral analysis and linear unmixing algorithm.
2.20. Statistical analysis
In vitro data expressed as mean ± s.d. In vivo data expressed as mean ± s.e.m. Statistical significance was determined using an unpaired t-test. Kaplan–Meier method was used to calculate survival curves. All statistical tests were two-sided. Differences between designated groups compared to control group (unless indicated otherwise) were considered significant at values of P < 0.05 (*), P < 0.03 (**) or P < 0.01 (***).
3. Results
3.1. Synthesis and chemical characterization of HPMA copolymer-ALN-TNP-470 conjugate
HPMA copolymer-Gly-Gly-Pro-Nle-ALN-TNP-470 conjugate was synthesized as previously described [38]. Briefly, an intermediate was synthesized by copolymerization of HPMA, ALN monomer (MA-Gly-Gly-Pro-Nle-ALN), and amino group containing monomer (MA-Gly-Gly-Pro-Nle-NH(CH2)2NH2). Optionally, for the evaluation of biodistribution, a polymerizable derivative of FITC, 5-[3-(methacryloylaminopropyl) thioureidyl] fluorescein (MA-FITC), was added to the monomer mixture. To achieve control of the copolymer Mw and PDI we used controlled/“living” free radical polymerization (RAFT) technique. In the second step, TNP-470 was linked to amino groups by nucleophilic substitution of the terminal chlorine of TNP-470 (Scheme 1). We have previously synthesized a 80 kDa HPMA copolymer-ALN-TNP-470 conjugate with a mean size distribution of 100 nm [38]. Here, the Mw and the PDI of the conjugate were estimated by SEC and exhibited apparent Mw of 30 kDa and PDI of 1.2. Additionally, the conjugate’s effective diameter and PDI were determined by dynamic light scattering (DLS) and were measured as 21.7 ± 0.7 nm and 0.13 ± 0.05 respectively (table in Scheme 1). The loading percentage of ALN and TNP-470 on FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate was measured as 6.3 and 7.7 mol% respectively. For biodistribution studies, we synthesized a non-targeted control conjugate of HPMA copolymer-FITC-Tyr (P-FITC-Tyr) with Mw of 27.7 kDa. The content of FITC and Tyr was measured as 0.7 mol% and 1.8 mol% respectively.
3.2. Hemolysis and WBC count following treatment
The clinical manifestation of chemotherapy-related toxicity includes hematological disorders such as decreased production of red blood cells (anemia), low levels of white blood cells (neutropenia or granulocytopenia), and low levels of platelets (thrombocytopenia), which may be life-threatening to the patient [53–55]. Preventing these side effects would be valuable for treating patients more effectively. To evaluate the biocompatibility of HPMA copolymer-ALN-TNP-470 conjugate, we performed red blood cell (RBC) lysis assay ex vivo and white blood cells (WBC) count following treatment with the conjugate in vivo. Briefly, rat red blood cell solution (2% w/w) was incubated with serial dilutions of free or conjugated ALN and TNP-470 for 1 h at 37 °C. Negative controls were PBS and dextran (Mw of 70 kDa), and positive controls were a 1% w/v solution of Triton X-100 (100% lysis) and PEI. The results show that HPMA copolymer-ALN-TNP-470 conjugate was not hemolytic ex vivo at concentrations up to 5 mg/ml (Fig. 1A). For peripheral WBC count, blood samples were drawn from the retro-orbital sinus. The cell counts obtained from blood sample drawn from mice in each group were compared. Mice treated with a combination of free ALN plus TNP-470 had a minor insignificant higher level of WBC compared with the control mice (p = 0.138) (Fig. 1B). In tumor-bearing mice the combination of free ALN plus TNP-470 had different effect on WBC levels as described comprehensively later in the Results Section. Mice treated with HPMA copolymer-ALN-TNP-470 conjugate had similar levels of WBC as mice in the control group (p = 0.303) (Fig. 1B). Overall, these results show that the conjugate did not induce depression of the peripheral WBC and found to be hemocompatible to Balb/c mice.
Fig. 1.

HPMA copolymer-ALN-TNP-470 conjugate is hemocompatible and does not cause leukopenia in non-tumor bearing mice. (A) Red Blood Cells Lysis (RBC) assay of free or conjugated ALN and TNP-470. HPMA copolymer-ALN-TNP-470 conjugate (closed triangles) did not induce hemolysis at all tested concentrations compared with free ALN plus TNP-470 (open triangles) that induced hemolytic effect at concentrations higher than 1 mg/ml. Positive control of PEI (closed squares) showed dose dependent hemolysis. Results are presented as % of hemoglobin release ± s.d. (B) Effect of free or conjugated ALN and TNP-470 on peripheral WBC counts in Balb/c mice. Free combination of ALN plus TNP-470 had insignificant effect on WBC levels similar to HPMA copolymer-ALN-TNP-470 conjugate as compared with mice treated with vehicle (control group). Data represent mean ± s.e.m.
3.3. Body weight change of treated non-tumor-bearing mice
An important indicator of a non-specific toxic effect following treatments with anticancer chemotherapy is body weight decrease. Therefore, we monitored the mice body weights following treatments with the conjugate. Free or conjugated ALN and TNP-470 (30 mg/kg s.c., q.o.d. TNP-470-equivalent dose) were injected to 28 days old non-tumor bearing Balb/c mice (Fig. 2A). Mice treated with HPMA copolymer-ALN-TNP-470 conjugate did not produce any observable side effect and gained weight similar to the control group. Mice treated with free ALN plus TNP-470 were found to be lethargic, and lost more than 15% of their initial body weight and had to be euthanized on day 24 according to the animal committee guideline. HPMA copolymer-ALN-TNP-470 conjugate showed an obvious reduction in non-specific systemic toxic effect compared with free ALN plus TNP-470, as evidenced by the difference in the weight change (p < 0.01 on day 24). Their body weight increased during the course of the treatment (Fig. 2A).
Fig. 2.

HPMA copolymer-ALN-TNP-470 conjugate does not cause body weight loss nor affects the neurological function of non-tumor bearing mice. (A) Treatment with free combination of ALN plus TNP-470 (open triangles) hindered weight gain in mice, whereas HPMA copolymer-ALN-TNP-470 conjugate (closed triangles) did not affect body weights similar to control mice (open circles). (B) Mice treated with HPMA copolymer-ALN-TNP-470 conjugate remained on the rotarod similar time as the control group, whereas mice treated with free combination of ALN plus TNP-470 remained significantly shorter period of times. Data represent mean ± s.e.m.
3.4. Neurological function following treatment
In clinical trials, TNP-470 treatment resulted in severe ataxia and other symptoms of cerebellar dysfunction in humans [28]. We therefore tested the effects of free and conjugated ALN and TNP-470 on the motor coordination and balance skills of mice using the rotarod test, a classic assay for ataxia in rodents. Mice were placed on a rotating rod and the time for which the mice remained on the rod was recorded. The performance of animals treated with HPMA copolymer-ALN-TNP-470 conjugate was similar to that of the control mice, whereas mice treated with a combination of free ALN plus TNP-470 remained on the rotating rod for a significantly (p = 0.0045) shorter time (average of 8 s) than mice in the other two groups (Fig. 2B). These results clearly indicate that HPMA copolymer-ALN-TNP-470 conjugate does not affect the motor coordination in mice whereas free combination of ALN plus TNP-470 cause ataxia and affect the neurological function.
3.5. Measurement of viable CEC and CEP levels in treated non-tumor-bearing mice
CEC are a mixed population of mature and non-mature endothelial cells. Recent studies suggested that apoptotic CEC correlated with the anti-angiogenic treatment outcome [48,56]. It has been postulated that apoptotic CEC are, in fact, sloughed off the vessel wall as a result of a tumor vascular damage [48] and therefore such cells have been suggested as a surrogate biomarker of treatment outcome of anti-angiogenic therapy [45]. In addition to CEC, also viable CEP have been shown to correlate with angiogenic activity in a variety of preclinical models [46]. Collectively, CEC and CEP can be used as surrogate (minimally-invasive) measurable biomarkers for anti-angiogenic activity [57]. To assess the anti-angiogenic effect of HPMA copolymer-ALN-TNP-470 conjugate, blood was collected from non-tumor bearing mice treated with free or conjugated ALN and TNP-470. Levels of viable CEP, viable CEC and apoptotic CEC were measured by flow cytometry (Fig. 3). The levels of viable CEP and viable CEC in blood collected from mice treated with a combination of free ALN plus TNP-470 and in blood collected from mice treated with HPMA copolymer-ALN-TNP-470 conjugate were similar to the levels measured in the blood collected from mice treated with vehicle (control group). However, apoptotic CEC levels in blood collected from mice treated with a combination of free ALN plus TNP-470 were significantly higher compared with control group (p < 0.03), whereas mice treated with HPMA copolymer-ALN-TNP-470 conjugate showed similar levels of apoptotic CEC to the control group. The high levels of apoptotic CEC originated from the vasculature and released into the blood circulation in the non-tumor-bearing mice suggest that the combination of free ALN plus TNP-470 may have toxic anti-angiogenic effects on the normal vasculature as these mice have no tumors.
Fig. 3.
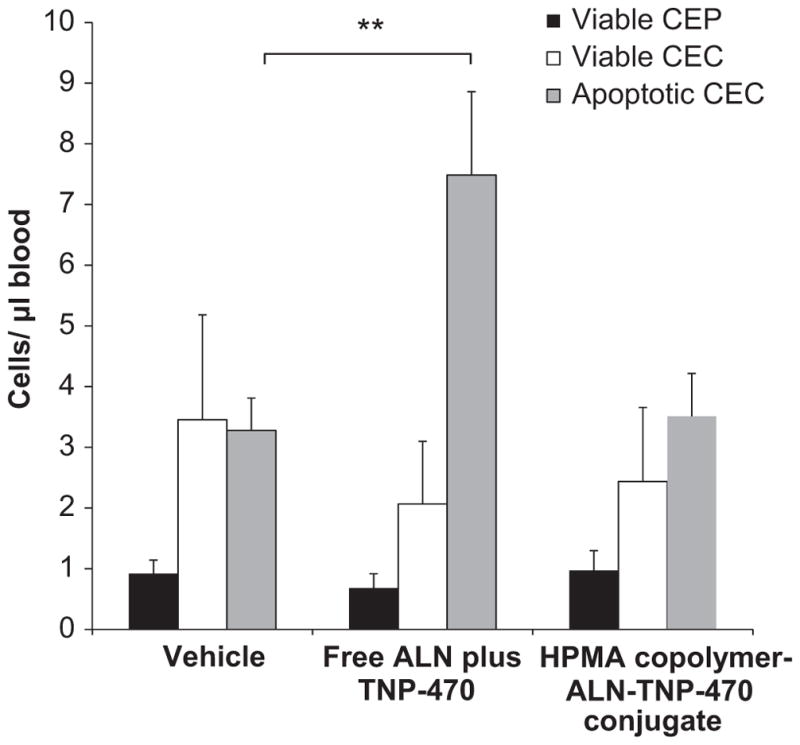
Assessment of viable CEP, viable CEC and apoptotic CEC in non-tumor bearing mice treated with free or conjugated ALN and TNP-470. Free combination of ALN plus TNP-470 had no effect on viable CEP (black bars) and viable CEC (white bars) levels. However, free combination of ALN plus TNP-470 caused a significant induction effect on apoptotic CEC (gray bars) levels, whereas HPMA copolymer-ALN-TNP-470 conjugate had no significant effect on the levels of viable CEP (black bars), viable CEC (white bars) and apoptotic CEC (gray bars) compared with the control group. Data represent mean ± s.e.m.
3.6. Cytotoxic effect of HPMA copolymer-ALN-TNP-470 conjugate on murine osteosarcoma cells
Having shown that HPMA copolymer-ALN-TNP-470 conjugate displays an acceptable safety profile when administrated to non-tumor bearing mice, we next evaluated the anti-tumor activity of the conjugate in vitro. We have previously shown that HPMA copolymer-ALN-TNP-470 conjugate has anti-tumor activity by inhibiting human osteosarcoma proliferation in vitro [38]. In this study we used syngeneic tumor model in immunocompetent mice instead of the human osteosarcoma xenograft in SCID mice. Therefore, prior to the efficacy and safety studies in vivo, we examined the ability of the conjugate to inhibit proliferation of murine osteosarcoma cells in vitro. K7M2 murine osteosarcoma cells were incubated for 72 h with serial dilutions of free combination of ALN plus TNP-470 or with HPMA copolymer-ALN-TNP-470 conjugate (at TNP-470-equivalent concentration). Viable cells were counted by a Z1 Coulter® Particle Counter (Beckman Coulter™). Both free and conjugated ALN-TNP-470 had cytotoxic effects by inducing growth inhibition on K7M2 cells at IC50 of 10 μM Fig. 4). HPMA copolymer alone was inert in vitro and in vivo (data not shown) in agreement with previously published data [58].
Fig. 4.

HPMA copolymer-ALN-TNP-470 conjugate inhibits the proliferation of K7M2 murine osteosarcoma cells. Free (open triangles) and polymer-conjugated ALN and TNP-470 (closed triangles) had similar effect on cell proliferation exhibiting retention of activity following polymer conjugation. Free and conjugated ALN and TNP-470 inhibited serum-induced proliferation of K7M2 murine osteosarcoma at IC50 of 10 μM. Data represent mean ± s.d.
3.7. FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate biodistribution
For biodistribution studies we used a rapid fluorescence-based method. This radioactive-free approach allows easy monitoring and quantitative measurements of fluorescence-labeled conjugates distributed in mice tissues [59]. Overlay images composed of brightfield and FITC components show the localization of targeted FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate and non-targeted FITC-labeled HPMA copolymer in resected organs (Fig. 5A). Targeted HPMA copolymer-ALN-TNP-470 conjugate showed high localization in the kidneys, bones, and tumor and relatively low accumulation in the heart, spleen and liver. Non-targeted HPMA copolymer-FITC-Tyr conjugate was accumulated mainly in the kidneys, liver and tumor. As expected, the non-targeted conjugate was localized in the kidneys and tumor but did not accumulate in the bones, spleen or heart. Quantification of FITC component revealed 1.5- and 19-fold increase in total signal (scaled counts/sec)/tissue weight of targeted conjugate over non-targeted conjugate at the tumor site and in the bones respectively (Fig. 5B and C). Conversely, in the liver and in the kidneys, the ratios of targeted and non-targeted conjugates were 0.3 and 0.4 respectively. There was no significant difference in the heart and spleen localization between the non-targeted and targeted conjugates (Fig. 5C). The targeting-dependent biodistribution profile of FITC-labeled conjugates in the heart, kidneys, lungs, liver, spleen and bones highlights the contribution of ALN as a targeting moiety, in agreement with previously published data [34].
Fig. 5.

Biodistribution of FITC-labeled HPMA copolymer-ALN-TNP-470 conjugate in K7M2 tumor-bearing Balb/c mice. (A) Overlay images of FITC-fluorescence spectrum and dissected organs (composed images of unmixed multispectral cubes) of mice treated with FITC-labeled targeted HPMA copolymer-ALN-TNP-470 conjugate (upper panel) and FITC-labeled non-targeted HPMA copolymer (lower panel). (B) Quantification of FITC-fluorescence component (total signal of photons/sec/tissue weight) showing targeting-dependent biodistribution profiles of targeted and non-targeted FITC-labeled conjugates at the tumor, liver, heart, bones, kidneys and spleen. (C) Table presenting the targeted/non-targeted ratio of the signals at each organ and the statistical analysis. Data represent mean ± s.e.m.
3.8. Body weight change of treated tumor-bearing mice
We next evaluated the toxicity profile of the conjugate in mice bearing K7M2 murine osteosarcoma. Free or conjugated ALN and TNP-470 (30 mg/kg s.c., q.o.d. TNP-470-equivalent dose) were injected to K7M2 osteosarcoma-bearing mice and body weights were monitored (Fig. 6A). Mice treated with HPMA copolymer-ALN-TNP-470 conjugate did not produce concomitant overt signs of toxicity and gained more than 10% weight up to experiment termination (day 30). The response of K7M2 tumor-bearing mice and non-tumor-bearing mice to treatment with HPMA copolymer-ALN-TNP-470 conjugate was similar in terms of body weight percent change and noticeable side effects. Mice treated with vehicle (control group) gained weight similar to mice treated with the conjugate. However, on day 16 mice had to be euthanized due to tumor size and the disability of the animals to walk properly. As opposed to the other two groups, mice treated with the combination of free ALN plus TNP-470 were found to be lethargic, experienced more than 15% weight loss, and had to be euthanized on day 20 according to the animal committee guideline. The body weight percent changes of mice in this group were significantly lower compared with mice treated with the conjugate (p < 0.01), indicating a severe toxic response to treatment with the combination of free ALN plus TNP-470. These findings indicate that conjugating ALN and TNP-470 to HPMA copolymer backbone considerably reduce the systemic side effects accompanied with the free drugs.
Fig. 6.

HPMA copolymer-ALN-TNP-470 conjugate does not cause weight loss, induce the levels of viable and apoptotic CEC and does not affect WBC levels in K7M2 tumor-bearing mice. (A) Treatment with free combination of ALN plus TNP-470 (open triangles) caused weight loss in mice whereas HPMA copolymer-ALN-TNP-470 conjugate (closed triangles) did not affect body weights similar to control mice (open circles). (B) Mice treated with HPMA copolymer-ALN-TNP-470 conjugate showed higher levels of viable CEC (white bars) and apoptotic CEC (gray bars) compared with mice treated with free combination of ALN plus TNP-470 and the control group. (C) Effect of free or conjugated ALN and TNP-470 on peripheral WBC counts in K7M2 tumor-bearing Balb/c mice. Mice treated with free combination of ALN plus TNP-470 had decreased levels of peripheral WBC, whereas mice treated with HPMA copolymer-ALN-TNP-470 conjugate had similar levels of WBC as mice treated with vehicle (control group). Data represent mean ± s.e.m.
3.9. Measurement of viable CEC and CEP levels in treated tumor-bearing mice
Having shown that HPMA copolymer-ALN-TNP-470 conjugate abrogates the toxicity effects caused by free ALN plus TNP-470 administration, we next measured the levels of viable CEP, viable CEC and apoptotic CEC in mice bearing K7M2 tumors in the tibia. In non-tumor bearing mice, as expected, the conjugate did not affect viable CEP, viable CEC or apoptotic CEC levels whereas the combination of free ALN plus TNP-470 increased the levels of apoptotic CEC suggesting that such treatment may affect the normal vascu-lature. Treatment with HPMA copolymer-ALN-TNP-470 conjugate in K7M2 tumor-bearing mice increased the levels of both viable and apoptotic CEC by 4-fold compared with control (Fig. 6B). Treatment with the combination of free ALN plus TNP-470 increased the levels of apoptotic CEC by 2-fold compared with control mice, but did not affect viable CEC levels. The elevated levels of apoptotic CEC found in the peripheral blood can be associated with anti-angiogenic activity in response to treatments. However, as opposed to treatment with free ALN plus TNP-470 which has shown increased apoptotic CEC levels in both non-tumor and tumor-bearing mice, the conjugate has shown a significant increase in apoptotic CEC only in tumor-bearing mice but not in non-tumor-bearing mice, suggesting that such therapy is mainly targeting the tumor vascu-lature, but not normal vasculature. Interestingly, elevated levels of viable CEC were detected in tumor-bearing mice treated with the conjugate but not in non-tumor-bearing mice. Therefore, our results suggest that the apoptotic CEC found in peripheral blood of K7M2-bearing mice following treatment with the conjugate, were released from the tumor vasculature and not from healthy blood vessels.
3.10. Effect of treatment on WBC levels of tumor-bearing mice
HPMA copolymer-ALN-TNP-470 conjugate did not affect the peripheral WBC of K7M2 tumor-bearing mice similar to its effect in the non-tumor bearing mice (Fig. 6C). In contrast, treatment with free combination of ALN plus TNP-470 decreased the peripheral WBC count by ~40%.
3.11. Antitumor efficacy of HPMA copolymer-ALN-TNP-470 conjugate
We have previously shown that HPMA copolymer-ALN-TNP-470 conjugate remarkably inhibited human osteosarcoma growth in SCID mice by 96% compared with 45% by free ALN plus TNP-470. It is well known that host–tumor interactions and the immune system play an important role in the angiogenic cascade and tumor progression. These crucial factors might influence the therapeutic efficacy. Therefore, we evaluated the potential of the conjugate to inhibit tumor growth in syngeneic model. Tumors were established by injection of mCherry-labeled K7M2 murine osteosarcoma cells into the tibia of Balb/c mice. Tumors were monitored by non-invasive intravital fluorescence imaging system. On day 16, HPMA copolymer-ALN-TNP-470 conjugate inhibited the growth of murine osteosarcoma by an average of 65% compared with 50% inhibition by the free ALN plus TNP-470 (Fig. 7A and B). Although the inhibitory effect of the conjugate was slightly superior to the effect of free ALN plus TNP-470 on K7M2 tumors, mice treated with the conjugate showed 50% increase in overall survival rate compared with 25% increase in mice treated with free ALN plus TNP-470 compared with control group (Fig. 7C). Taken together, in murine osteosarcoma model, the conjugate exhibited improved tumor inhibition activity with significantly reduced toxicity and side effects compared with the combination of free ALN plus TNP-470. Maximum tolerated dose (MTD) was not achieved for the conjugate.
Fig. 7.

HPMA copolymer-ALN-TNP-470 conjugate inhibits K7M2 murine osteosarcoma and prolong the survival rate of the mice. (A) On day 16 HPMA copolymer-ALN-TNP-470 conjugate (closed triangles) inhibited tumor growth by 64% (P = 0.003) compared with 45% (P = 0.027) of free ALN and TNP-470 (open triangles). Data represent mean ± s.e.m. (B) Intravital non-invasive fluorescence imaging of mCherry-labeled K7M2 tumor-bearing mice, treated with free or conjugated ALN and TNP-470 and taken on day 16 from best responses achieved. (C) Percent survival of mice treated with HPMA copolymer-ALN-TNP-470 conjugate (closed triangles), a combination of free ALN plus TNP-470 (open triangles) compared with control group (squares). Scale bar represents 10 mm (n = 5 mice per group).
4. Discussion
The present study describes a strategy of targeted combination therapy for the treatment of cancer-related bone diseases such as osteosarcoma and bone metastases. Using SPPS and RAFT polymerization techniques, we synthesized and characterized HPMA copolymer-based polymer–drug conjugate bearing ALN, TNP-470 and FITC (used as a detection moiety). ALN and TNP-470 were conjugated via Gly-Gly-Pro-Nle tetrapeptide. This cleavable linker allows liberation of the active agents in the presence of cathepsin K, a cysteine protease secreted from osteoclasts into the resorption lacuna, mainly in bone-related diseases with high absorption activity [60]. The resulting 30 kDa HPMA copolymer-ALN-TNP-470 conjugate showed biocompatiblity in vitro and in vivo, and bio-distribution studies revealed high localization of FITC-labeled conjugate at the tumor site. Finally, the conjugate increased the efficacy of the combination of free ALN and TNP-470 while abrogating its toxicity, selectively inhibited tumor-induced neo-vascularization and improved the survival rate of mice bearing orthotropic K7M2 murine osteosarcoma.
Comparable conjugate with Mw of 80 kDa was previously synthesized and its activity was evaluated in a model of human osteosarcoma xenograft in SCID mice [38]. The renal threshold in a healthy organism is in the range of 30–50 kDa [25]. To ensure elimination by glomerular filtration following parenteral administration, we aimed to synthesize a conjugate with a final size lower than the renal threshold. For that, we calculated the monomer/chain transfer agent ratio and the conversion of the polymerization prior to RAFT polymerization. This way we achieved a conjugate with Mw of 30 kDa and narrow PDI of 1.2. Although the renal clearance of conjugates is usually related to their Mw, the effective size might influence the glomerular permeation [61]. The size of a polymeric drug delivery system is dictated not only by its Mw, but also by its conformation, charge, and hydrophobicity. The conjugate described here, consists of two therapeutic agents (i.e. ALN and TNP-470), with different chemical characteristics influencing the effective size of HPMA copolymer-ALN-TNP-470 conjugate. Therefore, beside the Mw measured by SEC profiles, further appraisal was conducted using DLS. HPMA copolymer-ALN-TNP-470 conjugate effective diameter and PDI were measured as 18.1 ± 0.6 nm and 0.13 ± 0.05 respectively, warranting concomitant glomerular permeation.
NBP such as ALN inhibit the activity of farnesyl pyrophosphate synthetase, which directly suppresses the geranygeranylation and farensylation of small G-proteins such as Rab, Ras and RhoA. Suppression of these key proteins results in anti-neoplastic action including: induction of apoptosis, cell cycle perturbations, anti-invasive, anti-migration and anti-angiogenic effects [62]. One of the main characteristics of ALN, which makes the direct anti-angiogenic/anti-tumor activity difficult to demonstrate in vivo, is its pharmacokinetic profile which exhibits a strong affinity to bone mineral under physiological conditions [63]. Utilizing ALN qualities for targeting tumors not necessarily confined to bony tissues can be achieved by altering its pharmacokinetic profile. Alternatively, ALN can be used as both targeting moiety and therapeutic agent. Different drug-delivery systems for several applications bearing a bone targeting moiety have been previously reported. This includes, HPMA copolymer-doxorubicin-hydroxybisphosphonate conjugate [64], HPMA copolymer–alendronate–paclitaxel conjugate [65] and fluorescein-labeled bone-targeted model conjugates for detection purposes bearing 1% loading of ALN or D-aspartic acid (D-Asp8) oligopeptides [33,35,37,66–68]. The common denominator of these conjugates was that they all selectively accumulate in bone tissues regardless of relatively low loading percentage of their bone targeting moiety. Here, by combining SPPS and RAFT, we achieved high loading percentage of ALN (6.3 wt%) and TNP-470 (7.7 wt%) on the polymeric backbone. This offers both rapid accumulation in bone tumors and metastases (which produce calcified matter) and enhances its anti-tumor and anti-angiogenic activity.
In addition, ALN as a potent targeting moiety has a distinct advantage over classic receptor mediated targeting (active) moieties. The number of cell surface receptors and their availability determine how many molecules of a targeting compound can be specifically bound [69]. Since receptor capacity is limited, saturation of receptors could affect the specificity and efficiency of receptor-mediated targeting. ALN, in contrast, can be defined as a highly potent non-receptor mediated targeting moiety, therefore has the potential to be very effective as a targeting moiety for bone neoplasms.
In addition to the physical properties of targeted delivery systems and the loading percentage of the active agents, many efforts have been directed to the optimization of the active components combinations [70]. The rational of choosing the therapeutic agents for polymer-based combination delivery system for cancer therapy is based on several elements. The active agents will preferably have different toxicity, mechanism of action, and mechanism of resistance, as well as anti-angiogenic and anti-tumor activity, and synergistic effect [71]. In regards to our combined targeted delivery system, we implemented these principles when conjugated ALN and TNP-470 with HPMA copolymer backbone. Their chemical features, mechanism of action and molecular targets are dissimilar, and recent study has shown that combination treatment with ALN plus TNP-470 has anti-angiogenic and anti-tumor synergistic effect [38].
The biocompatibility and the toxicity profile of drug delivery systems is of fundamental importance for their possible therapeutic uses [72]. As a part of the evaluation hierarchy, we performed safety and toxicity studies prior to the determination of the efficacy profile. Taken into consideration the administration route and the known side effects associated with ALN and TNP-470, we first evaluated their compatibility with blood components. RBC hemolysis assay showed that the conjugate is hemocompatible up to a dose of 5 mg/ml whereas the combination of free ALN plus TNP-470 was hemolytic in concentration higher than 1 mg/ml. When administrated to Balb/c non-tumor bearing mice, the conjugate did not induce depression of the peripheral WBC similar to its effect in tumor-bearing mice. Treatment with the combination of free ALN plus TNP-470 did not affect the peripheral WBC levels of non-tumor-bearing mice but decreased the peripheral WBC levels by ~40% of tumor-bearing mice. The dissimilarity between the effect of free ALN plus TNP-470 on total WBC levels in tumor-bearing mice and in non-tumor-bearing mice could be partially explained by the effect of ALN on bone-active cytokines and high resorption activity of osteoclasts at tumor site. NBP such as ALN can stimulate the release of bone-active cytokines such as IL-1β or TNF-α that induce transendothelial migration of certain cells of the immune system from the peripheral blood into perivascular tissues [54]. This affects the redistribution of peripheral WBC and potentially cause transient leukopenia [73]. In non-tumor-bearing mice the osteoclasts have low resorption activity and administration of ALN does not induce leukopenia therefore the total WBC levels remain normal. In mice bearing K7M2 osteosarcoma tumors, there is high pathological bone resorption activity of osteoclasts and administration of ALN induce migration of immune system cells to peri-vascular tissues thus affecting the total WBC count. Further safety assessments were performed by rotarod test, evaluating the motor coordination and balance skills of non-tumor-bearing mice. In agreement with previously published data [21], the results clearly show that TNP-470-related neurotoxicity was avoided following conjugation to HPMA copolymer backbone.
The data obtained from biodistribution studies support the effective targeting of HPMA copolymer-ALN-TNP-470 conjugate to tumor site. The accumulation of FITC-labeled conjugate in different organs varied, while highest accumulation was measured at tumor site and bones. In kidneys and liver, the conjugate accumulation was higher than the heart and spleen, presumably due to the renal filtration and retrieving and the presence of sinusoidal blood vessel structures in liver. Similar to other detection moieties used for tracing, FITC was conjugated through a non-cleavable linker to HPMA copolymer backbone. Although we used advanced fluoresce technique to conduct biodistribution studies, the acquired data represent solely the localization of the complete conjugate, regardless to the fate of ALN and TNP-470. The crucial data on time and location of ALN and TNP-470 release from the conjugate could be extremely valuable particularly if acquired at real-time. This could be achieved by coupling latent fluorophore activation to the drug release event that will report the release by an on/off fluorescent signal [74]. Such a reporting drug-delivery system could potentially clarify the mechanisms of absorption and distribution, the chemical changes of the substance in the body (e.g. by enzymes) and the effects and routes of excretion of the metabolites of ALN and TNP-470.
Our next goal was to evaluate the ability of the conjugate to inhibit tumor-induced neovascularization and tumor growth. We aimed to examine the response to therapy in a mice model that will recapitulate many of the defining features of human osteosarcoma, and will resemble the clinical setting of the malignancy. For that reason, we chose an orthotropic model of K7M2 murine osteosarcoma inoculated in Balb/c mice tibia (syngeneic model). It has been shown that orthotropic tumor models better approximate the tumor microenvironment of naturally occurring tumors in humans, and thus serve as better models for tumor progression and therapy-response examination [75–77]. It has also been proposed that the use of syngeneic tumors in immunocompetent mice is superior to that of human xenografts in NOD/SCID mice [78,79]. The ability of the conjugate to inhibit angiogenesis was assessed by measurements of viable and apoptotic CEC and viable CEP levels in mice blood. In non-tumor-bearing mice, the conjugate did not affect viable CEP, viable CEC and apoptotic CEC levels, whereas in tumor-bearing mice the conjugate increased the levels of both viable and apoptotic CEC. It is plausible that in K7M2 tumor-bearing mice the vascular network was developed, and the anti-angiogenic effect of the conjugate resulted in shading of endothelial cells from tumor blood vessels into the circulation. However, in this study we cannot rule out the possibility that some of the viable CEC are derived from the bone-marrow compartment. On the other hand, in non-tumor bearing mice, no tumor-related neovascularization occurred, and the conjugate did not exhibit measurable anti-angiogenic activity, suggesting that such treatment is directed solely to the tumor endothelium. Administration of the combination of free ALN plus TNP-470 resulted in elevated levels of apoptotic CEC in both non-tumor and tumor-bearing mice. Overall, the conjugate not only exhibited improved anti-angiogenic activity but also enhanced specificity. These findings suggest that conjugating ALN and TNP-470 to HPMA copolymer backbone altered the non-specific anti-angiogenic activity of the free agents and reduced their undesired-associated side effects. Finally, the conjugate substantially enhanced the activity of ALN and TNP-470 in vivo in K7M2 tumor model and improved the overall survival rate of mice.
The promising early preclinical results presented in this study, encourage further evaluation of our therapy and open the possibility of using HPMA copolymers as platforms for delivery of a cocktail of active agents to tumors. The use of dual-targeted delivery system that co-delivers two therapeutic agents at a single administration is not confined to bone-related malignancies exclusively, and can be used as a powerful strategy to prevent the transition of dormant avascular tumors to fast-growing angiogenic tumors.
5. Conclusions
We demonstrated a concept of a narrowly-dispersed combined polymer therapeutic designed to target bone-related malignances such as osteosarcoma and bone metastases by co-delivery of two synergistic drugs at a single administration. A polymer–drug conjugate of HPMA copolymer-ALN-TNP-470 at the size of 30 kDa was synthesized and characterized in murine syngeneic model of osteosarcoma. The conjugate showed increased anti-tumor efficacy and decreased toxicity compared to the combination of free drugs, and specifically inhibited tumor-induced neovascularization. Potential outcome of the proposed therapy if successful is to improve the quality of life for individuals and their families living with cancer.
Acknowledgments
This study was supported (in part) by grant no. 5145-300000 from the Chief Scientist Office of the Ministry of Health, Israel, THE ISRAEL SCIENCE FOUNDATION (grant No. 1309/10), a grant from the Alon Foundation, the Israel Cancer Association, Israel Cancer Research Fund (RSF) and NIH grant RO1 GM069847 (JK). We thank the TAU Center for Nanoscience and Nanotechnology and The European Association for Cancer Research for their financial support (ES). This work is also supported by ISF and Marie Curie (under the FP7 EU program) given to YS. LB is supported by Fine, Jacobs and Israel Student Education Foundation studentships. Dr. Paula Ofek is acknowledged for the generation of mCherry-infected K7M2 cell line.
References
- 1.Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007;26:1–18. doi: 10.1089/dna.2006.0505. [DOI] [PubMed] [Google Scholar]
- 2.DuBois S, Demetri G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer. 2007;109:813–9. doi: 10.1002/cncr.22455. [DOI] [PubMed] [Google Scholar]
- 3.Bielack SS, Marina N, Ferrari S, Helman LJ, Smeland S, Whelan JS, et al. Osteosarcoma: the same old drugs or more? J Clin Oncol. 2008;26:3102–3. doi: 10.1200/JCO.2008.17.1108. [DOI] [PubMed] [Google Scholar]
- 4.Roodman GD. Role of cytokines in the regulation of bone resorption. Calcif Tissue Int. 1993;53(Suppl 1):S94–8. doi: 10.1007/BF01673412. [DOI] [PubMed] [Google Scholar]
- 5.Harvey HA, Cream LR. Biology of bone metastases: causes and consequences. Clin Breast Cancer. 2007;7(Suppl 1):S7–13. doi: 10.3816/cbc.2007.s.001. [DOI] [PubMed] [Google Scholar]
- 6.Berenson JR, Lipton A. Bisphosphonates in the treatment of malignant bone disease. Annu Rev Med. 1999;50:237–48. doi: 10.1146/annurev.med.50.1.237. [DOI] [PubMed] [Google Scholar]
- 7.Lipton A. Emerging role of bisphosphonates in the clinic-antitumor activity and prevention of metastasis to bone. Cancer Treat Rev. 2008;34(Suppl 1):S25–30. doi: 10.1016/j.ctrv.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Orcel P, Beaudreuil J. Bisphosphonates in bone diseases other than osteoporosis. Joint Bone Spine. 2002;69:19–27. doi: 10.1016/s1297-319x(01)00336-0. [DOI] [PubMed] [Google Scholar]
- 9.Hillner BE. The role of bisphosphonates in metastatic breast cancer. Semin Radiat Oncol. 2000;10:250–3. doi: 10.1053/s1053-4296(00)80042-9. [DOI] [PubMed] [Google Scholar]
- 10.Koka S, Salinas TJ, Kennel KA. Osteoporosis, fracture risk, and prosthodontic implications. Int J Prosthodont. 2009;22:505–6. [PubMed] [Google Scholar]
- 11.Leah E. Therapy: bisphosphonate users: cancer risk. Nat Rev Rheumatol. 2010;6:616. doi: 10.1038/nrrheum.2010.167. [DOI] [PubMed] [Google Scholar]
- 12.Kennel KA, Drake MT. Adverse effects of bisphosphonates: implications for osteoporosis management. Mayo Clin Proc. 2009;84:632–7. doi: 10.1016/S0025-6196(11)60752-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quan GM, Choong PF. Anti-angiogenic therapy for osteosarcoma. Cancer Metastasis Rev. 2006;25:707–13. doi: 10.1007/s10555-006-9031-1. [DOI] [PubMed] [Google Scholar]
- 14.Lee YH, Tokunaga T, Oshika Y, Suto R, Yanagisawa K, Tomisawa M, et al. Cell-retained isoforms of vascular endothelial growth factor (VEGF) are correlated with poor prognosis in osteosarcoma. Eur J Cancer. 1999;35:1089–93. doi: 10.1016/s0959-8049(99)00073-8. [DOI] [PubMed] [Google Scholar]
- 15.Kaya M, Wada T, Nagoya S, Sasaki M, Matsumura T, Yamashita T. The level of vascular endothelial growth factor as a predictor of a poor prognosis in osteosarcoma. J Bone Joint Surg Br. 2009;91:784–8. doi: 10.1302/0301-620X.91B6.21853. [DOI] [PubMed] [Google Scholar]
- 16.Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80:1588–94. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- 17.Folkman J. Endogenous angiogenesis inhibitors. APMIS. 2004;112:496–507. doi: 10.1111/j.1600-0463.2004.apm11207-0809.x. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 19.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez PM, Rickles FR. Tissue factor and angiogenesis in cancer. Curr Opin Hematol. 2002;9:401–6. doi: 10.1097/00062752-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Satchi-Fainaro R, Puder M, Davies JW, Tran HT, Sampson DA, Greene AK, et al. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat Med. 2004;10:255–61. doi: 10.1038/nm1002. [DOI] [PubMed] [Google Scholar]
- 22.Egerton N. Optimizing ixabepilone treatment schedules in patients with advanced or metastatic breast cancer. Cancer Chemother Pharmacol. 2010;66:1005–12. doi: 10.1007/s00280-010-1467-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 24.Wong AL, Chou N, Lee KM, Ang BW, Cheng CL, Lee SC. Subdural collections arising from calvarial metastases following discontinuation of anti-angiogenic therapy. Ann Oncol. 2009;20:1605–6. doi: 10.1093/annonc/mdp368. [DOI] [PubMed] [Google Scholar]
- 25.Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed Engl. 2006;45:1198–215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 26.Satchi-Fainaro R, Mann-Steinberg H. TNP-470: the resurrection of the first synthetic angiogenesis inhibitor. In: Figg W, Folkman J, editors. Angiogenesis: an integrative approach from science to medicine. Heidelberg, Germany: Springer-Verlag; 2008. pp. 387–406. [Google Scholar]
- 27.Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, et al. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature. 1990;348:555–7. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- 28.Bhargava P, Marshall JL, Rizvi N, Dahut W, Yoe J, Figuera M, et al. A phase I and pharmacokinetic study of TNP-470 administered weekly to patients with advanced cancer. Clin Cancer Res. 1999;5:1989–95. [PubMed] [Google Scholar]
- 29.Kruger EA, Figg WD. TNP-470: an angiogenesis inhibitor in clinical development for cancer. Expert Opin Investig Drugs. 2000;9:1383–96. doi: 10.1517/13543784.9.6.1383. [DOI] [PubMed] [Google Scholar]
- 30.Satchi-Fainaro R, Mamluk R, Wang L, Short SM, Nagy JA, Feng D, et al. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell. 2005;7:251–61. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 31.Convertine AJ, Ayres N, Scales CW, Lowe AB, McCormick CL. Facile, controlled, room-temperature RAFT polymerization of N-isopropylacrylamide. Bio-macromolecules. 2004;5:1177–80. doi: 10.1021/bm049825h. [DOI] [PubMed] [Google Scholar]
- 32.Segal E, Satchi-Fainaro R. Design and development of polymer conjugates as anti-angiogenic agents. Adv Drug Deliv Rev. 2009;61:1159–76. doi: 10.1016/j.addr.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Miller S, Sima M, Kopeckova P, Kopecek J. Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjug Chem. 2003;14:853–9. doi: 10.1021/bc034090j. [DOI] [PubMed] [Google Scholar]
- 34.Pan H, Sima M, Kopeckova P, Wu K, Gao S, Liu J, et al. Biodistribution and pharmacokinetic studies of bone-targeting N-(2-hydroxypropyl)methacrylamide copolymer-alendronate conjugates. Mol Pharmacol. 2008;5:548–58. doi: 10.1021/mp800003u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D, Miller SC, Shlyakhtenko LS, Portillo AM, Liu XM, Papangkorn K, et al. Osteotropic peptide that differentiates functional domains of the skeleton. Bioconjug Chem. 2007;18:1375–8. doi: 10.1021/bc7002132. [DOI] [PubMed] [Google Scholar]
- 36.Husmann K, Muff R, Bolander ME, Sarkar G, Born W, Fuchs B. Cathepsins and osteosarcoma: expression analysis identifies cathepsin K as an indicator of metastasis. Mol Carcinog. 2008;47:66–73. doi: 10.1002/mc.20362. [DOI] [PubMed] [Google Scholar]
- 37.Pan H, Kopeckova P, Wang D, Yang J, Miller S, Kopecek J. Water-soluble HPMA copolymer–prostaglandin E1 conjugates containing a cathepsin K sensitive spacer. J Drug Target. 2006;14:425–35. doi: 10.1080/10611860600834219. [DOI] [PubMed] [Google Scholar]
- 38.Segal E, Pan H, Ofek P, Udagawa T, Kopeckova P, Kopecek J, et al. Targeting angiogenesis-dependent calcified neoplasms using combined polymer therapeutics. PLoS One. 2009;4:e5233. doi: 10.1371/journal.pone.0005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170:793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Byrne KJ, Dalgleish AG, Browning MJ, Steward WP, Harris AL. The relationship between angiogenesis and the immune response in carcinogenesis and the progression of malignant disease. Eur J Cancer. 2000;36:151–69. doi: 10.1016/s0959-8049(99)00241-5. [DOI] [PubMed] [Google Scholar]
- 41.Schein PS, Scheffier B. Barriers to efficient development of cancer therapeutics. Clin Cancer Res. 2006;12:3243–8. doi: 10.1158/1078-0432.CCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 42.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin Exp Metastasis. 2000;18:261–71. doi: 10.1023/a:1006767007547. [DOI] [PubMed] [Google Scholar]
- 44.Bertolini F, Mancuso P, Braidotti P, Shaked Y, Kerbel RS. The multiple personality disorder phenotype(s) of circulating endothelial cells in cancer. Biochim Biophys Acta. 2009;1796:27–32. doi: 10.1016/j.bbcan.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Shaked Y, Bocci G, Munoz R, Man S, Ebos JM, Hicklin DJ, et al. Cellular and molecular surrogate markers to monitor targeted and non-targeted anti-angiogenic drug activity and determine optimal biologic dose. Curr Cancer Drug Targets. 2005;5:551–9. doi: 10.2174/156800905774574020. [DOI] [PubMed] [Google Scholar]
- 46.Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–7. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- 47.Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, et al. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis; implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005;7:101–11. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 48.Mancuso P, Colleoni M, Calleri A, Orlando L, Maisonneuve P, Pruneri G, et al. Circulating endothelial-cell kinetics and viability predict survival in breast cancer patients receiving metronomic chemotherapy. Blood. 2006;108:452–9. doi: 10.1182/blood-2005-11-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Omelyanenko V, Kopeckova P, Gentry C, Kopecek J. Targetable HPMA copolymer-adriamycin conjugates. Recognition, internalization, and subcellular fate. J Control Release. 1998;53:25–37. doi: 10.1016/s0168-3659(97)00235-6. [DOI] [PubMed] [Google Scholar]
- 50.Kuljanin J, Jankovic I, Nedeljkovic J, Prstojevic D, Marinkovic V. Spectrophotometric determination of alendronate in pharmaceutical formulations via complex formation with Fe(III) ions. J Pharm Biomed Anal. 2002;28:1215–20. doi: 10.1016/s0731-7085(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 51.Hashimoto K, Morishige K, Sawada K, Tahara M, Kawagishi R, Ikebuchi Y, et al. Alendronate inhibits intraperitoneal dissemination in in vivo ovarian cancer model. Cancer Res. 2005;65:540–5. [PubMed] [Google Scholar]
- 52.Duncan R, Ferruti P, Sgouras D, Tuboku-Metzger A, Ranucci E, Bignotti F. A polymer-Triton X-100 conjugate capable of PH-dependent red blood cell lysis: a model system illustrating the possibility of drug delivery within acidic intracellular compartments. J Drug Target. 1994;2:341–7. doi: 10.3109/10611869409015915. [DOI] [PubMed] [Google Scholar]
- 53.Prommer EE. Toxicity of bisphosphonates. J Palliat Med. 2009;12:1061–5. doi: 10.1089/jpm.2009.9936. [DOI] [PubMed] [Google Scholar]
- 54.Pietschmann P, Stohlawetz P, Brosch S, Steiner G, Smolen JS, Peterlik M. The effect of alendronate on cytokine production, adhesion molecule expression, and transendothelial migration of human peripheral blood mononuclear cells. Calcif Tissue Int. 1998;63:325–30. doi: 10.1007/s002239900535. [DOI] [PubMed] [Google Scholar]
- 55.Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91:1616–34. doi: 10.1093/jnci/91.19.1616. [DOI] [PubMed] [Google Scholar]
- 56.Dellapasqua S, Bertolini F, Bagnardi V, Campagnoli E, Scarano E, Torrisi R, et al. Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol. 2008;26:4899–905. doi: 10.1200/JCO.2008.17.4789. [DOI] [PubMed] [Google Scholar]
- 57.Schneider M, Tjwa M, Carmeliet P. A surrogate marker to monitor angiogenesis at last. Cancer Cell. 2005;7:3–4. doi: 10.1016/j.ccr.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 58.Hopewel JW, Duncan R, Wilding D, Chakrabarti K. Preclinical evaluation of the cardiotoxicity of PK2: a novel HPMA copolymer-doxorubicin-galactosamine conjugate antitumour agent. Hum Exp Toxicol. 2001;20:461–70. doi: 10.1191/096032701682693017. [DOI] [PubMed] [Google Scholar]
- 59.Lee MJ, Veiseh O, Bhattarai N, Sun C, Hansen SJ, Ditzler S, et al. Rapid pharmacokinetic and biodistribution studies using cholorotoxin-conjugated iron oxide nanoparticles: a novel non-radioactive method. PLoS One. 2010;5:e9536. doi: 10.1371/journal.pone.0009536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inaoka T, Bilbe G, Ishibashi O, Tezuka K, Kumegawa M, Kokubo T. Molecular cloning of human cDNA for cathepsin K: novel cysteine proteinase predominantly expressed in bone. Biochem Biophys Res Commun. 1995;206:89–96. doi: 10.1006/bbrc.1995.1013. [DOI] [PubMed] [Google Scholar]
- 61.Kopecek J, Kopeckova P. HPMA copolymers: origins, early developments, present, and future. Adv Drug Deliv Rev. 2010;62:122–49. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caraglia M, Santini D, Marra M, Vincenzi B, Tonini G, Budillon A. Emerging anti-cancer molecular mechanisms of aminobisphosphonates. Endocr Relat Cancer. 2006;13:7–26. doi: 10.1677/erc.1.01094. [DOI] [PubMed] [Google Scholar]
- 63.Gittens SA, Bansal G, Zernicke RF, Uludag H. Designing proteins for bone targeting. Adv Drug Deliv Rev. 2005;57:1011–36. doi: 10.1016/j.addr.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 64.Hrubý M, Etrych T, Kučka J, Forsterová M, Ulbrich K. Hydroxybisphosphonate-containing polymeric drug-delivery systems designed for targeting into bone tissue. J Appl Polym Sci. 2006;101:3192–201. [Google Scholar]
- 65.Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. Targeting bone metastases with a bispecific anticancer and antiangiogenic polymer--alendronate–taxane conjugate. Angew Chem Int Ed Engl. 2009;48:2949–54. doi: 10.1002/anie.200805133. [DOI] [PubMed] [Google Scholar]
- 66.Abdollahi A, Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat. 2010;13:16–28. doi: 10.1016/j.drup.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Miller SC, Pan H, Wang D, Bowman BM, Kopeckova P, Kopecek J. Feasibility of using a bone-targeted, macromolecular delivery system coupled with prostaglandin E(1) to promote bone formation in aged, estrogen-deficient rats. Pharm Res. 2008;25:2889–95. doi: 10.1007/s11095-008-9706-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang D, Miller SC, Sima M, Parker D, Buswell H, Goodrich KC, et al. The arthrotropism of macromolecules in adjuvant-induced arthritis rat model: a preliminary study. Pharm Res. 2004;21:1741–9. doi: 10.1023/b:pham.0000045232.18134.e9. [DOI] [PubMed] [Google Scholar]
- 69.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–68. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 71.Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 2010;70:6704–14. doi: 10.1158/0008-5472.CAN-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rihova B, Bilej M, Vetvicka V, Ulbrich K, Strohalm J, Kopecek J, et al. Biocompatibility of N-(2-hydroxypropyl) methacrylamide copolymers containing adriamycin. Immunogenicity, and effect on haematopoietic stem cells in bone marrow in vivo and mouse splenocytes and human peripheral blood lymphocytes in vitro. Biomaterials. 1989;10:335–42. doi: 10.1016/0142-9612(89)90075-6. [DOI] [PubMed] [Google Scholar]
- 73.Vasikaran SD, Khan S, McCloskey EV, Kanis JA. Sustained response to intravenous alendronate in postmenopausal osteoporosis. Bone. 1995;17:517–20. doi: 10.1016/8756-3282(95)00400-9. [DOI] [PubMed] [Google Scholar]
- 74.Weinstain R, Segal E, Satchi-Fainaro R, Shabat D. Real-time monitoring of drug release. Chem Commun (Camb) 2010;46:553–5. doi: 10.1039/b919329d. [DOI] [PubMed] [Google Scholar]
- 75.Dass CR, Choong PF. Zoledronic acid inhibits osteosarcoma growth in an orthotopic model. Mol Cancer Ther. 2007;6:3263–70. doi: 10.1158/1535-7163.MCT-07-0546. [DOI] [PubMed] [Google Scholar]
- 76.Ostrand-Rosenberg S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol. 2004;16:143–50. doi: 10.1016/j.coi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 77.Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Models Mech. 2008;1:78–82. doi: 10.1242/dmm.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams SS, Alosco TR, Mayhew E, Lasic DD, Martin FJ, Bankert RB. Arrest of human lung tumor xenograft growth in severe combined immunodeficient mice using doxorubicin encapsulated in sterically stabilized liposomes. Cancer Res. 1993;53:3964–7. [PubMed] [Google Scholar]
- 79.Croy BA, Linder KE, Yager JA. Primer for non-immunologists on immune-deficient mice and their applications in research. Comp Med. 2001;51:300–13. [PubMed] [Google Scholar]