

Summary
The ULK1 complex, consisting of the ULK1 protein kinase itself, FIP200, Atg13, and Atg101, controls the initiation of autophagy in animals. We determined the structure of the complex of the human Atg13 HORMA (Hop1, Rev7, Mad2) domain in complex with the full-length HORMA domain-only protein Atg101. The two HORMA domains assemble with an architecture conserved in the Mad2 conformational heterodimer and the S. pombe Atg13-Atg101 HORMA complex. The WF finger motif that is essential for function in human Atg101 is sequestered in a hydrophobic pocket, suggesting that the exposure of this motif is regulated. Benzamidine molecules from the crystallization solution mark two hydrophobic pockets that are conserved in, and unique to, animals, and are suggestive of sites that could interact with other proteins. These features suggest that the activity of the animal Atg13-Atg101 subcomplex is regulated and that it is an interaction hub for multiple partners.
Keywords: autophagy, Atg1, protein structure, hydrogen-deuterium exchange, X-ray crystallography
Graphical Abstract

Introduction
Macroautophagy (henceforward, “autophagy”) denotes a class of bulk and selective cellular self-consumptive processes that share conserved protein machinery (Mizushima and Komatsu, 2011; Reggiori and Klionsky, 2013). These processes include starvation-induced bulk autophagy, the destruction of damaged mitochondria by mitophagy, sequestration of cytosolic pathogens by xenophagy, and many others. There is intense interest in therapeutic targeting autophagy in human cells for both activation and inhibition in cancer (Galluzzi et al., 2015), infectious diseases (Manzanillo et al., 2013), and neurodegenerative diseases (Pickrell and Youle, 2015). The divergent upstream triggers of these various processes converge with the activation of the kinase complex known as ULK1 (Unc51-like kinase autophagy activating kinase 1) in animals and Atg1 in other eukaryotes (Mizushima, 2010).
In humans and other animals, the ULK1 kinase complex consists of ULK1 itself, FIP200, Atg13, and Atg101 (Mizushima, 2010). ULK1 (referred to in yeast and plants as Atg1) and Atg13 are conserved throughout eukaryotes. FIP200 is unique to animals and is thought to be functionally replaced by Atg11 and/or Atg17 in other eukaryotes (Mizushima, 2010). Atg101 (Hosokawa et al., 2009a; Mercer et al., 2009) is found throughout nearly all eukaryotes, with the exception of the budding yeasts (Fig. S1). The molecular weights of the subunits of the human ULK1 complex sum to 377 kDa, and the apparent molecular weight of the assembled complex is 3 MDa (Mizushima, 2010). Of this considerable mass, experimental structural data for human or other metazoan ULK1 components is available only for the catalytic kinase domain of ULK1 (Lazarus et al., 2015) and for 12 residues of Atg13 that bind to LC3 (Suzuki et al., 2013). These comprise just 33 kDa, or 8 % of the unique mass of the complex.
Most of what is known about the organization of the human ULK1 complex has been inferred from studies of its counterparts in yeast (Hurley and Schulman, 2014). We previously determined the structure of the Atg17 scaffold, and showed that it was a dimer with double crescent or S-shape (Ragusa et al., 2012). Atg17 is constitutively bound to two other subunits, Atg29 and Atg31 (Kabeya et al., 2009). Atg29 and Atg31 are unique to budding yeast, while Atg101 is absent. The tips of the double crescent bind to the intrinsically disordered C-terminal region of Atg13 (Fujioka et al., 2014). An adjacent region of Atg13 binds to the C-terminal EAT domain of Atg1 (Fujioka et al., 2014). The Atg1 EAT domain can be subdivided into two MIT domains, both of which are bind to short MIM motifs in the C-terminal part of Atg13 (Fujioka et al., 2014). The Atg1 EAT domain is a dimer (Stjepanovic et al., 2014). The entire yeast Atg1 assembly forms tetramers in solution, and a low resolution solution structure of the tetramer has been analyzed (Köfinger et al., 2015). The goal of this study is to begin building up a similar picture of the human ULK1 complex structure.
The N-terminus of Saccharomyces cerevisiae Atg13 (Jao et al., 2013) consists of a Hop1/Rev7/Mad2 (HORMA) domain (Aravind and Koonin, 1998), and this domain appears to present in all Atg13 orthologs. The S. cerevisiae HORMA domain has a putative phosphate binding site (Jao et al., 2013) that is important for recruiting the membrane protein Atg9 (Suzuki et al., 2015b), which recruits the autophagic PI 3-kinase complex (Jao et al., 2013; Suzuki et al., 2015b). More recently, Atg101 has been predicted to consist in its entirety of a single HORMA domain, and to dimerize with Atg13 via this domain (Hegedus et al., 2014). Both predictions have recently been confirmed (Suzuki et al., 2015a). HORMA domain structure has been intensively studied for the mitotic spindle checkpoint protein Mad2 (Chao et al., 2012; Luo et al., 2002; Mapelli et al., 2007; Sironi et al., 2001), as well as p31(comet) (Yang et al., 2007), Rev7 (Hara et al., 2010), and HIM-3 and HTP-2 (Kim et al., 2014). The Mad2 HORMA domain can interconvert between two different conformations, closed (C) and open (O). The conformation is controlled in part by the length and position of a loop known as the “safety belt”. C-Mad2 and O-Mad2 assemble into a 1:1 heterodimer, which has been crystallized (Mapelli et al., 2007).
In order to obtain insight into the Atg13-Atg101 portion of the human ULK1 complex, we co-expressed the two human proteins in insect cells and determined their structure ab initio using Br− SAD phasing. As we were preparing this manuscript, the structure of the fission yeast (S. pombe) Atg13-Atg101 subcomplex was reported at 3.0 Å resolution (Suzuki et al., 2015a), and used as a basis to make predictions about functional regions of the human proteins. This study identified a WF finger as a key interaction motif on Atg101, a feature conserved in humans. The sequences of the Atg13 and Atg101 portions of the structure are 21% and 25% identical, respectively, between S. pombe and human. As compared to the 3.0 Å resolution S. pombe structure, the 1.6 Å resolution human structure provides a detailed and accurate account of the interactions that hold the complex together, shows that the WF finger is subject to sequestration, and identifies potential animal-specific hydrophobic pockets at the subunit junction.
Results
Mapping the order/disorder boundaries of Atg13 and Atg101
Atg13 contains a mixture of ordered and intrinsically disordered regions. Human Atg13 has marginal sequence homology to its structurally characterized S. cerevisiae (Jao et al., 2013) and S. pombe (Suzuki et al., 2015a) orthologs (11.7% and 19% identity, respectively). Therefore, we considered the precise boundary between the ordered and intrinsically disordered portions of Atg13 to be uncertain. We found that it was not possible to express the Atg13 HORMA domain in isolation, suggesting that Atg13 is not stable in the absence of Atg101. We therefore co-expressed full-length Atg101 with an Atg13 construct 1–364, which includes the HORMA domain and additional ~150 residues beyond the expected C-terminal boundary of the HORMA domain (Fig. 1A, B).
FIGURE 1. Domain structure and ordered regions of Atg13 and Atg101.

(A) Schematic diagram of the domain structures of human Atg101 and Atg13. The HORMA domains of Atg101 and 13 are colored in green and cyan respectively. The boundaries of constructs we used in this paper are highlighted in red. The LIR and MIM motifs of Atg13, which interact with LC3 and ULK1, respectively, are colored blue. (B) The purified Atg101-Atg13 complexes were analyzed by SDS-PAGE. Lane 1: Atg101(1–218)-Atg13(1–364); Lane 2: Atg101(1–218)-Atg13(1–224), two bands were overlapped; Lane 3: Atg101(1–199)-Atg13(12–200). (C, D) Deuterium uptake data for the Atg13-Atg101 complex. HDX-MS data are shown in heat map format where peptides are represented using rectangular strips above the protein sequence. Absolute deuterium uptake after 10s and 30s is indicated by a color gradient below the protein sequence. See also Supplementary Figure 1.
We used hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) (Engen, 2009) to probe the flexibility of the proteins. We incubated the samples in D2O for 10 or 30 s, reasoning that intrinsically disordered regions exchange completely within 30 s. Identified peptic peptides from Atg13 (Fig. 1C) and from Atg101 (Fig. 1D) covered 83 and 97 % of the respective sequences. Ala188 of Atg13 was found to be the most C-terminal of the well-ordered residues. We also determined that the first ~11 residues of Atg13 were disordered (Fig. 1C). The analysis showed that the great majority of Atg101 was well ordered, except for the extreme C-terminus. We established Phe194 as the most C-terminal well-ordered residue. Residues 196–198 showed some protection at 10 s, but none at 30 s. The final residues 199–218 of Atg101, which includes the HORMA domain safety belt, appeared to be completely unstructured.
Crystal structure of the human Atg13-Atg101 complex
We determined the structure of the human Atg13-Atg101 complex by SAD from solvent Br− ions (Fig. 2A, B, C). We went on to refine the native Atg13-Atg101 complex structure to 1.6 Å resolution. The structures of the two proteins are well-defined, with both chains traceable from start to finish without breaks. Hydrogen positions were refined, as well as alternative conformations of side-chains where present. Every residue of Atg101 was visualized, and only the final five residues of Atg13 were absent from the electron density. The entire safety belt of the Atg13 HORMA domain was visualized, the first time this has been possible for any of the Atg13 HORMA structures.
FIGURE 2. Structure determination of the Atg13-Atg101 complex.

(A) Br− SAD synthesis displayed at a contour level of 2σ (magenta), with parts of Atg101 αA and Atg13 αC shown in green and cyan, respectively. (B) The omit map corresponding to the region shown in (A) is displayed at a contour level of 2σ (magenta). (C) The omit map in the safety belt region is displayed at a contour level of 1σ (magenta), with the safety belt colored red. (D) The overall structure of the Atg101 and Atg13 HORMA dimer shown in a ribbon representation colored as above. The αA-αB connector of Atg13, including β2′, is colored orange. (E) Side by side comparison of Atg101-Atg13, SpAtg101- Atg13 and C-Mad2-O-Mad2. From left to right: Atg101-Atg13 (green and cyan), SpAtg101- Atg13 (yellow and teal), and C-Mad2-O-Mad2 (purple and orange). See also Supplementary Figures 2 and 3.
The human Atg13 structure contains an extensive connector between αA and αB (residues 33–58; Fig. 2D, Supplementary Fig. 1A), which was not visualized in the yeast structures. This region was visualized in its entirety in the electron density despite its low degree of protection as assessed by HDX. This region includes the unique β2′ strand, which forms an inter-domain β-sheet with β2 and β3 of Atg101 (Fig. 2D).
The two HORMA domains dimerize in the same orientation as in the C-Mad2-O-Mad2 conformational dimer (Mapelli et al., 2007) and in the S. pombe Atg13-Atg101 complex (Suzuki et al., 2015a) (Fig. 2E). The entire dimer can be superimposed on the Mad2 conformational dimer with an r.m.s.d. of 2.75 Å for 274 Cα positions. In the case of the S. pombe Atg13-Atg101 dimer, the values are 1.74 Å for 270 residues. The interface buries a total of 1523 Å2 of surface area.
The Atg13 side of the interface is centered on αC and the αA-αB connector, including β2′ (Fig. 3A). The Atg101 side consists of both αA and αC and the small β2-β3 sheet. Buried polar interactions form a prominent part of the Atg13 αC-Atg101 αA interface (Fig. 3B, C, D). Atg13 Ser127 forms a 2.8 Å hydrogen bond with Nε of Atg101 His31 (Fig. 3C). These residues are conserved in S. pombe and the same interaction probably occurs in the S. pombe complex. The analysis of the 3.0 Å resolution assigned the corresponding His30 of Atg101 to interact with Thr183 of Atg13 (Suzuki et al., 2015a), which involves an unfavorable geometry with the Ser hydroxyl group at a 90° angle with respect to the plane of the His imidazole. This difference in the structures likely reflects the difference in resolution rather than a true difference between the species. A buried salt bridge is formed between Atg13 αC Arg133 and Atg101 Asp54 of the β2′- β2 loop. This interaction is bidentate and the shortest heteroatom distance is 2.64 Å (Fig. 3D). These residues are not present in S. pombe but are highly conserved amongst animals (Supplementary Fig. 1A, B). This suggests that this salt bridge is likely to contribute substantially to the stability and specificity of animal Atg13-Atg101 complexes.
FIGURE 3. The Atg13-Atg101 interface.

(A) Overall view of the interface in a surface and ribbon model. Insets indicate close-ups of the indicated regions of the interface. Key residues of the Atg101 surface are colored red while those of Atg13 are in blue. (B) Overall view of the interaction of the key residues on the interface. (C) Detail of the hydrophobic core and the hydrogen bond between Atg101 His31 and Atg13 Ser127. (D) Detail of hydrogen bond between Atg101 Asp54 and Atg13 Arg133, and packing of Atg13 β2′ against Atg101 β2. (E) HDX-MS heat map data at 10 s overlaid onto the crystal structure of the Atg13-Atg101 complex. Absolute deuterium uptake is indicated by the color gradient.
The WF finger was identified as a functionally important motif of Atg101 on the basis of its high solvent accessibility in the S. pombe structure. In the crystal lattice of the S. pombe complex, the WF motif is actually buried in a lattice contact, thus stabilizing what would otherwise be an unfavorable degree of solvent exposure for such hydrophobic residues. The structure of human Atg101 alone (Michel and Weiergraeber, unpublished RCSB entry 4WZG), confirms that the WF motif can exist in an “out” conformation in the human protein. In our structure, the Atg101 WF motif residues Trp110 and Phe112 are folded back onto the Atg101 HORMA domain. These two side-chains pack against one another, Ile117, and the aliphatic parts of Gln104, Lys107, and Arg109 (Fig. 4).
FIGURE 4. An inactive conformation of the WF finger.

(A) Upper panel: left to right: Cartoon model of the WF fingers from Atg101-Atg13 (closed conformation), Atg101 alone (Michel and Weiergraeber, unpublished RCSB entry 4WZG; unpublished RCSB entry)(open), and SpAtg101-Atg13 (4YK8)(open). Lower panel: left, comparison of the WF finger of Atg101-Atg13 and SpAtg101-Atg13; right, Atg101-Atg13 and Atg101 alone. The WF finger from human Atg101 is colored purple while that of fission yeast colored red. (B) The WF finger is partially buried by the residues around it. Cartoon (left) and surface representations (right) are shown here. Key residues are shown in a stick model.
The human Atg13-Atg101 interface
We probed the role of various parts of the Atg13-Atg101 interface using mutational analysis. Because Atg13 is not stable when expressed alone, experiments were designed in the context of co-expression of the HORMA fragments in HEK293 cells. Because the interfacial area is large, we expected that the interaction would be relatively robust to perturbation by single mutations. We therefore designed multiple mutants in order to overcome this potential obstacle. Mutants were assessed both by the ability of GST-Atg13-HORMA to pull-down Atg101, and the ability of Strep-Atg101-HORMA to pulldown Atg13 (Fig. 5A, B).
FIGURE 5. Mutational analysis of the Atg13-Atg101 interface.

HEK293 cells were cotransfected with mutant variants of GST-Atg13 and Strep-Atg101 HORMA domains. Cell lysates were precipitated with either glutathione sepharose (A) or Strep-Tactin (B) beads. The resulting precipitates were examined by SDS-PAGE.
The key polar residues of Atg13, Ser127 and Arg133, were mutated in a “role reversal” to the interaction partners in Atg101, His and Asp respectively. The S127H/R133D mutant completely blocked complex formation as judged by the absence of an Atg13 band in the Strep pull-down (Fig. 5B). In the GST pull-down, neither Atg13 nor Atg101 bands are visible, consistent with the expectation that breaking up the complex would destabilize Atg13 and so prevent its expression.
In order to test the role of the Atg13 αA-αB connector, the thirteen residues 37–49 were replaced with a tetra-Gly-Ser linker that was designed to be long enough to span the gap from residues 36–50. This loop was found to be non-essential for complex formation. This is consistent with the observation from HDX that this region exchanges rapidly, suggesting that it contributes marginally to overall stability.
The hydrophobic portion of the Atg13 αC interface was probed with the mutation I131D/V134D. This mutation sharply reduced complex formation by both assays (Fig. 5A, B). The S. pombe mutant T183R/L187R probed the same region of that structure, with the same consequence. When combined with the αC polar mutation S127H/R133D, or with both the polar mutation and the connector deletion, binding was eliminated completely (Fig. 5A, B). On the Atg101 side of the same contact, the Atg101 αC hydrophobic interface mutant I152D/I153D/V156D completely eliminated the complex, consistent with the central role for these residues in the heart of the interface.
On the Atg101 side of the polar core, the single polar mutations H31S and D54R had little impact on complex stability (Fig. 5A, B). The H31S effect is consistent with lack of effect of double large-to-small mutation F29A/H30A in the S. pombe complex. In S. pombe, the more drastic change F29R/H30R did disrupt the complex, consistent with an important role for these residues. Combining the D54R mutation with the nearby T49A/T52A pair had a marked effect on complex formation, consistent with an important role for this part of the interface. Likewise, the double mutant H31S/D54R markedly destabilized the complex, confirming the centrality of the polar core of the interface from the Atg101 side as well as the Atg13 side of the interface.
Benzamidine binding sites mark hydrophobic pockets
In the crystallographic mode of fragment-based drug discovery, targets are intentionally co-crystallized with very low molecular weight compounds to identify potential sites for inhibitor binding. This information can be built upon for drug discovery. Crystals of the human Atg13-Atg101 complex were obtained using 0.5 M benzamidine as an additive. This fortuitously revealed to us the presence of five hydrophobic pockets of potential functional interest (Fig. 6A). One benzamidine site is located near the “in” state WF finger (Fig. 6A) and participates in a lattice contact. This molecule is likely the one responsible for stabilizing the crystal. The finding that benzamidine interacts with a previously identified functional site, the WF finger, validates the concept that benzamidine binding is probing potentially significant sites.
FIGURE 6. Benzamidine binding sites mark hydrophobic pockets.

(A) Overall view of the locations of five benzamidine molecules. The complex of Atg101 and Atg13 is shown in a surface model and benzamidines are shown as a stick model. The WF finger is indicated. (B, C) Details of the environment of benzamidine molecules 1 and 2 are shown in cartoon (left) and surface (right) models. Benzamidines and highlighted residues are shown in a stick model. (D) Left: the electrostatic surface was generated at a saturating color level of ±5 kT/e. Right, residues located in the positive groove are shown in a stick model.
Of the remaining four sites, two caught our attention because they are surrounded by conserved residues in the animal sequences. Both of these sites are formed at the Atg13-Atg101 interface and are composed of residues from both proteins. Site 1 consists of Atg13 Ser114, Tyr115, and Tyr118, and Atg101 Tyr45 and Ile47 (Fig. 6B). One of the benzamidine nitrogens is bound by the main chain carbonyl of Atg101-Tyr45. Site 2 is formed by Atg13 Tyr138 and the aliphatic parts of Arg139, Arg142, and Lys143; and Atg101 Asp56 and Thr63 (Fig. 6C). Thr63 is not just part of the pocket. This Thr is also directly involved in the interface with Atg13 and as such the mutant T63R was tested previously. It shares this characteristic with most of the other benzamidine 1 and 2 pocket residues. The mutation T63R has a modest effect on both complex formation and autophagic function (Suzuki et al., 2015a). The dual role of Thr63 illustrates that it may be difficult to mutationally deconvolute functional roles of the pockets that are distinct from their roles in the interface.
Discussion
Adding the present structure of the human Atg13-Atg101 complex to the previously determined structure of the ULK1 catalytic domain (Lazarus et al., 2015), the field has now made substantial inroads into visualizing the ordered portions of the human ULK1 complex at high resolution. The intense interest in targeting autophagy in human cells in cancer, neurodegeneration, aging, and infectious diseases places a premium on accurate data on the structure of human complexes such as provided here.
The gross features of the Atg13-Atg101 HORMA heterodimer are similar in S. pombe and human, with both, in turn, similar to the C-Mad2-O-Mad2 dimer. In both S. pombe and human, Atg13 and Atg101 are in the C-Mad2 and O-Mad2 conformations, respectively. Both dimerize through an interface involving the αC helices of both subunits. The human Atg13-Atg101 complex has a polar core at its interface that includes a buried salt-bridge at its heart. The salt bridge is unique to the animal Atg13-Atg101 complex, which was not predicted, and perhaps could not have been predicted from the S. pombe structure. A buried Ser-His hydrogen bond is nearby in the polar core. While the corresponding residues are present in the S. pombe structure, their mutual interaction was not identified, probably because of the lower resolution of that structure. Thus, the present structure provides the first reliable account of the polar core at the heart of the animal Atg13-Atg101 interface.
The human complex structure suggests that the newly discovered WF finger of Atg101 (Suzuki et al., 2015a) is subject to regulation. In the S. pombe complex and in the isolated human Atg101 structure (Michel and Weiergraeber, unpublished RCSB entry 4WZG), the WF finger projects outwards from the rest of Atg101. In the present structure, it is folded back on the HORMA domain. This suggests that the WF finger is not always in a position to bind its as-yet unidentified interactions partners. The conformation seen in our structure would correspond an inactive state of the motif. Just as it remains to be determined what the WF finger binds to, it also remains to discover how the accessibility of the WF finger is switched on and off.
Several hydrophobic pockets were identified by benzamidine molecules, of which two caught our attention because of their high degree of conservation. These pockets are lined with residues that are well conserved in the Atg13 and Atg101 proteins of animals, but largely missing in fission yeast (Fig. 6D). The existence of these pockets could not have been predicted from the fission yeast structure. The high degree of conservation of the residues in these pockets and their hydrophobicity seems consistent with a functional role. It is intriguing that the pockets are formed at the junction between Atg13 and Atg101, which is consistent with the essential roles of both Atg13 (Chang and Neufeld, 2009; Ganley et al., 2009; Hosokawa et al., 2009b; Jung et al., 2009) and Atg101 (Hosokawa et al., 2009a; Mercer et al., 2009) in autophagy in animal cells.
Intrinsically disordered regions (IDRs) are abundant in autophagy proteins (Baskaran et al., 2014; Hurley and Schulman, 2014; Mei et al., 2014; Popelka et al., 2014). The presence of so much intrinsic disorder implies the need for a large number of IDR binding sites on the ordered components of the machinery. Those ordered components containing multiple binding pockets could function as interaction hubs, potentially bringing together multiple IDRs. Our observations suggest that in animal cells, the Atg13-Atg101 HORMA heterodimer is likely to function as just such a hub.
A number of HORMA domains have been shown to bind peptides in the C-conformation between the edge of the main β-sheet and the safety belt (Hara et al., 2010; Kim et al., 2014; Luo et al., 2002; Sironi et al., 2002). Atg13-HORMA has consistently been observed in the C-conformation in structures of the S. cerevisiae, S. pombe, and now human proteins. The S. cerevisiae structure contains a phosphate binding site (Jao et al., 2013) that is formed in part by the safety belt. The site is important for recruitment of Atg9 (Suzuki et al., 2015b). Atg9 is the sole integral membrane protein of the core autophagy machinery, and is important in the initiation of autophagy in budding yeast. It seems likely that in S. cerevisiae, a peptide region of Atg9 binds to Atg13-HORMA in the canonical mode. However, this binding occurs through a non-conserved part of the N-terminal domain of S. cerevisiae Atg9, and this interaction mode is probably confined to budding yeast.
In human Atg13-HORMA, the safety belt is only 13 residues long, as compared 25–35 residues in both budding and fission yeast Atg13, and other HORMA proteins. The structure shows that the human Atg13 safety belt is pinned to the rest of the domain through several large hydrophobic residues. This leads us to infer that the canonical peptide binding function of the safety belt region of HORMA domains has been lost in animal Atg13 proteins. With respect to Atg101, the S. pombe protein is locked in the O-conformation because its β5-αC linker contains only 5 residues, which is too short to reach the C-state. The linker in human Atg101 is 11 residues long and in principle might allow for conversion. However, human Atg101 is seen in the O-conformation whether bound to Atg13 or free. This is consistent with the idea that human Atg101 is also locked in the O-state. Thus is seems that the HORMA domains of autophagy in animals are outliers, in that they are unlikely to bind peptides in the canonical mode seen for other HORMA proteins.
The other notable property of HORMA domains, at least in the case of Mad2, is the ability to interconvert between C- and O- conformations, and so heterodimerize (Mapelli et al., 2007). It appears that the ability to form and C- and O–state heterodimer, and not safety belt peptide binding, is the property that is shared between animal Atg13 and Atg101 and other HORMA proteins. Given that budding yeasts are an outlying branch in the phylogeny of Atg13 and lack Atg101 entirely (Fig. S1), we speculate that the autophagy HORMA domains evolved from a single Mad2-like ancestor as follows. The first change would have been gene duplication, with a stable C- and O- heterodimer taking the place of the ancestral conformational heterodimer of chemically identical subunits. This would have led to an ancestral Atg13-Atg101 pair similar to that of S. pombe (Fig. 7). In the evolution of budding yeast, Atg101 was lost. Budding yeast Atg13 evolved to be stable in the absence of Atg101, and acquired a safety belt phosphate binding site. This additional interaction site might compensate in part for interactions that were lost with the disappearance of Atg101. The structurally unrelated Atg29 and Atg31 proteins of the budding yeast Atg1 complex may also have a role in functional compensation. In the evolution of the animal Atg13-Atg101 pair, the Atg13 safety belt peptide-binding site appears to have been lost when this loop was shortened to the minimum necessary to connect β6 to β7. More was gained than lost, however, with the acquisition of hydrophobic sites 1 and 2, and the electropositive nature of the groove between the two proteins.
FIGURE 7. Scheme for the evolution of Atg13 and Atg101.
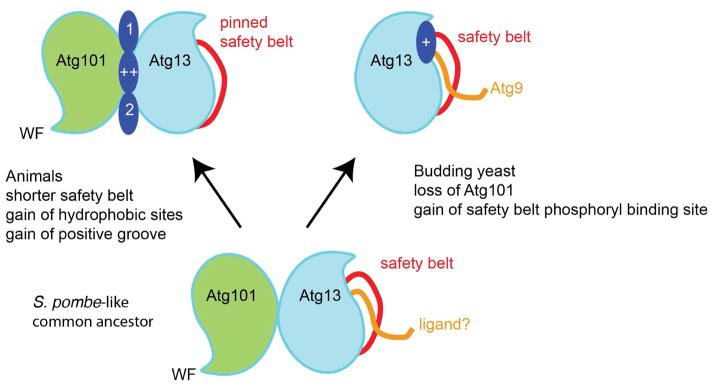
A speculative scheme for the evolution of animal Atg13 and Atg101, and budding Atg13 from an ancestral S. pombe- like Atg13-Atg101 pair.
The new structures of Atg13-Atg101 have revealed a number of novel interaction sites: the conserved WF finger and the animal-specific hydrophobic pockets. This begs the question as to what the interaction partners are. Obvious candidates include the C-terminal portion of Atg13 itself and other extended regions of the ULK1 complex, such as the long linker between the catalytic and EAT domains of ULK1 itself. Other members of the autophagy machinery would also be candidates, for example, the class III phosphatidylinositol 3-phosphate kinase complex I, which is rich in IDRs (Baskaran et al., 2014). The other obvious class of associating proteins would be the many substrates of ULK1. The catalytic domain of ULK1 preferentially phosphorylates sequences containing hydrophobic residues at the −3 and +1 positions (Egan et al., 2015). Clearly, this preference alone is not enough to confer the specificity needed for accurate ULK1 signaling. Interactions with other sites, such as provided by the Atg13-Atg101 HORMA dimer, would be an attractive mechanism for enhancing the specificity of substrate recognition.
Methods
Cloning and protein purification
Human Atg13-Atg101 constructs were expressed in Spodoptera frugiperda (Sf9) cells. DNAs coding for human Atg13 and Atg101 were subcloned into pFastBac Dual following the polyhedron and p10 promoters, respectively. Baculoviruses were generated in Sf9 cells with the bac-to-bac system (Life Technologies). Recombinant Atg13 was expressed with an N-terminal GST tag followed by a Tobacco Etch Virus (TEV) protease cleavage site, and Atg101 was expressed either with or without a His8 tag. Cells were infected and harvested after 48 to 72 hours. Cells were pelleted at 2000 x g for 20 min at 4°C. Cell pellets were lysed in 25 mM Tris-HCl pH 8.0, 150mM NaCl, 0.5 mM TCEP-HCl, and protease inhibitors (Roche) using ultrasonication. The lysate were centrifuged at 25, 000 x g for 1 hour at 4°C. The supernatant was bound to glutathione sepharose (GS4B) at 4°C overnight, eluted, and applied to a Hi Trap Q HP column. Hi Trap Q peak fractions were collected and digested with TEV protease at 4°C overnight. The TEV-treated protein solution was further purified with a Hi Trap S HP column and a Superdex 75 16/60 column equilibrated with 25 mM Tris-HCl pH 8.0, 150mM NaCl, 0.5 mM TCEP-HCl. Peak fractions were collected and flash-frozen in liquid N2 for storage.
HDX-MS Experiments
Amide HDX-MS was initiated by a 20-fold dilution of 40 μM Atg101-Atg13 complex (consisting of full-length Atg101 and residues 1–364 of Atg13) into a D2O buffer containing 25 mM Tris-HCl (pD 8.0), 150 mM NaCl, and 5 mM DTT at 30 °C. After intervals of 10 s and 30s, exchange was quenched at 0 °C with the addition of ice-cold quench buffer (400 mM KH2PO4/H3PO4, pH 2.2). Quenched samples were injected onto an HPLC (Agilent 1100) with in-line peptic digestion and desalting. Desalted peptides were eluted and directly analyzed by an Orbitrap Discovery mass spectrometer (Thermo Scientific). Peptide identification was done by running tandem MS/MS experiments. Peptides were identified using PEAKS Studio 7 (www.bioinfor.com). Initial mass analysis of the peptide centroids was performed using HDExaminer 1.4.3 (Sierra Analytics) followed by manual verification of every peptide. The deuteron content was adjusted for deuteron gain/loss during digestion and HPLC. Both nondeuterated and fully deuterated complex were analyzed. Fully deuterated samples were prepared by three cycles of drying and resolubilization in D2O and 6 M guanidinium hydrochloride.
Crystallization of the human Atg13-Atg101 complex
The Atg13 (residues 12–200) and Atg101 (residues 1–198) complex (Fig. 1A, B) was concentrated to 6 mg/ml with a 10 kD cut-off centrifugal filter (Millipore). Crystals were grown by hanging-drop vapor-diffusion at 19° C. The protein solution was mixed with well buffer composed of 19% PEG 3350, 0.5 M benzamidine, and 0.2 M lithium citrate. Crystals appeared in 2 days and grew to full size (0.05 × 0.1 × 0.3 mm) in 5 to 7 days. Crystals were flash-frozen with liquid nitrogen in the well solution.
Data collection and structure determination
Diffraction data were collected at BL8.3.1, Advanced Light Source (ALS), LBNL. Native crystals diffracted to 1.63 Å, and data were collected at λ = 1.07207 Å. Native crystals were soaked in cryo-protectant buffer supplemented with 0.5 M NaBr for 10 seconds, and then were flash frozen in liquid nitrogen. A Br− Single Anomalous Dispersion (SAD) data set (Dauter and Dauter, 2001) was collected at λ = 0.92000 Å. All data sets were processed with HKL2000 (HKL Research). Data collection and processing statistics are given in Table 1. Br- ion positions were identified and phases calculated using SHELXC/D/E (Schneider and Sheldrick, 2002). Model building and refinement were carried out with ARP/wARP (Langer et al., 2008), Coot (Emsley et al., 2010), REFMAC5 (Murshudov et al., 1997) and PHENIX (Adams et al., 2010).
Table 1.
Statistics of Crystallographic Data Processing and Refinement
| Native | Br SAD | |
|---|---|---|
| Data collection | ||
| Space group | C2 | C2 |
| Cell dimensions | ||
| A, b, c(Å) | 67.72, 64.45, 94.12 | 67.66, 67.76, 93.83 |
| α, β, γ(°) | 90.000, 97.54, 90.000 | 90.00, 97.48, 90.00 |
| Wavelength(Å) | 1.07207 | 0.92000 |
| Resolution(Å) | 50.00-1.63 (1.69-1.63) | 50.000-1.93 (2.00-1.93) |
| No. of reflections | 49908 | 29890 |
| Completeness (%) | 100.0 (100.0) | 100.0 (99.0) |
| Redundancy | 5.3 (3.9) | 4 (3.8) |
| Rsym | 0.092 (0.486) | 0.095 (0.942) |
| <I>/<σ(I)> | 12.89 (3.45) | 14.4 (1.37) |
| CC1/2 | 0.872 | 0.640 |
| Refinement | ||
| Resolution(Å) | 46.65–1.63 (1.66–1.63) | - |
| Rwork/Rfree (%) | 17.59/19.03 | - |
| Average B-factor(Å) | ||
| Atg101-Atg13 | 37.242/47.063 | - |
| Ligand (Benzamidine) | 56.457 | - |
| R. m. s. deviation from ideality | ||
| Bond length (Å) | 0.0059 | - |
| Bond angle (°) | 0.996 | - |
| Ramachandran Plot (%) | ||
| favored | 97.6 | - |
| allowed | 2.4 | - |
| outlier | 0 | - |
Pull-down assays
For pull-down experiments, Atg13 and Atg101 HORMA domains were subcloned into the pCAG vectors with appropriate affinity tags. Equal amounts of human Atg13 and Atg101 plasmids were transfected into HEK293 GnTI− cells adapted for suspension. Cells were grown in Freestyle media (Invitrogen, Grand Island, NY) supplemented with 1% FBS (Invitrogen) at 37°C, 80% humidity, 5% CO2, and rocked at 140 rpm. After three days, cells were harvested and lysed in ice-cold lysis buffer (25 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP-HCl, 1% Triton X-100, and one tablet of EDTA-free protease inhibitors (Roche) per 25 ml). The soluble fractions of cell lysates were isolated by centrifugation at 16,000 x g for 10 min. The lysates were added to 20 μl of pre-equilibrated Glutathione Sepharose 4B (GE Healthcare Life Sciences) or Strep-Tactin Superflow (IBA GmbH) beads, and incubated at 4°C for 30 min. The beads were then washed three times with 500 μl of wash buffer (25 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.5 mM TCEP-HCl), and the bound proteins were denatured by the addition of 20 μl of sample buffer and boiling for 5 min, resolved by SDS-PAGE.
Supplementary Material
Highlights.
Structure of human Atg13-Atg101 HORMA dimer refined at 1.6 Å resolution
Atg13 safety belt anchored, ordered, and consistent with stable heterodimer
WF finger is in a closed and inactive conformation
Hydrophobic pockets marked by benzamidines suggest function as interaction hub
Acknowledgments
This work was supported by the National Institutes of Health grant GM111730 (J. H. H). Beamline 8.3.1 at the Advanced Light Source, LBNL, is supported by the UC Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599 and the Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Coordinates have been deposited in the RCSB with accession code 5C50.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D-Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci. 1998;23:284–286. doi: 10.1016/s0968-0004(98)01257-2. [DOI] [PubMed] [Google Scholar]
- Baskaran S, Carlson LA, Stjepanovic G, Young LN, Kim DJ, Grob P, Stanley RE, Nogales E, Hurley JH. Architecture and Dynamics of the Autophagic Phosphatidylinostol 3-Kinase Complex. eLife. 2014 Dec;9:3. doi: 10.7554/eLife.05115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YY, Neufeld TP. An Atg1/Atg13 Complex with Multiple Roles in TOR-mediated Autophagy Regulation. Molecular Biology of the Cell. 2009;20:2004–2014. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao WCH, Kulkarni K, Zhang ZG, Kong EH, Barford D. Structure of the mitotic checkpoint complex. Nature. 2012;484:208–U289. doi: 10.1038/nature10896. [DOI] [PubMed] [Google Scholar]
- Dauter Z, Dauter M. Entering a new phase: Using solvent halide ions in protein structure determination. Structure. 2001;9:R21–R26. doi: 10.1016/s0969-2126(01)00565-2. [DOI] [PubMed] [Google Scholar]
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015 doi: 10.1016/j.molcel.2015.05.031. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D-Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen JR. Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y, Suzuki SW, Yamamoto H, Kondo-Kakuta C, Kimura Y, Hirano H, Akada R, Inagaki F, Ohsumi Y, Noda NN. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat Struct Mol Biol. 2014;21:513–521. doi: 10.1038/nsmb.2822. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1-ATG13-FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M. Crystal Structure of Human REV7 in Complex with a Human REV3 Fragment and Structural Implication of the Interaction between DNA Polymerase zeta and REV1. J Biol Chem. 2010;285:12299–12307. doi: 10.1074/jbc.M109.092403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus K, Nagy P, Gaspari Z, Juhasz G. The Putative HORMA Domain Protein Atg101 Dimerizes and Is Required for Starvation-Induced and Selective Autophagy in Drosophila. Biomed Research International. 2014 doi: 10.1155/2014/470482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009a;5:973–979. doi: 10.4161/auto.5.7.9296. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura SI, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 Association with the ULK1-Atg13-FIP200 Complex Required for Autophagy. Molecular Biology of the Cell. 2009b;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Schulman BA. Atomistic Autophagy: The Structures of Cellular Self-Digestion. Cell. 2014;157:300–311. doi: 10.1016/j.cell.2014.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Ragusa MJ, Stanley RE, Hurley JH. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013;110:5486–5491. doi: 10.1073/pnas.1220306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Molecular Biology of the Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Noda NN, Fujioka Y, Suzuki K, Inagaki F, Ohsumi Y. Characterization of the Atg17-Atg29-Atg31 complex specifically required for starvation-induced autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;389:612–615. doi: 10.1016/j.bbrc.2009.09.034. [DOI] [PubMed] [Google Scholar]
- Kim Y, Rosenberg SC, Kugel CL, Kostow N, Rog O, Davydov V, Su TY, Dernburg AF, Corbett KD. The Chromosome Axis Controls Meiotic Events through a Hierarchical Assembly of HORMA Domain Proteins. Dev Cell. 2014;31:487–502. doi: 10.1016/j.devcel.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köfinger J, Ragusa MJ, Lee IH, Hummer G, Hurley JH. Solution structure of the Atg1 complex: Implications for the architecture of the phagophore assembly site. Structure. 2015;23:809–818. doi: 10.1016/j.str.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus MB, Novotny CJ, Shokat KM. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS chemical biology. 2015;10:257–261. doi: 10.1021/cb500835z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XL, Tang ZY, Rizo J, Yu HT. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The mad2 conformational dimer: Structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–743. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Mei Y, Su M, Soni G, Salem S, Colbert CL, Sinha SC. Intrinsically disordered regions in autophagy proteins. Proteins. 2014;82:565–578. doi: 10.1002/prot.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5 doi: 10.4161/auto.5.5.8249. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D-Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popelka H, Uversky VN, Klionsky DJ. Identification of Atg3 as an intrinsically disordered polypeptide yields insights into the molecular dynamics of autophagy-related proteins in yeast. Autophagy. 2014;10:1093–1104. doi: 10.4161/auto.28616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 Complex as a Scaffold for Autophagosome Biogenesis. Cell. 2012;151:1501–1512. doi: 10.1016/j.cell.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194:341–361. doi: 10.1534/genetics.112.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr Sect D-Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of ’safety belt’ binding mechanism for the spindle checkpoint. EMBO J. 2002;21:2496–2506. doi: 10.1093/emboj/21.10.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A. Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for spindle checkpoint. EMBO J. 2001;20:6371–6382. doi: 10.1093/emboj/20.22.6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stjepanovic G, Davies CW, Stanley RE, Ragusa MJ, Kim DJ, Hurley JH. Assembly and dynamics of the autophagy initiating Atg1 complex. Proc Natl Acad Sci U S A. 2014;111:12793–12798. doi: 10.1073/pnas.1407214111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kaizuka T, Mizushima N, Noda NN. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struct Mol Biol. 2015a doi: 10.1038/nsmb.3036. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Tabata K, Morita E, Kawasaki M, Kato R, Dobson RC, Yoshimori T, Wakatsuki S. Structural Basis of the Autophagy-Related LC3/Atg13 LIR Complex: Recognition and Interaction Mechanism. Structure. 2013 doi: 10.1016/j.str.2013.09.023. [DOI] [PubMed] [Google Scholar]
- Suzuki SW, Yamamoto H, Oikawa Y, Kondo-Kakuta C, Kimura Y, Hirano H, Ohsumi Y. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A. 2015b;112:3350–3355. doi: 10.1073/pnas.1421092112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31(comet) blocks Mad2 activation through structural mimicry. Cell. 2007;131:744–755. doi: 10.1016/j.cell.2007.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.