

Abstract
The characterization of TDP-α-D-glucose dehydrogenase AtmS8, TDP-α-D-glucuronic acid decarboxylase AtmS9 and TDP-4-keto-α-D-xylose 2,3-dehydratase AtmS14 involved in Actinomadura melliaura AT2433 aminodideoxypentose is reported. This study provides the first biochemical evidence that both deoxypentose and deoxyhexose biosynthetic pathways share common strategies for sugar 2,3-dehydration/reduction and implicates the sugar nucleotide base specificity of AtmS14 as a potential mechanism for sugar nucleotide commitment to secondary metabolism. In addition, a re-evaluation of the AtmS9 homolog involved in calicheamicin aminodeoxpentose biosynthesis (CalS9) reveals CalS9 to catalyze UDP-4-keto-α-D-xylose as the predominant product rather than UDP-α-D-xylose as previously reported. Cumulatively, this work provides additional fundamental insights regarding the biosynthesis of novel pentoses attached to complex bacterial secondary metabolites.
Keywords: carbohydrate, dehydrogenase, decarboxylase, dehydratase, natural product, sugar nucleotide, biosynthesis
Functionalized deoxysugars attached to bacterial secondary metabolites are often critical to the given metabolite’s biological activity.[1] Yet, while many of the key sugar nucleotide-dependent transformations en route to the corresponding highly functionalized deoxyhexoses have been well-studied, less is known regarding the biogenesis of their deoxypentose counterparts. Examples of deoxypentose-substituted bacterial natural products include indolocarbazoles (AT2433[2]), enediynes (calicheamicin,[3] esperamicin[4] and maduropeptin[5]) and orthosomycins (avilamycin[6] and everninomicin[7]) (Scheme 1). Early studies regarding deoxypentose biosynthetic progenitors were inconsistent with classical esperamicin metabolic-labeling studies supporting a glucose-based precursor[8] while annotation of the avilamycin biosynthetic gene cluster implicated a ribose-based pathway.[9] Subsequent comparative genomics of the AT2433,[10] calicheamicin,[11] and rebeccamycin[12] biosynthetic loci provided the basis for a aminodideoxypentose biosynthetic pathway (Scheme 2) consistent with the esperamicin metabolic-labeling studies[8] bolstered further via biochemical characterization of CalS8 as a TDP-α-D-glucose dehydrogenase[13] and biosynthetic studies of aminopentose-containing Gram-negative exopolysaccharides.[14] To extend these studies, herein we report the biochemical characterization of AtmS8 (TDP-α-D-glucose dehydrogenase), AtmS9 (TDP-α-D-glucuronic acid decarboxylase) and AtmS14 (TDP-4-keto-α-D-xylose 2,3-dehydratase) and the NMR characterization of corresponding key reactants/products. This work highlights the early enzymes (AtmS8 and AtmS9) as base permissive and AtmS14 as TDP-sugar specific. Consistent with well-established precedent of 4-ketosugars as a key precursor to deoxyhexose 2,3-dehydration/reduction,[15] this study also confirms the keto-sugar nucleotide TDP-4-keto-α-D-xylose as the decarboxylase product which is in contrast to prior work implicating the AtmS9 homolog CalS9 (72% homology, 60% identity, calicheamicin biosynthesis) as a catalyst for both C5-decarboxylation and C4-reduction.[16] Cumulatively, this study provides further support for the pathway put forth in Scheme 2 and notably provides the first biochemical evidence that the biosynthetic strategies for 2,3-dehydration/reduction of deoxypentose and deoxyhexose are conserved. As such, this work lays a foundation for comparative mechanistic and structural studies to further probe points of biosynthetic convergence/divergence.
Scheme 1.

Representative natural products that contain deoxypentose moiety (highlighted in dark color).
Scheme 2.
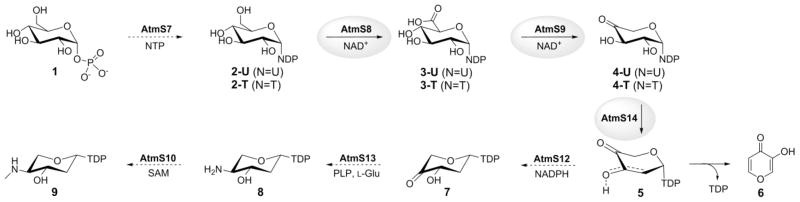
The proposed biosynthetic pathway for the AT2433 aminopentose precursor. Dotted arrows denote enzymes for which putative function is based upon sequence homology and the enzymes characterized in current study are highlighted by grey circles.
In the AT2433 producer Actinomadura melliaura, a total of seven enzymes are postulated to participate in TDP-2,4-dideoxy-4-methylamino-β-L-xylose (9) biosynthesis where putative function is based upon BLAST analysis: AtmS7, α-D-glucose-1-phosphate thymidyltransferase; AtmS8, TDP-α-D-glucose dehydrogenase; AtmS9, TDP-α-D-glucuronic acid decarboxylase; AtmS14, TDP-4-keto-α-D-xylose 2,3-dehydratase; AtmS12, TDP-2-deoxy-4-keto-α-D-pentos-2-ene 2,3-reductase; AtmS13, TDP-2-deoxy-4-keto-β-L-xylose 3-aminotransferase;[10b] and AtmS10, TDP-4-amino-2,4-dideoxy-β-L-xylose 3-N-methyltransferase (Scheme 2).[10] For this study, atmS8 and atmS9 were amplified directly from Actinomadura melliaura genomic template DNA while atmS14 was amplified from a pUC57 vector containing a codon-optimized synthetic gene using the polymerase chain reaction (PCR). The corresponding atmS8, atmS9 and atmS14 gene-containing PCR fragments were digested, subsequently cloned into pET28a and the resulting constructs used to transform Escherichia coli BL21(DE3) to provide overproduction strains of the corresponding target enzymes as N-terminal His6-fusion proteins (see Supporting Information).
While HPLC-based discontinuous assays are the most common strategy for the study of sugar nucleotide-dependent biosynthetic enzymes,[17] alternative NMR-based assays offer convenient real-time detection of reactants/products that are unstable or poorly resolved via HPLC.[18] The current study employed a combination of reverse phase (RP)-HPLC and in-situ NMR to assess enzyme-catalyzed turnover and support product characterization. The in-situ NMR reactions were conducted in a 5 mm NMR tube at 30°C using standard assay conditions (see Supporting Information) in 25 mM sodium phosphate buffer containing 50–70% D2O, pH 7.5. Reactions were initiated via the addition of enzyme and incubated at 30°C for 2–4 h before the acquisition of 1D-1H with water pre-saturation (referred simply as 1D-1H henceforth) spectra. Reaction progress was monitored by the appearance of new product-based resonances and consequent decrease in the intensity of substrate signals in the 1D-1H spectra and depending on the progress, the reactions were incubated at 30°C and 1D spectra were acquired at different time intervals until the reactions neared completion. Where applicable, corresponding correlative HPLC assays followed standard protocols.
To confirm the putative steps/enzymes involved in the conversion of 2 to 5, the reaction was initiated by the addition of 10 μM of AtmS8 to an assay mixture containing commercially available TDP-α-D-glucose (2-T) and excess of NAD+ in the absence or presence of an NAD+ regeneration system (see Supporting Information) to potentially help drive the equilibrium toward the desired product 3-T (Figures 1A and 1B). Nearly complete conversion of 2-T to 3-T in the presence of the NAD+ regeneration system was observed in 16 h at 30°C (Figures 1A and 1B) as calculated by the ratio of the intensity of anomeric proton signals of the product to substrate in the 1H NMR spectra (5.57 ppm and 5.60 ppm, respectively; see Table S1 in Supporting Information). Under these conditions, the HPLC retention times for 2-T and 3-T were 21.8 min and 26.6 min, respectively, with products also confirmed by HRMS (see Supporting Information). No reaction was observed via NMR or HPLC in controls lacking AtmS8.
Figure 1.

AtmS8 catalyzed reactions over 16 h at 30°C. (A) Anomeric signals in the 1H NMR spectra of an AtmS8 reaction with 2-T as substrate: (i) reaction with NAD+ regeneration system; (ii) reaction without NAD+ regeneration system; and (iii) control, without enzyme. (B) Correlative HPLC chromatograms of an AtmS8 reaction with 2-T as substrate: (i) reaction with NAD+ regeneration system; (ii) NAD+ standard; (iii) 3-T standard; and (iv) 2-T standard. (C) Correlative HPLC chromatograms of an AtmS8 reaction with 2-U as substrate: (i) reaction with NAD+ regeneration system; (ii) NAD+ standard; (iii) 3-U standard; and (iv) 2-U standard.
Upon near completion of the AtmS8 reaction to 3-T based upon NMR, the addition of 15 μM of AtmS9 led to quantitative conversion of 3-T to 4-T (Figure 2A) over 15 h at 30 °C as observed via HPLC (4-T, 17.9 min) and the signature 4-T anomeric proton in the 1H NMR spectra (5.57 ppm). The identity of 4-T as the product was also confirmed by 1H-13C-HSQC (Table S1 Supporting Information) and HRMS. No reaction was observed via NMR or HPLC in controls lacking AtmS9.
Figure 2.

Anomeric signals in the 1H NMR spectra of AtmS9 and CalS9-catalyzed reactions. (A) Representative AtmS9-catalyzed reaction with 3-T as the substrate. (B) Representative AtmS9-catalyzed reaction with 3-U as the substrate. (C) Representative CalS9-catalyzed reaction with 3-U as the substrate.
Following the completion of the AtmS9 reaction to 4-T, 14 μM of AtmS14 was added and the reaction monitored via HPLC and NMR at 30°C. HPLC revealed two new products (3-hydroxy-4H-pyran-4-one 6, 6.8 min; TDP, 20.9 min; Figure S3, Supporting Information). Consistent with this, the 6 Ha, Hb and Hc protons (7.90 ppm and 6.44 ppm) were also clearly observed via 1H NMR (Figure 3) and the presence of 6 also confirmed via HRMS. As the first pentose-based example, the degradative formation of 6 from 5-T is consistent with pioneering hexose-based precedent first observed by Floss and Liu. Specifically, within the context of assays for TDP-4-keto-6-deoxy-α-D-glucose 2,3-dehydratases (Gra Orf27[15b] and TylX3[15b]) the TDP-2,6-dideoxy-4-keto-α-D-hexos-2-ene product was found to rapidly degrade to TDP and 3-hydroxy-2-methyl-4H-pyran-4-one (i.e., 2-methyl-substituted 6). Thus, the current study provides the first biochemical validation of the proposed mechanistic conservation among sugar nucleotide-dependent enzymes involved in pentose/hexose 2-deoxygenation.
Figure 3.

1H-NMR spectra of the AtmS8/AtmS9/AtmS14 coupled reaction: (i) AtmS8/AtmS9/AtmS14 coupled reaction where AtmS14 was added after the completion of AtmS9-catalyzed transformation based upon NMR (spectra recorded 20 h post-AtmS9 addition); and (ii) AtmS8/AtmS9 coupled reaction. Proton signals corresponding degraded diketo-intermediate (6) and the 4-T anomeric protons are labelled.
To test the nucleotide specificity of AtmS8, AtmS9 and AtmS14 enzymes, similar reactions were performed in which TDP-α-D-glucose (2-T) was replaced with commercially available ADP-α-D-glucose, CDP-α-D-glucose, GDP-α-D-glucose or UDP-α-D-glucose (2-U). Of these, only 2-U led to ~10% product 3-U in the presence of AtmS8 and the NAD+ regeneration system in 16 h at 30°C (Figure 1C). In contrast, the subsequent AtmS9-catalyzed conversion of 3-U to 4-U was quantitative (Figure 2B) but could not proceed to 6 (via 5-U) even in the presence of excess AtmS14 over extended incubation times (data not shown). This implicates the sugar nucleotide base specificity of AtmS14, and to a lesser extent AtmS8, as a potential mechanism for the commitment of specific sugar nucleotides to secondary metabolite production. Consistent with this, the calicheamicin aminodideoxypentosyl transferase CalG4 has been demonstrated to be specific for TDP-sugars (including 9).[20]
Based upon well-established precedent,[17] the presence of the C4-carbonyl is anticipated to be critical to both 2,3-dehydration (AtmS14) and PLP-dependent 4-transamination (AtmS13) yet, the AtmS9 homolog CalS9 (72% homology, 60% identity, calicheamicin biosynthesis) was recently reported to catalyze C5-decarboxylation and C4-reduction to give C4-reduced UDP-α-D-xylose.[16] To re-examine this phenomenon, a standard reaction containing 3-U and 16 μM CalS9 led to quantitative conversion of 3-U to 4-U over 16 h at 30 °C as observed via HPLC (4-U, 12.6 min), the signature 4-U anomeric proton in the 1H NMR spectra (5.56 ppm, Figure 2C) and product HRMS (see Supporting Information). However, it is important to note that AtmS9 reactions with 3-U or 3-T as substrates conducted for longer periods of time (> 24 h, 37 °C), led to detectable (U/T)DP-α-D-xylose formation (see Supporting Information) as previously observed for CalS9.[16] Similar discrepancies have been noted with other bacterial UDP-GlcA decarboxylases.[19]
In summary, the demonstrated biochemical characterization of AtmS8, AtmS9 and AtmS14 importantly provides the first biochemical evidence that the strategies for 2,3-dehydration/reduction are common among deoxypentose and deoxyhexose biosynthesis and provides strong support for the original aminodeoxypentose biosynthetic postulations put forth based upon At2433, calicheamicin and rebeccamycin comparative genomics.[10] While AtmS8 and AtmS9 were found to display some base permissivity, the strict TDP-sugar specificity of AtmS14 is consistent with the previously reported TDP-sugar specificity of the calicheamicin aminodeoxypentosyltransferase CalG4[20] and may implicate a point of sugar nucleotide commitment to secondary metabolism in Actinomadura melliaura. Re-examination of the AtmS9 homolog involved in calicheamicin aminodeoxypentose biosynthesis (CalS9) also revealed the predominate CalS9 product as UDP-4-keto-α-D-xylose rather than UDP-α-D-xylose as previously reported[16] where the corresponding decarboxylase (AtmS9/CalS9) reaction conditions were demonstrated to influence the product 4-keto/hydroxyl ratio. Taken together this study supports the steps proposed for aminodeoxypentose pathway common to AT2433 and calicheamicin and lays a foundation for comparative mechanistic and structural studies among deoxyhexose and deoxypentose biosynthetic enzymes.
Experimental Section
General methods
High-resolution electrospray ionization (ESI) mass spectra (HRMS) were carried at the Small Molecule Mass Spectrometry Core Laboratory of University of Kentucky College of Medicine. The HRMS data were recorded on a AB Sciex Triple TOF 5600 instrument coupled with an Eksigent Ekspert micro LC 200 system with source temperature of 150°C, ion spray voltage floating (ISVF) of 5000 V in positive mode. Samples were infused at 20 μL min−1 and spectra collected for 3 min at a resolution greater than 31000. In negative mode ISVF of −4000 V was used. C17 lysophosphatidyl choline with a mass of 510.3554 and C17 lysophosphatidic acid with a mass of 423.2517 were used as internal references to calibrate the spectra in positive and negative modes, respectively. NMR experiments were carried on a 600 MHz Varian (Palo Alto, CA) VNMRS spectrometer equipped with a z-axis gradient 5 mm HCN cold probe at the National Magnetic Resonance Facility at Madison (NMRFAM). Prior to HPLC analysis, protein was removed using an Amicon® Ultra centrifugal filter device. Analytical reverse-phase high pressure liquid chromatography (RP-HPLC) was conducted with a Gemini NX C-18 (5 μm, 250 × 4.6 mm) column (from Phenomenex, Torrance, California, USA) with a gradient of 1% B to 50% B over 30 min, 50% B for 5 min, 50% B to 1% over 1 min, 1% B for 7 min (A = 50 mM phosphate buffer pH 6 with 5 mM tetrabutylammonium bisulfate; B = acetonitrile; flow rate = 1 mL min−1) and detection monitored at 254 nm.
Enzyme reactions (500–650 μL final volume) were performed in 25–50 mM sodium phosphate buffer, pH/pD 7.5 (10–20% D2O). NMR spectra were collected at 30ºC in 5 mm NMR tubes on a 600 MHz Varian (Palo Alto, CA) VNMRS spectrometers equipped with a z-axis gradient 5 mm HCN cold probe. The probe was tuned and shimmed before the addition of enzyme. 1D-proton with water pre-saturation (referred simply as 1D-1H henceforth) was used after the addition of the enzyme to monitor the appearance of new signals corresponding to product formation. Processing of the spectra was accomplished using MestReNova (Santiago de Compostela, SPAIN) and the proton axis was referenced to the water resonance at 4.766 ppm.
In-vitro characterization of AtmS8
The AtmS8 reaction was initiated directly in a 5 mm NMR tube in a volume of 500 μl (50 % D2O) with 2 mM unlabelled TDP-α-D-glucose or UDP-αD-glucose (Sigma-Aldrich), 6 mM of NAD+, 1 mM MgCl2, 1 mM DTT in 50 mM NaH2PO4 buffer, pH 7.5 at 30°C with the addition of AtmS8 (10 μM) in the presence and absence of an NAD+ regeneration system (6 mM pyruvate and 2.7 U μL−1 lactate dehydrogenase). 1D-1H NMR spectra were recorded at regular intervals and monitored for the changes in the intensity of the anomeric proton signal for the depletion of the substrate and subsequent formation of product. The reaction was ~97 % complete after the 16 h incubation at 30°C as calculated by the ratio of the product to substrate signals from RP-HPLC and 1D-1H (Figure 1) where the glucuronic acid NMR chemical shift signatures (Table S1) were also found to match those of commercial standard UDP-GlcA (Sigma-Aldrich). Reaction aliquots were also used for HRMS (Supplemental Section 4).
In-vitro characterization of AtmS9
The AtmS9 reaction employed the reaction mixture from above section and was initiated by adding 15 μM AtmS9. The reaction was monitored by 1D-1H NMR spectra. Complete conversion of the substrate to product was observed after the overnight incubation at 30°C as calculated by the ratio of the product to substrate signals from 1D-1H (Figures 2 and S2) where the NMR chemical shifts (Table S1) were found to be consistent with previously reported data.[19a] Reaction aliquots were also used for HRMS (Supplemental Section 4).
In-vitro characterization of AtmS14
To the above reaction mixture, 14 μM AtmS14 was added and the reaction monitored by 1D-1H NMR at regular intervals at 30°C. Complete degradation of 4-T to pyromicomic acid (6) and TDP was observed in 6 h after the initiation of the reaction as observed by 1D NMR spectra (Figure 3) and RP-HPLC (Figure S3). Reaction aliquots were also used for HRMS (Supplemental Section 4).
Supplementary Material
Acknowledgments
This work was supported by NIH RO1 CA84374 (JST), NCATS (UL1TR000117) and made use of the Small Molecule Mass Spectrometry Core Laboratory of University of Kentucky College of Medicine and UW NMRFAM [supported in part by NIH (P41RR02301, P41GM10399, RR02781, RR08438), the University of Wisconsin, NSF (DMB-8415048, OIA-9977486, BIR-9214394), DOE and the USDA].
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.20xxxxxxx
Contributor Information
Dr. Pauline Peltier-Pain, Pharmaceutical Sciences Division, University of Wisconsin-Madison, 777 Highland Avenue, Madison, WI 53705 (USA); Present address: Glycom A/S, Denmark.
Dr. Shanteri Singh, Center for Pharmaceutical Research and Innovation, Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone Street, Lexington, KY 40536 (USA).
Prof. Jon S. Thorson, Center for Pharmaceutical Research and Innovation, Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone Street, Lexington, KY 40536 (USA).
References
- 1.a) Rupprath C, Schumacher T, Elling L. Curr Med Chem. 2005;12:1637–1675. doi: 10.2174/0929867054367167. [DOI] [PubMed] [Google Scholar]; b) Cipolla L, Araújo AC, Bini D, Gabrielli L, Russo L, Shaikh N. Expert Opin Drug Discov. 2010;5:721–737. doi: 10.1517/17460441.2010.497811. [DOI] [PubMed] [Google Scholar]; c) Gantt RW, Peltier-Pain P, Thorson JS. Nat Prod Rep. 2011;28:1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]; d) Singh S, Phillips GN, Jr, Thorson JS. Nat Prod Rep. 2012;29:1201–1237. doi: 10.1039/c2np20039b. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lin CI, McCarty RM, Liu HW. Chem Soc Rev. 2013;42:4377–4407. doi: 10.1039/c2cs35438a. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Elshahawi SI, Shaaban KA, Kharel MK, Thorson JS. Chem Soc Rev. 2015 doi: 10.1039/c4cs00426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Golik J, Doyle TW, Krishnan B, Dubay G, Matson JA. J Antibiot. 1989;42:1784–1789. doi: 10.7164/antibiotics.42.1784. [DOI] [PubMed] [Google Scholar]; b) Matson JA, Claridge C, Bush JA, Titus J, Bradner WT, Doyle TW, Horan AC, Patel M. J Antibiot. 1989;42:1547–1555. doi: 10.7164/antibiotics.42.1547. [DOI] [PubMed] [Google Scholar]; c) Shaaban KA, Elshahawi SI, Wang X, Horn J, Kharel MK, Leggas M, Thorson JS. J Nat Prod. 2015 doi: 10.1021/acs.jnatprod.5b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lee MD, Dunne TS, Siegel MM, Chang CC, Morton GO, Borders DB. J Am Chem Soc. 1987;109:3464–3466. [Google Scholar]; b) Lee MD, Dunne TS, Chang CC, Ellestad GA, Siegel MM, Morton GO, McGahren WJ, Borders DB. J Am Chem Soc. 1987;109:3466–3468. [Google Scholar]; c) Lee MD, Dunne TS, Chang CC, Siegel MM, Morton GO, Ellestad GA, McGahren WJ, Borders DB. J Am Chem Soc. 1992;114:985–997. [Google Scholar]
- 4.Golik J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. J Am Chem Soc. 1987;109:3462–3464. doi: 10.7164/antibiotics.38.1605. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder DR, Colson KL, Klohr SE, Zein N, Langley DR, Lee MS, Matson JA, Doyle TW. J Am Chem Soc. 1994;116:9351–9352. [Google Scholar]
- 6.Wright DE. Tetrahedron. 1979;35:1207–1237. [Google Scholar]
- 7.a) Ganguly AK, Saksena AK. J Antibiot. 1975;28:707–709. [PubMed] [Google Scholar]; b) Ganguly AK, Sarre OZ, Greeves D, Morton J. J Am Chem Soc. 1975;97:1982–1985. doi: 10.1021/ja00840a078. [DOI] [PubMed] [Google Scholar]
- 8.Lam KS, Veitch JA, Golik J, Krishnan B, Klohr SE, Volk KJ, Forenza S, Doyle TW. J Am Chem Soc. 1993;115:12340–12345. [Google Scholar]
- 9.Weitnauer G, Muhlenweg A, Trefzer A, Hoffmeister D, Sussmuth RD, Jung G, Welzel K, Vente A, Girreser U, Bechthold A. Chem Biol. 2001;8:569–581. doi: 10.1016/s1074-5521(01)00040-0. [DOI] [PubMed] [Google Scholar]
- 10.a) Gao Q, Zhang C, Blanchard S, Thorson JS. Chem Biol. 2006;13:733–743. doi: 10.1016/j.chembiol.2006.05.009. [DOI] [PubMed] [Google Scholar]; b) Singh S, Kim Y, Wang F, Bigelow L, Endres M, Kharel MK, Babnigg G, Bingman CA, Joachimiak A, Thorson JS, Phillips GN. Proteins. 2015 doi: 10.1002/prot.24844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 12.a) Sánchez C, Butovich IA, Braña AF, Rohr J, Méndez C, Salas JA. Chem Biol. 2002;9:519–531. doi: 10.1016/s1074-5521(02)00126-6. [DOI] [PubMed] [Google Scholar]; b) Hyun CG, Bililign T, Liao J, Thorson JS. ChemBioChem. 2003;4:114–117. doi: 10.1002/cbic.200390004. [DOI] [PubMed] [Google Scholar]; c) Onaka H, Taniguchi S, Igarashi Y, Furumai T. Biosci Biotechnol Biochem. 2003;67:127–138. doi: 10.1271/bbb.67.127. [DOI] [PubMed] [Google Scholar]
- 13.Bililign T, Shepard EM, Ahlert J, Thorson JS. ChemBioChem. 2002;3:1143–1146. doi: 10.1002/1439-7633(20021104)3:11<1143::AID-CBIC1143>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 14.a) Williams GJ, Breazeale SD, Raetz CR, Naismith JH. J Biol Chem. 2005;280:23000–23008. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hamad MA, Di Lorenzo F, Molinaro A, Valvano MA. Mol Microbiol. 2012;85:962–974. doi: 10.1111/j.1365-2958.2012.08154.x. [DOI] [PubMed] [Google Scholar]; c) Laverty G, Gorman SP, Gilmore BF. Pathogens. 2014;3:596–632. doi: 10.3390/pathogens3030596. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lee M, Sousa MC. Biochemistry. 2014;53:796–805. doi: 10.1021/bi4015677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Draeger G, Park SH, Floss HG. J Am Chem Soc. 1999;121:2611–2612. [Google Scholar]; b) Chen H, Agnihotri G, Guo Z, Que LS, Chen XH, Liu HW. J Am Chem Soc. 1999;121:8124–8125. [Google Scholar]; c) González A, Remsing LL, Lombó F, Fernández mJ, Prado L, Braña AF, Künzel E, Rohr J, Méndez C, Salas JA. Mol Gen Genet. 2001;264:827–835. doi: 10.1007/s004380000372. [DOI] [PubMed] [Google Scholar]; d) Maharjan J, Liou K, Lee HC, Kim CG, Lee JJ, Yoo JC, Sohng JK. Biotechnol Lett. 2003;25:909–915. doi: 10.1023/a:1024075902143. [DOI] [PubMed] [Google Scholar]; e) Kubiak RL, Thoden JB, Holden HM. Biochemistry. 2013;52:2078–88. doi: 10.1021/bi400176n. [DOI] [PubMed] [Google Scholar]
- 16.Simkhada D, Oh TJ, Pageni BB, Lee HC, Liou K, Sohng JK. Appl Microbiol Biotechnol. 2009;83:885–895. doi: 10.1007/s00253-009-1941-8. [DOI] [PubMed] [Google Scholar]
- 17.a) Blanchard S, Thorson JS. Curr Opin Chem Biol. 2006;10:263–71. doi: 10.1016/j.cbpa.2006.04.001. [DOI] [PubMed] [Google Scholar]; b) Thibodeaux CJ, Melancon CE, Liu HW. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]; c) Oh TJ, Mo SJ, Yoon YJ, Sohng JK. J Microbiol Biotechnol. 2007;17:1909–1921. [PubMed] [Google Scholar]; d) Thibodeaux CJ, Melancon CE, Liu HW. Angew Chem Int Ed Engl. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) White-Phillip J, Thibodeaux CJ, Liu HW. Meth Enzymol. 2009;459:521–544. doi: 10.1016/S0076-6879(09)04621-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yu B, Sun J, Yang X. Acc Chem Res. 2012;45:1227–1236. doi: 10.1021/ar200296m. [DOI] [PubMed] [Google Scholar]
- 18.a) Wang ZY, Luo S, Sato K, Kobayashi M, Nozawa T. Anal Biochem. 1998;257:26–32. doi: 10.1006/abio.1997.2521. [DOI] [PubMed] [Google Scholar]; b) Weber H, Brecker L. Curr Opin Biotechnol. 2000;11:572–578. doi: 10.1016/s0958-1669(00)00146-4. [DOI] [PubMed] [Google Scholar]; c) Mesnard F, Ratcliffe RG. Photosynth Res. 2005;83:163–180. doi: 10.1007/s11120-004-2081-8. [DOI] [PubMed] [Google Scholar]; d) Furtig B, Richter C, Schell P, Wenter P, Pitsch S, Schwalbe H. RNA Biol. 2008;5:41–48. doi: 10.4161/rna.5.1.5704. [DOI] [PubMed] [Google Scholar]; e) Keshari KR, Wilson DM, Chen AP, Bok R, Larson PE, Hu S, Van Criekinge M, Macdonald JM, Vigneron DB, Kurhanewicz J. J Am Chem Soc. 2009;131:17591–17596. doi: 10.1021/ja9049355. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Guyett P, Glushka J, Gu X, Bar-Peled M. Carbohydr Res. 2009;344:1072–1078. doi: 10.1016/j.carres.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Gasmi-Seabrook GM, Marshall CB, Cheung M, Kim B, Wang F, Jang YJ, Mak TW, Stambolic V, Ikura M. J Biol Chem. 2010;285:5137–5145. doi: 10.1074/jbc.M109.064691. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Jiang Y, McKinnon T, Varatharajan J, Glushka J, Prestegard JH, Sornborger AT, Schüttler HB, Bar-Peled M. Biophys J. 2010;99:2318–2326. doi: 10.1016/j.bpj.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Eixelsberger T, Brecker L, Nidetzky B. Carbohydr Res. 2012;356:209–214. doi: 10.1016/j.carres.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Singh S, Peltier-Pain P, Tonelli M, Thorson JS. Org Lett. 2014;16:3220–3223. doi: 10.1021/ol501241a. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Zolghadr B, Gasselhuber B, Windwarder M, Pabst M, Kracher D, Kerndl M, Zayni S, Hofinger-Horvath A, Ludwig R, Haltrich D, Oostenbrink C, Obinger C, Kosma P, Messner P, Schäffer C. Extremophiles. 2015;19:451–67. doi: 10.1007/s00792-015-0730-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Gu X, Glushka J, Yin Y, Xu Y, Denny T, Smith J, Jiang Y, Bar-Peled M. J Biol Chem. 2010;285:9030–9040. doi: 10.1074/jbc.M109.066803. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Polizzi SJ, Walsh RM, Jr, Peeples WB, Lim JM, Wells L, Wood ZA. Biochemistry. 2012;51:8844–55. doi: 10.1021/bi301135b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]; b) Chang C, Bitto E, Goff RD, Singh S, Bingman CA, Griffith BR, Albermann C, Phillips GN, Jr, Thorson JS. Chem Biol. 2008;15:842–853. doi: 10.1016/j.chembiol.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chang A, Singh S, Helmich KE, Goff RD, Bingman CA, Thorson JS, Phillips GN., Jr Proc Natl Acad Sci U S A. 2011;108:17649–17654. doi: 10.1073/pnas.1108484108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.