

Abstract
A set of instruments and specialized equipment is necessary to equip a laboratory to work with DNA. Reducing the barrier to entry for DNA manipulation should enable and encourage new labs to enter the field. We present three examples of open source/DIY technology with significantly reduced costs relative to commercial equipment. This includes a gel scanner, a horizontal PAGE gel mold, and a homogenizer for generating DNA-coated particles. The overall cost savings obtained by using open source/DIY equipment was between 50 and 90%.
Keywords: DNA, DNA nanotechnology, open source, open source scientific hardware
INTRODUCTION
In order to set up an effective DNA nanotechnology laboratory, or to expand a laboratory into the DNA nanotechnology field, a minimum set of equipment is necessary. Much of this equipment is derived from the molecular biology/biotechnology equipment industry. As such, it can be very expensive. We present examples of open source and do-it-yourself (DIY) hardware that reduce the barriers to entry.
The “Open Source” approach greatly increased the accessibility of diverse, practical software. BSD Unix and Linux were early examples of free and open operating systems. Email is delivered by a distributed system running largely on open source server software. The most popular web server, Apache, is open source and free to all users. Many examples of open source software are well known and well used in scientific fields. This includes ImageJ and the GIMP (for analyzing and laying out figures), Numerical Python (for scientific computing with Python). More specialized examples include the simulation of electrophoretic stacking [1].
In addition to cost advantages, this approach has benefits relative to commercial and custom built hardware. Open source scientific hardware (OSSH) designs provide complete build instructions including a bill of materials, schematics, CAD designs and any other information necessary to reproduce the artifact. This makes customization easier. This high level of disclosure also facilitates collaboration and rapid innovation [2–4].
Exciting OSSH projects have emerged in the scientific literature. Academic authors including Pearce have introduced simple examples like the open source lab jack [5]. Complex electromechanical examples include the open source syringe pump. Wijnen et al. found that they could build a functional, programmable syringe pump for a fraction of the cost of commercially available options. Their paper discussed the flexibility of creating open source hardware: the user is fully capable of modifying any element of the design. This allows for custom laboratory instruments that can often not be purchased at retail [6]. The use of 3D printer technology allowed for rapid fabrication and iterative design improvements in these projects. 3D printers are themselves being released under open source hardware licenses [7].
The open source instrument designs we describe here will lower the barrier to entry for DNA nanotechnology research. Applications of DNA nanotechnology are diverse. Increasingly functional examples include precise nanometer scale devices such as an engineered nanopore [9] and a locked box that opens in response to an input oligonucleotide [10]. Some practical applications have also emerged including signal amplification [11] and transduction [12].
We present a gel scanner design for scanning electrophoretically separated DNA. It was derived from an inexpensive office scanner and achieves comparable sensitivity to a commercial instrument. In the second case, an open source design for a gel mold for horizontal polyacrylamide gel electrophoresis (PAGE) is presented and demonstrated. PAGE analysis and purification of DNA oligonucleotides is prerequisite for many applications of DNA. High purity DNA has been important for the performance of many DNA circuits. For instance, the stacked amplification circuitry published by Chen et al. [13] used sequence-verified DNA amplified with high fidelity polymerase followed by PAGE purification. Finally, we show an open source design for a homogenizer which can be attached to any reciprocating motor. This homogenizer can be used to generate colloidal particles from polyacrylamide which can be useful for multiple DNA-based applications including affinity chromatography, NexGen sequencing, or sample enrichment. This is considerably more customizable and less expensive than commercial sources of DNA-coated particles. Both designs can be downloaded, modified, and 3D printed in-house or ordered from a prototype manufacturer.
MATERIALS AND METHODS
Conversion of a color office scanner to fluorescence scanner
More detailed instructions on converting our Canon LiDE 110 scanner (Canon USA, Melville, NY) to fluorescence detection are given in Supporting Information. Briefly, the scanner was disassembled and a Kodak number 15 filter was carefully taped across a region of the scanner bed. The control pins on the ribbon cable for the green and red illumination sources were covered with tape in order to insulate them from their contacts. Conductive ink was then used to bridge the blue control contact to the red and green control contacts so that blue LEDs were eliminated irrespective of the connection to which current was applied. The white background and the portion of the glass panel not covered by the yellow filter were blacked out with flat black spray paint. We added additional side on illumination with a string of high intensity blue LEDs. These LEDs were connected to a separate, 12 v external power source during scanning.
Agarose gel electrophoresis for establishing limit of detection
Low melting point PCR-grade agarose was purchased from Biorad (Hercules, CA). This was dissolved to 3% w/v in hot SB buffer (10 mM boric acid, 10 mM sodium borate all from Sigma-Aldrich, St. Louis, MO). For detection of dsDNA, Approximately 5 μl of GelGreen dye (Biotium, Hayward, CA) was added to the molten gel. This was cast in a Biorad Mini electrophoresis rig. A dilution series of NEB’s 2-log dsDNA ladder (New England Biolabs, Ipswich, MA) or a dilution series of a mixture of fluorescein-modified synthetic ssDNA (IDT, Coralville, IA) was added to the wells. This was run at 180 V for approximately 20 min. This gel was placed on the scanner bed, the scanner bed was closed and covered with black felt to prevent stray external light. The scanner was connected to a PC computer running GIMP and the 12 volt side-on LED illumination (TH Marine, Huntsville, AL) was turned on. The gel was scanned. The image was analyzed using ImageJ.
Horizontal gel casting
Acrylamide/bis-acrylamide (19:1) solution (30%) was purchased from Biorad. A prepolymer solution was prepared from this stock solution to a final concentration of 6%. This prepolymer solution also contained 5% w/v glycerol (Sigma Aldrich) and 10 mM sodium borate buffer, pH 8.5. The gel casting system should accommodate 25 ml of this solution but an excess prepolymer was used (50 ml) to compensate for any leakage and to help float bubbles out of the gel casting mold. A conical vial containing 50 ml of prepolymer was mixed with 44 μl of TEMED (Biorad) and 168 μl of 10% APS (Sigma Aldrich). This mixture was poured into the gel casting tray. The 3D printed lid/comb was slowly lowered into the prepolymer; excess prepolymer was displaced onto the top of the lid. This was allowed to polymerize for 1 h. The lid was then carefully removed to reveal the gel and wells.
Horizontal PAGE purification
A cast polyacrylamide gel was loaded with fluorescein-DNA (GTC TCT GTG CCG CTA TAA T-3’fluorescein) and quencher-DNA (5’Iowa black quencher-ATT ATA GCG GCA CAG AGA CTA AGG TCG G). The DNA was separated for 30 min at 160 v. Fluorescent or colored bands were cut from the gel with a clean razor blade. This small gel slice was then crushed in a 1.7 ml Eppendorf microtube. To this gel slice was added 100 μl nuclease free water and subjected to a freezethaw cycle by placing in a −20°C freezer for 30 min and then placing in an incubator at ~37°C. The crushed gel slice in water was allowed to incubate overnight. The suspension was then filtered using a spin filter (.45 um Costar Spin-X filter, Corning Inc., Corning NY) to remove any residual polyacrylamide particulates.
Titration of fluorescein-DNA with quencher-DNA
A 100 μl sample of fluorescein-DNA from IDT (desalted only, used as received) was diluted to 167 nM (Nanodrop UV/VIS, Thermo-fisher, Waltham, MA). This sample was then titrated with the PAGE purified quencherDNA. Briefly, 1 μl of Quencher-DNA (containing 5.4 pM) was added to the fluorescein-DNA, vortexed, centrifuged, and allowed to incubate for 5 min. The fluorescence of this sample was then quantified using a Quantifluor fluorimeter (Promega, Fitchburg, WI). The process was repeated until the fluorescence values no longer decreased. The titration was repeated with an approximately equally fluorescent sample of purified fluorescein-DNA (UV-Vis indicated a final concentration of 490 nM).
Fluorogenic OSD reaction
Using the titration data above, a FQ complex was prepared by incubating purified or unpurified fluorescein-DNA with sufficient quencher complex to achieve minimal fluorescence. We prepared 50 μl of the quenched FQ complex containing 27 pM of the quencher each. The background fluorescence was measured in three samples prior to the addition of displacer. These samples were then treated with 10 μl of 5 μm displacer DNA (5’ CCG ACC TTA GTC TCT GTG CCG CTA TAA T) which was fully complementary to the quencher-DNA. After 1 h incubation at room temperature with the displacer, the fluorescence of the three samples was quantified.
Generation of Polymer hydrogel particles
One milliliter of mineral oil (Sigma) was mixed to 1% w/v with Span-80 (Sigma). This was added to a 2 ml conical tube with a steel ball bearing. A separate tube was prepared with 100 μl of an aqueous solution of 20% acrylamide, sodium borate buffer (10 mM boric acid, 10 mM sodium borate), 10 μM DNA, 1% TEMED. A third tube was prepared with 8 μl of 10% APS. Once all reagents were chilled to 4 °C, the aqueous solutions were rapidly mixed then rapidly transferred to the tube containing the oil phase. This was then homogenized with ten 1-second pulses with the homogenizer at ~20 Hz (digital tachometer, Tondaj 6234P+, Shenzhen, China). The reaction tube was purged 5 times with flowing nitrogen gas to help remove dissolved oxygen. This was then allowed to stand at room temperature for 30 min. The resulting particles in oil were centrifuged to remove most of the oil. The remaining oil was removed with 5 washes with 70% ethanol. The resulting pellet was dried under a flow of compressed air. The final pellet was resuspended in SB buffer.
Generation of surface modified particles for imaging localization of DNA
The standard operating procedure for generating particles was followed with the following modifications: the acrylamide prepolymer mixture includes 100 nM acrydite- and fluorescein-DNA. In the experimental case, this DNA was incubated with one equivalent of a complementary, cholesterol modified oligonucleotide. In the control case, the cholesterol-DNA was omitted. After the oil was washed away, the particles were resuspended in SB buffer. The particles were then imaged using a fluorescence microscope (Lumascope 620, Etaluma Inc., Carlsbad, CA).
Generation of surface modified particles for DNA capture
The standard operating procedure for generating particles above was followed with the following modifications: the acrylamide prepolymer mixture includes 100 nM acrydite-DNA annealed with one equivalent of a complementary, cholesterol modified oligonucleotide (in the control case the cholesterol-DNA was omitted). Oil is thoroughly removed from the particles. The particles were then resuspended in buffer. Particles were washed once with SB buffer containing 10 μM of the reverse-complement of the cholesterol-DNA (IDT) and 1% triton X-100 (Sigma Aldrich). This removes the cholesterol-DNA from the surface of the particles. Particles were then washed 4 times with SB to remove residual surfactant and any free DNA. Experimental and control particles were then incubated with 10 μM fluorescein-DNA complementary to the acrydite-DNA for 10 min at room temperature. Unbound fluorescein-DNA was removed by washing three times with SB. The fluorescence intensity of the resulting particles was measured using a fluorescence microscope (Lumascope 620). The fluorescence was quantified using ImageJ and compared to the fluorescence of equally sized control particles (which were also imaged using brightfield microscopy).
RESULTS AND DISCUSSION
Conversion of a color office scanner to fluorescence scanner
A number of different methods have been used to capture images of agarose and polyacrylamide gels. The most common are photographic gel imagers and horizontal gel scanners. Traditionally, fluorescently stained or silver stained gels were imaged with a transilluminator and a digital or film camera. Another option is a gel scanner that raster scans a gel with laser excitation source and collects the emitted photons. Although they are highly effective, they are expensive. We converted an inexpensive (< $100) commercial office scanner into a comparable fluorescence gel scanner.
Figure 1 summarizes the conversion of the commercial $100 color scanner to fluorescence detection. The basic technology for flatbed color office scanners is presented in Figure 1A. The scanner illuminates the sample sequentially with three colors of LED illumination. A black and white detector collects reflected light from each illumination source. The three reflected light intensity maps are combined to produce a color image. As such, both blue light and a light sensor are present for fluorescence detection. In practice, it proved advantageous to use side on illumination rather than the internal blue LED array (Fig. 1B).
Figure 1. Design and performance of the converted gel scanner.

A. Diagram shows the operation of the original office scanner. Red, green, and blue LEDs illuminate a subject and a color image is reconstructed by a sensor. B. Diagram shows how a modified fluorescence scanner operates with the blue LED only and a filtered sensor to collect green and red light. C. The elements of the gel scanner are shown including a digital photograph of the filter, the absorption spectrum of the filter, the position of the filter on the underside of the scanner bed, and the assembled scanner with side-on illumination. D. Representative gel scans are shown (top) of an a agarose gel run with a 100 bp ladder stained with GelGreen and (bottom) a PAGE gel with a dilution series (100 pM, 10 pM, 5 pM) of two species of fluorescein-DNA. The inset shows a contrast-enhanced, magnified region containing the faint 5 pM bands.
Although the original scanner itself is commercial, the “source code” for the conversion is presented. Supporting information online contains step-by-step instructions and a detailed list of materials required. The brief description below summarizes these instructions. Green and red illumination sources were disabled. Green or red light would pass through the filter and cause unacceptable levels of background live reflected light. The blue light emitted by the scanner was preserved so that the scanner would pass its internal self-checks. A yellow wratten filter (Fig. 1C) is then taped to the underside of the scanner bed. Side-on blue illumination (rather than the scanner’s internal blue LED) was then added for efficient exultation to increase the signal-to-noise.
The sensitivity of our blue light scanner is demonstrated in Figure 1D and E. A dilution series of a ladder was loaded into an agarose gel with GelGreen stain (Biotium). A band containing as little as 3.7 ng was clearly visible; based on the SNR of this sample, our limit of detection (LOD) is approximately 1 ng. The manufacturer reports that in the best case scenario, as little as 0.1 ng of DNA can be resolved with gel green. This $100 device is nearly within one order of magnitude of the manufacturer’s ideal reported LOD. In a separate experiment, a dilution series of fluorescein-modified ssDNA was also separated by gel electrophoresis; a sample of 5 pM each of 2 species of covalently labeled DNA could be easily resolved. This was superior to the performance of the UVP Multi Doc-it Digital Imaging System, a commercial UV transilluminator imaging station (Fig. S1).
Horizontal adapter for polyacrylamide gel electrophoresis
Agarose gels are suitable for molecular biology and for large molecular weight DNA fragments. DNA nanotechnology is usually built on the basis of smaller DNA fragments (under 100 bases). This requires the use of polyacrylamide gel electrophoresis (PAGE). PAGE gels are generally run using a vertical gel rig rather than a horizontal gel rig. This is due to two related facts. First: acrylamide will not polymerize in the presence of oxygen. This is usually overcome by sandwiching the polymerizing acrylamide between two glass plates. Second: because the gel is positioned between solid pieces of glass, the comb can only be inserted into the gel from the side. This means that gel must be mounted vertically in order to load the samples.
With the advent of 3D printing, a gel casting form can be easily manufactured that allows for the polymerization of an acrylamide gel in the exact format necessary for horizontal gel rigs. Other techniques for creating gels appropriate for horizontal PAGE have been published [15]. Ours has the distinction of using an open source, 3D printed gel mold which can be ordered or modified as needed.
The design was drawn in an open source CAD program, FreeCAD. The design fits Biorad’s Mini-Sub® Cell GT Systems. The design has two parts: (1) a hybrid lid and comb and (2) a tray. 3D renderings of the casting chamber lid with integrated comb are shown in Figure 2A and B. The tray is shown in Figure 2C. A digital photograph of the final, printed parts is shown in Figure 2D. The precise specifications for this casting tray and lid are available in the supporting information online and in the NIH 3D print repository (model ID 3DPX-001228 and 3DPX-001229). Additionally, our prototypes were printed by shapeways. com and additional prints can be ordered directly from them.
Figure 2. Methods for casting PAGE gels for use in horizontal electrophoresis setup.

A and B. 3D renderings of the top part of a casting mold for horizontal PAGE gels. C. 3D rendering of the bottom part of the casting mold. D. Digital photograph of the 3D printed casting mold for PAGE gels.
The casting mold is designed to exclude oxygen and air pockets and allow efficient polymerization. Using this device requires three steps. First, tape is applied to the tray (Fig. 2C) in order to retain ~50 ml of the liquid acrylamide solution. Second, 50 mL of the liquid acrylamide solution is treated with radical initiator and then added to the taped tray. Third, the lid and comb are lowered slowly into the tape mold. This displaces the acrylamide solution and any air pockets to the top of the lid. In general (due perhaps to the thickness of the gel slabs that we pour) degassing has not been necessary. However, because the mold is opaque, trapped air pockets are not apparent until after the gel is ruined. Special care must therefore be taken to allow any air pockets to escape while lowering the lid and comb. The internal, enclosed portion of the mold is approximately 25 ml. The additional volume acts as a barrier to isolate the mold from oxygen and to compensate for any leakage. After approximately 40 min, the tape is removed and the lid carefully withdrawn from the tray.
DNA circuit performance improves with horizontal PAGE purification
Synthetic DNA must often be purified before downstream use [13]. Applications include toehold-mediated strand exchange [16] reaction or hairpin opening [17]. Synthesis errors such as single base eliminations and branch impurities can hurt reaction performance. Oligonucleotides can be purified using horizontal PAGE. The gel is loaded as with a standard agarose gel and voltage is applied. When the DNA has migrated to an appropriate distance, the band is carefully cut from the gel using a clean razor blade or scalpel. This gel is then crushed, subjected to a freeze-thaw cycle, and allowed to incubate in water overnight. The suspension is then filtered to remove any acrylamide chunks. Because the quencher oligonucleotide was HPLC purified by the vendor, it is likely that most impurities were already removed. To test this hypothesis, we mixed equimolar quantities of all possible combinations of purified and unpurified fluorescein- and quencher- modified DNA (see Fig. S3). PAGE purification of the fluorescein-DNA improved quencher performance, but PAGE purification of the HPLC-purified quencher did not further improve performance.
The performance of functional nucleic acid reactions such as a one-step strand displacement (OSD) reaction [18] is greatly increased by using purified nucleic acids. We purified fluorescein-DNA and quencher-DNA with PAGE in a horizontally-cast gel as described above (gel image is shown in supplementary information as Fig. S4). When adding a complementary quencher to the fluorescein-DNA to form the fluorescein-quencher (FQ) complex, we expect nearly complete quenching when the molar concentrations are equal (see schematic Fig. 3A). We carefully controlled the concentration of the two fluorescein-modified DNA samples using UV/Vis spectrophotometry. Although initial concentrations of the fluorescent DNA were equal, their initial fluorescent intensities were slightly different. It seems likely that the impurities that were removed during purification have a higher molecular weight which affects the calculation of molar concentration by UV/Vis. Nonetheless, purified DNA was quenched efficiently to leave only <5% of the original fluorescence remaining after titration (Fig. 3B, dark blue diamonds). When the unpurified DNA was used (standard desalting from IDT) the fluorescence plateaued at ~15% of the original fluorescence intensity (Fig. 3B, red squares). Clearly, hybridization based quenching is more efficient with PAGE purified DNA.
Figure 3. Horizontal PAGE purification effects on performance of DNA-DNA reactions.

A. Diagram shows a simple hybridization-mediated quenching reaction. B. Fluorescence titration curve shows purified fluorescein-DNA titrated with purified or unpurified quencher-DNA. C. Diagram shows a strand displacement reaction. D. Fluorescence data show de-quenching by displacer DNA in the case of a PAGE purified complex and a desalted complex.
Likewise, the purified FQ complex responds more favorably to toehold mediated strand displacement than its unpurified counterpart. The reaction schematic is shown in Figure 3C. The fully complementary displacer can form a more stable, complete duplex with the quencher strand by displacing the shorter fluorophore-modified strand. This displacement relieves the quenched condition and restores fluorescence. The signal to noise ratio increases from 6 to 9 with purification. The signal to background ratio increases from 3 to 5.5 with purification.
3D printed homogenizer for reciprocating motor
In order to create hydrogel microparticles, we required a method for generating water-in-oil emulsions. In previous work [19] such particles were generated using a commercial homogenizer, the Tissuelyzer LT by Qiagen. This device rapidly oscillates a tube containing a dispersing element like a steel ball bearing. We rationalized that a similar, though less precise, instrument could be constructed for generating polymer microspheres. The design and operation of our “open source homogenizer” is shown in Figure 4.
Figure 4. 3D printed homogenizer design and operation.
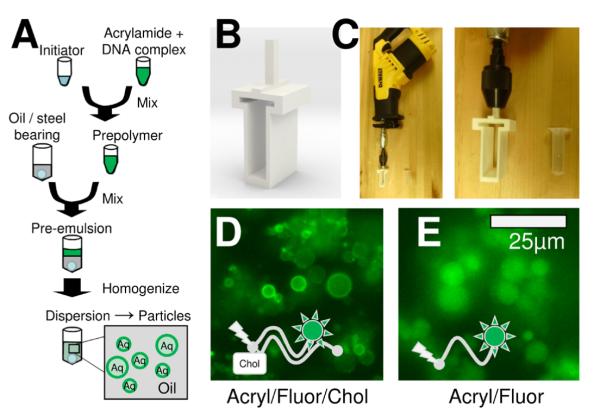
A. Diagram shows how dispersion polymerization particles were generated. B. 3D rendering shows the design of the 3D printed homogenizer. C. Digital photographs show how the 3-D printed homogenizer is assembled using a reciprocating motor. D. Fluorescence micrograph shows particles generated with DNA bearing the acrydite, cholesterol, and fluorescein modifications. E. Fluorescence micrograph shows particles generated with DNA bearing the acrydite and fluorescein modifications (i.e. without cholesterol). Scale bar is 25 μm for both images.
We generated particles by dispersion polymerization. This is shown in Figure 4A. A mixture containing acrylamide, DNA, and buffer were mixed with an initiator. This prepolymer was then transferred to a vial containing oil, surfactant, and a steel ball bearing. This was then rapidly oscillated using the homogenizer shown in Figure 4B and C. The homogenizer consisted of a 3-D printed tube holder (Fig. 4B) and reciprocating saw with appropriate adapters. This tube holder was designed in FreeCAD and then printed by shapeways.com. The designs are included in supplementary material online and at the NIH 3-D print repository (Model ID 3DPX-001227). Once homogenized, the mixture is allowed to polymerize under nitrogen. This produced hydrogel microparticles which were washed from the oil and resuspended in buffer. The design as shown can accommodate two sample tubes. We also tested a design capable of accommodating six sample tubes simultaneously. This design is shown in supporting material Figure S2. The 3D CAD files are available for download in supporting material online or at the NIH 3D print repository (Model ID 3DPX-001748).
The resulting microparticles are shown in Figure 4D and E. These microparticles were generated containing acrydite-DNA. The acrydite modification polymerizes into the growing polyacrylamide chain. This covalently links the DNA to the hydrogel polymer matrix [20]. Depending on the additional modifications, the DNA can be located to the interior or surface of the particle. If the DNA bears a cholesterol modification, it will tend to be distributed at the aqueous-oil interface prior to polymerization. After polymerization, the DNA is locked in place (Fig. 4D). If the DNA does not have a cholesterol modification, it is evenly distributed throughout the particle (Fig. 4E).
Surface modified DNA is effective for hybridization-based capture
We expected that the DNA immobilized on the surface of our hydrogel particles would be effective at capturing complementary DNA. We prepared surface modified hydrogel particles that lacked any fluorophores. We incubated them with complementary, fluorescein-DNA. As expected, particles bearing DNA on their surface effectively captured the fluorescent DNA. Particles with DNA polymerized within the internal structure were ineffective at capturing significant quantities of fluorescein-DNA. These results are shown in Figure 5.
Figure 5. Hydrogel particles for affinity capture.
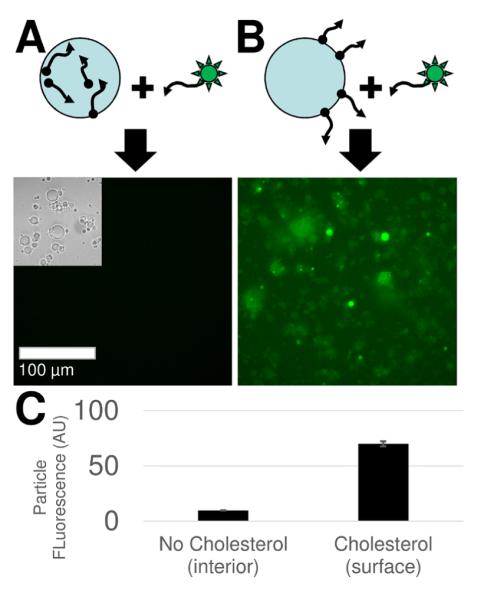
A. Diagram and corresponding fluorescence micrograph show how particles generated without cholesterol show little affinity for complementary, fluorescein-DNA (inset shows brightfield). The scale bar shows 100 μm and is consistent throughout. B. Particles generated with cholesterol show significant affinity for complementary, fluorescein-DNA. C. Graph shows fluorescence intensity values from representative particles in both cases.
CONCLUSIONS
There are many reasons for exploring an open source approach to laboratory equipment. Open source equipment is more flexible and usually less expensive than the equivalent commercially produced experimental apparatus. While cost savings is an important consideration in and of itself, it also allows for researchers to take a smaller risk in trying a new procedure or exploring a new research interest.
Functional microparticles represent an excellent example of an adaptable research tool that can be enhanced with the OSSH approach. Functional microparticles can be useful in multiple diverse applications. They can be used for capture, purification [21], aggregation assays [22], chromatography [23], flow cytometry assays [24], and much more. With the equipment presented here, a modest investment would allow a researcher to accomplish much more than by buying a single type of commercial microparticles. Researchers can produce their own microparticles in multiple chemistries, and coat them with purified DNA with many different sequences and chemical modifications. Equipment can be easily customized for the needs of the particular research project or lab. Furthermore, shipping and customs can cause delays for researchers in some countries and the generation of reagents and tools in-house can greatly speed progress.
Other examples of OSSH in the literature can also yield savings of about 90%. A comprehensive review has been published elsewhere [25]. An in-house built autosampler saves 95% of the cost of a commercial alternative. The open source syringe pump previously mentioned [6] can save 50-90% (depending on the commercial syringe pump replaced). An open source colorimeter [26] is reported to save 97% relative to a commercial instrument.
This paper aims to contribute to a growing mass of OSSH with a large return on investment. The cost savings of building rather than buying a piece of equipment can range from 50 to 90%. For our three examples the savings are about 90% in each case. In the case of the scanner (cost ~$200), it replaces a gel document system that costs several thousand dollars (greater than $2,000, used). In the case of the horizontal gel casting mold (cost ~$30), it replaces a second gel rig that costs several hundred dollars (typical cost ~$500-$1000 depending on size). Even in the case that a single initial apparatus must be purchased, the cost savings would be ~50%. In the case of the homogenizer (cost ~$200 including motor and speed controller), it replaces a commercial tissue homogenizer that costs several thousand dollars (> $5000). We used it for the generation of microparticles; the homogenizer could also be used for disrupting cells or tissue. The ability to save funds and time through OSSH should help spare funds for labor and expand the number of researchers engaged in productive experiments. The purification, analysis and immobilization of nucleic acids all require substantial capital investment. These designs are designed to help researchers enter the growing field of DNA nanotechnology by reducing this barrier to entry.
Supplementary Material
Acknowledgments
We gratefully acknowledge the University of Idaho for seed funding for this project. A.S. received summer support though NIH/NIGMS INBRE fellowship P20GM103408. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH."
Footnotes
Competing interests: The authors have declared that no competing interests exist.
Supplementary information
Figure S1. Comparison of sensitivity of converted gel scanner with commercial transilluminator.
Figure S2. A version of the tube holder that can accommodate six tubes is also printable.
Figure S3. A par graph shows a comparison of the quencher efficiency (percentage of fluorescence remaining after adding equimolar quencher DNA).
Figure S4. Digital photograph of the gel used for PAGE purification.
File S1. Construction instructions and parts lists for the devices described.
File S2. A zip file containing 3D cad files for the 3D printed parts used in this study.
Supplementary information of this article can be found online at http://www.jbmethods.org.
References
- 1.Bercovici M, Lele SK, Santiago JG. Open source simulation tool for electrophoretic stacking, focusing, and separation. J Chromatogr A. 2008;1216:1008–1018. doi: 10.1016/j.chroma.2008.12.022. doi: 10.1016/j.chroma.2008.12.022. PMID: 19124132. [DOI] [PubMed] [Google Scholar]
- 2.Pearce JM. Building Research Equipment with Free, Open-Source Hardware. Science. 2012;337:1303–1304. doi: 10.1126/science.1228183. doi: 10.1126/science.1228183. PMID: 22984059. [DOI] [PubMed] [Google Scholar]
- 3.Hienerth C, von Hippel E, Berg Jensen M. User community vs. producer innovation development efficiency: A first empirical study. Res Policy. 2014;43:190. doi: 10.1016/j.respol.2013.07.010. [Google Scholar]
- 4.Pearce JM. Laboratory equipment: Cut costs with open-source hardware. Nature. 2014;505:618. doi: 10.1038/505618d. doi: 10.1038/505618d. PMID: 24476879. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C, Anzalone NC, Faria RP, Pearce JM. Open-source 3D-printable optics equipment. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059840. doi: 10.1371/journal.pone.0059840. PMID: 23544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijnen B, Hunt EJ, Anzalone GC, Pearce JM. Open-source syringe pump library. PLoS One. 2014;9 doi: 10.1371/journal.pone.0107216. doi: 10.1371/journal.pone.0107216. PMID: 25229451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones R, Haufe P, Sells E, Iravani P, Olliver V, et al. RepRap – the replicating rapid prototyper. Robotica. 2011;29:177. doi: 10.1017/S026357471000069X. [Google Scholar]
- 8.Macdonald J, Li Y, Sutovic M, Lederman H, Pendri K, et al. Medium Scale Integration of Molecular Logic Gates in an Automaton. Nano Lett. 2006;6:2598. doi: 10.1021/nl0620684. doi: 10.1021/nl0620684. [DOI] [PubMed] [Google Scholar]
- 9.Langecker M, Arnaut V, Martin TG, List J, Renner S, et al. Synthetic lipid membrane channels formed by designed DNA nanostructures. Science. 2012;338:932–936. doi: 10.1126/science.1225624. doi: 10.1126/science.1225624. PMID: 23161995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459:73–76. doi: 10.1038/nature07971. doi: 10.1038/nature07971. PMID: 19424153. [DOI] [PubMed] [Google Scholar]
- 11.Allen PB, Arshad SA, Li B, Chen X, Ellington AD. DNA circuits as amplifiers for the detection of nucleic acids on a paperfluidic platform. Lab Chip. 2012;12:2951–2958. doi: 10.1039/c2lc40373k. doi: 10.1039/c2lc40373k. PMID: 22729075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Ellington AD, Chen X. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkr504. doi: 10.1093/nar/gkr504. PMID: 21693555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Briggs N, McLain JR, Ellington AD. Stacking nonenzymatic circuits for high signal gain. Proc Natl Acad Sci U S A. 2013;110:5386–5391. doi: 10.1073/pnas.1222807110. doi: 10.1073/pnas.1222807110. PMID: 23509255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen YJ, Dalchau N, Srinivas N, Phillips A, Cardelli L, et al. Programmable chemical controllers made from DNA. Nat Nanotechnol. 2013;8:755–762. doi: 10.1038/nnano.2013.189. doi: 10.1038/nnano.2013.189. PMID: 24077029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellomy GR, Record MT. A method for horizontal polyacrylamide slab gel electrophoresis. BioTechniques. 1989;7:16. PMID: 2629829. [PubMed] [Google Scholar]
- 16.Chen X. Expanding the rule set of DNA circuitry with associative toehold activation. J Am Chem Soc. 2012;134:263–271. doi: 10.1021/ja206690a. doi: 10.1021/ja206690a. PMID: 22129141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin P, Choi HMT, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451:318–322. doi: 10.1038/nature06451. doi: 10.1038/nature06451. PMID: 18202654. [DOI] [PubMed] [Google Scholar]
- 18.Yurke B, Turberfield AJ, Mills AP, Jr, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406:605–608. doi: 10.1038/35020524. doi: 10.1038/35020524. PMID: 10949296. [DOI] [PubMed] [Google Scholar]
- 19.Allen PB, Khaing Z, Schmidt CE, Ellington AD. 3D Printing with Nucleic Acid Adhesives. ACS Biomater Sci Eng. 2014;1:19–26. doi: 10.1021/ab500026f. doi: 10.1021/ab500026f. PMID: 25984570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehman FN, Audeh M, Abrams ES, Hammond PW, Kenney M, et al. Immobilization of acrylamide-modified oligonucleotides by co-polymerization. Nucleic Acids Res. 1999;27:649–655. doi: 10.1093/nar/27.2.649. doi: 10.1093/nar/27.2.649. PMID: 9862993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo T. Streamlined method for purifying single-stranded DNA from PCR products for frequent or high-throughput needs. Biotechniques. 2005;38:700. doi: 10.2144/05385BM04. PMID: 15945367. [DOI] [PubMed] [Google Scholar]
- 22.Rogers PH, Michel E, Bauer CA, Vanderet S, Hansen D, et al. Selective, Controllable, and Reversible Aggregation of Polystyrene Latex Microspheres via DNA Hybridization. Langmuir. 2005;21:5562–5569. doi: 10.1021/la046790y. doi: 10.1021/la046790y. PMID: 15924490. [DOI] [PubMed] [Google Scholar]
- 23.Arndt-Jovin DJ, Jovin TM, Bähr W, Marquardt M, Frischauf AM. Covalent Attachment of DNA to Agarose. Eur J Biochem. 1975;54:411–418. doi: 10.1111/j.1432-1033.1975.tb04151.x. doi: 10.1111/j.1432-1033.1975.tb04151.x. PMID: 1100376. [DOI] [PubMed] [Google Scholar]
- 24.Tang H, Deschner R, Allen P, Cho Y, Sermas P, et al. Analysis of DNA-guided self-assembly of microspheres using imaging flow cytometry. J Am Chem Soc. 2012;134:15245–15248. doi: 10.1021/ja3066896. doi: 10.1021/ja3066896. PMID: 22938015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearce J. The Open-Source Lab: How to Build Your Own Hardware and Reduce Research Costs. 1st Elsevier; Amsterdam: 2014. [Google Scholar]
- 26.Anzalone GC, Glover AG, Pearce JM. Open-source colorimeter. Sensors (Basel) 2013;13:5338–5346. doi: 10.3390/s130405338. doi: 10.3390/s130405338. PMID: 23604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.