

Background: In the plant sulfur assimilation pathway, APS kinase is a redox-regulated branch point enzyme.
Results: Structural and biochemical analysis of the cyanobacterial APSK reveals an unregulated precursor of the plant enzyme.
Conclusion: Protein engineering of cyanobacterial APSK recapitulates the structural development of redox control in the plant enzyme.
Significance: Understanding the evolution of biochemical regulation provides insight for engineering metabolic controls.
Keywords: Arabidopsis, plant biochemistry, protein structure, redox regulation, sulfur
Abstract
In plants, adenosine 5′-phosphosulfate (APS) kinase (APSK) is required for reproductive viability and the production of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as a sulfur donor in specialized metabolism. Previous studies of the APSK from Arabidopsis thaliana (AtAPSK) identified a regulatory disulfide bond formed between the N-terminal domain (NTD) and a cysteine on the core scaffold. This thiol switch is unique to mosses, gymnosperms, and angiosperms. To understand the structural evolution of redox control of APSK, we investigated the redox-insensitive APSK from the cyanobacterium Synechocystis sp. PCC 6803 (SynAPSK). Crystallographic analysis of SynAPSK in complex with either APS and a non-hydrolyzable ATP analog or APS and sulfate revealed the overall structure of the enzyme, which lacks the NTD found in homologs from mosses and plants. A series of engineered SynAPSK variants reconstructed the structural evolution of the plant APSK. Biochemical analyses of SynAPSK, SynAPSK H23C mutant, SynAPSK fused to the AtAPSK NTD, and the fusion protein with the H23C mutation showed that the addition of the NTD and cysteines recapitulated thiol-based regulation. These results reveal the molecular basis for structural changes leading to the evolution of redox control of APSK in the green lineage from cyanobacteria to plants.
Introduction
Advances in genomics and sequence analysis now allow the exploration of the evolutionary history of protein structure and function. The use of ancestral protein reconstruction (i.e. paleoenzymology) and the synthesis of genes encoding proteins that are potential precursors to those found in different evolutionary lineages can provide insight into the original properties that were the basis for current specialized proteins (1–4). Evolutionary comparisons to understand enzyme function largely focus on catalysis and substrate recognition; however, three-dimensional structural studies combined with phylogenetic comparisons can also identify core scaffolds and elucidate how the adaptation of new domains leads to the development of regulatory features (5–7). For example, crystallographic and biochemical examination of adenosine 5′-phosphosulfate kinase (APSK)3 from Arabidopsis thaliana has identified a redox control element in the N-terminal domain (NTD) of the enzyme (8–11). Phylogenetic sequence comparisons suggest that evolutionary precursors of the Arabidopsis enzyme (i.e. APSK from cyanobacteria) lack this structural domain and that adoption of a disulfide in the NTD of APSK from the green lineage occurred as the sulfur assimilation pathway evolved for use in plastids of plants and mosses (8). Here we aimed to reconstruct the structural evolution of redox regulation in the APSK from cyanobacteria to plants.
The assimilation of sulfur from the environment and its incorporation into a diverse set of metabolites, including cysteine, methionine, glutathione, iron-sulfur clusters, vitamin cofactors, and multiple sulfur-containing specialized molecules, is common to eukaryotes and prokaryotes (12–19). Interestingly, plants and microbes differ in their approach to converting inorganic sulfate to more usable forms. Once sulfate is transported into the cell, it is incorporated into adenosine 5′-phosphosulfate (APS) by ATP sulfurylase (20–22). The high energy phosphosulfate bond in APS provides an energetic driving force in sulfur assimilation. In plants, the next metabolic transformations form a branch point (Fig. 1A). Reduction of APS to sulfite by APS reductase provides the primary route for incorporation of sulfur into various metabolites (23). APS can also be phosphorylated by APSK into the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) (Fig. 1A). The partitioning of sulfur into PAPS by APSK is essential for plant growth and development, as the enzyme is required for fertility and seed production (24–27). A similar branching of sulfur metabolism has been observed in some pathogenic bacteria such as Mycobacterium tuberculosis (28, 29). In contrast, other bacteria, fungi, and humans sequentially use both APSK and APS reductase in the primary sulfur assimilation pathway to convert APS into PAPS, which is then further metabolized into sulfite by PAPS reductase (12, 13, 17).
FIGURE 1.
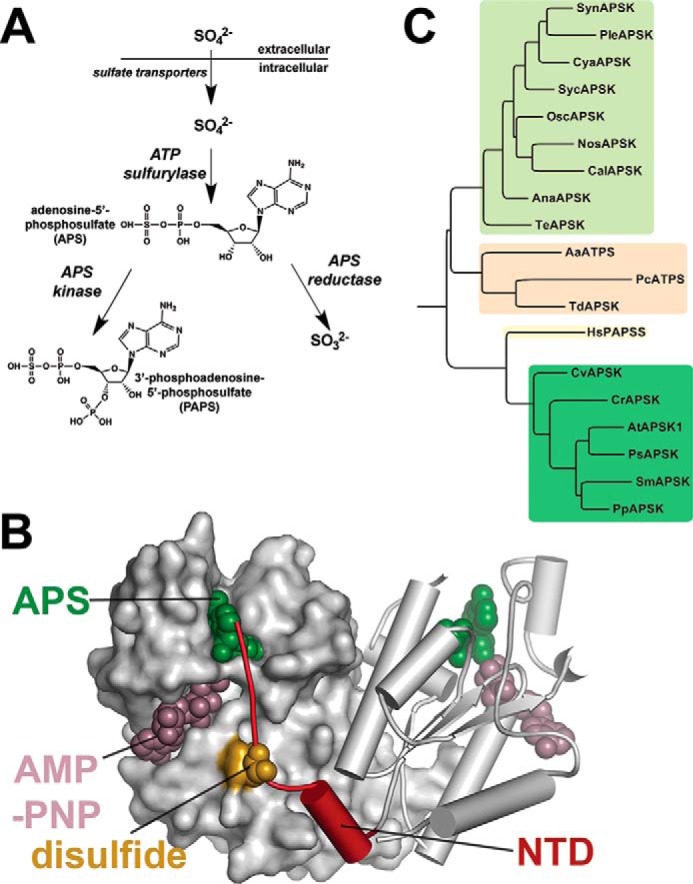
Overview of plant sulfur assimilation and APSK. A, the metabolic branch point from APS in the sulfur assimilation pathway of plants. B, structural overview of the NTD (red) and regulatory disulfide in AtAPSK. One monomer is shown as a surface rendering and the other as a ribbon diagram. Bound AMP-PNP (rose color) and APS (green) are represented by space-filling models. The position of the disulfide bond (gold) formed between Cys-86 of the right dimer and Cys-119 of the left dimer is shown by a space-filling model. C, phylogenetic comparison of APSK. Representative species in the clades for cyanobacteria (light green) and the algae, mosses, and plants (dark green) were chosen as a broad sample of the green lineage. Sequences of other structurally characterized APSK from bacteria and fungi (tan) and human (yellow) were included for comparison. The following sequences were used for comparison: SynAPSK (P72940.1, Synechocystis sp. PCC 6803), PleAPSK (WP_019504653.1, Pleurocapsa sp. PCC 7319), CyaAPSK (WP_013322291.1, Cyanothece sp. PCC 7822), SycAPSK (WP_012307108.1, Synechococcus sp. PCC 7002), OscAPSK (WP_017716042.1, Oscillatoria sp. PCC 10802), NosAPSK (WP_044500498.1, Nostoc sp. PCC 7107), CalAPSK (WP_035174174.1, Calothrix sp. PCC 7103), AnaAPSK (WP_016950032.1, Anabaena sp. PCC 7108), TeAPSK (WP_011611551.1, T. erythraeum), CvAPSK (XP_005843477.1, C. variabilis), CrAPSK (BAF46286.1, C. reinhardtii), AtAPSK1 (NP_179082.1, A. thaliana), PsAPSK (ABR17904.1; P. sitchensis), SmAPSK (XP_002976147.1, S. moellendorffii), PpAPSK (XP_001755205.1, P. patens), AaATPS (O67174.1, Aquifex aeolicus), PcATPS (Q12650.1, P. chrysogenum), TdAPSK (PDB 3CR8, T. denitrificans), and HsPAPSS (O43252.2, Homo sapiens).
In plants, the branch point from APS into either sulfite or PAPS may be regulated by redox conditions. Thiol-based redox switches differentially modulate the activity of APS reductase and APSK (8–11, 30). Reduction attenuates APS reductase activity and enhances APSK activity, whereas oxidation accelerates APS reductase and attenuates PAPS production by APSK (8–11, 30). This reciprocal redox regulation provides a means of directing sulfur flux into either primary sulfur metabolism (APS reductase) or specialized metabolism (APSK) depending on the cellular conditions.
APSK from A. thaliana (AtAPSK) is biochemically well characterized (8–11, 31). AtAPSK exhibits an ordered kinetic mechanism in which the binding of ATP increases affinity for APS; however, this enzyme also exhibits severe substrate inhibition by APS (8–11, 31). AtAPSK functions as a homodimer and has a 19-amino acid NTD that contains a cysteine residue, which reversibly forms a disulfide with another cysteine on the adjacent monomer (Fig. 1B). The disulfide may alter the flexibility of the NTD, as the reduced form of AtAPSK is nearly 20-fold more efficient than the oxidized form and is 15-fold less susceptible to substrate inhibition by APS (8–11). Structure and sequence comparisons show that the APSK from cyanobacteria are missing the NTD found in the enzyme from algae, mosses, gymnosperms, and angiosperms and lack the disulfide cysteines found in the homologs from mosses, gymnosperms, and angiosperms (8) (Figs. 1C and 2).
FIGURE 2.

Multiple sequence alignment of APSK from cyanobacteria, algae, mosses, and plants. Protein abbreviations are the same as given for Fig. 1C. Conserved residues are highlighted in green with variations indicated by gray text. Residues in the APS binding site and ATP binding site are highlighted in gold and rose, respectively. The invariant catalytic aspartate is highlighted in orange. Amino acid positions corresponding to residues in the disulfide linkage are highlighted in red. The tan box delineates residues corresponding to the NTD of AtAPSK. The light blue box indicates residues in the β1e-α7 loop. Residues corresponding to the sulfate binding site at the dimer interface of SynAPSK are indicated by light blue text. The multiple sequence alignment was generated with MultAlin.
To understand the molecular evolution of the redox control feature, we investigated the redox-insensitive APSK (SynAPSK) from the cyanobacterium Synechocystis sp. PCC 6803. Crystallographic analysis of SynAPSK in complex with either APS and a non-hydrolyzable ATP analog or APS and sulfate reveals the overall structure of the enzyme, which lacks the NTD found in homologs from mosses and plants. A series of engineered SynAPSK variants reconstructed the structural evolution of the plant APSK. Biochemical analysis of SynAPSK, SynAPSK H23C mutant, SynAPSK fused to the AtAPSK NTD (SynAPSKfus), and SynAPSKfus with the H23C mutation (SynAPSKfus/H23C) showed that the addition of the NTD did not drastically affect the steady-state kinetic behavior of the enzyme and that introduction of the disulfide between the NTD and core altered the redox sensitivity of SynAPSK both in terms of catalytic efficiency and substrate inhibition by APS. Overall, these studies suggest the molecular basis for structural changes leading to the evolution of redox control in APSK in the green lineage from cyanobacteria to plants.
Experimental Procedures
Materials
Generation of the pET-28a-AtAPSK1Ä77 and pET-28a-SynAPSK bacterial expression vectors was described previously (8, 20). The gene encoding the SynAPSKfus protein was synthesized (Genewiz, Inc.) to place the NTD of AtAPSK1 (STNIKWHECSVEKVDRQRL) in-frame with the coding region of SynAPSK and included NdeI and EcoRI restriction sites at the 5′- and 3′-ends of the gene, respectively. The SynAPSKfus coding region was inserted into pET-28a using the NdeI and EcoRI restriction sites. Oligonucleotides for generating the H23C and T61E SynAPSK mutants were from Integrated DNA Technology. APS and AMP-PNP were purchased from Sigma-Aldrich.
Protein Expression and Purification
All proteins were expressed and purified using a common protocol. Escherichia coli BL21(DE3) cells were transformed with the appropriate expression vector. Cells were grown in Terrific broth containing 50 μg ml−1 kanamycin at 37 °C (250 rpm) until an A600 nm ∼ 0.8 was reached. Protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and temperature was reduced to 18 °C overnight. Cells were pelleted by centrifugation (10,000 × g for 15 min), resuspended in lysis buffer (50 mm Tris, pH 8.0, 500 mm NaCl, 20 mm imidazole, 1 mm β-mercaptoethanol, 10% (v/v) glycerol, and 1% (v/v) Tween 20), and sonicated. The lysed cells were centrifuged (30,000 × g for 60 min), and the supernatant was passed over a Ni2+-nitrilotriacetic acid (Qiagen) column equilibrated with wash buffer (lysis buffer minus Tween 20). Once the column was washed with 20 column volumes of wash buffer, the protein was eluted with elution buffer (wash buffer with 250 mm imidazole). To remove the N-terminal hexahistidine tag, thrombin (1/2000th of total protein) was added to the eluent, and the mixture was dialyzed overnight at 4 °C against 25 mm HEPES, pH 7.5, 200 mm KCl, 5% (v/v) glycerol, and 2 mm DTT. Following dialysis, the sample was passed over a mixed Ni2+-nitrilotriacetic acid/benzamidine-Sepharose column to remove thrombin and uncleaved His-tagged protein. The flow-through was then loaded onto a Superdex-200 26/60 HiLoad FPLC size-exclusion column equilibrated with dialysis buffer. Peak fractions were collected and concentrated using centrifugal concentrators (Amicon) with protein concentration determined using molecular extinction coefficients calculated in ProtParam. For oxidized proteins, DTT was omitted from the dialysis and the final purification steps. All proteins were flash-frozen in liquid nitrogen and stored at −80 °C.
Protein Crystallography
Crystals of the SynAPSK·AMP-PNP·Mg2+·APS complex grew at 4 °C in hanging drops (2 μl) from a 1:1 ratio of protein (10 mg ml−1) preincubated with 5 mm APS, 5 mm AMP-PNP, and 10 mm MgCl2 and mixed with crystallization buffer (0.1 m sodium cacodylate trihydrate, pH 6.5, 0.2 m magnesium acetate tetrahydrate, and 20% (w/v) PEG 8000). Crystals of the SynAPSK·APS·sulfate complex were obtained with a similar crystallization set-up but using protein preincubated with 5 mm APS and 10 mm K2SO4 and mixed with 0.1 m CAPS/KOH, pH 10.5, 2 m ammonium sulfate, and 0.2 m LiSO4. All crystals were cryoprotected in mother liquor containing 20% glycerol before freezing in liquid nitrogen. X-ray diffraction data (0.5° oscillations, 360 images, and 100 K) were collected at Structural Biology Center beamlines 19-ID (SynAPSK·AMP-PNP·Mg2+·APS complex) and 19-BM (SynAPSK·APS·sulfate complex) of the Argonne National Laboratory Advanced Photon Source. HKL3000 (32) was used to integrate, merge, and scale diffraction intensities. The structure of SynAPSK in complex with AMP-PNP, Mg2+, and APS was solved by molecular replacement using the structure of AtAPSK (8) with coordinates of the NTD deleted. Molecular replacement was implemented with PHASER (33) and yielded six molecules in the asymmetric unit. The structural model of SynAPSK was built in COOT (34) and refined using PHENIX (35). The initial model building and refinement used noncrystallographic symmetry restraints, which were released in later rounds of refinement. TLS (translation-libration-screen) modeling was used throughout refinement. Iterative rounds of model building and refinement were continued until the R-factors converged to those reported in Table 1. The final model includes residues 4–176 of each chain, 1 sodium ion, 2 cacodylate molecules, and 88 waters. The structure of the SynAPSK·APS·sulfate complex was solved by molecular replacement using the SynAPSK structure determined above. Two molecules were found in the asymmetric unit with model building and refinement performed as described above (see Table 1). The final model includes residues 3–176 of chain A and residues 2–177 of chain B, 4 sulfates, and 557 waters. Coordinates and structure factors for the SynAPSK·AMP-PNP·Mg2+·APS and SynAPSK·APS·sulfate complexes were deposited in the Protein Data Bank with accession codes 5CB6 and 5CB8, respectively.
TABLE 1.
Summary of crystallographic statistics
| AMP-PNP·Mg2+·APS | APS·sulfate | |
|---|---|---|
| Crystal | ||
| Space group | C2 | I23 |
| Cell dimensions | a = 136.9 Å, b = 109.7 Å, c = 100.8 Å; β = 90.7° | a = b = c = 140.8 Å |
| Data collection | ||
| Wavelength (Å) | 0.979 | 0.979 |
| Resolution range (Å) (highest shell resolution) | 40.4–2.79 (2.84–2.79) | 31.5–1.88 (1.91–1.88) |
| Reflections (total/unique) | 130,683/37,195 | 810,057/37,779 |
| Completeness (%) (highest shell) | 99.9 (99.8) | 100 (100) |
| 〈I/σ〉 (highest shell) | 14.8 (2.0) | 50.0 (5.0) |
| Rsym (%) (highest shell) | 17.7 (82.3) | 7.8 (84.9) |
| Model and Refinement | ||
| Rcryst/Rfree | 16.4/21.8 | 15.5/17.6 |
| No. of protein atoms | 8,135 | 2,791 |
| No. of water molecules | 88 | 557 |
| No. of ligand atoms | 365 | 82 |
| r.m.s.d., bond lengths (Å) | 0.010 | 0.006 |
| r.m.s.d., bond angles (°) | 1.235 | 1.038 |
| Avg B-factor (Å2): protein, waters, ligands | 55.0, 45.1, 59.1 | 23.1, 37.1, 22.9 |
| Stereochemistry (%): favored, allowed, outliers | 97.4, 2.6, 0 | 98.3, 1.7, 0 |
Site-directed Mutagenesis
The H23C and T61E SynAPSK point mutants and SynAPSKfus with the mutation corresponding to H23C were generated using the QuikChange PCR method (Agilent) and confirmed by sequencing (Washington University DNA Sequencing Facility). Protein expression and purification of the mutants was performed as described above.
Enzyme Assays
Steady-state kinetic assays were performed as described previously with the resulting initial velocity data fit to either the Michaelis-Menten equation, v = Vmax × [S]/(Km +[S]), or the equation for substrate inhibition, v = Vmax × [S]/(Km + [S] × (1 + [S]/Ki)), using KaleidaGraph (8, 20) For determination of kinetic parameters with APS, ATP was held constant at 0.1 mm. Determination of kinetic parameters with varied ATP was performed at 5 μm APS. Sulfate inhibition assays were performed at 0.1 mm ATP and 5 μm APS with varied K2SO4 (0–100 mm). Steady-state kinetic parameters for reduced and oxidized protein used the standard conditions with either 5 mm DTT (reduced) or 5 mm trans-4,5-dihydroxy-1,2-dithiane (oxidized DTT) added to the assay.
Results
X-ray Crystal Structure of the SynAPSK·AMP-PNP·Mg2+·APS Complex
SynAPSK was overexpressed in E. coli as an N-terminal His-tagged protein and purified using Ni2+ affinity and size-exclusion chromatography. Purified SynAPSK eluted from the gel filtration column as a dimer of ∼40 kDa (monomer molecular weight, ∼20 kDa). Steady-state kinetic parameters for APS and ATP were determined (Table 2) for the purified protein, which was subsequently used for protein crystallization with β,γ-imidoadenosine 5′-triphosphate (AMP-PNP) and APS to obtain the structure of a Michaelis complex.
TABLE 2.
Steady-state kinetic parameters
Average values ± S.E. (n = 3) are shown. Assays were performed in the presence of either 5 mm DTT (RED, reduced) or 5 mm trans-4,5-dihydroxy-1,2-dithiane (OX, oxidized), as indicated.
| kcat | bKmAPS | KiAPS | kcat/KmAPS | KmATP | bkcat/KmATP | |
|---|---|---|---|---|---|---|
| s−1 | μm | μm | m−1 s−1 × 106 | μm | m−1 s−1 × 106 | |
| SynAPSKRED | 28.1 ± 1.6 | 0.30 ± 0.05 | 14.7 ± 2.4 | 93.67 | 0.16 ± 0.07 | 175.6 |
| SynAPSKOX | 19.3 ± 0.5 | 0.23 ± 0.02 | 18.2 ± 2.1 | 83.91 | 0.13 ± 0.06 | 148.5 |
| T61ERED | 23.5 ± 0.8 | 0.28 ± 0.03 | 19.5 ± 2.6 | 83.93 | 0.20 ± 0.08 | 117.5 |
| H23CRED | 19.0 ± 0.9 | 0.22 ± 0.03 | 12.8 ± 1.7 | 86.36 | 0.15 ± 0.05 | 126.7 |
| H23COX | 17.6 ± 1.1 | 0.20 ± 0.05 | 16.6 ± 3.2 | 88.00 | 0.16 ± 0.08 | 110.0 |
| SynAPSKfusRED | 26.3 ± 1.3 | 0.32 ± 0.05 | 14.8 ± 2.0 | 82.19 | 0.27 ± 0.11 | 97.41 |
| SynAPSKfusOX | 18.7 ± 1.1 | 0.24 ± 0.06 | 12.0 ± 2.4 | 77.92 | 0.19 ± 0.04 | 98.42 |
| SynAPSKfus/H23CRED | 11.0 ± 0.5 | 0.11 ± 0.02 | 20.4 ± 3.1 | 100.0 | 0.15 ± 0.06 | 73.33 |
| SynAPSKfus/H23COX | 12.8 ± 1.1 | 0.49 ± 0.09 | 2.6 ± 0.4 | 26.12 | 0.28 ± 0.04 | 45.71 |
| AtAPSKREDa | 272 ± 39 | 0.48 ± 0.41 | 37.5 ± 6.9 | 567 | 0.18 ± 0.09 | 1,511 |
| AtAPSKOXa | 14.1 ± 2.3 | 0.43 ± 0.26 | 2.5 ± 0.8 | 0.328 | 0.21 ± 0.04 | 67.1 |
a Values for AtAPSK were reported previously (8) and are included here for comparison.
The 2.79 Å resolution x-ray crystal structure of SynAPSK in complex with AMP-PNP, a non-hydrolyzable ATP analog, Mg2+, and APS was solved by molecular replacement using AtAPSK (8) as a search model (Table 1). Six molecules were found in the asymmetric unit. Chains A/D and B/F form noncrystallographic dimers with chains C and E sharing crystallographic symmetry mates that complete the dimer. The core structure of the SynAPSK monomer is a canonical α/β-purine nucleotide-binding domain with a central parallel β-sheet (β1b-β1c-β1a-β1d-β1e) surrounded by α1 and α7 on one face of the β-sheet and α2, α3, and α4 on the other side (Fig. 3A). Clear electron density for AMP-PNP, Mg2+, and APS (Fig. 3B) identified the nucleotide binding sites as situated along the top of the core α/β-domain. Two additional α-helices (α5 and α6) are linked to the central scaffold via flexible loops and provide a cap over the bound nucleotides. The two monomers in the SynAPSK dimer are related by a 2-fold symmetry through an interface formed by α2 and α3 of each chain.
FIGURE 3.

Structure of the SynAPSK·AMP-PNP·Mg2+·APS complex. A, ribbon diagram of the dimer. Secondary structure features are labeled in the left monomer with α-helices and β-strands colored in gold and rose, respectively. The positions of AMP-PNP and APS are shown as stick molecules. The lower view shows a 90° rotation of the view in the top panel. B, 2Fo − Fc omit map (1.5 σ) for AMP-PNP, Mg2+, and APS in the SynAPSK active site. C, stereoview of the SynAPSK active site.
A comparison of the SynAPSK monomer with other structures in the Protein Data Bank (PDB) using DALI (36) reveals conservation of a three-dimensional fold between the APSK domain of human PAPS synthetase (37) (PDB code: 2PEY; 50% sequence identity; Z = 30.3; 0.8 Å r.m.s.d. for 176 Cα atoms), Penicillium chrysogenum APSK (38) (PDB code: 1M7G; 44% sequence identity; Z = 29.6; 1.0 Å r.m.s.d. for 171 Cα atoms), and AtAPSK (8) (PDB code: 4FXP; 47% sequence identity; Z = 28.4; 1.1 Å r.m.s.d. for 172 Cα atoms), and the APSK domains of the bifunctional ATP sulfurylases from P. chrysogenum (39) (PDB code: 1I2D; 42% sequence identity; Z = 28.3; 1.0 Å r.m.s.d. for 172 Cα atoms), Aquifex aeolicus (40) (PDB code: 2GKS; 51% sequence identity; Z = 26.9; 1.1 Å r.m.s.d. for 166 Cα atoms), and Thiobacillus denitrificans (41) (PDB code: 3CR8; 43% sequence identity; Z = 21.0; 1.6 Å r.m.s.d. for 143 Cα atoms).
SynAPSK binds APS to position the ribose hydroxyl groups for Asp42-mediated phosphoryl group transfer from ATP to the 3′-OH group (Fig. 3C). The adenosine ring of APS is sandwiched between Phe-54 and Phe-134 with the amine group of the ring forming a hydrogen bond to the carbonyl of Gly-133. Arg-45 and Arg-59 provide electrostatic interactions with the sulfophosphate moiety. Additional hydrogen bonds with the mixed anhydride are contributed by the carbonyl group of Ser-86 and the amine group of Ile-85. With the exception of Gly-133, all of the residues that form interactions with APS are invariant in the APSK from plants, mosses, algae, and cyanobacteria (Fig. 2).
The ATP binding site is defined through interactions with the AMP-PNP analog through the nucleotide phosphate groups (Fig. 3C). Multiple interactions are mediated through the main-chain carbonyls and amide nitrogens of Gly-15, Gly-17, Lys-18, Thr-19, and Thr-20 in the canonical P-loop and the side chains of Thr-20 and Lys-120. The side-chain group of Arg-117 provides contacts with the ribose and adenosine groups of ATP. The Mg2+ ion is bound by contacts with the β- and γ-phosphate groups, the hydroxyl group of Thr-19, the carboxylates of Asp-40 and Asp-42, and a water molecule. As with the APS binding site, the ATP binding site is generally conserved in the APSK from cyanobacteria, mosses, and plants (Fig. 2), but there are conservative substitutions between the P-loop of the cyanobacterial APSK (GAGKTT) and that of the plant, moss, and algal APSK (GSGKST). Moreover, the AMP-PNP binding site displays both sequence and structural variability in the residues surrounding the adenosine moiety of the nucleotide (Figs. 2 and 4). As noted above, a comparison of the SynAPSK and AtAPSK monomers highlights their overall structural conservation but also shows differences in the β1e-α7 loop that caps the adenosine group of bound ATP (Fig. 4A). In SynAPSK, the β1e-α7 loop differs in sequence (RTDLEEL) compared with the corresponding loop of AtAPSK (REGGTSP) (Fig. 4B). These changes shift the position of the loop backbone and alter the interactions with ATP in each protein. In SynAPSK, the backbone carbonyl of Glu-156 and the side-chain hydroxyl group of Ser-161 hydrogen bond to N1 and N6, respectively, of the adenosine ring. In contrast, only the backbone carbonyl of Thr-255 in AtAPSK hydrogen bonds to N5 of the adenosine ring. A sequence comparison of the APSK from plants, mosses, algae, and cyanobacteria shows that this loop region, as well as the C-terminal tail, displays greater variability than other regions of the enzyme.
FIGURE 4.
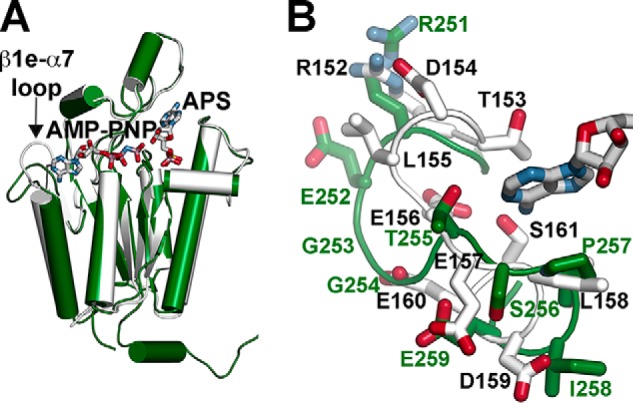
Structural comparison of SynAPSK and AtAPSK monomers. A, an overlay of SynAPSK (white) and AtAPSK (green) is shown. The difference in position of the β1e-α7 loop is indicated. B, close-up view of differences in the β1e-α7 loop of SynAPSK (white) and AtAPSK (green). The adenosine and ribose portions of AMP-PNP bound to SynAPSK are shown in gray.
X-ray Crystal Structure of the SynAPSK·APS·Sulfate Complex
Within the APSK active site, the P-loop is responsible for the discrimination of nucleotides containing a phosphate at the β-position (i.e. ADP and ATP) versus those with a sulfate at this position (i.e. APS) (9). The 1.88 Å resolution x-ray crystal structure of SynAPSK in complex with APS and sulfate was determined to examine where sulfate binds in the ATP binding site (Table 1 and Fig. 5A). The overall structure is nearly identical to the SynAPSK·AMP-PNP·Mg2+·APS complex with a 0.3 Å r.m.s.d. for 174 Cα atoms. Unambiguous electron density for APS and sulfate in the active site was observed (Fig. 5B). As reported in other APSK structures (8, 42), the sulfate molecule is bound by multiple amide nitrogen contacts with residues in the P-loop (Gly-15, Gly-17, Lys-18, and Thr-19) and charge-charge interaction with the side-chain amine of Lys-18.
FIGURE 5.
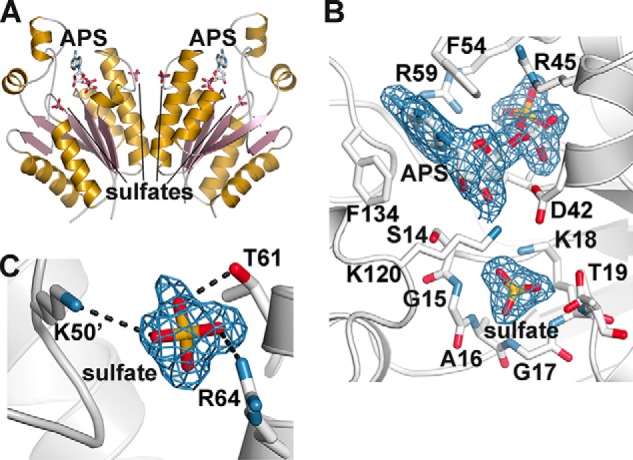
Structure of the SynAPSK·APS·sulfate complex. A, ribbon diagram of the dimer. Secondary structure features are colored as described in the legend for Fig. 3A. The positions of APS and sulfates are indicated. B, view of APS and sulfate binding in the SynAPSK active site. Electron density of the 2Fo − Fc omit map (1.5 σ) for APS and sulfate is shown. C, the sulfate binding site at the dimer interface of SynAPSK. The 2Fo − Fc omit map (1.5 σ) for the sulfate molecule is shown.
In addition to the sulfate in the active site of SynAPSK, a second sulfate binding site was identified in the dimer interface (Fig. 5, A and C). This molecule interacts with Thr-61 and Arg-64 of one monomer and Lys-50 from the adjacent monomer. To test whether this binding site was functionally relevant, the SynAPSK T61E mutant was generated, and the resulting mutant was expressed, purified, and kinetically analyzed. Substitution of Thr-61 with a glutamate, which is the preferred amino acid at this position in the plant, moss, algal, and cyanobacterial APSK (Fig. 2), did not significantly alter the steady-state kinetic parameters of the mutant (Table 2). Moreover, the effects of sulfate on the activity of SynAPSK (IC50 = 17.0 ± 0.4 mm) and the SynAPSK T61E mutant (IC50 = 15.7 ± 0.2 mm) were comparable. Because the residue ancestrally corresponding to Thr-61 of SynAPSK is typically a glutamate, the binding of a sulfate in the dimer interface of SynAPSK appears to be a crystallization artifact related to the sequence of SynAPSK. Overall, these results are consistent with sulfate inhibition occurring through binding at the P-loop in the active site.
Structural Evolution of the N-terminal Domain and Redox Control Element in APSK
Crystallographic and biochemical studies identified the NTD of AtAPSK as a redox control feature that modulates enzymatic activity (8–11). In contrast to plant APSK, the cyanobacterial enzymes lack both the N-terminal extension (as well as a chloroplast localization sequence) and the cysteine residues that form the regulatory disulfide (Figs. 1C and 2). To test the structural evolution of the thiol-based switch in APSK, a series of engineered variants were constructed for kinetic analysis (Fig. 6A). Using SynAPSK as the structural core, a synthetic gene was used to place the 19-amino acid NTD of AtAPSK (STNIKWHECSVEKVDRQRL) in-frame with the coding region of SynAPSK to yield SynAPSKfus. The fused NTD contains one cysteine of the regulatory disulfide switch. A comparison of the sequence and structure of AtAPSK with SynAPSK identified His-23 as the residue for mutagenesis to cysteine, the second residue in the disulfide linkage. Site-directed mutagenesis of this residue yielded the SynAPSK H23C mutant and the SynAPSKfus/H23C variant.
FIGURE 6.
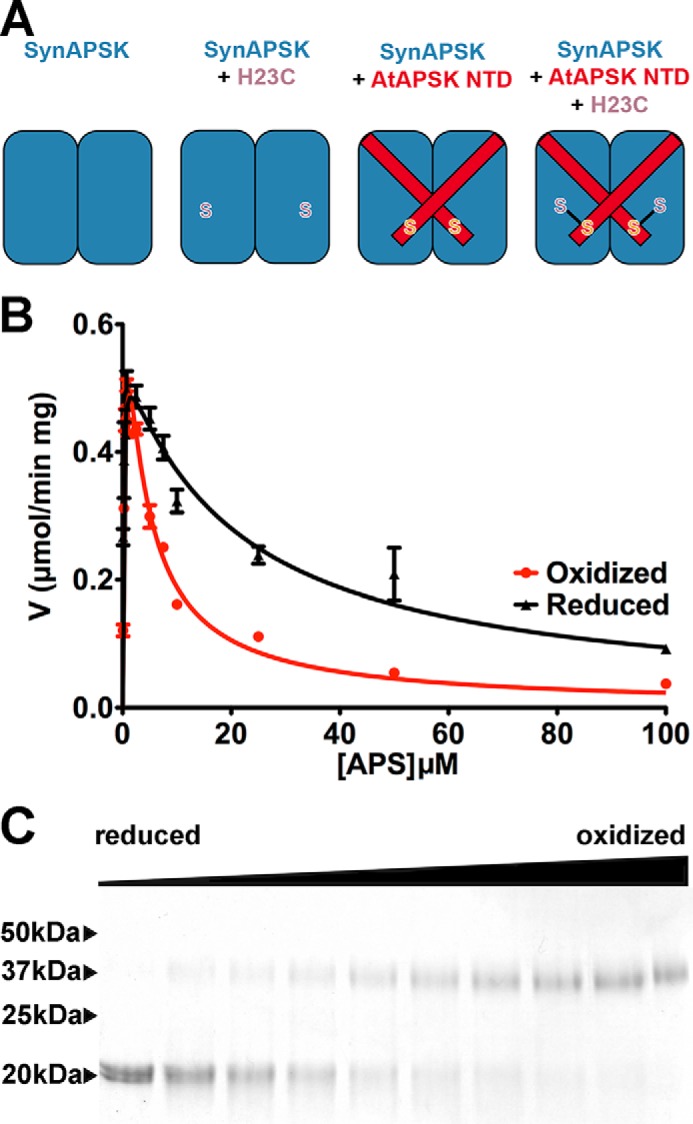
Structural evolution of the NTD and redox control element in APSK. A, schematic of SynAPSK variants used for reconstructing the structural evolution of redox control. B, steady-state kinetic comparison of the SynAPSKfus/H23C protein in assays performed under either reducing (5 mm DTT, black) or oxidizing (5 mm trans-4,5-dihydroxy-1,2-dithiane, red) conditions. Data for V versus APS concentration are shown with the fits to substrate inhibition displayed. C, SDS-PAGE analysis of SynAPSKfus/H23C protein incubated with varied ratios of DTT to trans-4,5-dihydroxy-1,2-dithiane. The leftmost lane contains 5 mm DTT, and the rightmost lane contains 5 mm trans-4,5-dihydroxy-1,2-dithiane, with in-between lanes containing increasing ratios of oxidized trans-4,5-dihydroxy-1,2-dithiane to reduced DTT. Arrowheads on the left side of the gel indicate the positions of the molecular weight markers.
Steady-state kinetic analysis of SynAPSK, SynAPSK H23C, SynAPSKfus, and SynAPSKfus/H23C demonstrated that engineering of the NTD and disulfide leads to differences in redox sensitivity (Table 2 and Fig. 6B). Analysis of SynAPSK under reducing and oxidizing conditions yielded comparable kcat and Km values. Likewise, the SynAPSK H23C mutant displayed kinetic parameters similar to the wild type under either reducing or oxidizing conditions. Fusion of the NTD of AtAPSK to SynAPSK resulted in slight (i.e. less than 2-fold) increases in the Km values for ATP. Under oxidizing conditions, SynAPSKfus/H23C displayed a 4.5-fold lower kcat/Km for APS and an 8-fold lower Ki for APS substrate inhibition (Fig. 6B). In addition, a less than 2-fold change in Km for ATP was observed under oxidizing conditions. Formation of the engineered disulfide linkage in the NTD of SynAPSK was also confirmed by nonreducing SDS-PAGE analysis of SynAPSKfus/H23C incubated in changing ratios of reduced DTT: trans-4,5-dihydroxy-1,2-dithiane (oxidized DTT) (Fig. 6C). Under reducing conditions, the unlinked monomer was observed. With increasing oxidation, the formation of the disulfide cross-linked dimer was apparent.
Discussion
The uptake and assimilation of sulfur for a variety of metabolic purposes is a common feature in prokaryotes and eukaryotes alike. In particular, the use of APS (Fig. 1A), a high energy molecule with twice the energy of the pyrophosphate linkage of ATP (43–45), for incorporation of sulfur into compounds of primary and specialized metabolism places constraints on the protein structure required for recognizing this special metabolite. The enzymes responsible for producing APS (ATP sulfurylase) and its conversion to useable forms (APS kinase) are highly conserved in sequence and core structure across a wide range of organisms (18, 46); however, these enzymes display a remarkable variety in domain organization/architecture and regulatory features.
The structures of SynAPSK in complex with either AMP-PNP, Mg2+, and APS (Fig. 3) or APS and sulfate (Fig. 5) reveal a compact homodimeric structure with a monomer fold that is retained in APSK from prokaryotes and eukaryotes. Although there are minor sequence variations in the P-loop and larger differences in the sequence and positioning of the β1e-α7 loop between cyanobacterial and plant APSK (Fig. 4), the positioning of the conserved catalytic aspartate, a nearly invariant APS binding site, and a well conserved ATP binding site (Fig. 3C) suggests a reaction sequence for SynAPSK that is shared with other enzymes of the family (31, 47, 48). For phosphoryl group transfer, ATP·Mg2+ binds first followed by APS. The Mg2+ ion is required for catalysis and organization of the active site with Asp-42 acting as a general base for the abstraction of a proton from the ribose 3′-OH group of APS, which leads to its nucleophilic attack on the γ-phosphate of ATP. Following catalysis, PAPS is released followed by ADP. Presumably, the active site “lid” domain, which includes α5 and α6, moves between the open and closed forms to allow for binding and release of the nucleotides (42).
The x-ray crystal structure of SynAPSK and the amino acid sequence comparisons with representative members of different cyanobacterial families (i.e. Anabaena sp. PCC 7108, Nostoc sp. PCC 7107, Calothrix sp. PCC 7103, Trichodesmium erythraeum, Cyanothece sp. PCC 7822, Pleurocapsa sp. PCC 7319, Synechococcus sp. PCC 7002, and Oscillatoria sp. PCC 10802) indicate that these organisms use highly homologous (61–83% amino acid sequence identity) enzymes for the formation of PAPS (Fig. 2). With the exception of the APSK from Anabaena, which has a short N-terminal extension, the cyanobacterial APSK lack the NTD found in the homologs from algae, mosses, and plants (Fig. 2). This major difference, along with other sequence variations, leads to a clear phylogenetic split between the cyanobacterial APSK and the APSK from organisms along the evolutionary lineage from cyanobacteria to green algae and chlorophytes (Chlorella variabilis and Chlamydomonas reinhardtii), mosses (Physcomitrella patens and Selaginella moellendorffii), gymnosperms (Picea sitchensis), and angiosperms (A. thaliana) (Fig. 1C). Nonetheless, the APSK in the green lineage share 46–57% amino acid sequence identity with SynAPSK and likely retain a homodimeric stucture as observed in both SynAPSK and AtAPSK (8).
A comparison of the three-dimensional structure ofSynAPSK with other APSK in the Protein Data Bank highlights the various ways in which its core scaffold, along with that of ATP sulfurylase, has been duplicated, recombined into multifunctional enzymes, and modified to different configurations and/or functions. Phylogenetic comparisons indicate that the APSK domain of the human PAPS synthetase shares a closer similarity to the enzymes from algae, mosses, and plants than that of cyanobacteria (Fig. 1C), but the APSK domains from fungi (P. chrysogenum) and the bacteria A. aeolicus and T. denitrificans are more related to the cyanobacterial APSK (Fig. 1C). However, each of these organisms links APSK and ATP sulfurylase in different ways. For example, the human PAPS synthetase is a bifunctional enzyme with an N-terminal APSK domain linked to a C-terminal ATP sulfurylase domain (37). Each domain forms a dimeric structure connected by a linker region. The bifunctional enzyme from A. aeolicus swaps the order of the two domains and uses the C-terminal APSK domain for homodimer formation (40). In yeast/fungi (P. chrysogenum and Saccharomyces cerevisiae), ATP sulfurylase functions as a homohexamer but contains a nonfunctional C-terminal APSK-like domain (39). In the enzyme from P. chrysogenum, the APSK-like domain functions as a regulatory subunit of ATP sulfurylase activity (39) with a monofunctional APSK providing for PAPS synthesis (38). With the hexameric APSK from T. denitrificans, it is an N-terminal ATP sulfurylase-like domain that lost catalytic function to serve as an oligomerization domain for the catalytic C-terminal APSK domain (41). The evolutionary differences in the organization of APSK and ATP sulfurylase likely reflect the different needs and uses of these organisms for sulfur in their metabolisms. This also appears to be the situation in the evolution of the NTD in APSK from cyanobacteria to plants.
Previous studies demonstrate that AtAPSK is redox-regulated (8–11). The oxidized form of AtAPSK exhibited a 20-fold reduction in catalytic efficiency and was 15-fold more sensitive to substrate inhibition by APS than the reduced protein (Table 2) (8). Structural and functional analyses defined the role of the NTD and a disulfide linkage in modulating these differences (8–11) (Fig. 1B). The redox-sensitive APSK in plants appears to have evolved after bifurcation of the sulfur assimilation pathway in the green plant lineage to provide a control mechanism for partitioning sulfur flow via APS into primary and specialized thiol metabolic routes in plastids (8–11). Earlier biochemical studies on the APSK from Arabidopsis demonstrated similar redox regulation that could be mediated by E. coli thioredoxin (31), which is consistent with a common target recognition mechanism for the reduction of disulfides by thioredoxin (49). In plants, oxidative stresses increase the demand for cysteine and glutathione and activate two key enzymes (APS reductase and glutamate-cysteine ligase) in the primary pathway leading to these molecules (40, 50–53). Oxidizing conditions attenuate APSK activity to limit APS use in the secondary route. Thus, reciprocal regulation of the APS branch point can be controlled by the cellular redox state.
As cyanobacteria represent the evolutionary origins of the chloroplast in plants (54, 55), structural studies of SynAPSK provide a foundation for investigating the structural development of redox regulation in APSK from the green plant lineage. Using a series of SynAPSK variants, we reconstructed the evolutionary path of a regulatory feature from cyanobacteria to plants (Fig. 6A). As with SynAPSK, both the SynAPSK H23C mutant and SynAPSK with an AtAPSK NTD fusion (i.e.SynAPSKfus) displayed minimal differences in steady-state kinetic parameters under either reducing or oxidizing conditions (Table 2). Only the version with both an NTD and an H23C substitution recapitulated the effect of oxidation and disulfide formation on APSK activity (Fig. 6, B and C). However, the 4.5-fold reduction in catalytic efficiency and 8-fold stronger effect of substrate inhibition by APS on SynAPSKfus/H23C suggests that other changes are required to complete the evolutionary path and tighter redox control observed with AtAPSK.
Although the monomer structures of SynAPSK and AtAPSK are similar (DALI Z score = 28.4 with a 1.1 Å r.m.s.d. for 172 Cα atoms), superimposition of the dimeric structures of each enzyme (4.2 Å r.m.s.d. for 172 Cα atoms) reveals differences in the positioning of secondary structural features within each monomer (Fig. 7). Along the dimer interface, α2 and α3 are shifted 3.8° and 9.6°, respectively, toward the exterior of the dimer in AtAPSK compared with their positions in SynAPSK. Similarly, α1 and α7, which buttress the ATP binding site, are tilted 10.5 and 15°, respectively, into the dimer interface of AtAPSK compared with SynAPSK. Although the general position of His-23 of SynAPSK and Cys-119 of AtAPSK are comparable and redox effects can be conferred to SynAPSKfus/H23C, additional structural changes likely contribute to the more effective thiol switch in AtAPSK.
FIGURE 7.
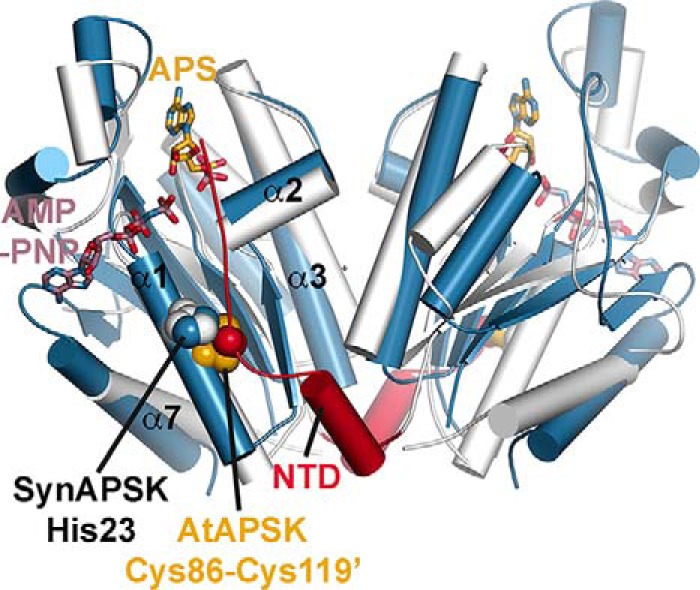
Structural comparison of SynAPSK and AtAPSK dimers. The overlay of SynAPSK (white) and AtAPSK (blue) highlights the shifts in secondary structure between each dimer. The positions of AMP-PNP (rose) and APS (gold) in the SynAPSK structure are shown as stick drawings. The positions of the NTD of AtAPSK (red), the disulfide residues of AtAPSK (gold/red space-filling models), and His-23 in SynAPSK (blue/white space-filling models) are indicated.
Previous studies done on AtAPSK and the APSK domain of PAPS synthetase from humans also suggest that slight structural variations with SynAPSK may lead to biochemical differences. Deletion of the NTD in AtAPSK and PAPS synthetase abolishes substrate inhibition by APS (9, 56). In contrast, SynAPSK is inhibited by APS even though it lacks the corresponding NTD (Table 2). Mutations leading to shifts in the dimeric structure of APSK may optimize disulfide formation and the positioning of the NTD (11). Flexibility in the NTD of APSK has been observed in multiple crystal structures (8, 9, 38, 40–42, 57). For example, ligand binding studies of reduced and oxidized AtAPSK indicate that the NTD and disuflide bond formation alters ligand binding preference to modulate substrate inhibition by APS (9, 11). Localized structural movements can create active site conformations that favor or disfavor catalysis. Given the mobility of structural features in APSK from all organisms, it is plausible that conformational dynamics contribute to the distinct biochemical properties of various APSK (58, 59).
Another potential regulatory feature was also examined (Fig. 5 and Table 2). The structure of the SynAPSK·APS·SO42+ complex shows an ordered sulfate bound to residue Thr-61. As this residue appeared to be at a potential allosteric site, the T61E point mutant was generated. No difference in sulfate inhibition was observed between the wild type and the T61E mutant, indicating that this ordered sulfate is a crystallographic artifact resulting from the sequence of SynAPSK.
As genomes become available for various organisms across the “tree of life,” such information provides fertile ground for the examination and reconstruction of the molecular evolution of protein structure and function, biochemical regulation, and the organization of metabolic pathways (1–7). Evolutionary biochemistry offers the opportunity to explore and compare proteins as they adapt to various systems-level demands. Unraveling how regulatory features, such as thiol-based redox switches, develop promises to have both protein and metabolic engineering applications.
Author Contributions
J. H. and J. M. J. designed the research; J. H., D. N., S. G. L., T. S., and J. M. J. performed the research; J. H., D. N., and J. M. J. analyzed the data; J. H., T. S., and J. M. J. wrote the paper; and all of the authors edited the paper.
Acknowledgments
Portions of this research were carried out atthe Argonne National Laboratory Structural Biology Center of the Advanced Photon Source, a national user facility operated by the University of Chicago for the Department of Energy Office of Biological and Environmental Research (DE-AC02-06CH11357).
This work was supported by Grant MCB-0904215 from the National Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5CB6 and 5CB8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- APSK
- adenosine 5′-phosphosulfate kinase
- APS
- adenosine 5′-phosphosulfate
- NTD
- N-terminal domain
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- AtAPSK
- APSK from A. thaliana
- SynAPSK
- APSK from Synechocystis sp. PCC 6803
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- fus
- fusion.
References
- 1. Gaucher E. A., Thomson J. M., Burgan M. F., Benner S. A. (2003) Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature 425, 285–288 [DOI] [PubMed] [Google Scholar]
- 2. Perez-Jimenez R., Inglés-Prieto A., Zhao Z. M., Sanchez-Romero I., Alegre-Cebollada J., Kosuri P., Garcia-Manyes S., Kappock T. J., Tanokura M., Holmgren A., Sanchez-Ruiz J. M., Gaucher E. A., Fernandez J. M. (2011) Single-molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat. Struct. Mol. Biol. 18, 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harms M. J., Thornton J. W. (2013) Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev. Genet. 14, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carter C. W. (2014) Urzymology: experimental access to a key transition in the appearance of enzymes. J. Biol. Chem. 289, 30213–30220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vogel C., Bashton M., Kerrison N. D., Chothia C., Teichmann S. A. (2004) Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 14, 208–216 [DOI] [PubMed] [Google Scholar]
- 6. Allen A., Kwagh J., Fang J., Stanley C. A., Smith T. J. (2004) Evolution of glutamate dehydrogenase regulation of insulin homeostasis is an example of molecular exaptation. Biochemistry 43, 14431–14443 [DOI] [PubMed] [Google Scholar]
- 7. Ballicora M. A., Dubay J. R., Devillers C. H., Preiss J. (2005) Resurrecting the ancestral enzymatic role of a modulatory subunit. J. Biol. Chem. 280, 10189–10195 [DOI] [PubMed] [Google Scholar]
- 8. Ravilious G. E., Nguyen A., Francois J. A., Jez J. M. (2012) Structural basis and evolution of redox regulation in plant adenosine-5′-phosphosulfate kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ravilious G. E., Jez J. M. (2012) Nucleotide binding site communication in Arabidopsis thaliana adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 287, 30385–30394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang M., Ravilious G. E., Hicks L. M., Jez J. M., McCulla R. D. (2012) Redox switching of adenosine-5′-phosphosulfate kinase with photoactivatable atomic oxygen precursors. J. Am. Chem. Soc. 134, 16979–16982 [DOI] [PubMed] [Google Scholar]
- 11. Ravilious G. E., Westfall C. S., Jez J. M. (2013) Redox-linked gating of nucleotide binding by the N-terminal domain of adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 288, 6107–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Ploeg J. R., Eichhorn E., Leisinger T. (2001) Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch. Microbiol. 176, 1–8 [DOI] [PubMed] [Google Scholar]
- 13. Mendoza-Cózatl D., Loza-Tavera H., Hernández-Navarro A., Moreno-Sánchez R. (2005) Sulfur assimilation and glutathione metabolism under cadmium stress in yeast, protists and plants. FEMS Microbiol. Rev. 29, 653–671 [DOI] [PubMed] [Google Scholar]
- 14. Yi H., Galant A., Ravilious G. E., Preuss M. L., Jez J. M. (2010) Sensing sulfur conditions: simple to complex protein regulatory mechanisms in plant thiol metabolism. Mol. Plant 3, 269–279 [DOI] [PubMed] [Google Scholar]
- 15. Yi H., Ravilious G. E., Galant A., Krishnan H. B., Jez J. M. (2010) From sulfur to homoglutathione: thiol metabolism in soybean. Amino Acids 39, 963–978 [DOI] [PubMed] [Google Scholar]
- 16. Takahashi H., Kopriva S., Giordano M., Saito K., Hell R. (2011) Sulfur assimilation in photosynthetic organisms: molecular functions and regulations of transporters and assimilatory enzymes. Annu. Rev. Plant Biol. 62, 157–184 [DOI] [PubMed] [Google Scholar]
- 17. Hatzios S. K., Bertozzi C. R. (2011). The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ravilious G. E., Jez J. M. (2012) Structural biology of plant sulfur metabolism: from assimilation to biosynthesis. Nat. Prod. Rep. 29, 1138–1152 [DOI] [PubMed] [Google Scholar]
- 19. Koprivova A., Kopriva S. (2014) Molecular mechanisms of regulation of sulfate assimilation: first steps on a long road. Front. Plant Sci. 5, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M., Krishnan H. B. (2006) Soybean ATP sulfurylase, a homodimeric enzyme involved in sulfur assimilation, is abundantly expressed in roots and induced by cold treatment. Arch. Biochem. Biophys. 450, 20–29 [DOI] [PubMed] [Google Scholar]
- 21. Ravilious G. E., Herrmann J., Lee S. G., Westfall C. S., Jez J. M. (2013) Kinetic mechanism of the dimeric ATP sulfurylase from plants. Biosci. Rep. 33, e00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herrmann J., Ravilious G. E., McKinney S. E., Westfall C. S., Lee S. G., Baraniecka P., Giovannetti M., Kopriva S., Krishnan H. B., Jez J. M. (2014) Structure and mechanism of soybean ATP sulfurylase and the committed step in plant sulfur assimilation. J. Biol. Chem. 289, 10919–10929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin M. N., Tarczynski M. C., Shen B., Leustek T. (2005) The role of 5′-adenylylsulfate reductase in controlling sulfate reduction in plants. Photosynth. Res. 86, 309–323 [DOI] [PubMed] [Google Scholar]
- 24. Mugford S. G., Yoshimoto N., Reichelt M., Wirtz M., Hill L., Mugford S. T., Nakazato Y., Noji M., Takahashi H., Kramell R., Gigolashvili T., Flügge U. I., Wasternack C., Gershenzon J., Hell R., Saito K., Kopriva S. (2009) Disruption of adenosine-5′-phosphosulfate kinase in Arabidopsis reduces levels of sulfated secondary metabolites. Plant Cell 21, 910–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mugford S. G., Matthewman C. A., Hill L., Kopriva S. (2010) Adenosine-5′-phosphosulfate kinase is essential for Arabidopsis viability. FEBS Lett. 584, 119–123 [DOI] [PubMed] [Google Scholar]
- 26. Yatusevich R., Mugford S. G., Matthewman C., Gigolashvili T., Frerigmann H., Delaney S., Koprivova A., Flügge U. I., Kopriva S. (2010) Genes of primary sulfate assimilation are part of the glucosinolate biosynthetic network in Arabidopsis thaliana. Plant J. 62, 1–11 [DOI] [PubMed] [Google Scholar]
- 27. Kopriva S., Mugford S. G., Baraniecka P., Lee B. R., Matthewman C. A., Koprivova A. (2012) Control of sulfur partitioning between primary and secondary metabolism in Arabidopsis. Front. Plant Sci. 3, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hatzios S. K., Bertozzi C. R. (2011) The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schnell R., Schneider G. (2010) Structural enzymology of sulphur metabolism in Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 396, 33–38 [DOI] [PubMed] [Google Scholar]
- 30. Bick J. A., Setterdahl A. T., Knaff D. B., Chen Y., Pitcher L. H., Zilinskas B. A., Leustek T. (2001) Regulation of the plant-type 5′-adenylyl sulfate reductase by oxidative stress. Biochemistry 40, 9040–9048 [DOI] [PubMed] [Google Scholar]
- 31. Lillig C. H., Schiffmann S., Berndt C., Berken A., Tischka R., Schwenn J. D. (2001) Molecular and catalytic properties of Arabidopsis thaliana adenylyl sulfate (APS)-kinase. Arch. Biochem. Biophys. 392, 303–310 [DOI] [PubMed] [Google Scholar]
- 32. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 33. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harjes S., Bayer P., Scheidig A. J. (2005) The crystal structure of human PAPS synthetase 1 reveals asymmetry in substrate binding. J. Mol. Biol. 347, 623–635 [DOI] [PubMed] [Google Scholar]
- 38. MacRae I. J., Segel I. H., Fisher A. J. (2000) Crystal structure of adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry 39, 1613–1621 [DOI] [PubMed] [Google Scholar]
- 39. MacRae I. J., Segel I. H., Fisher A. J. (2001) Crystal structure of ATP sulfurylase from Penicillium chrysogenum: insights into the allosteric regulation of sulfate assimilation. Biochemistry 40, 6795–6804 [DOI] [PubMed] [Google Scholar]
- 40. Yu Z., Lansdon E. B., Segel I. H., Fisher A. J. (2007) Crystal structure of the bifunctional ATP sulfurylase-APS kinase from the chemolithotrophic thermophile Aquifex aeolicus. J. Mol. Biol. 365, 732–743 [DOI] [PubMed] [Google Scholar]
- 41. Gay S. C., Segel I. H., Fisher A. J. (2009) Structure of the two-domain hexameric APS kinase from Thiobacillus denitrificans: structural basis for the absence of ATP sulfurylase activity. Acta Crystallogr. D Biol. Crystallogr. 65, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lansdon E. B., Segel I. H., Fisher A. J. (2002) Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry 41, 13672–13680 [DOI] [PubMed] [Google Scholar]
- 43. Asahi T. (1964) Sulfur metabolism in higher plants. IV. Mechanism of sulfate reduction in chloroplasts. Biochim. Biophys. Acta 82, 58–66 [Google Scholar]
- 44. Osslund T., Chandler C., Segel I. (1982) ATP sulfurylase from higher plants: purification and preliminary kinetics studies of the cabbage leaf enzyme. Plant Physiol. 70, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mueller J. W., Shafqat N. (2013) Adenosine-5′-phosphosulfate: a multifaceted modulator of bifunctional 3′-phospho-adenosine-5′-phosphosulfate synthases and related enzymes. FEBS J. 280, 3050–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prioretti L., Gontero B., Hell R., Giordano M. (2014) Diversity and regulation of ATP sulfurylase in photosynthetic organisms. Front. Plant Sci. 5, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Renosto F., Seubert P. A., Knudson P., Segel I. H. (1985) Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum: determining ligand dissociateion constants of binary and ternary complexes from the kinetics of enzyme inactivation. J. Biol. Chem. 260, 11903–11913 [PubMed] [Google Scholar]
- 48. Satishchandran C., Markham G. D. (2000) Mechanistic studies of Escherichia coli adenosine-5′-phosphosulfate kinase. Arch. Biochem. Biophys. 378, 210–215 [DOI] [PubMed] [Google Scholar]
- 49. Palde P. B., Carroll K. S. (2015) A universal entropy-driven mechanism for thioredoxin-target recognition. Proc. Natl. Acad. Sci. U.S.A. 112, 7960–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jez J. M., Cahoon R. E., Chen S. (2004) Arabidopsis thaliana glutamate-cysteine ligase: functional properties, kinetic mechanism, and regulation of activity. J. Biol. Chem. 279, 33463–33470 [DOI] [PubMed] [Google Scholar]
- 51. Hothorn M., Wachter A., Gromes R., Stuwe T., Rausch T., Scheffzek K. (2006) Structural basis for the redox control of plant glutamate cysteine ligase. J. Biol. Chem. 281, 27557–27565 [DOI] [PubMed] [Google Scholar]
- 52. Hicks L. M., Cahoon R. E., Bonner E. R., Rivard R. S., Sheffield J., Jez J. M. (2007) Thiol-based regulation of redox-active glutamate-cysteine ligase from Arabidopsis thaliana. Plant Cell 19, 2653–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gromes R., Hothorn M., Lenherr E. D., Rybin V., Scheffzek K., Rausch T. (2008) The redox switch of γ-glutamylcysteine ligase via a reversible monomer-dimer transition is a mechanism unique to plants. Plant J. 54, 1063–1075 [DOI] [PubMed] [Google Scholar]
- 54. Reyes-Prieto A., Weber A. P., Bhattacharya D. (2007) The origin and establishment of the plastid in algae and plants. Annu. Rev. Genet. 41, 147–168 [DOI] [PubMed] [Google Scholar]
- 55. Gould S. B., Waller R. F., McFadden G. I. (2008) Plastid evolution. Annu. Rev. Plant Biol. 59, 491–517 [DOI] [PubMed] [Google Scholar]
- 56. Sekulic N., Konrad M., Lavie A. (2007) Structural mechanism for substrate inhibition of the adenosine 5′-phosphosulfate kinase domain of human 3′-phosphoadenosine 5′-phosphosulfate synthetase 1 and its ramifications for enzyme regulation. J. Biol. Chem. 282, 22112–22121 [DOI] [PubMed] [Google Scholar]
- 57. Sekulic N., Dietrich K., Paarmann I., Ort S., Konrad M., Lavie A. (2007) Elucidation of the active conformation of the APS-kinase domain of human PAPS synthetase 1. J. Mol. Biol. 367, 488–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Henzler-Wildman K. A., Thai V., Lei M., Ott M., Wolf-Watz M., Fenn T., Pozharski E., Wilson M. A., Petsko G. A., Karplus M., Hübner C. G., Kern D. (2007) Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844 [DOI] [PubMed] [Google Scholar]
- 59. Henzler-Wildman K., Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]