

Background: Signaling by IL-31 involves OSMRβ and IL-31Rα, which is expressed in different isoforms.
Results: Four of five membrane-bound isoforms activate IL-31 signaling, which is counter-regulated by SOCS3.
Conclusion: IL-31 responsiveness is controlled by the expression of specific IL-31Rα isoforms and is negatively regulated by SOCS proteins.
Significance: This study provides new insights into the regulation of IL-31 signaling.
Keywords: interleukin, receptor, signal transduction, STAT3, suppressor of cytokine signaling 3 (SOCS3), IL-31 signaling, IL-31R α isoforms, OSMR β, feedback inhibition, suppressors of cytokine signaling 1 (SOCS1)
Abstract
Interleukin-31 (IL-31) is a T helper type 2 cell-derived cytokine tightly associated with inflammatory skin disorders. IL-31-induced signaling is mediated by a receptor complex composed of oncostatin M receptor β and the cytokine-specific receptor subunit IL-31Rα, of which there are several isoforms. The latter can be classified as long or short isoforms with respect to their intracellular domain. At present, the signaling capabilities of the different isoforms remain inchoately understood, and potential mechanisms involved in negative regulation of IL-31Rα signaling have so far not been studied in detail. Here, we show that both the long and short isoforms of IL-31Rα are capable of inducing STAT signaling. However, the presence of a functional JAK-binding box within IL-31Rα is an essential prerequisite for functional IL-31-mediated STAT3 signaling. Moreover, both the long and short isoforms require oncostatin M receptor β for their activity. We also show that IL-31 induces expression of four suppressor of cytokine signaling family members and provide evidence that SOCS3 acts as a potent feedback inhibitor of IL-31-induced signaling. Taken together, this study identifies crucial requirements for IL-31 signaling and shows its counter-regulation by SOCS3.
Introduction
Cytokine-dependent immune responses are induced by the interplay of specific cytokines and their cognate receptors. Among the different types of cytokine receptors, gp130 is a widely expressed subunit that can associate with several cytokine-specific receptor chains to form functional receptor complexes for numerous cytokines, including interleukin-6 (IL-6), IL-11, IL-27, leukemia inhibitory factor, oncostatin M (OSM),2 ciliary neurotrophic factor, cardiotrophin 1, and cardiotrophin-like cytokine (1). Engagement of gp130 typically activates the transcription factors STAT1, STAT3, and STAT5, but it can also activate MAPK signaling and the PI3K/Akt pathway (2). Several years ago, a novel cytokine receptor that displays certain similarities to gp130 was described as the gp130-like receptor (GPL) (3) or gp130-like monocyte receptor (4). Initially, four different isoforms of the GPL receptor were isolated from myelomonocytic and glioblastoma cells (3). Based on the identification of its ligand IL-31, this receptor was given the name IL-31 receptor α (IL-31Rα) (5). In the same study, four splice variants named IL-31RA-v1 to IL-31RA-v4 (splice variants x1 to x4, GenBankTM accession numbers AY499339, AY499340, AY499341, and AY499342, respectively), which slightly differ from the previously described GPL isoforms, were identified (5). IL-31Rα chains encoded by IL-31RA-v1, IL-31RA-v3, and IL-31RA-v4 possess typical features of type I cytokine receptors, including a cytokine-binding domain (CBD), three type III fibronectin (FnIII) domains, a transmembrane domain, and a JAK-binding motif (3, 5, 6). In contrast, IL-31RA-v2 contains only the CBD motif and one FnIII domain and is thus considered to act as a soluble receptor. IL-31RA-v3 is the longest isoform and has three tyrosine residues located in the intracellular part of the receptor (5). This isoform shows the highest similarities to the sequences first described for gp130-like monocyte receptor (4) and GPL745 (3). The study describing the ligand for IL-31Rα further identified OSMRβ as the second receptor unit in the heterodimeric IL-31 receptor (IL-31R) (5). The signaling capabilities of this novel receptor complex have been investigated mainly using cell lines expressing long isoforms of IL-31Rα/GPL that contain intracellular tyrosine residues. Those studies reported that IL-31 activates the Janus kinases JAK1 and JAK2, which subsequently stimulate the phosphorylation of STAT3 and, to a lesser degree, STAT5 and STAT1. Moreover, IL-31 was shown to activate p38 MAPK, ERK1/2, PI3K/Akt, and JNK signaling (7–10).
In contrast, the role(s) of the short IL-31Rα isoforms (devoid of intracellular tyrosines) remains controversial. BaF3 transfectants overexpressing either the long variant IL-31RA-v3 or the short isoform encoded by IL-31RA-v4 in combination with OSMRβ were tested for STAT activation, and both receptors were found to be capable of inducing STAT1, STAT3, and STAT5 phosphorylation in response to IL-31, suggesting that STAT activation can be induced by these receptors, independent of the three intracellular tyrosines (5). However, several other short variants of IL-31Rα have been described, of which at least one may behave as a decoy receptor, because overexpression of this isoform dampens IL-31-induced STAT3 signaling (9).
In general, detailed information on the expression of different IL-31Rα isoforms in specific cell types is scarce. Diveu et al. (9) investigated the presence of long and short IL-31RA in different tumor cell lines. They reported expression of long isoforms in different glioblastoma cell lines and the pulmonary tumor cell line A549, whereas the short isoforms were expressed mainly in the melanoma cell line A375, the mammary tumor cell line MCF-7, and the myelomonocytic cell line U937 (9). In addition, we showed previously that human primary dendritic cells stimulated with IFN-γ predominantly up-regulate the short transcript variants 3 and 4 of IL-31RA (11).
Cytokine-mediated activation of the JAK/STAT signaling pathway is not only essential for protective immune responses but also acts as an important factor in the pathogenesis of autoimmune diseases. Thus, tight regulation of cytokine signaling is a prerequisite to avoid exaggerated cytokine responses that may have detrimental consequences for the host. Among the variety of mechanisms that serve to limit excessive cytokine signaling, the suppressor of cytokine signaling (SOCS) family of proteins is a highly potent group of feedback inhibitors that has been shown to play an important role in regulating numerous pathways, including gp130 signaling. Eight SOCS proteins have been described, SOCS1–SOCS7 and CISH, and all share a central Src homology 2 domain, a SOCS box located at the C terminus, and an N-terminal domain that varies in length and function. SOCS proteins can exert their inhibitory functions by targeting the receptor complex and associated proteins for proteasome-mediated degradation. Additionally, individual SOCS proteins directly interact with phosphorylated tyrosines of activated receptors, and SOCS1 and SOCS3 inhibit the activation of JAKs by an N-terminal kinase inhibitory region (12, 13). Although signals induced by receptors of the IL-6 family are mainly regulated by SOCS3 (14), it remains completely unknown whether SOCS3 or other SOCS proteins are involved in the regulation of IL-31-mediated signaling.
In this study, we investigated functional requirements of different human membrane-associated IL-31Rα isoforms and provided novel insights into the mechanisms involved in negative regulation of IL-31 signaling. Comparative analyses of the signaling capacities of long and short IL-31Rα isoforms revealed a clear difference in the signal strength of STAT activation but not of MAPK activation.
We further show that OSMRβ is essential for functional IL-31 signaling. Thus, IL-31Rα, unlike its highly related receptor gp130 (15), is not capable of acting as a homodimer. In addition, we show an increase in SOCS1, SOCS2, SOCS3, and CISH expression upon IL-31 treatment and report that IL-31-induced STAT3 activation is strongly inhibited by SOCS3.
Experimental Procedures
Cloning of Expression Plasmids
The coding sequences (CDS) of IL-31RA transcript variants 1–5 (NM 139017,NM 001242636, NM 001242637, NM 001242638, and NM 001242639) and OSMRB (NM 003999) were amplified using cDNA from human IFN-γ-stimulated monocyte-derived dendritic cells (11) and THP1 cells activated by IFN-γ and LPS, respectively, serving as templates. For the PCR, Phusion polymerase (SBI, Fermentas, Germany) was used according to the supplier's recommendations. The amplicons were ligated into the pCDH-CMV-CDS-EF1-copGFP plasmid (SBI, System Biosciences, Mountain View, CA). To detect IL-31Rα and OSMRβ by Western blotting, the CDS were additionally cloned into the tagged expression vectors pcDNA 3.1+/myc-His A (Invitrogen, Life Technologies, Inc., Vienna, Austria) and the pCMV-3TagA (Agilent Technologies, Santa Clara, CA), respectively. Mutations were introduced by reverse PCR. The CDS encoding human CIS (CISH, NM 013324) was amplified from cDNA derived from LPS-stimulated monocyte-derived dendritic cells (16) and cloned into the pcDNA 3.1+ expression vector (Invitrogen). Sanger sequencing was carried out to verify the sequence of all created constructs, and efficient protein expression was analyzed by Western blotting. Sequences of the primers with attached restriction sites (underlined) or mutated bases (lowercase) are as follows: IL-31RA transcript variants 1 + 3 sense (pCDH), 5′-AGTCGCTAGCGAATGTGCATCAGGCAACTCA-3′, and IL-31RA transcript variants 1 + 3 sense (pcDNA), 5′-AGTCGGATCCGAATGTGCATCAGGCAACTCA-3′; IL-31RA transcript variants 2 + 4 sense (pCDH), 5′-AGTCGCTAGCATGAAGCTCTCTCCCCAGCCTTCATGTGTTA-3′, and IL-31RA transcript variants 2 + 4 sense (pcDNA), 5′-AGTCGGATCCATGAAGCTCTCTCCCCAGCCTTCATGTGTTA-3′; IL-31RA transcript variant 5 sense (pCDH), 5′-AGTCGCTAGCATGACATACTGGAGATTAGAGAACATAGCGAA-3′, and IL-31RA transcript variant 5 sense (pcDNA), 5′-AGTCGGATCCATGACATACTGGAGATTAGAGAACATAGCGAA-3′; IL-31RA transcript variants 1/2/5 antisense (pCDH), 5′-AGTCGCGGCCGCGGTCGCATTTAGACTTCTCCCTT-3′, and IL-31RA transcript variants 1/2/5 antisense (pcDNA), 5′-AGTCCTCGAGGACTTCTCCCTTGGTGTGCTCTGGA-3′; IL-31RA transcripts variants 3 + 4 antisense (pCDH), 5′-AGTCGCGGCCGCCACTTATATTGAAGTTGGGCAGGAA-3′, IL-31RA transcripts variants 3 + 4 antisense (pcDNA), and 5′-AGTCCTCGAGTATTGAAGTTGGGCAGGAAGACAGAATT-3′; OSMRB (pCDH) sense, 5′-AGTCGAATTCGATGGCTCTATTTGCAGTCTTTCAG-3′, and antisense, 5′-AGTCTTCGAATGCTGGTTAGCAGTAGTGTTCACC-3′;OSMRB (pCMV-3Tag) sense, 5′-AGTCGAATTCGATGGCTCTATTTGCAGTCTTTCAG-3′, and antisense, 5′-AGTCAAGCTTGCAGTAGTGTTCACCTGGATCTAAAAGGGTAAT-3′; CISH sense, 5′-AGTCGAATTCATGTACCTAGAACACACCAGCCACTGTC-3′, and antisense, 5′-AGTCGCGGCCGCTCAGAGCTGGAAGGGGTACTGTCG-3′; IL-31Rα Y671F sense, 5′-tTGTGACCTGCCCCTTCAGGCC-3′, and antisense, 5′-ACCCATTCTTTTCCCCTCCTAAATTGTTTTC-3′; IL-31Rα Y702F sense, 5′-tCCTACGTTCGAGGATGCCAGAGG-3′, and antisense 5′-ATTGGGATTTTCTGGGCGGAATCTC-3′; IL-31Rα Y740F sense, 5′-tTTTGAAAAATTCAGTGACAGCCAGGGAAT-3′, and antisense, 5′-ATGGATTTGGGGCTCCTTCCTCACA-3′; IL31Rα W586A sense, 5′-cGCCCACCGTTCCCAACC-3′, and antisense, 5′-cACACAGATGAGTCAATTTGTTGGGTTT-3′; IL31Rα P590A/P592A sense, 5′-ACgCTGCTGAAAGTAGTATAGCCACATG-3′, and antisense, 5′-TGGcAACGGTGGGCCAAC-3′; IL31Rα W586A/P590A/P592A sense, 5′-cGCCCACCGTTgCCAACg-3′, and antisense, 5′-cACACAGATGAGTCAATTTGTTGGGTTT-3′; IL31Rα K580stop sense, 5′-AATTGACTCATCTGTGTTGGCCCAC-3′, and antisense, 5′-aGTTGGGTTTTTTGAGACCATATGCC-3′; OSMRβ Y917F sense, 5′-tTGTGTCCCAGTTGGCTTCACCC-3′, and antisense, 5′-AATTCAAACTTGTTTCTCCAGCTGGGA-3′; OSMRβ Y945F sense, 5′-tTAAAATGCAAATGGCAGTCTCCCTG-3′, and antisense, 5′-ACTCTGAACAGTGTGGTGCCTCTACTGG-3′.
Cell Culture
HEK293 and HeLa cells were maintained in high glucose DMEM, supplemented with 2 mm glutamine, 1× nonessential amino acids, 100 units/ml penicillin, 100 μg/ml streptomycin (all from Sigma, Vienna, Austria), and 10% FCS (PAA, Pasching, Austria). Hep3B cells were cultured in RPMI 1640 medium supplemented with 2 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (all from Sigma, Vienna, Austria), 50 μm 2-mercaptoethanol (Gibco, Life Technologies, Inc., Vienna, Austria) and 10% FCS (PAA, Pasching, Austria).
Generation and Stimulation of Human Monocyte-derived Dendritic Cells
All studies involving human cells were conducted in accordance with the guidelines of the World Medical Association's Declaration of Helsinki. Human PBMCs were isolated from buffy coats by density gradient centrifugation using Histopaque (Sigma-Aldrich, Vienna, Austria). Monocyte-derived DCs (moDCs) were generated according to standard protocols. In brief, adherent monocytes were cultured in RPMI 1640 medium supplemented with 2 mm glutamine, 100units/ml penicillin, 100 μg/ml streptomycin (all from Sigma, Vienna, Austria), and 10% heat-inactivated FCS (PAA, Pasching, Austria) and differentiated into moDCs by stimulation with 50 ng/ml GM-CSF and 50 ng/ml IL-4 (kind gift from Novartis, Vienna, Austria) for 6 days.
After differentiation, cells were stimulated for 24 h with 10 ng/ml recombinant human IFN-γ (Immunotools, Friesoythe, Germany) to induce the expression of IL-31Rα (11) and rested overnight in fresh medium. For stimulation, cells were induced with 100 ng/ml rhIL-31.
Transient Transfection and Reporter Gene Assays
Transient transfection was carried out using Lipofectamine 2000 (Invitrogen, Life Technologies, Inc., Vienna, Austria) according to the manufacturer's recommendations. The day before transfection, 1.5·105 HEK293 cells, 5·104 Hep3B cells, or 5·104 HeLa cells were seeded into 24-well plates in 0.5 ml of cell culture medium without antibiotics. Cells were transfected with 800 ng of a STAT3 luciferase reporter (kindly provided by Prof. Xinmin Cao, Singapore), 25 ng of IL-31Rα expression plasmid, 25 ng of OSMRβ expression plasmid, or an equal amount of empty vector. For inhibition studies, 200 ng of pcDNA3.1+ containing the CDS of SOCS family members (17) were co-transfected. The day after the transfection, cells were induced with IL-31 (30 ng/ml if not stated otherwise). Luciferase activity was assessed after 1 day of stimulation as described previously (18). As an internal control for transfection efficiency, in some experiments a Renilla-luciferase expression plasmid was co-transfected, and cell lysates were analyzed using the dual-luciferase assay reporter system (Promega, Mannheim, Germany).
Flow Cytometry
HeLa, HEK293, and Hep3B cells were suspended by treatment with Accutase (eBioscience, Vienna, Austria), transferred into ice-cold assay buffer (PBS containing 3% FCS), and stained for IL-31Rα and OSMRβ using fluorescence-labeled antibodies (IL-31RA PE, FAB2769P; OSMRB APC FAB 4389A; R&D Systems, Biomedica, Vienna, Austria). To control for unspecific staining, an aliquot of the cells was incubated with appropriate isotype control antibodies (IC108P, IC002A; R&D Systems). After 45 min of incubation on ice, cells were washed with and taken up in assay buffer and analyzed with a FACS Canto II flow cytometer and FACSDiva software (BD Biosciences).
Western Blotting
1.5·105 HeLa, 1.5·105 Hep3B, or 5·105 HEK293 cells per well were seeded into 6-well plates and transfected with 100 ng of IL-31Rα expression plasmids, 100 ng of OSMRβ expression plasmid, or an equal amount of empty vector using Lipofectamine 2000. The day after the transfection, cells were induced with IL-31 for 15 min. Lysates were prepared using Nonidet P-40 lysis buffer (Invitrogen, Life Technologies, Inc., Vienna, Austria) according to the manufacturer's recommendations and supplemented with 2× SDS sample buffer (Bio-Rad, Vienna, Austria). Protein lysates were denatured by 7 min of incubation at 95 °C, separated on precast 4–12% NuPAGE gradient gels (Invitrogen, Life Technologies, Inc., Vienna, Austria), and blotted onto nitrocellulose membranes in a Trans-blot semi-dry blotting chamber (both Bio-Rad, Vienna, Austria). The membrane was blocked by incubation in TBS, containing 0.1% Tween 20 and 5% nonfat dry milk for 1 h. The primary antibodies (anti-β-actin (13E5) catalog no. 4970; anti-His tag (D3I10) catalog no. 12698; anti-pERK1/2 (E10) catalog no. 9106; anti-ERK1/2 catalog no. 9102; anti-pSTAT1 catalog no. 9171; anti-STAT1 catalog no. 9171; anti-pSTAT3 (D3A7) catalog no. 9145; anti-STAT3 catalog no. 9132; anti-pSTAT5 catalog no. 9351; anti-STAT5 catalog no. 9363, anti-CISH catalog no. 8731, anti-SOCS1 catalog no. 3957, anti-SOCS2 catalog no. 2779, and anti-SOCS3 catalog no. 2923) and HRP-linked secondary antibodies (anti-mouse IgG catalog no. 7076 and anti-rabbit IgG catalog no. 7074) used were purchased from Cell Signaling Technology (Danvers, MA) and used according to the manufacturer's instructions. Detection was carried out using Supersignal enhanced chemiluminescence substrate (West Pico, Pierce) or Signal Fire Plus ECL reagent (Cell Signaling Technology, Danvers, MA) and Kodak x-ray film.
RNA Isolation and Quantitative Real Time PCR
Total RNA from cells was isolated using TRI®Reagent (Sigma, Vienna, Austria) and reverse-transcribed with RevertAid H Minus M-MulV reverse transcriptase (Thermo Scientific, St. Leon-Roth, Germany) according to the manufacturer's instructions. Quantitative real time PCR was carried out on a Rotorgene 3000 (Corbett Research) using the iQ SYBR Green Supermix (Bio-Rad, Vienna, Austria) and the primers listed below. The transcript for the large ribosomal protein P0 (RPL P0) was used as a reference. The specificity of the PCR was verified by recording a melting curve for the PCR products. Relative mRNA expression levels were calculated using the following formula: x = 2−ΔCt, where Ct represents the threshold cycle of a given gene, and ΔCt signifies the difference between the Ct values of the gene in question and the Ct value of the reference gene RPL P0. Induction ratios were calculated using the formula x = 2−ΔΔCt. ΔΔCt represents the difference between the ΔCt values of induced and uninduced samples.
Primer sequences are as follows: RPL P0 sense, 5′-GGCACCATTGAAATCCTGAGTGATGTG-3′, and antisense, 5′-TTGCGGACACCCTCCAGGAAG-3′; MCP-1 sense, 5′-GCTCATAGCAGCCACCTTCA-3′, and antisense, 5′-ACAATGGTCTTGAAGATCACAGC-3′; SOCS1 sense, 5′-TTGGAGGGAGCGGATGGGTGTAG-3′, and antisense, 5′-AGAGGTAGGAGGTGCGAGTTCAGGTC-3′; SOCS2 sense, 5′-CCAAATCAACCAAAAAAAGTGACCATGAAGTCCTG-3′, and antisense, 5′-CGGGGATTGAGTTGCACCTGTATAGCATGATATTC-3′; SOCS3 sense, 5′-ATACTATACCTTCCTGTACCTGGGTGGATGGAGCG-3′, and antisense, 5′-TGAGTATGTGGCTTTCCTATGCTGGGTCCCTCT-3′; SOCS4 sense, 5′-AGACTGATGGCGATGGTGATGGC-3′, and antisense, 5′-GTTTCTTCTGGGCACTTTCTGGATGTA-3′; SOCS5 sense, 5′-AAACAGGCGTTTGGAATAGCTGCTGCAATGTAGTC-3′, and antisense, 5′-CACAAAATCATCCTGGGCATAGGAACAGATCCAAC-3′; SOCS6 sense, 5′GCTGAAAAAACTTGCAAAGCAAGGATGGTACTGGG-3′, and antisense, 5′-CGAACAAGAAAAGAACCATCTGGCACGTTTGC-3′; SOCS7 sense, 5′-TCTAAAGGAAGCGCAGCTCATTTCCAAACAGAAGC-3′, and antisense, 5′-TCTTCAAAGCTGGAGCTTGGCAACCAAATGC-3′; CISH sense, 5′-ACCCCTACTGGCAGCCAGCCTCTGTTTCTG-3′, and antisense, 5′-TCCCCTAGCCTGGCTTACCCCTCAACAAGG-3′; gp80 sense, 5′-GCACCCCATCCCTGACGACAAAG-3′, and antisense, 5′-GAACTTGCTCCCGACACTACTGGCG-3′; gp130 sense, 5′-ATTGAAGCCATAGTCGTGCCTGTTTGC-3′, and antisense, 5′-TTGTGCCTTGGAGGAGTGTGAGGTGA-3′; OSMRB sense, 5′-AAAAGGCATTGATTGTGGACAACCTAAAGCC-3′, and antisense, 5′-ATGTCAGGATAACAGGTCTCCTTGATCCACTGAC-3′; IL-31RA isoforms 1–5 sense, 5′-GGCATGGAGATGATTTCAAGGATAAGCTAAACCTG-3′, and antisense, 5′-CTGGCTTCATCTGTGAAAATTTCTTGCAGAAC-3′; IL-31RA isoforms 3 + 4 sense, 5′-TTCTGTCTTCCTGCCCAACTTCAATA-3′, and antisense, 5′-TTGCATCCGTAGGTGGTCCATG-3′; IL-31RA isoforms 1/2/5 sense, 5′-GGACCCGCCCAGAAGCCAA-3′, and antisense, 5′-CTTCTCCCTTGGTGTGCTCTGGA-3′; IL-31RA isoform 5 sense, 5′-CTTGCCCAGCGGTTGAGGAATCC-3′, and antisense, 5′- GGTCCCATGCACCAAGGTCATCTG-3′; IL-31RA isoforms 6 + 7 sense, ′-CATATTCCATCCAGGCTTATGCCAAAGA-3′, and antisense, 5′-ACTTCGCTATGGGCGTGCTTACAGA-3′.
siRNA-based silencing of SOCS3
The day before transfection, 1.5·105 HeLa cells were seeded into 6-well plates. Cells were transfected with 100 ng of IL-31Rα isoform 1 expression plasmid and either 20 pmol of siRNA targeting SOCS3 (Invitrogen Stealth RNA, forward, 5′-CCCAGAAGAGCCUAUUACAUCUACU-3′, and reverse, 5′-AGUAGAUGUAAUAGGCUCUUCUGGG-3′) or control oligonucleotide (AllStars Negative Control siRNA; Qiagen) using Lipofectamine 2000 according to the manufacturer's recommendations. 24 h post-transfection, the medium was changed, and cells were induced with 30 ng/ml rhIL-31 for different time periods.
Results
IL-31 Receptor Complex Transmits Signals Independent of Intracellular Tyrosine Residues within IL-31Rα
To date, the NCBI database lists seven main transcript variants of human IL-31RA along with their conceptually translated protein isoforms (IL-31Rα). These isoforms are derived from alternative splicing and differ from each other at the C and N termini. Isoforms 1–4 feature the typical characteristics of a type I cytokine receptor, including a cytokine binding domain (CBD) with four conserved cysteines, FnIII domains, a WSXWS motif, a transmembrane domain (TMD), a box motif and a variant intracellular domain. Isoforms 1 and 2 harbor three tyrosine residues in the intracellular domain that are absent in the intracellular portion of isoforms 3 and 4. Isoforms 1, 3, and 6 and isoforms 2 and 4, respectively, share the same signal peptide. Isoform 5 and the recently identified isoform 7 lack a signal peptide and parts of the CBD. Transcript variants 6 and 7 do not contain the TMD and box motif present in all the other isoforms and probably encode soluble variants of IL-31Rα (Fig. 1A). To compare the signaling competence of the different IL-31Rα isoforms, we cloned the five human membrane-associated IL-31Rα isoforms into appropriate expression vectors.
FIGURE 1.

STAT3 activation by human IL-31Rα isoforms. A, schematic representation of the domain architecture of the seven human IL-31Rα isoforms. The variable portion of the intracellular domain is shown in gray. Gray marks indicate cysteine residues in the cytokine binding domain. Tyrosine residues implicated in STAT3 recruitment are highlighted in red. TMD, transmembrane domain; box, JAK-binding box. B, flow cytometric analysis of human OSMRβ expression in HeLa cells. The white histogram indicates the isotype control. C, luciferase reporter gene assay. HeLa cells were transfected with a STAT3 luciferase reporter, a Renilla-luciferase expression plasmid, and expression plasmids encoding human IL-31Rα isoforms. As a control an equal amount of the empty vector (pCDH) was co-transfected. One day post-transfection, cells were treated with rhIL-31 (30 ng/ml). Luciferase activity was assessed after 24 h of cytokine stimulation. Data represent mean values of five independent experiments carried out in duplicate, normalized to the corresponding Renilla luciferase signals. White bars, uninduced (ui); black bars, IL-31. Error bars indicate standard deviations. RLU, relative light units. D, flow cytometric analysis of different IL-31Rα isoforms transfected into HeLa cells. White histograms display the isotype control. E, Western blotting. HeLa cells were transfected with expression plasmids encoding His-tagged human IL-31Rα isoforms or empty pcDNA-His expression vector. IL-31Rα protein expression was detected with His tag-specific antibodies. Expression of β-actin was monitored to control for equal loading. F and G, mRNA and protein expression of IL-31Rα isoforms 1 and 5 in HeLa cells. Cells were transiently transfected with increasing amounts of plasmids encoding human IL-31Rα isoforms 1 and 5 (gray triangles). As a control, the empty vector (pcDNA-His) was transfected. mRNA expression of IL-31Rα isoforms 1 and 5 was analyzed by qRT-PCR (F). Protein expression of IL-31Rα isoform 1 and isoform 5 is shown by Western blotting using an anti-His tag antibody (G). β-Actin expression was monitored to control for equal loading. H, STAT3 reporter gene assay. HeLa cells were transiently transfected with a STAT3 luciferase reporter and IL-31Rα isoform 5 at the concentration leading to detectable protein expression as shown in G. 24 h post-transfection, cells were stimulated with rhIL-31 (30 ng/ml). Luciferase activity was assessed 24 h after cytokine stimulation. Error bars indicate standard deviations. ALU, arbitrary light units.
IL-31 was shown to activate STAT3 downstream of a heterodimeric receptor complex composed of IL-31Rα and OSMRβ (5). To analyze potential differences in the signaling strength mediated by various IL-31Rα isoforms, we performed STAT3 luciferase reporter gene assays in the OSMRβ+ cervical cancer cell line HeLa (Fig. 1B). We observed activation of the STAT3 reporter by cells transfected with isoforms 1–4, whereas cells transfected with isoform 5 did not respond to IL-31 treatment (Fig. 1C). To test whether the inability of isoform 5 to activate STAT3 is caused by its truncated CBD or low expression of this isoform, we expressed all isoforms as His-tagged proteins and analyzed HeLa transfectants for equal protein expression of IL-31Rα isoforms by flow cytometry (Fig. 1D) and Western blotting (Fig. 1E). Although similar expression of the IL-31Rα variants 1–4 was observed on the cell surface as well as in whole cell lysates, no signals were obtained for isoform 5. Hence, HeLa cells were transfected with rising concentrations of expression plasmids encoding the human IL-31Rα isoform 1 or 5, starting with the concentration used so far (100 ng per 1.5·105 plated cells), which then was increased to 800 ng per 1.5·105 plated cells. IL-31Rα expression was analyzed by qRT-PCR and Western blotting. Although the mRNA of the two isoforms does express equally (Fig. 1F), there is a big difference in protein expression, as isoform 5 protein is detected only in cells transfected with the highest concentration of the expression vector (Fig. 1G). This indicates that splicing to isoform 5 limits its translation. Although we do detect IL-31Rα isoform 5 protein when expressed at high amounts, this isoform is still not able to respond to IL-31, as shown by STAT3 reporter gene assay (Fig. 1H). This may be explained by the fact that surface expression of this isoform, as well as cytokine binding to this isoform, may be impaired, because isoform 5 does not possess a signal peptide and lacks a part of the CBD. We next aimed to investigate the role of intracellular tyrosine residues within IL-31Rα for IL-31 signaling. Despite different signal peptides, isoforms 1 and 2 as well as isoforms 3 and 4 show similar levels of IL-31-induced STAT3 activation (Fig. 1C). Therefore, we studied IL-31 signaling using isoforms 1 and 3 as examples. These two proteins share the same 52-amino acid signal peptide but differ in having (isoform 1) or not having (isoform 3) three intracellular tyrosines (Fig. 1A).
As shown in Fig. 1, isoform 3 seems to be a less potent activator of STAT3 than isoform 1. To test whether this is due to the tyrosine residues within IL-31Rα isoform 1, the three intracellular tyrosine residues Tyr-671, Tyr-702, and Tyr-740 were mutated to phenylalanine, and STAT3 activation was analyzed in reporter gene assays and by Western blotting. Indeed, in terms of STAT3 activation, mutation of the three tyrosines within IL-31Rα isoform 1 led to a phenotype similar to that of isoform 3 (Fig. 2, A and B), indicating that intracellular tyrosines within IL-31Rα determine its capacity to activate STAT3.
FIGURE 2.

Substitution of tyrosines in the intracellular domain of IL-31Rα isoform 1 results in diminished responsiveness to IL-31. A, STAT3 reporter gene assay. Wild-type IL-31Rα-isoform 1 (WT), a mutated version thereof in which all three intracellular tyrosine residues were replaced by phenylalanine (Y671F/Y702F/Y740F), and IL-31Rα isoform 3 was transiently transfected together with a STAT3 luciferase reporter into HeLa cells. One day post-transfection, cells were treated with rhIL-31 (30 ng/ml). Luciferase activity was assessed after 24 h of cytokine stimulation. Data represent mean values of three independent experiments carried out at least in duplicate. Error bars indicate standard deviations. (ANOVA, Tukey's multiple comparison test: **, p ≤ 0.01; ***, p ≤ 0.001); ALU, arbitrary light units. n.s., not significant. B, equal expression of all transfected IL-31Rα isoforms and total STAT3 expression was monitored by Western blotting using anti-His tag and anti-STAT3 antibodies. For the detection of IL-31-dependent STAT3 phosphorylation, an anti-pSTAT3 antibody was used.
To study the differences in IL-31 responsiveness observed for IL-31Rα isoforms 1 and 3 in more detail, we transiently transfected HeLa cells with the corresponding expression plasmids together with a STAT3-luciferase reporter, and we assessed luciferase activity upon administration of rising IL-31 concentrations. The signals obtained from cells expressing the long isoform 1 of IL-31Rα were higher than those from cells expressing the short isoform 3. Nevertheless, we observed dose-dependent activation of the STAT3 reporter in both types of transfectant (Fig. 3A), indicating that the IL-31 receptor complex (IL-31R) can transmit IL-31-induced signals independent of intracellular tyrosine residues in IL-31Rα. Because the CC-chemokine CCL2/MCP-1 is activated by IL-31 (19) and is a well described STAT3 target gene (20), we tested whether the differences in STAT3 activation downstream of the long and short IL-31Rα isoforms affect CCL2/MCP-1 expression. As shown by qRT-PCR (Fig. 3B) and ELISA (Fig. 3C), IL-31 stimulation leads to a dose-dependent increase in CCL2/MCP-1 by both transfectants, which clearly correlated with STAT3 activation.
FIGURE 3.

Long and short isoforms of IL-31Rα exhibit similar signaling competences. A, luciferase reporter gene assay. HeLa cells were transiently transfected with a STAT3 luciferase reporter and expression plasmids encoding either the short isoform 3 or long isoform 1 of human IL-31Rα. As a control, an equal amount of the empty vector (pCDH) was co-transfected. One day post-transfection, cells were treated with rhIL-31 at rising concentrations as indicated. Luciferase activity was assessed after 24 h of cytokine stimulation. Data represent mean values of three independent experiments carried out in duplicate. Error bars indicate standard deviations. B and C, IL-31-dependent CCL2/MCP-1 expression by transfected HeLa cells. Ccl2/Mcp1 mRNA expression was analyzed after 4 h of IL-31 stimulation by qRT-PCR (B), and CCL2/MCP-1 secretion was measured by ELISA 24 h post IL-31 induction. Data represent mean values of two independent experiments carried out in duplicate. Error bars indicate standard deviations. D, Western blotting. HeLa cells transiently transfected with IL-31Rα isoforms 3 and 1 in pCDH expression vector were treated for 15 min with rhIL-31 at the indicated concentrations. Tyrosine phosphorylation of STAT3, STAT1, STAT5, and ERK1/2 was detected by specific antibodies. To control for equal loading, nonphosphorylated protein was detected. One out of three independent experiments is shown. E, HeLa cells transiently transfected with IL-31Rα isoforms 3 and 1 in pcDNA-His expression vector were treated for 15 min with the indicated concentrations of rhIL-31, and phosphorylation of STAT3, STAT1, STAT5, and ERK1/2 was detected by specific antibodies. To control for equal loading, nonphosphorylated protein and β-actin is shown. IL-31Rα expression was monitored by a His tag-specific antibody. One representative experiment is shown.
Besides STAT3 (5), STAT1, STAT5, and the MAPKs ERK1/2 are reportedly phosphorylated downstream of IL-31R signaling (10). To test the ability of the short and long IL-31Rα isoforms to activate these signaling pathways, Western blot analyses of transfected HeLa cells were carried out. We observed dose-dependent phosphorylation of STAT3, STAT1, STAT5, and ERK1/2 following treatment with IL-31. Similar to the results from the reporter gene assays, cells expressing the long isoform of the receptor exhibited greater activation of STATs. In contrast, the phosphorylation grade of ERK1/2 did not differ between the two types of transfectant (Fig. 3D). These observations are independent of the expression plasmid encoding IL-31Rα, because the results were the same whether we used His-tagged receptors or untagged proteins (Fig. 3E).
OSMRβ Expression Is a Prerequisite for Functional IL-31 Signaling
Phosphorylated tyrosine residues located close to JAK-binding regions within the cytoplasmic portion of cytokine receptors serve as docking sites for STAT molecules and are central for STAT activation. The fact that the short IL-31Rα isoforms, devoid of any intracellular tyrosines, still substantially stimulated STAT phosphorylation (Figs. 1–3) led us to hypothesize that OSMRβ, the second receptor chain, provides the required STAT-recruiting sites. To analyze the contribution of OSMRβ to IL-31 signaling, we screened different cell lines for endogenous OSMRβ expression by flow cytometry and qRT-PCR and selected OSMRβ+ HeLa cells, OSMRβdim HEK293 cells, and OSMRβ− Hep3B cells for further experiments. These cells differ also in their endogenous expression of IL-31Rα and the related IL-6 receptor chains gp130 and gp80 (Fig. 4, A and B). Functionality of the respective receptors was analyzed by STAT3 reporter gene assays (Fig. 4C).
FIGURE 4.
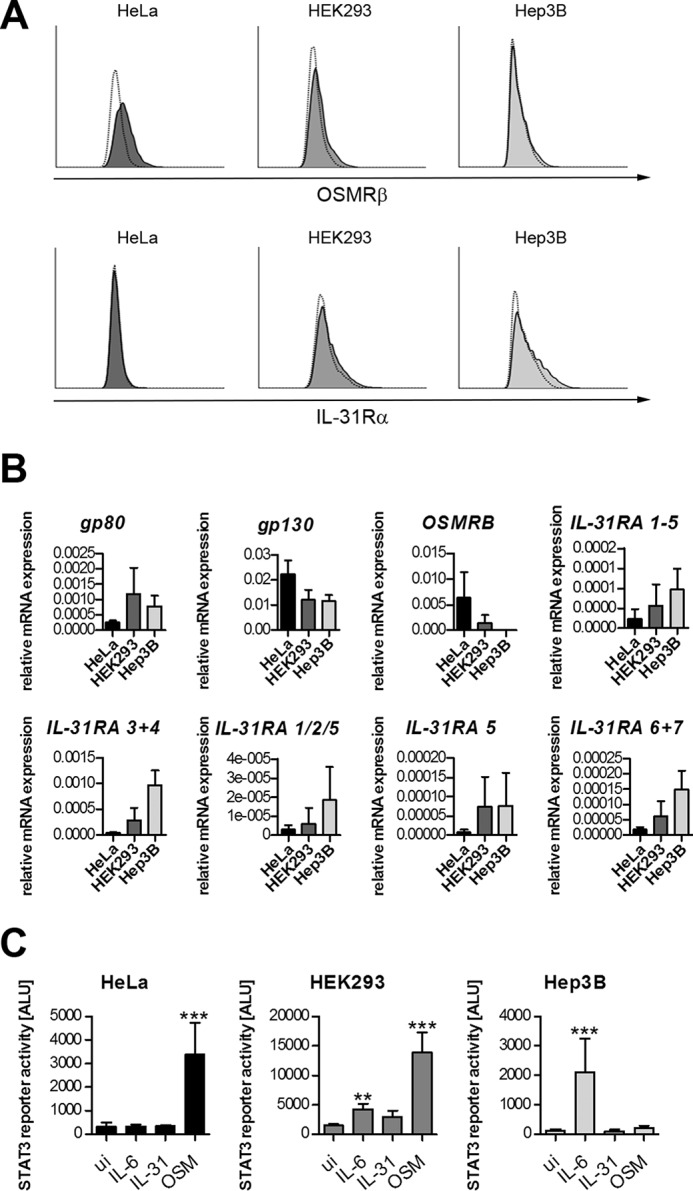
The human cell lines HeLa, HEK293, and Hep3B differ in OSMRβ expression and their responsiveness to different IL-6 family cytokines. A, HeLa, HEK293, and Hep3B cells were stained with fluorescence-labeled antibodies directed against human OSMRβ (upper panel) and IL-31Rα (lower panel) and analyzed by flow cytometry. White histograms represent the isotype control. B, HeLa, HEK293, and Hep3B cells were analyzed for mRNA expression of receptor subunits involved in signaling by IL-6, OSM, and IL-31 by qRT-PCR. Mean values of five independent experiments are shown. Error bars indicate standard deviations. C, luciferase reporter gene assay. HeLa, HEK293, and Hep3B cells were transiently transfected with a STAT3 luciferase reporter. 24 h post-transfection, cells were stimulated with 30 ng/ml of the indicated cytokines. Data represent mean values of two independent experiments carried out in triplicate. Error bars indicate standard deviations. ANOVA, Dunnett's multiple comparison test: **, p ≤ 0.01; ***, p ≤ 0.001.
To investigate the contribution of OSMRβ to IL-31 signaling, Western blot analyses of HeLa, HEK293, and Hep3B cells transfected with the long IL-31Rα isoform 1 or the short variant 3 were performed. In general, OSMRβdim HEK293 cells overexpressing IL-31Rα displayed markedly reduced IL-31-induced STAT3 phosphorylation compared with OSMRβ+ HeLa transfectants, whereas Hep3B cells failed to phosphorylate STAT3 (Fig. 5A). Similar results were obtained when we compared the ability of those three cell types to activate a STAT3 reporter. However, ectopic overexpression of OSMRβ restored the capacity of both IL-31Rα isoforms to activate STAT3 in Hep3B cells and significantly enhanced STAT3 activation in HEK293 and HeLa cells (Fig. 5, B and C). This indicates that STAT3 signaling by IL-31Rα requires OSMRβ expression.
FIGURE 5.

OSMRβ is required for functional IL-31 signaling. A, HeLa, HEK293, and Hep3B cells, transfected to transiently express the short isoform 3 or the long isoform 1 of IL-31Rα, were stimulated with 30 ng/ml rhIL-31 for 15 min. STAT3 phosphorylation (p-STAT3) was detected by Western blotting. To control for equal loading, total STAT3 protein was measured. B, luciferase reporter gene assay. HeLa, HEK293, and Hep3B cells were transiently transfected with a STAT3 luciferase reporter and expression plasmids encoding either isoform 1 or 3 of IL-31Rα. As a control, the empty vector (pCDH) was used. Additionally, OSMRβ or empty vector (−OSMRβ) was co-transfected. 24 h post-transfection, cells were stimulated with 30 ng/ml rhIL-31. Empty bars show the unstimulated samples (ui), and black bars indicate IL-31-induced samples. Data represent mean values of three independent experiments carried out in duplicate. Error bars indicate standard deviations. C, Western blot showing expression of the IL-31 receptor chains. HeLa, HEK293, and Hep3B cells were transiently transfected with expression plasmids encoding either isoform 1 or 3 of IL-31Rα with an N-terminal His tag. As a control, the empty vector (pcDNA-His) was used. Additionally, FLAG-tagged OSMRβ (+OSMR β) or the empty pCMV-FLAG vector (−OSMRβ) was co-transfected. 48 h post-transfection, cells were lysed and expression of IL-31Rα and OSMRβ was monitored by Western blotting. D, IL-31-induced STAT3 activation in HEK293 cells expressing OSMRβ mutants. Cells were transfected with a STAT3 reporter, IL-31Rα isoforms 3, 1, or the empty vector control (pCDH) in combination with wild-type (WT) or mutated versions of human OSMRβ. Filled bars represent IL-31-treated samples, and empty bars represent the corresponding un-induced samples. Data represent mean values of five independent experiments carried out in duplicate. The expression efficiency of the IL-31Rα isoforms and all variants of OSMRβ was examined by Western blot detection of His- and FLAG-tagged proteins, respectively (E). Error bars indicate standard deviations. ANOVA, Tukey's multiple comparison test: **, p ≤0.01; ***, p ≤0.001.
Human OSMRβ has seven intracellular tyrosine residues, which are potential sites for the recruitment of STAT molecules. Indeed, tyrosine residues at positions 917 (Tyr-917) and 945 (Tyr-945) serve as docking sites for STAT3 in OSM signaling (21). To analyze the role of Tyr-917 and Tyr-945 in STAT3 activation by a receptor complex composed of OSMRβ and IL-31Rα, we created single and double mutants of OSMRβ in which the tyrosines were substituted by phenylalanine (Y917F, Y945F, and Y917F/Y945F). These were co-transfected with a STAT3 reporter and either the long or the short isoform of IL-31Rα into OSMRβdim HEK293 cells. The single mutations Y917F and Y945F had only a small effect on IL-31-induced STAT3 activation. However, overexpression of the double mutant OSMRβ Y917F/Y945F resulted in a clear decrease in STAT3 activation, to a level comparable with that obtained upon co-expression of the empty vector (Fig. 5, D and E).
Identification of Critical Residues within the Box Motif of IL-31Rα
Thus far, our data show that tyrosine residues within IL-31Rα are dispensable for IL-31R downstream signaling. Nevertheless, a functional type I cytokine receptor must be able to bind at least two molecules of the JAK family, which allows for reciprocal phosphorylation of these receptor-associated kinases (22). To analyze the role of the JAK-binding box, we introduced a stop codon into the sequence of IL-31Rα (K580Stop), which results in a polypeptide that ends three amino acids after the transmembrane region and hence lacks the JAK-binding box. As a consequence, STAT3 signaling was completely abrogated, as determined by STAT3 luciferase reporter gene assays in HEK293 cells (Fig. 6A). For a detailed analysis of the kinase recruitment domain, we substituted conserved amino acids within the box motif (22–24) of the short IL-31Rα isoform by alanines and tested the responsiveness to IL-31 in STAT3 reporter gene assays. The mutation W586A resulted in a diminished signal. Even more severe effects were observed upon mutation of the proline doublet (P590A/P592A). The triple mutation W586A/P590A/P592A resulted in complete loss of STAT3 activation (Fig. 6B). In contrast, ERK1/2 phosphorylation, which is independent of Janus kinases, is not affected by the triple mutation (Fig. 6C). Taken together, these results indicate that the three conserved amino acids, Trp-586, Pro-590, and Pro-592, within the box motif of IL-31Rα are essential elements in STAT activation but are dispensable for MAPK signaling.
FIGURE 6.
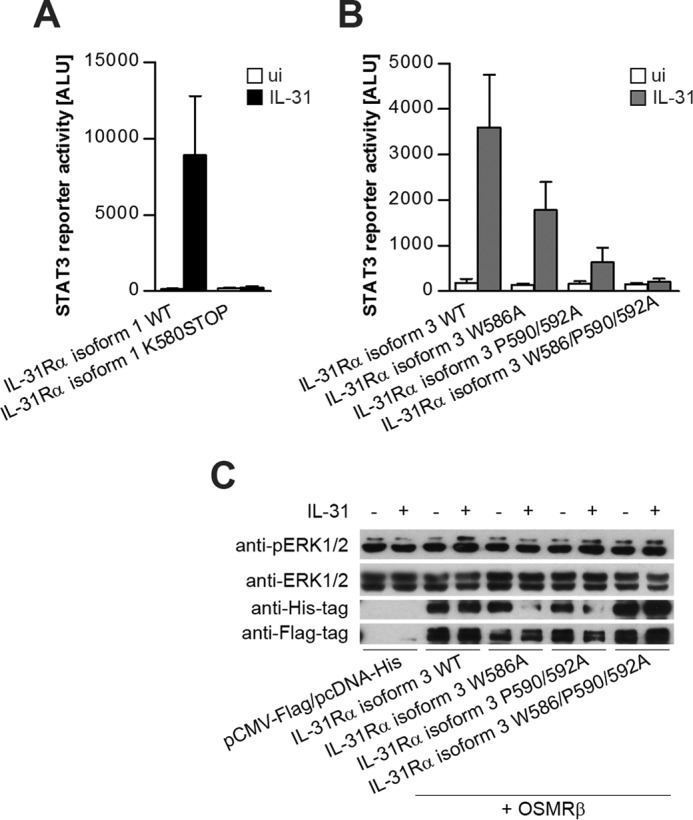
Conserved box motif within IL-31Rα is required for functional IL-31-dependent STAT3 signaling. STAT3 reporter gene assay of HEK293 cells co-expressing OSMRβ and the indicated IL-31Rα expression plasmids. Luciferase activity was measured after 24 h of stimulation with 30 ng/ml rhIL-31. Mean values of three independent experiments are shown. Error bars indicate standard deviations (A and B). C, Western blot to show ERK1/2 activation induced by IL-31. HEK293 cells were transiently transfected with FLAG-tagged OSMRβ and the His-tagged variants of IL-31Rα isoform 3. Expression efficiency of all transfected constructs was monitored using antibodies directed against FLAG and His tags. To control for equal loading, total ERK1/2 was detected.
IL-31 Induces Expression of Its Feedback Inhibitor SOCS3
Inflammation is commonly controlled by different negative regulatory mechanisms, including inhibition by SOCS proteins. Because IL-31 is strongly involved in skin inflammation (5, 10), we asked whether IL-31 signaling induces a regulatory feedback loop that employs SOCS proteins. Hence, HeLa cells transfected with the short isoform 3 or the long isoform 1 of IL-31Rα were induced with IL-31 for different time periods, and mRNA expression of all human SOCS family members was analyzed by qRT-PCR. Both types of transfectants exhibited up-regulation of SOCS2, SOCS3, and CISH, whereas SOCS1 was induced only in cells expressing the long variant of IL-31Rα (isoform 1), and the expression levels of the other SOCS proteins remained unaffected (Fig. 7A). A similar pattern was observed when we analyzed IL-31-induced SOCS expression in moDCs. To induce IL-31Rα expression, moDCs were stimulated with IFN-γ for 24 h prior to IL-31 treatment. Because IFN-γ itself is able to induce SOCS expression (25), cells were washed and then rested in fresh medium until SOCS expression returned to the level of untreated cells, before IL-31 was administered. Upon IL-31 stimulation, we observed a clear induction of CISH and SOCS3, whereas the expression of SOCS1 and SOCS2 was not affected (Fig. 7B).
FIGURE 7.

IL-31 induces the expression of SOCS1, SOCS2, SOCS3, and CISH. A, HeLa cells were transfected to transiently express either isoform 3 or isoform 1 of IL-31Rα. The day after transfection, cells were stimulated with 30 ng/ml rhIL-31 for the indicated periods. mRNA expression of SOCS family members was measured by qRT-PCR. Mean values of three independent experiments are shown. Error bars indicate standard deviations. B, monocyte-derived dendritic cells were stimulated with IL-31 (100 ng/ml) for the indicated periods, and expression of SOCS family members was detected by qRT-PCR. Mean values of six independent experiments using cells from individual donors are shown. Error bars indicate standard deviations.
To determine whether the IL-31-induced SOCS family members SOCS1, SOCS2, SOCS3, and CISH counteract IL-31-mediated STAT3 activation, expression vectors encoding the respective SOCS proteins were transfected, and STAT3 activation was analyzed by reporter gene assays and Western blot. Of the four co-expressed SOCS proteins, only SOCS1 and SOCS3 were able to block IL-31-induced STAT3 activation (Fig. 8, A and B). However, the fact that SOCS1 was not expressed in primary IL-31-responsive cells indicates that SOCS3 may be the only family member that acts as a negative feedback regulator in IL-31 signaling. In addition, our data suggest that IL-31 not only induces SOCS family members that are involved in a direct negative feedback loop (SOCS1 and SOCS3) but might also control IL-31-independent signals.
FIGURE 8.

SOCS3 is a feedback inhibitor of IL-31-dependent STAT3 activation. A, STAT3 reporter gene assay of HeLa cells co-expressing the long isoform 1 of IL-31Rα and expression plasmids encoding SOCS family members as indicated. As a control, empty vector (pcDNA) was co-transfected. B, Western blot showing IL-31-dependent STAT3 activation in HEK293 cells transiently transfected with IL-31Rα isoform 1 (His-tagged), OSMRβ (FLAG-tagged), and the indicated SOCS proteins. As a control, the empty expression vector (pcDNA) was transfected. STAT3 activation was detected with an anti-pSTAT3-antibody. To control for equal loading, total STAT3 was monitored. The expression efficiency of all transfected components was analyzed by incubation with antibodies as indicated. C, HeLa cells were transiently transfected with IL-31Rα isoform 1 (His-tagged) and either siRNA targeting SOCS3 (siSSOCS3) or a nontargeting control RNA-oligonucleotide (siCTRL). 24 h post-transfection, cells were stimulated with IL-31. Cells were harvested for Western blot analyses of pSTAT3, STAT3 and His-tagged IL-31Rα after the indicated periods. Efficiency of SOCS3-silencing was analyzed by qRT-PCR (D).
To confirm the role of SOCS3 as a negative feedback regulator of IL-31 signals, HeLa cells were transfected with the long IL-31Rα isoform 1 and SOCS3-specific siRNA to knock down SOCS3 protein. Western blot analyses showed that silencing of SOCS3 led to an extended STAT3 activation upon IL-31 treatment. Whereas in control samples phospho-STAT3 was only detected up to 24 h post-IL-31 stimulation, STAT3 activation in SOCS3-silenced samples lasted for 72 h. Of note, STAT3 activation directly correlated with IL-31Rα expression, indicating that SOCS3 counteracts IL-31 signaling by promoting IL-31Rα degradation (Fig. 8C).
Discussion
IL-31 belongs to the IL-6 family of cytokines, but unlike other members of this cytokine family, IL-31 does not signal via the gp130 receptor subunit. Instead, a related protein called gp130-like (GPL) or IL-31Rα combines with the OSMRβ chain to form the functional receptor for IL-31. For human IL-31Rα, several splice variants and different predicted protein sequences have been described. The membrane-anchored isoforms 1–5 mainly differ in the presence of conserved cytoplasmic tyrosine residues to which STAT molecules can be recruited (Fig. 1A). As the short IL-31Rα isoforms 3 and 4 lack the intracellular tyrosine residues, we aimed to investigate the consequences of this deficit for IL-31 receptor signaling. Long and short isoforms of human IL-31Rα (isoforms 1–5) were cloned and transfected into the human cell line HeLa, which endogenously expresses high levels of OSMRβ. Although mRNA encoding isoform 5 was detectable in human cell lines, overexpression of isoform 5 resulted in only weak IL-31Rα protein expression, and reporter gene assays showed that this isoform is not able to mediate IL-31-dependent STAT3 activation. In contrast, cells transfected with expression plasmids encoding isoforms 1–4 showed robust IL-31Rα protein expression and thus were able to induce STAT3 activation upon IL-31 treatment. Nevertheless, long receptor variants were more potent activators of STAT3 than the short IL-31Rα isoforms. Mutation of the intracellular tyrosines indicates that the tyrosine residues within IL-31Rα are not essential but have the potential to enhance IL-31 signaling. These observations are in line with a previous study showing that mutating the tyrosines within IL-31Rα to phenylalanines partially diminishes (but does not completely abolish) IL-31 signaling in A549 cells (7). We performed a detailed comparative analysis of IL-31Rα signaling using isoforms 1 and 3 representing long and short variants, respectively. In contrast to STAT activation, the extent of IL-31-induced ERK1/2 activation did not differ between the two transfectants. This implies that the tyrosines within IL-31Rα do not play a role in ERK phosphorylation and is in line with the previous observation that OSMRβ mediates IL-31-induced ERK signaling, whereas IL-31Rα is unable to recruit adapter molecules involved in MAPK activation (26). Our data suggest that OSMRβ may be the key element in IL-31 signal transmission, because cells lacking OSMRβ are unresponsive to IL-31. Furthermore, overexpression of OSMRβ strongly enhances IL-31 signaling via both the short and long IL-31Rα isoforms. It was previously shown that, of the seven intracytoplasmic tyrosine residues of human OSMRβ, Tyr-917 and Tyr-945 both bind to STAT3 with similar high affinities and thus contribute to OSM-mediated STAT3 activation (21). In our study, substitution of these residues significantly reduced STAT3 activation, suggesting that Tyr-945 and Tyr-917 are crucial for IL-31-mediated STAT3 activation as well.
Membrane-proximal regions with highly conserved proline-rich domains, so-called box motifs, were shown to efficiently contribute to the binding of JAKs (2, 23, 27, 28). Introduction of a stop codon that prevents the expression of a functional box motif within IL-31RA resulted in a complete loss of IL-31-induced STAT3 activation. Similar effects were observed when we mutated the highly conserved amino acids Trp-586, Pro-590, and Pro-592 within the box motif, yet this triple mutation did not affect the phosphorylation of ERK1/2. This clearly shows that, although intracellular tyrosines are dispensable for signal transduction via the short IL-31 receptor, a fully functional box motif is essential for effective trans-phosphorylation of receptor-associated kinases and subsequent STAT activation. The previously reported high importance of JAK-binding motifs within cytokine receptors (29) may also help explain the negative effects described for a particular IL-31Rα isoform that is truncated immediately after the trans-membrane domain and thus lacks a functional box motif (9). This isoform is capable of ligand binding but it fails to recruit receptor-associated kinases, which results in a blockade of STAT3 phosphorylation.
Recapitulating our data on IL-31R signaling, we propose that the tasks of the short membrane-anchored isoforms are limited to ligand recognition and kinase recruitment, whereas the long variants of IL-31Rα may have extended signaling capacities such as STAT binding and activation. Hence, cells may regulate their responsiveness to IL-31 by expression of specific IL-31Rα isoforms and their OSMRβ expression. However, there is still a lack of knowledge regarding the specific expression of particular IL-31Rα isoforms in primary cells.
In humans, enhanced IL-31 expression is strongly correlated with inflammatory skin diseases, allergic contact dermatitis, and allergic asthma (10). In particular, skin inflammation is strongly regulated by SOCS1 and SOCS3, as SOCS3 deficiency in keratinocytes causes severe skin inflammation, whereas keratinocyte-specific SOCS1 overexpression down-regulates the expression of specific pro-inflammatory mediators in the skin (30). In the context of type 2 inflammation, the SOCS family member cytokine-inducible SH2-containing protein (CIS) has recently been shown to regulate pro-allergic T helper cell development and to control allergic inflammation (31). In this study, we demonstrated that IL-31 is able to enhance the expression of SOCS1, SOCS2, SOCS3, and CISH in a time-dependent manner. However, of the four SOCS members up-regulated upon IL-31 stimulation, only SOCS1 and SOCS3 acted as potent feedback inhibitors in overexpression studies. Similar observations were reported for signaling by the related cytokine IL-6. Overexpression of SOCS1 and SOCS3 inhibits IL-6-dependent STAT3 activation in vitro (32–34). Yet, in vivo studies indicate that SOCS3 is more important to the negative regulation of IL-6 signaling than SOCS1, although SOCS1 is also able to block STAT3 signaling downstream of gp130 when overexpressed (35). When we investigated IL-31-mediated SOCS induction in human moDCs, we did not observe up-regulation of SOCS1, indicating that, similar to IL-6 signaling, SOCS3 is more likely to act as a feedback inhibitor of IL-31 than is SOCS1. The role of SOCS3 for the inhibition of IL-31 signaling was confirmed by siRNA-based knockdown experiments. Silencing of SOCS3 resulted in a prolonged and stronger STAT3 phosphorylation in response to IL-31, which correlated with the stability of IL-31Rα protein expression. This suggests that SOCS3 regulates IL-31 signaling at least in part by promoting degradation of the receptor as shown previously for gp130 (36). The fact that IL-31, in addition to up-regulating its potential feedback regulator SOCS3, also enhanced the expression of CISH and SOCS2, neither of which interfered with IL-31 signaling even when overexpressed, suggests that IL-31 might control signaling ofIL-31-independent pathways as well.
Author Contributions
E. M. planned and conducted the experiments, analyzed the data, and wrote the manuscript. M. M., S. E., A. S., and T. N. carried out experiments and performed data analyses. A. D. was involved in data analyses and manuscript preparation. J. H. H. planned and supervised the study, analyzed the data, and wrote the manuscript.
Acknowledgment
We thank Emerita Professor Xinmin Cao (Institute of Molecular and Cell Biology, A*Star, Singapore) for providing the STAT3 reporter plasmid.
This work was supported by Austrian Science Fund FWF Grants P 23933 and P 25696. The authors declare that they have no conflicts of interest with the contents of this article.
- OSM
- oncostatin M
- OSMRβ
- oncostatin M receptor β subunit
- LIF
- leukemia inhibitory factor
- CBD
- cytokine binding domain
- CISH
- human cytokine-inducible Src homology 2-containing protein
- FnIII
- fibronectin type III
- GPL
- gp130-like
- IL-31Rα
- interleukin 31 receptor α subunit
- SOCS
- suppressor of cytokine signaling
- STAT
- signal transducer and activator of transcription
- TMD
- transmembrane domain
- moDC
- monocyte-derived dendritic cell
- qRT-PCR
- quantitative RT-PCR
- ANOVA
- analysis of variance
- CDS
- coding sequence.
References
- 1. Taga T., Kishimoto T. (1997) Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 15, 797–819 [DOI] [PubMed] [Google Scholar]
- 2. Heinrich P. C., Behrmann I., Haan S., Hermanns H. M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Diveu C., Lelièvre E., Perret D., Lak-Hal A. H., Froger J., Guillet C., Chevalier S., Rousseau F., Wesa A., Preisser L., Chabbert M., Gauchat J. F., Galy A., Gascan H., Morel A. (2003) GPL, a novel cytokine receptor related to GP130 and leukemia inhibitory factor receptor. J. Biol. Chem. 278, 49850–49859 [DOI] [PubMed] [Google Scholar]
- 4. Ghilardi N., Li J., Hongo J. A., Yi S., Gurney A., de Sauvage F. J. (2002) A novel type I cytokine receptor is expressed on monocytes, signals proliferation, and activates STAT-3 and STAT-5. J. Biol. Chem. 277, 16831–16836 [DOI] [PubMed] [Google Scholar]
- 5. Dillon S. R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S. R., Haugen H. S., Maurer M., Harder B., Johnston J., Bort S., Mudri S., Kuijper J. L., Bukowski T., Shea P., et al. (2004) Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 5, 752–760 [DOI] [PubMed] [Google Scholar]
- 6. Boulay J. L., O'Shea J. J., Paul W. E. (2003) Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 19, 159–163 [DOI] [PubMed] [Google Scholar]
- 7. Chattopadhyay S., Tracy E., Liang P., Robledo O., Rose-John S., Baumann H. (2007) Interleukin-31 and oncostatin-M mediate distinct signaling reactions and response patterns in lung epithelial cells. J. Biol. Chem. 282, 3014–3026 [DOI] [PubMed] [Google Scholar]
- 8. Dambacher J., Beigel F., Seiderer J., Haller D., Göke B., Auernhammer C. J., Brand S. (2007) Interleukin 31 mediates MAP kinase and STAT1/3 activation in intestinal epithelial cells and its expression is upregulated in inflammatory bowel disease. Gut 56, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diveu C., Lak-Hal A. H., Froger J., Ravon E., Grimaud L., Barbier F., Hermann J., Gascan H., Chevalier S. (2004) Predominant expression of the long isoform of GP130-like (GPL) receptor is required for interleukin-31 signaling. Eur. Cytokine Netw. 15, 291–302 [PubMed] [Google Scholar]
- 10. Cornelissen C., Lüscher-Firzlaff J., Baron J. M., Lüscher B. (2012) Signaling by IL-31 and functional consequences. Eur. J. Cell Biol. 91, 552–566 [DOI] [PubMed] [Google Scholar]
- 11. Horejs-Hoeck J., Schwarz H., Lamprecht S., Maier E., Hainzl S., Schmittner M., Posselt G., Stoecklinger A., Hawranek T., Duschl A. (2012) Dendritic cells activated by IFN-γ/STAT1 express IL-31 receptor and release proinflammatory mediators upon IL-31 treatment. J. Immunol. 188, 5319–5326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubo M., Hanada T., Yoshimura A. (2003) Suppressors of cytokine signaling and immunity. Nat. Immunol. 4, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 13. Yoshimura A., Naka T., Kubo M. (2007) SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465 [DOI] [PubMed] [Google Scholar]
- 14. Kershaw N. J., Murphy J. M., Liau N. P., Varghese L. N., Laktyushin A., Whitlock E. L., Lucet I. S., Nicola N. A., Babon J. J. (2013) SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20, 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murakami M., Hibi M., Nakagawa N., Nakagawa T., Yasukawa K., Yamanishi K., Taga T., Kishimoto T. (1993) IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 260, 1808–1810 [DOI] [PubMed] [Google Scholar]
- 16. Posselt G., Schwarz H., Duschl A., Horejs-Hoeck J. (2011) Suppressor of cytokine signaling 2 is a feedback inhibitor of TLR-induced activation in human monocyte-derived dendritic cells. J. Immunol. 187, 2875–2884 [DOI] [PubMed] [Google Scholar]
- 17. Hebenstreit D., Luft P., Schmiedlechner A., Duschl A., Horejs-Hoeck J. (2005) SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol. Immunol. 42, 295–303 [DOI] [PubMed] [Google Scholar]
- 18. Schwarz H., Schmittner M., Duschl A., Horejs-Hoeck J. (2014) Residual endotoxin contaminations in recombinant proteins are sufficient to activate human CD1c+ dendritic cells. PLoS One 9, e113840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shiao Y. M., Chung H. J., Chen C. C., Chiang K. N., Chang Y. T., Lee D. D., Lin M. W., Tsai S. F., Matsuura I. (2013) MCP-1 as an effector of IL-31 signaling in familial primary cutaneous amyloidosis. J. Invest. Dermatol. 133, 1375–1378 [DOI] [PubMed] [Google Scholar]
- 20. Buryzek L., Syrovets T., Simmet T. (2002) The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and Janus kinase (JAK)/STAT signaling pathways. J. Biol. Chem. 277, 33509–33517 [DOI] [PubMed] [Google Scholar]
- 21. Hintzen C., Evers C., Lippok B. E., Volkmer R., Heinrich P. C., Radtke S., Hermanns H. M. (2008) Box 2 region of the oncostatin M receptor determines specificity for recruitment of Janus kinases and STAT5 activation. J. Biol. Chem. 283, 19465–19477 [DOI] [PubMed] [Google Scholar]
- 22. Tanner J. W., Chen W., Young R. L., Longmore G. D., Shaw A. S. (1995) The conserved box 1 motif of cytokine receptors is required for association with JAK kinases. J. Biol. Chem. 270, 6523–6530 [DOI] [PubMed] [Google Scholar]
- 23. Haan C., Hermanns H. M., Heinrich P. C., Behrmann I. (2000) A single amino acid substitution (Trp(666)→Ala) in the interbox1/2 region of the interleukin-6 signal transducer gp130 abrogates binding of JAK1, and dominantly impairs signal transduction. Biochem. J. 349, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pezet A., Buteau H., Kelly P. A., Edery M. (1997) The last proline of Box 1 is essential for association with JAK2 and functional activation of the prolactin receptor. Mol. Cell. Endocrinol. 129, 199–208 [DOI] [PubMed] [Google Scholar]
- 25. Krebs D. L., Hilton D. J. (2000) SOCS: physiological suppressors of cytokine signaling. J. Cell Sci. 113, 2813–2819 [DOI] [PubMed] [Google Scholar]
- 26. Dreuw A., Radtke S., Pflanz S., Lippok B. E., Heinrich P. C., Hermanns H. M. (2004) Characterization of the signaling capacities of the novel gp130-like cytokine receptor. J. Biol. Chem. 279, 36112–36120 [DOI] [PubMed] [Google Scholar]
- 27. Murakami M., Narazaki M., Hibi M., Yawata H., Yasukawa K., Hamaguchi M., Taga T., Kishimoto T. (1991) Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc. Natl. Acad. Sci. U.S.A. 88, 11349–11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radtke S., Hermanns H. M., Haan C., Schmitz-Van De Leur H., Gascan H., Heinrich P. C., Behrmann I. (2002) Novel role of Janus kinase 1 in the regulation of oncostatin M receptor surface expression. J. Biol. Chem. 277, 11297–11305 [DOI] [PubMed] [Google Scholar]
- 29. Heldin C. H. (1995) Dimerization of cell surface receptors in signal transduction. Cell 80, 213–223 [DOI] [PubMed] [Google Scholar]
- 30. Kubo M. (2013) Therapeutic hope for psoriasis offered by SOCS (suppressor of cytokine signaling) mimetic peptide. Eur. J. Immunol. 43, 1702–1705 [DOI] [PubMed] [Google Scholar]
- 31. Yang X. O., Zhang H., Kim B. S., Niu X., Peng J., Chen Y., Kerketta R., Lee Y. H., Chang S. H., Corry D. B., Wang D., Watowich S. S., Dong C. (2013) The signaling suppressor CIS controls proallergic T cell development and allergic airway inflammation. Nat. Immunol. 14, 732–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naka T., Narazaki M., Hirata M., Matsumoto T., Minamoto S., Aono A., Nishimoto N., Kajita T., Taga T., Yoshizaki K., Akira S., Kishimoto T. (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924–929 [DOI] [PubMed] [Google Scholar]
- 33. Starr R., Willson T. A., Viney E. M., Murray L. J., Rayner J. R., Jenkins B. J., Gonda T. J., Alexander W. S., Metcalf D., Nicola N. A., Hilton D. J. (1997) A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921 [DOI] [PubMed] [Google Scholar]
- 34. Schmitz J., Weissenbach M., Haan S., Heinrich P. C., Schaper F. (2000) SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 275, 12848–12856 [DOI] [PubMed] [Google Scholar]
- 35. Croker B. A., Krebs D. L., Zhang J. G., Wormald S., Willson T. A., Stanley E. G., Robb L., Greenhalgh C. J., Förster I., Clausen B. E., Nicola N. A., Metcalf D., Hilton D. J., Roberts A. W., Alexander W. S. (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 4, 540–545 [DOI] [PubMed] [Google Scholar]
- 36. Kershaw N. J., Laktyushin A., Nicola N. A., Babon J. J. (2014) Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: identification of the active components and JAK2 and gp130 as substrates. Growth Factors 32, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]