

Abstract
We describe the molecular and clinical characterization of nine individuals with recurrent, 3.4-Mb, de novo deletions of 3q13.2q13.31 detected by chromosomal microarray analysis. All individuals have hypotonia and language and motor delays and also variably express mild to moderate cognitive delays (8/9), abnormal behavior (7/9), and autism spectrum disorders (3/9). Common facial features include down-slanting palpebral fissures with epicanthal folds, a slightly bulbous nose, and relative macrocephaly. Twenty-eight genes map to the deleted region, including four strong candidate genes, DRD3, ZBTB20, GAP43, and BOC, with important roles in neural and/or muscular development. Analysis of the breakpoint regions based on array data revealed directly oriented human endogenous retrovirus (HERV-H) elements ∼5kb in size and of >95% DNA sequence identity flanking the deletion. Subsequent DNA sequencing revealed different deletion breakpoints and suggested non-allelic homologous recombination (NAHR) between HERV-H elements as a mechanism of deletion formation, analogous to HERV-I-flanked and NAHR-mediated AZFa deletions. We propose that similar HERV elements may also mediate other recurrent deletion and duplication events on a genome-wide scale. Observation of rare recurrent chromosomal events such as these deletions helps to further the understanding of mechanisms behind naturally occurring variation in the human genome and its contribution to genetic disease.
Keywords: 3q13, microdeletion, hypotonia, NAHR, recurrent, developmental delay, microarray, HERV-H
Introduction
Deletions of the proximal long arm of chromosome 3 are rare, with less than 30 cases reported in the literature to date. These deletions vary considerably in size, location, breakpoints, gene content, and associated clinical phenotypes. Various molecular or cytogenetic techniques have been used to characterize them [Arai et al., 1982; Jenkins et al., 1985; McMorrow et al., 1986; Okada et al., 1987; Fujita et al., 1992; Genuardi et al., 1994; Mackie Ogilvie et al., 1998; Hou, 2004; Kosaki et al., 2005; Lawson-Yuen et al., 2006; Sato et al., 2007; Simovich et al., 2008; Shimojima et al., 2009; Molin et al., 2012; Wisniowiecka-Kowalnik et al., 2013]. This variability, in addition to the relative case rarity, has made it difficult to make clear genotype-phenotype correlations.
Recently, a notable milestone was reached by Molin et al. [2012], who found a smallest region of overlapping deletion (SRO) of about 580 kb at 3q13.31 in 24 of 27 individuals reported with proximal 3q deletions. Features of this novel 3q13.31 microdeletion syndrome included global developmental delay, hypotonia, postnatal overgrowth, hypoplastic male genitals, and characteristic craniofacial features that included epicanthal folds, hypertelorism, down-slanting palpebral fissures, protruding lips with full lower lip and thin upper lip, and a high arched palate. The authors hypothesized that the observed global developmental delay may be caused by haploinsufficiency of DRD3 (OMIM #126451) and ZBTB20 (OMIM #606025), two genes within the SRO [Molin et al., 2012]. In this report, we present clinical and molecular findings for nine patients harboring recurrent 3.4-Mb deletions at 3q13.2q13.31 that completely encompass the previously reported SRO.
Recurrent copy-number variants (CNVs) are generally described as being of virtually identical size as a result of non-allelic homologous recombination (NAHR) between directly-oriented low-copy repeats (LCRs, also called segmental duplications) [Stankiewicz and Lupski, 2010]. These LCRs are defined as segments of the genome that are 1 kb or longer and at least 90% identical [Bailey et al., 2001], with those that mediate NAHR usually longer than 10 kb with over 95% DNA sequence identity [Sharp et al., 2005; Liu et al., 2012]. Although repetitive elements such as SINEs, LINEs and human endogenous retroviruses (HERVs) are not classically thought of as LCRs, some of them fulfill NAHR criteria and can mediate NAHR events [Beck et al., 2011]. Indeed, HERV-I (also named HERV15) repetitive elements ∼10kb in size and of 94% DNA sequence identity have been shown to mediate the recurrent ∼800-kb deletions at Yq11.2 in patients with complete germ cell aplasia (Sertoli Cell Only syndrome, OMIM 415000) [Kamp et al., 2000; Sun et al., 2000; Turner et al., 2008]. Likewise, HERV-H and L1MA4 elements have also been implicated in mediating recurrent deletions and translocations of autosomes [Rosenfeld et al., 2011; Hermetz et al., 2012; Lamb et al., 2012].
Here, we describe nine identically-sized deletions observed in patients with 3q13.31 microdeletion syndrome. Our molecular characterization of the breakpoints provides insight into the mechanisms of recurrent CNV formation and further supports the role of repetitive elements as substrates for NAHR. Moreover, the consistent genotype among these individuals allows for a clearer correlation of a phenotypic pattern with deletion of a specific set of genes.
Materials & Methods
Patient ascertainment
Patients 1-4 were ascertained by Signature Genomic Laboratories following referral for clinical microarray-based comparative genomic hybridization (aCGH) testing. Patients 5 and 6 were ascertained following referral for clinical genetic evaluation by Nemours Children's Clinic and Brigham and Women's Hospital, respectively. Patient 7 was ascertained by ARUP Laboratories following referral for clinical SNP microarray testing. Patients 8 and 9 were ascertained by Baylor College of Medicine (BCM) Medical Genetics Laboratories following referral for aCGH testing. Either de-identified clinical information was supplied by clinicians or informed consent was obtained to publish clinical information and images using an Institutional Review Board-approved protocol from Spokane or BCM.
Microarray analysis
Oligonucleotide-based aCGH analysis was performed on DNA from patient 1 using a 105K-feature, whole-genome microarray (SignatureChipOS version 1, custom-designed by Signature Genomics, Spokane, WA; manufactured by Agilent Technologies, Santa Clara, CA) as previously described [Ballif et al., 2008]. DNA from patients 2-5 was analyzed using a 135K-feature, whole-genome microarray (SignatureChipOS version 2, custom-designed by Signature Genomics; manufactured by Roche NimbleGen, Madison, WI) as previously described [Duker et al., 2010]. Results were analyzed and visualized using custom aCGH analysis and web-based data visualization software (Genoglyphix, Signature Genomics). DNA from patient 6 was studied using 44K-feature, whole-genome, oligonucleotide-based aCGH (GenomeDx, version 1.0, GeneDx, Gaithersburg, MD). DNA from patient 7 was analyzed using a 2.7 million-feature SNP array, consisting of ∼1.95 million non-polymorphic oligonucleotide probes for evaluating copy number and ∼750K SNP probes (Affymetrix CytoScan HD, Affymetrix, Santa Clara, CA) according to the manufacturer's instructions. DNA from patients 8 and 9 was analyzed using 105K and 180K-feature oligonucleotide-based microarrays custom-designed by BCM Medical Genetics Labs, CMA v7.4 and 8.1 (exon-targeted), respectively, each manufactured by Agilent Technologies. Data were analyzed using custom web-based software as described previously [Cheung et al., 2005].
Fluorescence in situ hybridization
Copy-number abnormalities detected by microarray in patients 1-5 and 8-9 were visualized by metaphase or interphase fluorescence in situ hybridization (FISH) using one or more BAC clones located within the abnormal regions as previously described [Traylor et al., 2009]. When available, parental samples were also analyzed using FISH.
Long-range PCR and DNA sequencing
Long-range PCR primers were designed to sequence across a 3,137 bp region of high sequence identity harbored within two HERV-H elements to amplify the deletion-specific breakpoint fragment. Primer sequences are provided in the online supporting information (Supp. Table S1). Amplification was achieved using Takara LA Taq polymerase (Takara Bio, Otsu, Japan) according to the manufacturer's protocol. PCR products were treated with ExoSAP-IT (USB, Cleveland, OH, USA) to degrade unconsumed dNTPs and primers. Sanger sequencing with primers used for amplification was performed on the deletion-specific amplicon for each patient (Lone Star Labs, Houston, TX, USA). The breakpoint region for each patient was then determined by aligning the Sanger sequencing reads to the reference sequence using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA). Data are accessible at the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp/, accession numbers AB811796 through AB811844).
Results
Molecular analysis
Microarray analysis showed all nine patients to have apparently identical, 3.4-Mb 3q13.2q13.31 deletions (Figure 1). No patients had any additional, clinically significant CNVs. Parental FISH showed the deletions to be apparently de novo in patients 1-4, 6, and 8-9 (Supp. Figure S1); additionally, the mother of patient 5 did not carry the deletion, while the father was not available for study.
Figure 1. Microarray characterization of recurrent 3q13.2q13.31 deletions.

(A) Schematic of molecularly characterized deletions in the literature overlapping the recurrently deleted region from this report. The purple bars represent the minimum size of the reported deletions, and, where available, horizontal black lines extend to show the maximum deletion size. The vertical dashed lines and light blue bar represent the recurrently deleted region. (B) Zoomed-in view of the recurrently deleted region with a representative microarray plot showing the deletion in patient 4, as characterized by a 135K whole-genome oligonucleotide-based array. Probes are ordered on the x-axis according to physical mapping positions, with the most proximal 3q13.2 probes to the left and the most distal 3q13.31 probes to the right. Values along the y-axis represent log2 ratios of patient:control signal intensities. Results are visualized using custom aCGH analysis software (Genoglyphix, Signature Genomics). Genes in the region are represented by yellow boxes. All coordinates shown are according to the hg19 build of the human genome, and the shaded gray region extending through the figure represents the SRO previously defined by Molin et al [2012].
Six of these recurrent deletions were identified among 51,000 routine postnatal clinical specimens submitted to Signature Genomics and BCM tested on oligonucleotide-based arrays (0.01%). No such deletions were identified among two adult control cohorts of 19,584 [a filtered set from Rosenfeld et al., 2013] and 3,181 [International Schizophrenia Consortium, 2008], among a pediatric control cohort of 2,026 [Shaikh et al., 2009], nor among 4,706 parents tested by oligonucleotide arrays at Signature or BCM. Additionally, no similar large deletions are present in the Database of Genomic Variants (http://projects.tcag.ca/variation/). The largest deletion among controls was in a single adult, overlapping 800kb of the proximal end of the region. Due to the rarity of the deletion, a case-control comparison does not reach significance (6/51,000 vs. 0/29,497, one-tailed p=0.06, Fisher exact test).
Genomic sequences were downloaded from the UCSC genome browser (http://genome.ucsc.edu). Bioinformatic analyses of the shared deletion breakpoints revealed no LCRs; however, HERV-H elements were identified at both breakpoints. Each element is annotated in Repeat Masker (http://www.repeatmasker.org) as two long terminal repeat-7 (LTR7) sequences bracketing two central HERVH-int annotations totaling approximately 5.7 kb. These repeats lie in direct orientation with reference to each other and share three stretches of greater than 96% sequence identity spanning a total of approximately 4.8 kb. DNA for further testing was available for eight patients (1-5 and7-9). Long range PCR amplification of deletion-specific amplicons followed by Sanger sequencing analysis revealed that each patient has breakpoints occurring within a 1,340 base pair stretch of 97.6% sequence homology that is contained within both HERV-H elements (chr3:112,138,686-112,140,025 and chr3:115,514,625-115,515,964, hg19) (Figure 2).
Figure 2. Breakpoint characterization of recurrent 3q13.2q13.31 deletions.

(A) Representative Sanger sequencing trace of the breakpoint region of patient 1. The patient's breakpoint sequence is presented between the proximal and distal chromosome 3 reference sequences. The informative cis-morphisims that define the breakpoint uncertainty region are highlighted in yellow. (B) Schematic representation of the breakpoints of 8 patients that were available for further studies. The uncertainty region for each patient is depicted in grey. The structures of the HERV-H elements that contain the breakpoints of the patients are shown below as grey rectangles. All genomic coordinates are shown below in the GRCh37/hg19 assembly. (C) Structures of the 3q13.2q13.31 HERV elements compared to the consensus HERV-H sequence from RepBase. Gaps in the consensus represent insertions in the 3q13.2q13.31 HERVs. Gaps in the 3q13.2q13.31 HERVs represent deletions compared to the consensus. The color of the HERV elements denotes identity at that position when aligned with the other HERV over a 50 base pair window. Blue represents 0% sequence identity (i.e. caused by a 50 bp or larger insertion or deletion present in one 3q13.2q13.31 HERV but not the other) while orange represents perfect identity. The region of the cross-over in each patient is presented as an X with the size of the X representing the uncertainty bounded by informative cis-morphisms. The purple X represents the breakpoints in patients 2 and 7 that occur between the same two cis-morphisms.
Because of the high DNA sequence identity, exact determination of the breakpoints was not possible; however, single nucleotide and indel differences between the viral elements (cis-morphisms or paralogous sequence variants) enabled narrowing the regions containing the breakpoints to 9-121 bp segments (Table 1; Figure 2). Overall, six different breakpoint regions were identified. Patients 2 and 7 both had breakpoints that mapped between the informative cis-morphisms at 112,138,807 / 115,514,746 and 112,138,859 / 115,514,798. The remaining patients had unique breakpoint regions.
Table 1. Breakpoint regions for 8 patients with recurrent 3q13.2q13.31 deletions.
| Patient | Proximal Min | Proximal Max | Distal Min | Distal Max | Uncertainty (bp) |
|---|---|---|---|---|---|
| 1 | 112,139,023 | 112,139,077 | 115,514,961 | 115,515,016 | 55 |
| 2 | 112,138,807 | 112,138,859 | 115,514,746 | 115,514,798 | 52 |
| 3 | 112,139,311 | 112,139,338a | 115,515,251 | 115,515,278a | 27 |
| 4 | 112,139,553 | 112,139,611 | 115,515,494 | 115,515,552 | 58 |
| 5 | 112,138,686 | 112,138,807 | 115,514,625 | 115,514,746 | 121 |
| 7 | 112,138,807 | 112,138,859 | 115,514,746 | 115,514,798 | 52 |
| 8 | 112,139,369 | 112,139,435 | 115,515,310 | 115,515,376 | 66 |
| 9 | 112,139,977 | 112,139,986a | 115,515,918 | 115,515,927a | 9 |
In these cases, the informative cis-morphisms are 1 bp deletions. All coordinates are provided in the GRCh37/hg19 assembly.
Clinical analysis
An overview of the clinical features of our nine patients is presented in Table 2, and detailed summaries are provided in the online supporting information. Neurologically, motor and language delays are observed in all patients. Cognitive delay is present in 8/9; patient 1 has gross motor delays, stuttering, anxiety, and short attention span but no definitive cognitive problems. Patient 3 displayed loss of skills at 2 years following stroke-like episodes. Seven patients exhibit abnormal behaviors, including stereotypic behaviors, sensory issues, and aggression. Patients 4 and 9 have diagnoses of pervasive developmental delay, and patient 3 has high-functioning autism, with improvements in her behavior with age. Hypotonia is found in all nine patients and is likely a major contributing factor to their prevalent motor delays. Brain abnormalities include agenesis of the corpus callosum (patients 3 & 9), Chiari malformation (patient 5), and probable benign communicating hydrocephalus (patient 8). Neurologic exam was suggestive of cerebellar dysfunction in patient 6, though brain imaging has not been performed. Patients 3 and 8 had multiple seizure episodes without need for medication, and EEG abnormalities were documented in patients 1 and 6. A characteristic craniofacial appearance is also present (Figure 3). Notably, epicanthal folds, down-slanting palpebral fissures, broad or depressed nasal bridge, bulbous nasal tip, and a prominent lower lip are present to varying degrees in many of our patients. All nine patients have a head circumference above the mean, with three demonstrating frank macrocephaly. Ocular abnormalities include myopia or hyperopia in three patients and strabismus or nystagmus in four other patients. Other congenital anomalies were uncommon, including hydrocele and unilateral renal agenesis (patient 8), unilateral cryptorchidism (patient 5), and spontaneously resolving patent ductus arteriosus (patient 4). Patient 5 also demonstrated nonspecific mild-to-moderate type 2 myofiber atrophy on muscle biopsy.
Table 2. Clinical features in individuals with recurrent 3q13.2q13.31 deletions.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Total | Previous reportsa | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inheritance | dn | dn | dn | dn | Not mat | dn | Unk | dn | dn | 7/7 dn | 3/3 dn | |
| Demographics | Age | 8y | 3y | 9y 8m | 3y 4m | 16m | 42y | 16y | 10y | 5y | 10-13y | |
| Sex | F | M | F | M | M | F | F | M | F | 2M, 1F | ||
| Growth | Height standard deviations | +1.7 | +2.4 | -0.4 | WNL | +1.5 | -0.9 | 0 | +1.7 | +0.7 | 1/9>+2 | 1/3 >+2 |
| Weight standard deviations | +2.2 | +0.7 | -0.4 | WNL | -0.7 | obese | 0 | +1.0 | +1.2 | 2/9>+2 | 1/3 >+2 | |
| OFC standard deviations | +2.1 | +0.7 | +0.7 | +0.9 | +2.3 | +1.1 | +2.1 | +1.6 | +1.1 | 3/9>+2 | 0/3 >+2 | |
| Neurological | Cognitive delay | - | + | + | + | + | + | + | + | + | 8/9 | 3/3 |
| Motor delay | + | + | + | + | + | + | + | + | + | 9/9 | 3/3 | |
| Language delay/disability | + | + | + | + | + | + | + | + | + | 9/9 | 2/3 | |
| Brain abnormality | - | NA | + | - | + | NA | - | + | + | 4/7 | NS | |
| Seizures/EEG abnormalities | + | - | + | - | - | + | - | + | - | 4/9 | 0/3 | |
| Hypotonia | + | + | + | + | + | + | + | + | + | 9/9 | 0/1 | |
| Reduced reflexes | + | - | - | - | + | NS | - | - | + | 3/8 | NS | |
| Abnormal behaviors | + | + | + | + | - | + | - | + | + | 7/9 | 1/3 | |
| Autism spectrum disorder | - | - | + | + | - | - | - | - | + | 3/9 | 0/3 | |
| Enuresis | + | - | + | - | NA | + | - | + | - | 4/8 | NS | |
| Musculoskeletal | Lordosis/scoliosis | + | - | + | - | - | - | + | - | - | 3/9 | 0/3 |
| Foot abnormalities | + | + | - | - | - | + | + | - | - | 4/9 | 0/3 | |
| Craniofacial | Absent eyebrows | - | - | - | - | - | + | + | - | - | 2/9 | 0/2 |
| Epicanthal folds | - | + | - | + | + | + | - | + | + | 6/9 | 1/2 | |
| Down-slanting PF | + | + | + | - | + | + | - | + | - | 6/9 | 0/3 | |
| Ptosis | + | - | + | - | - | + | + | - | - | 4/9 | 0/2 | |
| Broad nasal bridge | + | + | - | + | + | - | - | - | + | 5/9 | 1/2 | |
| Bulbous nasal tip | - | - | + | + | + | - | - | + | - | 4/9 | 1/2 | |
| High/abnormal palate | + | - | - | - | - | + | - | + | - | 3/9 | NS | |
| Prominent lower lip | + | + | - | + | + | - | + | - | - | 5/9 | 1/2 | |
| Ocular | Myopia/hyperopia | + | - | - | - | - | - | + | + | - | 3/9 | 0/2 |
| Strabismus/nystagmus | - | + | + | - | - | + | - | - | + | 4/9 | 1/2 | |
| Urogenital | Genital abnormalities | - | - | - | - | + | - | - | + | - | 2/4 M | 1/2 M |
| Renal abnormalities | - | - | - | - | - | - | - | + | - | 1/9 | 0/3 |
dn, de novo; F, female; M, male; m, months; mat, maternal; NA, not applicable; NS, not specified; OFC, occipitofrontal circumference; PF, palpebral fissures; Unk, unknown; WNL, within normal limits; y, years
Three patients reported by Molin et al. (cases 9-11) (14) were approximately of the same size and may represent this recurrent deletion.
Figure 3. Facial features of individuals with 3q13.2q13.31 deletions.
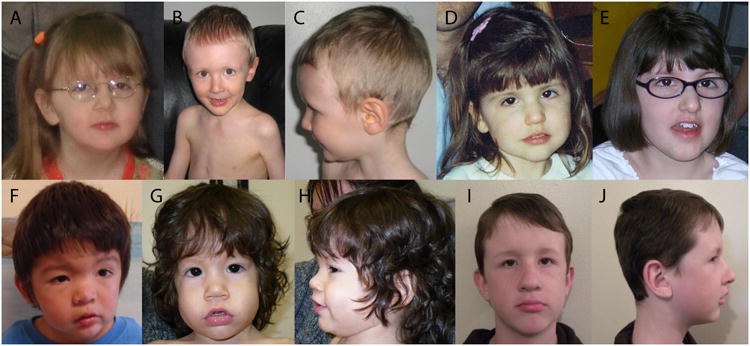
(A) Patient 1 at 5 years of age. (B-C) Patient 2 at 3 years of age. (D-E) Patient 3 at 2.5 years (D) and 10 years of age (E). (F) Patient 4 at 3 years of age. (G-H) Patient 5 at 16 months of age. (I-J) Patient 8 at 10 years of age. (K-L) Patient 9 at 5 years of age. Characteristic facial features include bulbous nasal tip, broad or depressed nasal bridge, prominent lower lip, and characteristically shaped eyes with mildly down-slanting palpebral fissures and mild epicanthal folds.
Discussion
We report nine patients with a recurrent 3.4-Mb deletion at 3q13.2q13.31, all apparently occurring de novo when parents were tested. Our patients presented with a phenotypic spectrum, but a pattern could nevertheless be seen, including motor and language delays, hypotonia, variable cognitive impairment, abnormal behaviors, and dysmorphic features (Table 2, Figure 3). The large head sizes in our patients are in agreement with postnatal growth above the mean as a key characteristic of the 3q13.31 microdeletion syndrome [Molin et al., 2012]. In our cohort, this trend is most apparent with respect to head size, with a mean head circumference of +1.4 standard deviations (SD), while mean height is +0.8 SD, and mean weight is +0.6 SD. Two of the four patients with brain abnormalities had agenesis of the corpus callosum, a feature that has been previously reported in at least seven patients with 3q13 deletions [McMorrow et al., 1986; Genuardi et al., 1994; Mackie Ogilvie et al., 1998; Lawson-Yuen et al., 2006; O'Driscoll et al., 2010; Molin et al., 2012; Wisniowiecka-Kowalnik et al., 2013]. Genital anomalies were present in only two of the four male patients in this study, possibly indicating a lower frequency of the abnormal male genitalia that were previously identified as a key characteristic of the 3q13.31 microdeletion syndrome [Molin et al., 2012]. Genital anomaliesmay be more prevalent in patients whose 3q13 deletion breakpoints extend beyond the boundary of the recurrent deletion reported in this study, although a male with a smaller deletion within this region has been reported with micropenis and cryptorchidism [Molin et al., 2012].
Twenty-eight RefSeq genes have been mapped within the recurrently deleted region at 3q13.2q13.31 (Figure 1). At least five genes of these genes, ZBTB20, DRD3, GAP43 (OMIM #162060), BOC (OMIM #608708), and ZDHHC23 (Entrez Gene #254887) have been implicated in neurodevelopment or nervous system function, making them candidates for the neurodevelopmental phenotypes in these patients. Two of these genes, ZBTB20 and DRD3, are within the currently defined SRO for the 3q13.31 microdeletion syndrome [Molin et al., 2012]. Zbtb20 encodes a transcriptional repressor that is a cell fate determinant for hippocampal neurons [Nielsen et al., 2007; Xie et al., 2010]. Interestingly, Zbtb20 has been shown to regulate several genes whose haploinsufficiency results in overlapping neurodevelopmental phenotypes to this syndrome [Nielsen et al., 2013], including Foxp2 [MacDermot et al., 2005], Mef2c [Le Meur et al., 2010], Satb2 [Rosenfeld et al., 2009], and Sox5 [Lamb et al., 2012]. Further support for the role of this gene in the microdeletion syndrome's pathogenesis is provided by its de novo disruption in a male with pervasive developmental delay due to a chromosome 3 inversion [Talkowski et al., 2012]. DRD3 encodes a dopamine receptor present in the limbic system [Sokoloff et al., 1990]. Both heterozygous and homozygous knockout mice show hyperactivity [Accili et al., 1996], and homozygous knockout mice have deficits in spatial working memory [Glickstein et al., 2002]. GAP43 encodes a phosphoprotein with roles in axonal growth, learning, and memory [Cammarota et al., 1997; Routtenberg et al., 2000; Pascale et al., 2004]. Heterozygous knockout mice demonstrate learning delays and autistic-like features, including resistance to change, stress-induced behavioral withdrawal, and anxiety [Zaccaria et al., 2010]. BOC encodes a co-receptor for SHH [Sanchez-Arrones et al., 2012], and targeted knockout in mice results in misguidance of commissural axons [Okada et al., 2006]. Finally, ZDHHC23 encodes a palmitoyl transferase that modifies large conductance calcium-activated potassium channels [Tian et al., 2012], whose altered function can result in epileptic pathology [N'Gouemo, 2011], a feature seen in 4/9 of our patients.
Given their roles in neural development, haploinsufficiency of some of these genes may also contribute to the corpus callosum and cerebellar malformations in these and previously reported patients. Mice lacking Gap43 display an absence of the corpus callosum, hippocampal commissure, and anterior commissure, due to failure of commissural axons to cross the midline during brain development [Shen et al., 2002]. Heterozygous mice exhibit features intermediate between those of wild type and Gap43 knockout mice [Donovan et al., 2002; Shen et al., 2002]. Given its role in the SHH pathway, BOC is another attractive candidate for the corpus callosum abnormalities. Homozygous Boc knockout mice are viable and do not have holoprosencephaly (HPE)-related phenotypes, as the gene has overlapping roles with other Shh co-receptors [Allen et al., 2011; Zhang et al., 2011]. However, the gene has been shown to modify the HPE phenotype in mice lacking Cdon, which encodes another Shh co-receptor related to and a binding partner of Boc [Zhang et al., 2011]. Furthermore, one reported HPE-associated mutation impairs the ability of CDON to bind to BOC, although other HPE CDON mutations do not [Bae et al., 2011]. Finally, Zbtb20 is also expressed in the cerebellum and corpus callosum [Mitchelmore et al., 2002]. Thus, it is possible that haploinsufficiencyof any, or a combination of, these genes may be involved in the brain abnormalities observed in patients with 3q13 deletions, though additional factors are likely required for manifestation, as penetrance is not complete for this phenotype.
The hypotonia and reduced reflexes present in these individuals are suggestive of neuromuscular dysfunction, and patient 5 showed myofiber atrophy. In addition to its role in the SHH pathway, BOC is also a cell adhesion molecule with roles in proper myogenesis [Kang et al., 2002], making it a candidate for these phenotypes. GAP43 may play important roles in skeletal muscle development and function [Guarnieri et al., 2013], and mice lacking the orthologue demonstrated muscle incoordination, reduced strength, and altered reflexes, with heterozygotes demonstrating moderate impairments [Metz and Schwab, 2004]. Finally, the dopamine receptor Drd3 helps regulate spinal reflexes [Clemens and Hochman, 2004], so its deletion may contribute to the depressed reflexes observed.
The deletion breakpoints of each of our patients were mapped within HERV-H elements. HERVs comprise approximately 8% of the reference human genome and are the remnants of viral infections in the germline cells of our distant ancestors [Lander et al., 2001; Paces et al., 2004]. The vast majority of HERVs contain single nucleotide variations, deletions or insertions that render them incapable of transposition or infection. Nonetheless, phylogenetic studies reveal HERVs undergo extensive inter-element recombination [Hughes and Coffin, 2001]. Thus, HERV elements scattered throughout the genome could provide a substrate for NAHR. Previous studies have identified HERV-H elements at the breakpoints of recurrent 1q41q42 deletions [Rosenfeld et al., 2011] and recurrent t(4;18) (q35.1;q22.3) translocations [Hermetz et al., 2012], each in two unrelated patients. Similarly, a single patient has been identified with a deletion at 8q13.3 involving EYA1 apparently mediated by HERV-H elements [Sanchez-Valle et al., 2010].
Evidence from cell culture suggests that NAHR events require stretches of extremely high homology or identity shared between two non-allelic loci known as minimal efficient processing segments (MEPSs) [Gu et al., 2008]. In human meiosis, minimum MEPS length was estimated at 300 to 500 base pairs [Reiter et al., 1998]. However, in a single sperm/cell assay, Lam and Jeffreys [2006] identified both meiotic and mitotic NAHR events between human alpha-globin genes mediated by matching fragments smaller than 50 bp. Interruption of the MEPSs with one or more base mismatches is known to deleteriously affect the efficiency of NAHR; in mice, the presence of as few as two nucleotide mismatches results in an ∼20 fold reduction in NAHR [Waldman and Liskay, 1988]. Based on the short length of uncertainty identified in our patients' breakpoints, when comparing to the reference haploid human genome, 300 bases of 100% identity are not present. Moreover, recent studies suggest that efficiency of NAHR is correlated with LCR length [Cooper et al., 2011; Liu et al., 2011; Dittwald et al., 2013]. However, the homologous regions of the HERV-H elements identified at the breakpoints in our patients totaled only 4.8 kb. Despite the apparent factors opposing NAHR between these HERV-H elements, we have identified nine such recurrent deletions.
Previous studies have identified evidence for NAHR events mediated by HERV elements [Kamp et al., 2000; Sun et al., 2000; Turner et al., 2008]. Sperm PCR analysis estimated the de novo mutation rate of the HERV-mediated AZFa deletion in Yq11.2 to be ∼2 × 10-3 per generation, much higher than the average locus-specific CNV mutation rate throughout the genome. The observation that the deletions were present in the sperm, but not in blood, suggests that the rearrangements occurred during meiosis, strengthening the NAHR hypothesis [Turner et al., 2008].
Alternatively, the deletions identified in our patients might be best explained by errors in DNA replication, such as those proposed in fork stalling and template switching (FoSTeS) [Lee et al., 2007] or microhomology-mediated break-induced replication (MMBIR) [Hastings et al., 2009] mechanisms. Under such a model, the HERV-H sequences would provide the microhomology substrate that facilitates a template switch during DNA replication. However, to date, no recurrent CNVs mediated by FoSTeS or MMBIR have been described. Of note, the same sperm PCR analysis estimated the de novo deletion to duplication ratio at Yq11.2 to be 4.11 to 1, considerably higher than the ∼2 to 1 ratio observed at autosomal loci, suggesting a difference between these HERV-mediated events on the Y chromosome and other, more canonical NAHR events [Turner et al., 2008].
Our cohort confirms many of the features previously attributed to the deletion of the SRO within 3q13.31, including global developmental delay, hypotonia, and dysmorphic features. In addition, the recurrent deletions in our cohort are associated with a high prevalence of behavioral and brain abnormalities. The molecular characterization of our patients' breakpoints revealed that HERV-mediated structural rearrangements may be more common than previously thought. Additional studies will be required to elucidate the exact mechanism of formation of CNVs associated with retroviral elements and determine their importance to human disease.
Supplementary Material
Acknowledgments
We thank Erin Dodge (Signature Genomics) for assistance in figure creation. We also thank Evan Eichler and Bradley Coe (University of Washington) for supplying control data. IMC is a fellow of the BCM Medical Scientist Training Program (5T32GM007330-34).AS, JWE, JBR, and JAR are employees of Signature Genomic Laboratories, a subsidiary of PerkinElmer, Inc. SWC, AP, and PS are employees of the Medical Genetics Laboratory at the Department of Molecular and Human Genetics at Baylor College of Medicine, which derives revenue from molecular diagnostic testing, including aCGH.
Contract grant sponsors: Polish National Science Center (2011/01/B/NZ2/00864) and the European Social Fund (UDA-POKL.04.01.01-00-072/09-00)
References
- Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, Gauda EB, Lee EJ, Cool MH, Sibley DR, Gerfen CR, Westphal H, et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci U S A. 1996;93:1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, Krauss RS, McMahon AP. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20:775–787. doi: 10.1016/j.devcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Matukiyo H, Takazawa H. A case report of partial deletion of the long arm of the no 3 chromosome. Med Genet Res. 1982;4:1–4. [Google Scholar]
- Bae GU, Domene S, Roessler E, Schachter K, Kang JS, Muenke M, Krauss RS. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. 2011;89:231–240. doi: 10.1016/j.ajhg.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, McDonald-McGinn DM, Zackai EH, Hersh JH, Bejjani BA, Shaffer LG. Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet. 2008;74:469–475. doi: 10.1111/j.1399-0004.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Paratcha G, Levi de Stein M, Bernabeu R, Izquierdo I, Medina JH. B-50/GAP-43 phosphorylation and PKC activity are increased in rat hippocampal synaptosomal membranes after an inhibitory avoidance training. Neurochem Res. 1997;22:499–505. doi: 10.1023/a:1027324214060. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- Clemens S, Hochman S. Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci. 2004;24:11337–11345. doi: 10.1523/JNEUROSCI.3698-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, Rodriguez Rojas LX, Elton LE, Scott DA, Schaaf CP, Torres-Martinez W, Stevens AK, et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013 doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan SL, Mamounas LA, Andrews AM, Blue ME, McCasland JS. GAP-43 is critical for normal development of the serotonergic innervation in forebrain. J Neurosci. 2002;22:3543–3552. doi: 10.1523/JNEUROSCI.22-09-03543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, Thompson R, Traylor R, Bejjani BA, Shaffer LG, Rosenfeld JA, Lamb AN, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet. 2010;18:1196–1201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Meng J, Kawamura M, Tozuka N, Ishii F, Tanaka N. Boy with a chromosome del (3)(q12q23) and blepharophimosis syndrome. Am J Med Genet. 1992;44:434–436. doi: 10.1002/ajmg.1320440409. [DOI] [PubMed] [Google Scholar]
- Genuardi M, Calvieri F, Tozzi C, Coslovi R, Neri G. A new case of interstitial deletion of chromosome 3q, del (3q)(q13.12q21.3), with agenesis of the corpus callosum. Clin Dysmorphol. 1994;3:292–296. [PubMed] [Google Scholar]
- Glickstein SB, Hof PR, Schmauss C. Mice lacking dopamine D2 and D3 receptors have spatial working memory deficits. J Neurosci. 2002;22:5619–5629. doi: 10.1523/JNEUROSCI.22-13-05619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri S, Morabito C, Paolini C, Boncompagni S, Pilla R, Fano-Illic G, Mariggio MA. Growth associated protein 43 is expressed in skeletal muscle fibers and is localized in proximity of mitochondria and calcium release units. PLoS ONE. 2013;8:e53267. doi: 10.1371/journal.pone.0053267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermetz KE, Surti U, Cody JD, Rudd MK. A recurrent translocation is mediated by homologous recombination between HERV-H elements. Mol Cytogenet. 2012;5:6. doi: 10.1186/1755-8166-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou JW. Congenital arhinia with de novo reciprocal translocation, t(3;12)(q13.2;p11.2) Am J Med Genet A. 2004;130A:200–203. doi: 10.1002/ajmg.a.30268. [DOI] [PubMed] [Google Scholar]
- Hughes JF, Coffin JM. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat Genet. 2001;29:487–489. doi: 10.1038/ng775. [DOI] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MB, Stang HJ, Davis E, Boyd L. Deletion of the proximal long arm of chromosome 3 in an infant with features of Turner syndrome. Ann Genet. 1985;28:42–44. [PubMed] [Google Scholar]
- Kamp C, Hirschmann P, Voss H, Huellen K, Vogt PH. Two long homologous retroviral sequence blocks in proximal Yq11 cause AZFa microdeletions as a result of intrachromosomal recombination events. Hum Mol Genet. 2000;9:2563–2572. doi: 10.1093/hmg/9.17.2563. [DOI] [PubMed] [Google Scholar]
- Kang JS, Mulieri PJ, Hu Y, Taliana L, Krauss RS. BOC, an Ig superfamily member, associates with CDO to positively regulate myogenic differentiation. EMBO J. 2002;21:114–124. doi: 10.1093/emboj/21.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaki R, Fukuhara Y, Kosuga M, Okuyama T, Kawashima N, Honna T, Ueoka K, Kosaki K. OEIS complex with del(3)(q12.2q13.2) Am J Med Genet A. 2005;135:224–226. doi: 10.1002/ajmg.a.30733. [DOI] [PubMed] [Google Scholar]
- Lam KW, Jeffreys AJ. Processes of copy-number change in human DNA: the dynamics of {alpha}-globin gene deletion. Proc Natl Acad Sci U S A. 2006;103:8921–8927. doi: 10.1073/pnas.0602690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb AN, Rosenfeld JA, Neill NJ, Talkowski ME, Blumenthal I, Girirajan S, Keelean-Fuller D, Fan Z, Pouncey J, Stevens C, Mackay-Loder L, Terespolsky D, et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. 2012;33:728–740. doi: 10.1002/humu.22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lawson-Yuen A, Berend SA, Soul JS, Irons M. Patient with novel interstitial deletion of chromosome 3q13.1q13.3 and agenesis of the corpus callosum. Clin Dysmorphol. 2006;15:217–220. doi: 10.1097/01.mcd.0000220609.17284.a9. [DOI] [PubMed] [Google Scholar]
- Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, Guichet A, Barth M, Charollais A, Journel H, Auvin S, Boucher C, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47:22–29. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet. 2011;89:580–588. doi: 10.1016/j.ajhg.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, Vernes SC, Vargha-Khadem F, McKenzie F, Smith RL, Monaco AP, Fisher SE. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–1080. doi: 10.1086/430841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie Ogilvie C, Rooney SC, Hodgson SV, Berry AC. Deletion of chromosome 3q proximal region gives rise to a variable phenotype. Clin Genet. 1998;53:220–222. doi: 10.1111/j.1399-0004.1998.tb02681.x. [DOI] [PubMed] [Google Scholar]
- McMorrow LE, Reid CS, Coleman J, Medeiros A, D'Andrea M, Santucci T, McCormack MK. A new interstitial deletion of the long arm of chromosome 3. Am J Hum Genet. 1986;39:124. [Google Scholar]
- Metz GA, Schwab ME. Behavioral characterization in a comprehensive mouse test battery reveals motor and sensory impairments in growth-associated protein-43 null mutant mice. Neuroscience. 2004;129:563–574. doi: 10.1016/j.neuroscience.2004.07.053. [DOI] [PubMed] [Google Scholar]
- Mitchelmore C, Kjaerulff KM, Pedersen HC, Nielsen JV, Rasmussen TE, Fisker MF, Finsen B, Pedersen KM, Jensen NA. Characterization of two novel nuclear BTB/POZ domain zinc finger isoforms. Association with differentiation of hippocampal neurons, cerebellar granule cells, and macroglia. J Biol Chem. 2002;277:7598–7609. doi: 10.1074/jbc.M110023200. [DOI] [PubMed] [Google Scholar]
- Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, Cormier-Daire V, David A, de Leeuw N, Delobel B, Duban-Bedu B, Fischetto R, et al. A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet. 2012;49:104–109. doi: 10.1136/jmedgenet-2011-100534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N'Gouemo P. Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets. 2011;15:1283–1295. doi: 10.1517/14728222.2011.620607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JV, Nielsen FH, Ismail R, Noraberg J, Jensen NA. Hippocampus-like corticoneurogenesis induced by two isoforms of the BTB-zinc finger gene Zbtb20 in mice. Development. 2007;134:1133–1140. doi: 10.1242/dev.000265. [DOI] [PubMed] [Google Scholar]
- Nielsen JV, Thomassen M, Mollgard K, Noraberg J, Jensen NA. Zbtb20 Defines a Hippocampal Neuronal Identity Through Direct Repression of Genes That Control Projection Neuron Development in the Isocortex. Cereb Cortex. 2013 doi: 10.1093/cercor/bhs400. [DOI] [PubMed] [Google Scholar]
- O'Driscoll MC, Black GC, Clayton-Smith J, Sherr EH, Dobyns WB. Identification of genomic loci contributing to agenesis of the corpus callosum. Am J Med Genet A. 2010;152A:2145–2159. doi: 10.1002/ajmg.a.33558. [DOI] [PubMed] [Google Scholar]
- Okada A, Charron F, Morin S, Shin DS, Wong K, Fabre PJ, Tessier-Lavigne M, McConnell SK. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 2006;444:369–373. doi: 10.1038/nature05246. [DOI] [PubMed] [Google Scholar]
- Okada N, Hasegawa T, Osawa M, Fukuyama Y. A case of de novo interstitial deletion 3q. J Med Genet. 1987;24:305–308. doi: 10.1136/jmg.24.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paces J, Pavlicek A, Zika R, Kapitonov VV, Jurka J, Paces V. HERVd: the Human Endogenous RetroViruses Database: update. Nucleic Acids Res. 2004;32:D50. doi: 10.1093/nar/gkh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascale A, Gusev PA, Amadio M, Dottorini T, Govoni S, Alkon DL, Quattrone A. Increase of the RNA-binding protein HuD and posttranscriptional up-regulation of the GAP-43 gene during spatial memory. Proc Natl Acad Sci U S A. 2004;101:1217–1222. doi: 10.1073/pnas.0307674100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Hastings PJ, Nelis E, De Jonghe P, Van Broeckhoven C, Lupski JR. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Lucas A, Spence EJ, Powell C, Aylsworth AS, Torchia BA, Shaffer LG. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE. 2009;4:e6568. doi: 10.1371/journal.pone.0006568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15:478–481. doi: 10.1038/gim.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Lacassie Y, El-Khechen D, Escobar LF, Reggin J, Heuer C, Chen E, Jenkins LS, Collins AT, Zinner S, Babcock M, Morrow B, et al. New cases and refinement of the critical region in the 1q41q42 microdeletion syndrome. Eur J Med Genet. 2011;54:42–49. doi: 10.1016/j.ejmg.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Routtenberg A, Cantallops I, Zaffuto S, Serrano P, Namgung U. Enhanced learning after genetic overexpression of a brain growth protein. Proc Natl Acad Sci U S A. 2000;97:7657–7662. doi: 10.1073/pnas.97.13.7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Arrones L, Cardozo M, Nieto-Lopez F, Bovolenta P. Cdon and Boc: Two transmembrane proteins implicated in cell-cell communication. Int J Biochem Cell Biol. 2012;44:698–702. doi: 10.1016/j.biocel.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Sanchez-Valle A, Wang X, Potocki L, Xia Z, Kang SH, Carlin ME, Michel D, Williams P, Cabrera-Meza G, Brundage EK, Eifert AL, Stankiewicz P, et al. HERV-mediated genomic rearrangement of EYA1 in an individual with branchio-oto-renal syndrome. Am J Med Genet A. 2010;152A:2854–2860. doi: 10.1002/ajmg.a.33686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Shimokawa O, Harada N, Olsen OE, Hou JW, Muhlbauer W, Blinkenberg E, Okamoto N, Kinoshita A, Matsumoto N, Kondo S, Kishino T, et al. Congenital arhinia: molecular-genetic analysis of five patients. Am J Med Genet A. 2007;143:546–552. doi: 10.1002/ajmg.a.31613. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M, Frackelton EC, Geiger EA, et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Mani S, Donovan SL, Schwob JE, Meiri KF. Growth-associated protein-43 is required for commissural axon guidance in the developing vertebrate nervous system. J Neurosci. 2002;22:239–247. doi: 10.1523/JNEUROSCI.22-01-00239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojima K, Saito K, Yamamoto T. A de novo 1.9-Mb interstitial deletion of 3q13.2q13.31 in a girl with dysmorphic features, muscle hypotonia, and developmental delay. Am J Med Genet A. 2009;149A:1818–1822. doi: 10.1002/ajmg.a.32963. [DOI] [PubMed] [Google Scholar]
- Simovich MJ, Bland SD, Peiffer DA, Gunderson KL, Cheung SW, Yatsenko SA, Shinawi M. Delineation of the proximal 3q microdeletion syndrome. Am J Med Genet A. 2008;146A:1729–1735. doi: 10.1002/ajmg.a.32292. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Sun C, Skaletsky H, Rozen S, Gromoll J, Nieschlag E, Oates R, Page DC. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum Mol Genet. 2000;9:2291–2296. doi: 10.1093/oxfordjournals.hmg.a018920. [DOI] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, McClafferty H, Knaus HG, Ruth P, Shipston MJ. Distinct acyl protein transferases and thioesterases control surface expression of calcium-activated potassium channels. J Biol Chem. 2012;287:14718–14725. doi: 10.1074/jbc.M111.335547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traylor RN, Fan Z, Hudson B, Rosenfeld JA, Shaffer LG, Torchia BS, Ballif BC. Microdeletion of 6q16.1 encompassing EPHA7 in a child with mild neurological abnormalities and dysmorphic features: case report. Mol Cytogenet. 2009;2:17. doi: 10.1186/1755-8166-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman AS, Liskay RM. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol. 1988;8:5350–5357. doi: 10.1128/mcb.8.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniowiecka-Kowalnik B, Kastory-Bronowska M, Bartnik M, Derwinska K, Dymczak-Domini W, Szumbarska D, Ziemka E, Szczaluba K, Sykulski M, Gambin T, Gambin A, Shaw CA, et al. Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders. Eur J Hum Genet. 2013;21:620–625. doi: 10.1038/ejhg.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Ma X, Ji W, Zhou G, Lu Y, Xiang Z, Wang YX, Zhang L, Hu Y, Ding YQ, Zhang WJ. Zbtb20 is essential for the specification of CA1 field identity in the developing hippocampus. Proc Natl Acad Sci U S A. 2010;107:6510–6515. doi: 10.1073/pnas.0912315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccaria KJ, Lagace DC, Eisch AJ, McCasland JS. Resistance to change and vulnerability to stress: autistic-like features of GAP43-deficient mice. Genes Brain Behav. 2010;9:985–996. doi: 10.1111/j.1601-183X.2010.00638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hong M, Bae GU, Kang JS, Krauss RS. Boc modifies the holoprosencephaly spectrum of Cdo mutant mice. Dis Model Mech. 2011;4:368–380. doi: 10.1242/dmm.005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.