

Abstract
The self-assembly of hybrid diblock copolymers composed of poly(HPMA) and β-sheet peptide P11 (CH3CO-QQRFQWQFEQQ-NH2) blocks was investigated. Copolymers were synthesized via thiol-maleimide coupling reaction, by conjugation of semitelechelic poly(HPMA)-SH with maleimide-modified β-sheet peptide. As expected, CD and CR binding studies showed that the peptide block imposed its β-sheet structural arrangement on the structure of diblock copolymers. TEM and AFM proved that peptide and these copolymers had the ability to self-assemble into fibrils.
Keywords: diblock copolymers, conjugated polymers, reversible addition fragmentation chain transfer (RAFT), self-assembly, HPMA, β-sheet peptide
Introduction
Molecular self-assembly of synthetic block copolymers has been lately the focus of research activity of numerous investigators.[1–3] While synthetic copolymers are still of interest, the possibility to prepare hybrid block copolymers based on combination of proteins or peptides and synthetic polymers has become an attractive way to expand the diversity of structures and functions in synthetic materials. Peptide/protein-polymer hybrid copolymers are able to overcome some of the disadvantages of the individual components. For example, a structural protein or peptide block (such as coiled-coil or β-sheet) may control the nanostructure of the synthetic component,[4–6] whereas a synthetic polymer block may increase the stability[7] and the biocompatibility of the protein/peptide segment.[8,9]
A series of interesting reports has described the synthesis, as well as the self-assembly[10–15] of different β-sheet peptide/poly(ethylene glycol) (PEG) hybrid diblock copolymers. Two main approaches to obtain hybrid copolymers can be identified: polymerization strategies, using a peptide macroinitiator to synthesize the polymer block, and coupling strategies, using a preformed synthetic polymer to react with selective functionalities on the peptide.[16,17] However, when analyzing the self-assembly mechanism, a common trend arises: the hierarchical organization of the β-sheet motifs in the peptide blocks mediates the self-organization of the conjugates. As a result, fibrils can be formed by stacking of the β-sheets on top of each other.[18] Studies showed not only that the self-organization of different β-sheet peptides into fibrils was preserved after conjugation with PEG, but moreover, the lateral aggregation of these fibrils was prevented.[10,14] It seems that the PEG coating provides solubility and facilitates self-assembly into uniform fibrils with no lateral aggregation. This is particularly important in case of PEG-coated amyloid fibrils that exhibit a strong reduction in the formation of amyloid plaques,[14] associated with e. g. Alzheimer’s disease.
While various β-sheet peptides have been successfully conjugated with PEG, there is no report on their conjugation with poly[N-(2-hydroxypropyl)methacrylamide] [poly-(HPMA)], a non-immunogenic, neutral, hydrophilic polymer currently employed in the delivery of anticancer drugs. Compared to PEG conjugates, imposing a certain secondary structure on a poly(HPMA)-peptide conjugate might be a challenging task, since in water, poly(HPMA) has a random coil conformation, while PEG adopts an extended conformation.[19] However, previous work on HPMA copolymers containing coiled-coil peptide grafts demonstrated that self-assembly of the hybrid poly(HPMA)-peptide copolymer systems is possible, and moreover, it can even trigger the formation of a hydrogel.[20,21]
In this article, the synthesis and the self-assembly of hybrid diblock copolymers, based on poly(HPMA) and β-sheet peptide blocks, are described. We hypothesized that the β-sheet-forming peptide block will impose its structural arrangement on the overall structure of the diblock copolymers. The β-sheet peptide was synthesized using solid-phase methodology and manual 9-fluorenyl-methoxycarbonyl (Fmoc)/tert. butyl ester (tBu) strategy, and purified by reverse phase HPLC. The sequence P11-2 (CH3CO-Gln-Gln-Arg-Phe-Gln-Trp-Gln-Phe-Glu-Gln-Gln-NH2, CH3CO-QQRFQWQFEQQ-NH2, herein termed P11), previously designed by Aggeli et al.,[22] was used. We chose this well-characterized, short peptide sequence for the fact that in aqueous solutions, it can form antiparallel β-sheets self-organized into fibrils,[23] as well as for the easiness of its synthesis. A novel approach to prepare the hybrid diblock copolymers was developed. Well-defined semitelechelic poly(HPMA) polymers terminated with thiol functional groups were prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization in methanol followed by aminolysis. Then, the N-terminus of the P11 peptide was modified with a maleimide group, and finally, in the last step, the conjugation was accomplished via the maleimide-thiol coupling reaction. Circular dichroism spectroscopy (CD) and Congo Red (CR) binding studies were performed to characterize the secondary structure of the peptide and of the peptide domain in diblock copolymers. The morphology of the materials was investigated by transmission electron microscopy (TEM), and atomic force microscopy (AFM).
Experimental Part
Materials
Side chain-protected Fmoc-amino acids, rink amide 4-methyl-benzhydrylamine (MBHA) resin were from Novabiochem (San Diego, CA). 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (HATU, >98%) and 1-hydroxybenzotriazole (HOBt) were purchased from AK Scientific (Mountain View, CA). N,N-dimethylformamide (DMF, 99.8%), sodium hydrosulfite (Na2S2O4, 79%), piperidine (99.5+%, Biotech grade), triisopropylsilane (TIS, 99%), N,N-diisopropylethylamine (DIPEA; 99%), acetic anhydride (99+%) and methanol were from Sigma-Aldrich (St. Louis, MO). Diethyl ether and dichloromethane (DCM) were from Maleinckrodt Baker (Philipsburg, NJ). Trifluoroacetic acid (TFA, 99%) was purchased from Acros Organics (Morris Plains, NJ). Succinimidyl trans-4-(maleimidylmethyl)cyclohexane-1-carboxylate (SMCC) was from Soltech Ventures (Beverly, MA). 4,4′-Azobis(4-cyanopentanoic acid) (V-501), N,N′-diisopropylcarbodiimide (DIC, >98%), hexylamine (>98%), and tris(2-carboxyethyl)phosphine (TCEP) were from Fluka (Milwaukee, WI). Thiol modified poly(ethylene glycol) (CH3-PEG-SH, weight-average molecular weight M̄w = 1 892 g · mol−1, polydispersity M̄w/M̄n = 1.21, SH amount = 498 μmol · g−1) was purchased from Rapp Polymere (Tübingen, Germany). CR (dye content ≈ 80%) was from Alfa Aesar (Ward Hill, MA). The chain transfer agent (CTA), 4-cyanopentanoic acid dithiobenzoate,[24] and HPMA[25] were synthesized as previously described.
Peptide Synthesis and Purification
P11 peptide, CH3CO-Gln-Gln-Arg-Phe-Gln-Trp-Gln-Phe-Glu-Gln-Gln-NH2,[22] was synthesized using solid-phase methodology and manual Fmoc/tBu strategy on rink amide resin, similar to a previously described protocol.[4] After swelling of the resin beads (200 mg, 0.14 mmol) in DCM (5 mL), and deprotection with 20% piperidine in DMF (2.5 mL, 2 × 5 min), the first amino acid, Fmoc-Gln(Trt)-OH (0.35 mmol; Trt = triphenylmethyl), was attached to the resin in DMF, in the presence of HOBt and DIC. The rest of the amino acids (each 0.175 mmol) were dissolved in DMF/HOBt solution and attached to the resin-bound peptide in DMF after deprotection, one at a time, in the presence of HATU and DIPEA (each 0.175 mmol). The completion of each coupling step was verified by Kaiser test. The final 11th residue was deprotected, and the peptide-resin was washed with DMF, DCM, and methanol, and dried under vacuum. Part of the resin-bound peptide was treated with a solution of acetic anhydride and DIPEA in DCM for acetylation of N-terminus, and then with TFA/TIS/H2O cocktail solution (95:2.5:2.5 vol.-%) for cleavage of the peptide from the resin beads and deprotection of the side chains. Crude P11 peptide was precipitated with diethyl ether. The rest of the resin-bound peptide was kept in DMF and further used for coupling with semitelechelic poly(HPMA) and PEG. Peptide purification was carried out by RP-HPLC using a semipreparative Zorbax 300SB-CN column (9.4 × 250 mm, 5 μm particle size, 300 Å pore size) from Agilent Technologies. The peptide was eluted with a linear gradient at a flow rate of 2 mL · min−1, using buffer A, H2O with 0.1% TFA, and buffer B, acetonitrile with 0.1% TFA. The purity of the collected fractions was verified with analytical RP-HPLC, using an Eclipse XDB-C8 column (4.6 × 150 mm, 5 μm particle size, 80 Å pore size). Collected fractions were lyophilized. The identity of the P11 peptide obtained after lyophilization was confirmed by MALDI-TOF mass spectrometry (MS; Voyager-DE STR Biospectrometry Workstation, PerSeptive Biosystems, Framingham, MA), showing a single main peak corresponding to the expected molecular weight at M+ + 1 = 1593.76 m/z.
Synthesis and Purification of β-Sheet Peptide-Poly(HPMA) Diblock Copolymers
Poly(HPMA) Synthesis by RAFT Polymerization
Two RAFT polymerizations of HPMA were performed following a procedure described by McCormick and co-workers,[26] employing 4,4′-azobis(4-cyanopentanoic acid; V-501) as initiator, and 4-cyanopentanoic acid dithiobenzoate as CTA. HPMA polymerizations were conducted at 60 °C in methanol, using initial monomer concentration ([Mo]) as 1 M, and constant ratio of CTA to initiator as 5, in sealed ampoules that were purged with nitrogen for 30 min prior to reaction. Monomer to CTA ratios ([Mo]/[CTA]) of 25 and 40 were used. Polymerization reactions were allowed to proceed for 24 h. The resulting polymers were precipitated into cold diethyl ether and washed with acetone for removal of the unreacted HPMA monomer. After freeze-drying of the products, the polymerization yields were estimated as being 48.5% and 98.4% respectively. The molecular weights of poly(HPMA) were analyzed by size exclusion chromatography (SEC) on an Äkta FPLC system (Amersham Pharmacia Biotech) equipped with UV and RI detectors, using a Superdex Peptide column, previously calibrated with HPMA fractions, and phosphate buffer solution (PBS, pH = 7.2) as eluent at a flow rate of 0.4 mL · min−1. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS was also employed for the evaluation of the polymers molecular weight. Both techniques confirmed that two low-molecular weight poly(HPMA) polymers [poly(HPMA)2k with M̄w = 1 990 g · mol−1 and M̄w/M̄n = 1.41, and poly(HPMA)5k with M̄w = 4 890 g · mol−1 and M̄w/M̄n = 1.14] were synthesized.
Modification of P11 β-Sheet Peptide with Maleimide Group
HPLC experiments and MALDI-TOF MS showed that P11 peptide even in crude form was pure (ca. 95%), which enabled the use of the peptide-bound resin for the synthesis of the diblock copolymers. For each coupling, part of the peptide-bound resin batch (0.046 mmol, 72 mg) was modified with maleimide group. A solution of SMCC (0.07 mmol, 23.4 mg) and DIPEA (0.138 mmol, 24.4 μL) was added to the peptide-bound resin beads in 2 mL DMF and kept at room temperature for 20 h. The identity of the maleimido-modified P11 peptide was verified by MALDI-TOF MS, after a small amount of the product was cleaved from the resin by using TFA/TIS/H2O cocktail solution (95:2.5:2.5 vol.-%), precipitated with cold diethyl ether, isolated by filtration, dried, dissolved in deionized (DI) water, and lyophilized. A single peak corresponding to the expected molecular weight at M+ = 1770.84 m/z was obtained.
Poly(HPMA) with Thiol Groups
The thiocarbonate end groups of poly(HPMA)2k (160 mg, M̄w = 1 990 g · mol−1, M̄w/M̄n = 1.41) and poly(HPMA)5k (160 mg, M̄w = 4 890 g · mol−1, M̄w/M̄n = 1.14) were transformed into thiol groups by aminolysis.[27] Each polymer was dissolved in 2 mL DMF and bubbled with nitrogen for 30 min. Aqueous Na2S2O4 solution (0.5 M, 50 μL) and hexylamine (0.1 g, 1.0 mmol) were added to the mixture. The reaction was allowed to proceed for 18 h at room temperature, under nitrogen atmosphere and magnetic stirring. Discoloration of the solutions was observed. The polymers were precipitated into diethyl ether, isolated by centrifugation, dried, dissolved in DI water, and lyophilized. Ellman test was performed on the thiol-terminated semitelechelic poly(HPMA) for quantification of thiol groups[28] as being 0.57 mmol · g−1 for poly(HPMA)2k, and 0.22 mmol · g−1 for poly(HPMA)5k, respectively.
Conjugation of the Poly(HPMA) with P11 Peptide
Thiol-terminated poly(HPMA)2k and 5k (130 mg each) were incubated in DMF in the presence of TCEP (4.8 mg) for 2 h, to eliminate the formation of the polymeric disulfides.[29] The maleimide-modified P11 peptide bound to the resin beads and the corresponding thiol-modified poly(HPMA) were mixed in 2 mL DMF. The maleimide-thiol coupling reactions took place in 24 h at room temperature. Following coupling, the products were cleaved from the beads by using a TFA/TIS/H2O cocktail solution (95:2.5:2.5 vol.-%), precipitated with diethyl ether, collected by filtration, dried in air, dissolved in DI water, and lyophilized. HPLC analysis of the products showed that a mixture of free P11 peptide and P11-poly(HPMA) conjugate was obtained after each coupling reaction. The conjugates were purified by dialysis (molecular weight cut-off 1 000 Da) against water for 72 h. Analytical reversed phase (RP)-HPLC confirmed that pure peptide-polymer conjugates [P11-poly(HPMA)2k and P11-poly(HPMA)5k] were obtained after dialysis. The yield of the coupling reaction was calculated as being 8% for P11-poly(HPMA)2k, and 6.66% for P11-poly(HPMA)5k with respect to the pure conjugates. For comparison, a similar coupling approach was employed for the conjugation of thiol-modified PEG (M̄w = 1 892 g · mol−1, M̄w/M̄n = 1.21, SH content = 0.49 mmol · g−1) with the maleimide-modified P11 peptide bound to the resin beads. The yield of the reaction was 14.5% with respect to the pure P11-PEG2k diblock copolymer.
CD Spectroscopy
CD spectra were collected on an Aviv 62DS CD spectrometer at 25 °C. The samples were prepared by dissolving a known mass of lyophilized product in water. The pH of the solutions was adjusted under pH-meter monitoring (Corning pH-meter 440) to the desired values (2, 7 or 11) by addition of appropriate amounts of 1 N HCl or 1 N NaOH solution, unless otherwise stated. Solutions with concentrations determined by UV spectroscopy (typically, 1 mg · mL−1, 0.6 × 10−3 M, with respect to the peptide) were incubated at room temperature for 3–10 d or longer prior to CD measurements. The pH of the solutions was checked periodically and adjusted when required. Wavelength scans were recorded at 1 nm intervals from 250 to 200 nm using a 0.1 cm path length quartz cuvette. Spectra were averaged from three consecutive scans after background was subtracted. Mean residue ellipticity ([θ], in deg · cm2 · dmol−1) was calculated from Equation (1) [21]
| (1) |
where [θ]obs is the ellipticity measured in millidegrees, MRW is the mean residue molecular weight of the peptide (molecular weight of the unacetylated peptide, 1 551 Da, divided by the number of amino acid residues), l is the optical path length of the cell in cm (0.1 cm), and c is the peptide concentration in mg · mL−1.
CR Binding Studies
Spectrophotometric analysis of CR binding to P11 and to P11-poly(HPMA)2k and P11-poly(HPMA)5k diblock copolymers was performed on a Cary 400 Bio UV-vis spectrophotometer (Varian, Palo Alto, CA), using a 0.1 cm path length quartz cuvette. Spectra were recorded in the scan mode, between 700 and 390 nm, with a 1 nm sampling interval. A 150 μM stock solution of CR was prepared in PBS (50 × 10−3 M Na2HPO4/NaH2PO4, 100 × 10 −3 M KCl, pH = 7.0) and 10% ethanol to prevent CR micelle formation.[30] The acidic pH of P11 peptide solutions and the basic pH of peptide-polymer conjugates solutions in water (all 1 mg · mL−1) were adjusted to neutral pH by dilution with PBS (50:50 vol.-%). Consequently, the secondary structure of the new solutions was verified by CD and confirmed as being β-sheet. CR stock solution was added to these solutions for a final concentration of 3 × 10−6 M CR. The resulting mixtures were incubated for 30 min at room temperature before the spectral analysis.
CR birefringence studies were performed on an Olympus IX70 microscope equipped with a digital camera, at a magnification of 40×. Drops (ca. 5 μL) of the mixture of P11 peptide and CR dye, and of the mixtures of P11-poly(HPMA)2k conjugate and CR dye, and P11-poly(HPMA)5k and CR dye, in PBS/ethanol 10%, used for CR binding investigation by UV-vis spectrophotometry, were applied to glass slides and left to dry in air for 30 min. The samples were observed under bright field illumination and then between crossed polarizers.
TEM
Samples were prepared on copper specimen grids coated with a carbon support film (CF200-Cu; Electron Microscopy Sciences, Fort Washington, PA). The solutions of P11 at pH = 2, and P11-poly(HPMA)2k and P11-poly(HPMA)5k at pH = 11, having a β-sheet secondary structure as confirmed by CD, were used for sample preparation. The grids were placed with the carbon-coated face on top of a 5 μL drop of aqueous peptide solution (0.1 mg · mL−1; sonicated for 15 min in advance), for 30 s. The same procedure was followed for the preparation of the conjugates samples. Negative staining of the samples was done by placing the grids with the wet side down on top of a 5 μL uranyl acetate solution 4% (w/v) for 20 s. The specimens were allowed to dry overnight, and then examined using a Philips Tecnai transmission electron microscope at 100 kV accelerating voltage. Random fields were photographed with magnifications ranging from 30 000× to 150 000×.
AFM
AFM observations were carried out on a modified Explorer AFM (Topometrix Inc., Santa Clara, CA) operating in contact mode, in air, at room temperature. Height images were recorded with silicon cantilevers (Mikromasch, Wilsonville, OR) at a resolution of 512 ×512 pixels (2 ×2 μm2). One drop of P11 peptide at pH = 2 or conjugate solution at pH = 11 (0.01 mg · mL−1), possessing β-sheet structure as determined by CD spectroscopy, was sonicated for 15 min and placed onto the center of a freshly cleaved mica disc (Pelco, Ted Pella, Redding, CA). The resulting films were allowed to dry overnight, and then, their morphology was investigated. DI water with pH = 2 and 11 (adjusted with either HCl or NaOH) were used as controls. AFM images obtained were processed using the WSxM software.
Results and Discussion
Synthesis of P11 Peptide and P11-Poly(HPMA) Conjugates
P11 β-sheet peptide, designed by Aggeli et al.,[22] is a sequence rich in glutamine (Gln) residues. The distribution of Gln, phenylalanine and tryptophan residues in P11 was designed to provide a hydrophilic face and a hydrophobic face that promote intermolecular recognition between adjacent P11 β-strands. Our interest in this well-characterized peptide was due to the fact that in acidic aqueous solutions (pH = 2), it forms antiparallel β-sheets, which with increasing concentration, self-assemble into tapes, ribbons and fibrils.[22,31] Secondly, P11 has a short amino acids sequence, which makes it easier to be synthesized by solid-phase method using manual Fmoc/tBu strategy. Indeed, a 96% yield of the P11 β-sheet peptide synthesis reaction was obtained, a good result, especially when considering the drawbacks of the process, associated with the aggregation of the β-sheet peptides on the resin. Part of P11 was acetylated, cleaved from the resin, purified and used for comparative studies with the P11-poly(HPMA) conjugates. The rest of the peptide-bound resin was modified at its N-terminus with maleimide group and used for the conjugation with the polymers. The poly (HPMA) polymers [poly(HPMA)2k, M̄w = 1 990 g · mol−1, M̄w/M̄n = 1.41, and poly(HPMA)5k, M̄w = 4 890 g · mol−1 M̄w/M̄n = 1.14] with thiol end groups were attached to P11 peptide via a thioether bond, according to the scheme illustrated in Figure 1. The copolymers were purified by dialysis and their purity was verified by HPLC (no free peptide was detected). Comparison of MALDI-TOF MS spectra of poly(HPMA) precursor and P11-poly(HPMA) conjugates, as well as their HPLC and SEC results (data not shown), proved that the copolymers were obtained, although the yield of the coupling reactions was low [8% for P11-poly(HPMA)2k, and 6.7% for P11-poly(HPMA)5k, with respect to the pure conjugates], presumably due to a low accessibility of the thiol reactive end-groups as a result of their location inside the polymer chains.[12,16]
Figure 1.

Synthesis of β-sheet P11 peptide-poly(HPMA) diblock copolymers via thiol-maleimide coupling reaction.
The peptide blocks in the copolymers were expected to form β-sheets under suitable conditions, and therefore, to impose their organization on the overall structure of the conjugates. Our hypothesis was verified by the results of the characterization studies presented below.
CD Spectra of P11 Peptide and P11-Poly(HPMA) Conjugates
The secondary structures of P11 peptide and P11-poly(HPMA) conjugates were evaluated by CD spectroscopy. P11 peptide (1 mg · mL−1) in water at pH = 2, incubated 3 d at room temperature had a β-sheet secondary structure, as described in literature.[31,32] The intense negative maximum near 218 nm on P11 CD spectrum (Figure 2a) is characteristic for peptides adopting a β-sheet conformation. As suggested by Aggeli et al.,[31] the slow kinetics of the anti-parallel β-sheet formation was speeded up from days to only hours by adding seeds of preformed β-sheets to a newly prepared P11 solution at acidic pH (data not shown).
Figure 2.

CD spectra of P11 (circles), P11-poly(HPMA)2k (squares) and P11-poly(HPMA)5k (triangles) conjugates. a) In water at pH = 2, after 3 d of incubation at room temperature, b) in water at pH = 7, after 18 d of incubation at room temperature, c) in water at pH = 11, after 10 d of incubation at room temperature.
However, CD spectra of aqueous solutions of P11-poly(HPMA)2k and P11-poly (HPMA)5k conjugates (2 and 4 mg · mL−1 respectively; the concentration of the peptide in the conjugate being thus kept approximately 1 mg · mL−1, 0.6 × 10−3 M) at pH = 2, after incubation at room temperature for 3 d, indicated a random coil structure (Figure 2a). Even after incubation of the conjugates for one month, their secondary structure at pH = 2 was unchanged. The same result was obtained at pH = 7 (Figure 2b). On the contrary, aqueous solutions of P11-poly(HPMA)2k and P11-poly(HPMA)5k at pH = 11 (adjusted with NaOH) showed β-sheet secondary structures after being incubated for 10 d at room temperature, whereas P11 peptide in water at pH = 11, after 10 d, had only a random coil structure (Figure 2c). It seems that the concentration of the copolymers, the incubation time, the pH, and the presence of Na+ were essential for the formation of β-sheet structure. This observation was sustained by a series of simple experiments. As in the case of peptide solutions, the self-assembly could be speeded up; when the concentration of the copolymer was doubled, a β-sheet structure was obtained in water at pH = 11, after only 8 d of incubation at room temperature. Interestingly, when the pH of an aqueous conjugate solution was adjusted to pH = 11 using NH4OH instead of NaOH, in two attempts, the CD investigations evidenced only a random coil structure. This result is in agreement with data published by Zhang et al., according to which Na+ plays an important role in the self-assembly, whereas does not induce the formation of β-sheets.[33] However, control experiments that were done on aqueous solutions of P11 and of its conjugates with HPMA in the presence of NaCl showed that Na+ alone, without the pH factor, is not able to induce the self-assembly. In order to explain the β-sheet formation in the P11-poly(HPMA) conjugates in aqueous samples at pH = 11 adjusted with NaOH, the behavior of ionizable groups on the peptide, as well as the influence of polymer conformation, should be analyzed. At pH = 11, free glutamic acid (Glu) in solution is negatively charged (pKa 4.1), whereas free arginine (Arg) is positively charged (pKa 12.5); however, the pKa values of these amino acids in the β-sheet self-assemblies may be different due to the interactions with neighboring charged side chains.[31] Moreover, when, for comparison purpose, aqueous solutions of 2 mg · mL−1 P11-PEG2k at pH = 2 and 11, prepared following the same procedure as for P11-poly(HPMA) conjugates, were investigated by CD, β-sheets were detected at both pH values. However, for the same concentration of solutions, the intensity of the negative maximum near 218 nm largely increased as pH varied from 2 to 11, suggesting that in a conjugate, more P11 β-sheets are obtained in basic conditions. Therefore, given the random coil conformation of poly(HPMA) blocks versus the extended conformation of PEG blocks in the copolymers in water, P11 peptide was able to impose its β-sheet structure on the structure of the P11-poly(HPMA) copolymers only when strong electrostatic interactions were present. Favorable electrostatic interactions between positive side chains of Arg and negative side chains of Glu, conformation of poly(HPMA) block, as well as an active involvement of Na+, by binding to the γ-carboxylate of Glu, are presumably main factors responsible for the stabilization of the diblock copolymer β-sheets at pH = 11.
Possible structural changes of the poly(HPMA) block at pH = 11 in the low molecular weight copolymers P11-poly(HPMA)2k and P11-poly(HPMA)5k were also considered. Semitelechelic poly(HPMA) and P11-poly(HPMA) solutions (0.5 mg · mL−1) were prepared in water at pH = 11 and incubated 10 d at room temperature. Comparison of HPLC spectra and SEC chromatograms of these samples before and after incubation showed no difference in the position of the peaks. Apparently, hydrolysis of HPMA side-chains, if any, was minor. Even though the hydrolysis of the maleimide to maleamic acid would occur in the conjugates samples at pH = 11, this modification would not destroy the peptide-polymer linkage, but, instead, it would transform it into a thiol-maleamic acid bond.
CR Binding Study
CR is a sulfonated azo dye that preferentially binds to proteins and peptides that are rich in β-sheets, similar to amyloids found in prion diseases. Given that the color of the CR changes from blue to red at pH = 3–5.2, CR binding experiments were conducted at pH = 7 in PBS buffer, only after CD investigations showed that the β-sheet structure was preserved in the new solutions after the pH change. CR binding indicated that peptide and the conjugates, P11-poly(HPMA)2k and P11-poly(HPMA)5k, bound to the dye, confirming the existence of β-sheet structure in these samples. CR binding to the β-sheets of P11 peptide resulted in a shift of the CR characteristic intense peak from 489 nm to 492 nm, whereas binding of CR to the conjugates produced a larger shift: to 500 nm for P11-poly(HPMA)2k, and to 508 nm for P11-poly(HPMA)5k (Figure 3a). The point of maximal spectral difference (λmax) was estimated using the differential spectrum obtained by subtracting the absorption spectrum of CR from the absorption spectrum of P11 peptide/CR mixture, of P11-poly(HPMA)2k/ CR mixture, and respectively, of P11-poly(HPMA)5k/CR mixture (Figure 3b). A maximum spectral difference at 524 nm was obtained for the peptide, whereas 540 nm was calculated for both diblock copolymers. These values are a clear indication of the CR binding to the β-sheets in both cases, peptide and diblock copolymers, since λmax at 520–540 nm is a characteristic feature of β-sheets presence, as evidenced in β-amyloid fibrils.[34,35]
Figure 3.

a) Congo Red binding assay: absorbance spectra of CR alone (stars) and bound to β-sheets of P11 peptide (circles), P11-poly(HPMA)2k (squares) and P11-poly(HPMA)5k (triangles) conjugates. b) Differential spectra of (P11/CR) – (CR), (P11-poly(HPMA)2k/CR) –(CR) and (P11-poly(HPMA)5k/CR) – (CR) showing the points of maximum absorption.
Green birefringence was detected under polarized light in peptide and both conjugates samples weakly stained by CR (samples from UV-vis CR binding studies were used). In bright field, peptide/CR and conjugates/CR samples appeared yellow or red, depending on the amount of the CR dye. On the contrary, under crossed polarizers, same samples exhibited a green birefringence, an indication that the dye molecules bound to the β-sheets with a preferential orientation in respect to the β-sheet fibril axis (data not shown).[36,37] Visualization of the samples edges, where CR was not bound to the fibrils, showed no change in the red color under crossed-polarized light.
Morphology of the P11 Peptide and P11-Poly(HPMA) Conjugates Self-Assemblies
The self-assembly of P11 peptide and low molecular weight P11-poly(HPMA)2k and P11-poly(HPMA)5k conjugates was studied first by TEM. Micrometer-long fibrils were constantly observed by TEM imaging for P11 peptide (Figure 4A), and P11-poly(HPMA)2k (Figure 4B), and P11-poly(HPMA)5k (Figure 4C) dried samples from aqueous solutions. The contact between the hydrophobic residues in the P11 peptide block and the hydrophobic surface of the carbon film apparently stabilized the preformed β-sheets, resulting in copolymer fibrils visible in TEM images. This observation underlines the importance of the environment in the formation and stabilization of the β-sheet structures in hybrid materials.[38] Moreover, while P11 peptide formed fibrils of around 20–25 nm in width, the P11-poly(HPMA) copolymers formed thinner (10–15 nm in width), longer fibrils. Even though the existence of fibrils is certain, it is important to mention that the negative staining with uranyl acetate, used in the preparation of the TEM samples, might play an important role in hindering the measurement of the fibrils width. However, our observations are consistent with those reported by Messersmith and coworkers for PEG-conjugated β-sheet peptides.[14] No attempt to formulate a conclusion regarding the minimization of the lateral aggregation in poly(HPMA)-coated fibrils had been made, since individual fibrils were difficult to distinguish because of the dense coverage of the grid.
Figure 4.
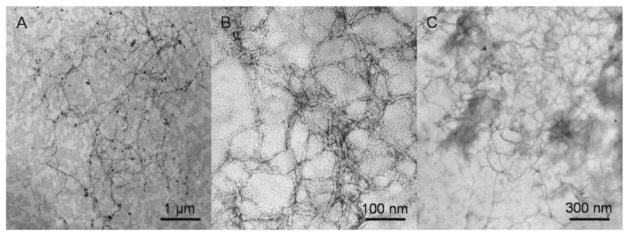
TEM images of negatively stained; A) P11 peptide sample in water at pH = 2; B) P11-poly(HPMA)2k conjugate sample in water at pH = 11; C) P11-poly(HPMA)5k conjugate sample in water at pH = 11.
AFM investigation of the self-assembly of low concentration solutions of P11 in water at pH = 2 confirmed the TEM results. Before sonication, mica surface exposed to P11 solutions at pH = 2 showed globular aggregates with variable diameters (100–200 nm) (data not shown). After sonication, mica surface exposed to the same solution was decorated with fibrils with an average width of around 50 nm (Figure 5A). This size was larger than the value estimated based on TEM observations of the fibrils. The finite radius of AFM tip may have contributed to the overestimation of fibril width. Moreover, high resolution contact mode AFM imaging revealed that on mica hydrophilic surface, two peptide fibrils interacted laterally, therefore resulting in a double value of the width, when compared to TEM results from peptide samples on hydrophobic carbon-coated copper grids. It seems that, as previously shown for various proteins,[39] sonication of the P11 aqueous sample resulted in an increase in the β-sheet content accompanied by an increase in the stability of the self-assemblies, which is consistent with the difference in the morphology of the P11 aggregates before (globules) and after the sonication (fibrils). In contrast, when solutions of P11-poly(HPMA)2k and P11-poly(HPMA)5k conjugates in water at pH = 11 (possessing a β-sheet structure) were brought in contact with the surface of freshly cleaved mica, even after sonication, the AFM imaging revealed only the presence of non-fibrillar, plaque-like aggregates with diameters of 75–150 nm (Figure 5B and C). This behavior, not seen in TEM imaging, is probably due to the amphiphilic nature of the P11-poly(HPMA) conjugates. The hydrophobic peptide block and the hydrophilic polymer block may interact differently with dissimilar substrates. While the fibrils of the peptide were adhering on mica without suffering major changes in their morphology, in case of the peptide-polymer conjugates, deformation of the fibrillar structure was probably induced by the immobilization of lower stability peptide-polymer conjugate fibrils on the high free energy surface of mica.[40] The copolymer fibrils, presumably formed in solution, being thinner and shorter than the peptide fibrils, lost their morphology when they came in contact with the mica surface. We hypothesize that the peptide block formed the plaques that were seen on AFM images of the copolymers, while the amorphous, soft polymer chains formed a featureless layer that covered mica surface. A concentration dependent fibril formation might also offer an explanation of the different TEM and AFM results. The concentration of the block copolymer solutions used in AFM imaging was 10× lower than the one used in TEM imaging. At low concentration, the copolymers might self-assemble only into protofibrils that appear as two-dimensional plaque-like structures on mica. As suggested by Jun et al.,[41] a critical aggregation concentration (CAC) threshold needs to be reached for fibril formation. Unfortunately, attempts to determine the exact threshold concentration for the fibrillation process to occur, and to visualize the copolymer morphology at higher concentrations were unsuccessful due to the thick coverage of the mica disc. However, based on the TEM and AFM results, one may roughly estimate that CAC of P11 peptide necessary for fibril formation in the diblock copolymers is between 0.005 and 0.01 mg · mL−1.
Figure 5.

AFM images; A) P11 peptide sample in water at pH = 2; B) P11-poly(HPMA)2k conjugate sample in water at pH = 11; C) P11-poly(HPMA)5k conjugate sample in water at pH = 11, on mica surface. Height scale was 5 nm.
Conclusion
In conclusion, novel peptide-polymer conjugates were created by coupling a β-sheet-forming peptide, P11, to semitelechelic poly(HPMA), via a thioether bond. The ability of the peptide to form β-sheet structures was retained upon conjugation to low molecular weight poly(HPMA). CD experiments indicated pH-dependent β-sheet formation in aqueous solutions of P11, P11-poly(HPMA)2k and P11-poly(HPMA)5k diblock copolymers. The binding of the P11 peptide to the semitelechelic poly(HPMA) resulted in a shift in folding conditions. Whereas the free peptide formed β-sheets at pH = 2, the polymer conjugates formed β-sheets at pH = 11. The presence of β-sheets in these compounds was confirmed by CR binding and CR birefringence studies. Moreover, TEM and AFM investigations indicated that hierarchically organized structures, such as fibrils, were formed by the peptide sequence after the conjugation with poly(HPMA). The results of the study confirmed our hypothesis according to which peptide-guided organization of conjugate materials can be achieved when using poly(HPMA). This contribution broadens the area of research on hybrid block copolymers, which traditionally uses PEG as the polymer of choice. Furthermore, poly(HPMA) conjugates whose nanostructure is mediated by the self-assembly of the peptide component can have potential applications as biocompatible, biomimetic materials with tunable mechanical properties.
Acknowledgments
This research was supported in part by NIH grant EB 005288. We thank Dr. Pavla Kopečková, Dr. Vladimir Hlady, and Dr. Amalia Aggeli for valuable discussions, Dr. Bryon Wright for help in taking the AFM images, and Dr. Christopher Rodesch for help in taking the CR birefringence images.
Contributor Information
Larisa Cristina Radu, Department of Bioengineering, University of Utah, Salt Lake City, Utah 84112 USA.
Jiyuan Yang, Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112 USA.
Jindřich Kopeček, Email: jindrich.kopecek@utah.edu, Department of Bioengineering, University of Utah, Salt Lake City, Utah 84112 USA. Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112 USA.
References
- 1.Klok HA, Lecommandoux S. Adv Mater. 2001;13:1217. [Google Scholar]
- 2.Segalman RA. Mater Sci Eng. 2005;48:191. [Google Scholar]
- 3.Krausch G, Magerle R. Adv Mater. 2002;14:1579. [Google Scholar]
- 4.Pechar M, Kopečková P, Joss L, Kopeček J. Macromol Biosci. 2002;2:199. [Google Scholar]
- 5.Hentschel J, Krause E, Börner HG. J Am Chem Soc. 2006;128:7722. doi: 10.1021/ja060759w. [DOI] [PubMed] [Google Scholar]
- 6.Eckhardt D, Groenewolt M, Krause E, Börner HG. Chem Commun. 2005:2814. doi: 10.1039/b503275j. [DOI] [PubMed] [Google Scholar]
- 7.Hamley IW, Ansari IA, Castelletto V, Nuhn H, Rösler A, Klok HA. Biomacromolecules. 2005;6:1310. doi: 10.1021/bm049286g. [DOI] [PubMed] [Google Scholar]
- 8.Vandermeulen GWM, Klok HA. Macromol Biosci. 2004;4:383. doi: 10.1002/mabi.200300079. [DOI] [PubMed] [Google Scholar]
- 9.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 10.Burkoth TS, Benzinger TLS, Jones DNM, Hallenga K, Meredith SC, Lynn DG. J Am Chem Soc. 1998;120:7655. [Google Scholar]
- 11.Rathore O, Sogah DY. J Am Chem Soc. 2001;123:5231. doi: 10.1021/ja004030d. [DOI] [PubMed] [Google Scholar]
- 12.Rösler A, Klok HA, Hamley IW, Castelletto V, Mykhaylyk OO. Biomacromolecules. 2003;4:859. doi: 10.1021/bm034058s. [DOI] [PubMed] [Google Scholar]
- 13.Smeenk JM, Otten MBJ, Thies J, Tirrell DA, Stunnenberg HG, van Hest JCM. Angew Chem Int Ed. 2005;44:1968. doi: 10.1002/anie.200462415. [DOI] [PubMed] [Google Scholar]
- 14.Collier JH, Messersmith PB. Adv Mater. 2004;16:907. [Google Scholar]
- 15.Pechar M, Brus J, Kostka L, Koňák Č, Urbanová M, Šlouf M. Macromol Biosci. 2007;7:56. doi: 10.1002/mabi.200600196. [DOI] [PubMed] [Google Scholar]
- 16.Börner HG, Schlaad H. Soft Matter. 2007;3:394. doi: 10.1039/b615985k. [DOI] [PubMed] [Google Scholar]
- 17.Börner HG. Macromol Chem Phys. 2007;208:124. [Google Scholar]
- 18.König HM, Kilbinger AFM. Angew Chem, Int Ed. 2007;46:8334. doi: 10.1002/anie.200701167. [DOI] [PubMed] [Google Scholar]
- 19.Kamei S, Kopeček J. Pharm Res. 1995;12:663. doi: 10.1023/a:1016247206531. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Xu C, Wang C, Kopeček J. Biomacromolecules. 2006;7:1187. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Xu C, Kopečková P, Kopeček J. Macromol Biosci. 2006;6:201. doi: 10.1002/mabi.200500208. [DOI] [PubMed] [Google Scholar]
- 22.Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TCB, Pitkeathly M, Radford SE. Nature. 1997;386:259. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 23.Aggeli A, Nyrkova IA, Bell M, Harding R, Carrick L, McLeish TCB, Semenov AN, Boden N. Proc Natl Acad Sci USA. 2001;98:11857. doi: 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Macromolecules. 2001;34:2248. [Google Scholar]
- 25.Kopeček J, Bažilová H. Eur Polym J. 1973;9:7. [Google Scholar]
- 26.Scales CW, Vasilieva YA, Convertine AJ, Lowe AB, McCormick CL. Biomacromolecules. 2005;6:1846. doi: 10.1021/bm0503017. [DOI] [PubMed] [Google Scholar]
- 27.You YZ, Oupický D. Biomacromolecules. 2007;8:98. doi: 10.1021/bm060635b. [DOI] [PubMed] [Google Scholar]
- 28.Ellman GL. Arch Biochem Biophys. 1959;82:70. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 29.Scales CW, Convertine AJ, McCormick CL. Biomacromolecules. 2006;7:1389. doi: 10.1021/bm060192b. [DOI] [PubMed] [Google Scholar]
- 30.Goeden-Wood NL, Keasling JD, Muller SJ. Macromolecules. 2003;36:2932. [Google Scholar]
- 31.Aggeli A, Bell M, Carrick LM, Fishwick CWG, Harding R, Mawer PJ, Radford SE, Strong AE, Boden N. J Am Chem Soc. 2003;125:9619. [Google Scholar]
- 32.Aggeli A, Bell M, Boden N, Keen JN, McLeish TCB, Nyrkova I, Radford SE, Semenov A. J Mater Chem. 1997;7:1135. [Google Scholar]
- 33.Zhang S, Holmes T, Lockshin C, Rich A. Proc Natl Acad Sci USA. 1993;90:3334. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiti F, Bucciantini M, Capanni C, Taddei N, Dobson CM, Stefani M. Protein Sci. 2001;10:2541. doi: 10.1110/ps.10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shahi P, Sharma R, Sanger S, Kumar I, Jolly RS. Biochemistry. 2007;46:7365. doi: 10.1021/bi7001136. [DOI] [PubMed] [Google Scholar]
- 36.Ganesh S, Prakash S, Jayakumar R. Biopolymers. 2003;70:346. doi: 10.1002/bip.10493. [DOI] [PubMed] [Google Scholar]
- 37.Koga T, Taguchi K, Kobuke Y, Kinoshita T, Higuchi M. Chem Eur J. 2003;9:1146. doi: 10.1002/chem.200390132. [DOI] [PubMed] [Google Scholar]
- 38.Kowalewski T, Holtzman DM. Proc Natl Acad Sci USA. 1999;96:3688. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stathopulos PB, Scholz GA, Hwang YM, Rumfeldt JAO, Lepock JR, Meiering EM. Protein Sci. 2004;13:3017. doi: 10.1110/ps.04831804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webber GB, Wanless EJ, Armes SP, Baines FL, Biggs S. Langmuir. 2001;17:5551. [Google Scholar]
- 41.Jun S, Hong Y, Imamura H, Ha BY, Bechhoefer J, Chen P. Biophys J. 2004;87:1249. doi: 10.1529/biophysj.103.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]