

ABSTRACT
The influence of the skin microbiota on host susceptibility to infectious agents is largely unexplored. The skin harbors diverse bacterial species that may promote or antagonize the growth of an invading pathogen. We developed a human infection model for Haemophilus ducreyi in which human volunteers are inoculated on the upper arm. After inoculation, papules form and either spontaneously resolve or progress to pustules. To examine the role of the skin microbiota in the outcome of H. ducreyi infection, we analyzed the microbiomes of four dose-matched pairs of “resolvers” and “pustule formers” whose inoculation sites were swabbed at multiple time points. Bacteria present on the skin were identified by amplification and pyrosequencing of 16S rRNA genes. Nonmetric multidimensional scaling (NMDS) using Bray-Curtis dissimilarity between the preinfection microbiomes of infected sites showed that sites from the same volunteer clustered together and that pustule formers segregated from resolvers (P = 0.001, permutational multivariate analysis of variance [PERMANOVA]), suggesting that the preinfection microbiomes were associated with outcome. NMDS using Bray-Curtis dissimilarity of the endpoint samples showed that the pustule sites clustered together and were significantly different than the resolved sites (P = 0.001, PERMANOVA), suggesting that the microbiomes at the endpoint differed between the two groups. In addition to H. ducreyi, pustule-forming sites had a greater abundance of Proteobacteria, Bacteroidetes, Micrococcus, Corynebacterium, Paracoccus, and Staphylococcus species, whereas resolved sites had higher levels of Actinobacteria and Propionibacterium species. These results suggest that at baseline, resolvers and pustule formers have distinct skin bacterial communities which change in response to infection and the resultant immune response.
IMPORTANCE
Human skin is home to a diverse community of microorganisms, collectively known as the skin microbiome. Some resident bacteria are thought to protect the skin from infection by outcompeting pathogens for resources or by priming the immune system’s response to invaders. However, the influence of the skin microbiome on the susceptibility to or protection from infection has not been prospectively evaluated in humans. We characterized the skin microbiome before, during, and after experimental inoculation of the arm with Haemophilus ducreyi in matched volunteers who subsequently resolved the infection or formed abscesses. Our results suggest that the preinfection microbiomes of pustule formers and resolvers have distinct community structures which change in response to the progression of H. ducreyi infection to abscess formation.
INTRODUCTION
The skin microbiome is dominated by members of four phyla, Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes. Within these groups, consistently present and abundant genera include Propionibacterium, Corynebacterium, Staphylococcus, Micrococcus, Streptococcus, and Brevibacterium (1–4). Human skin is characterized by three physiologically distinct microenvironments: sebaceous, moist, and dry. Similar skin niches tend to support comparable bacterial communities. Compared to sebaceous and moist sites, dry skin appears to be inhabited by a more mixed population (1). The volar forearm supports a highly rich bacterial community (high number of bacterial operational taxonomic units [OTUs]) and one of the most highly diverse communities (relatively even distribution of bacterial OTUs) (1, 5). The inherent richness and diversity observed in dry skin make it difficult to define a “core” skin microbiome for a given anatomic site. For example, one study detected a greater abundance of Betaproteobacteria and Flavobacteriales (1), whereas another study found similar numbers of Betaproteobacteria, Firmicutes, and Actinobacteria on the volar forearms of healthy volunteers (2). Furthermore, although resampling of the same site over time showed higher intrapersonal than interpersonal community consensus, minimal longitudinal stability was observed in the volar forearm microbiome (1, 2).
Despite the challenge of defining a “normal” skin microbiome, comparing the microbiomes of diseased and normal skin could further our understanding of the many factors that contribute to, or result from, infection and autoimmune disease. The skin harbors diverse bacterial species that may promote or antagonize the growth of an invading pathogen (6). Recent studies have correlated the presence of certain bacteria with specific disease states. Compared to healthy skin, Firmicutes are relatively more abundant and Actinobacteria and Proteobacteria are relatively less abundant in psoriatic lesions (3). In patients with the inflammatory skin disorder atopic dermatitis, increasing disease severity correlates with decreased microbial diversity overall and an increased prevalence of staphylococci, including both S. epidermidis and S. aureus (7). These studies indicate that several skin disorders are characterized by shifts in the skin microbiome, most notably, loss of protective bacteria and outgrowth of pathogenic bacteria. However, whether the skin microbiome influences or is influenced by infection with a skin pathogen is unknown.
H. ducreyi is a fastidious, Gram-negative facultative anaerobe that causes sexually transmitted genital ulcers (GU) called chancroid. Chancroid is endemic in resource-poor areas in Africa, Asia, and Latin America and is a public health problem because it facilitates the transmission of human immunodeficiency virus type 1 (HIV-1) (8). Due to syndromic management of GU, the prevalence of chancroid is now undefined but appears to have declined in several regions of endemicity (9).
Chronic cutaneous ulcers (CU) in the South Pacific islands and equatorial Africa are usually attributed to yaws, an infection caused by Treponema pallidum subsp. pertenue, which is spread by nonsexual personal contact. However, H. ducreyi has recently emerged as a major etiological agent of CU (10–12). In an area of Papua New Guinea where yaws is endemic, the community prevalence of CU that contained H. ducreyi DNA was 3%; H. ducreyi DNA was detected in nearly 60% of CU lesions, while T. pallidum subsp. pertenue DNA was detected in only 31% of lesions (10). In the Solomon Islands and in Ghana, H. ducreyi sequences are present in 32% and 9% of CU lesions, respectively; in both of these studies, H. ducreyi was the only pathogen detected (11, 12). Although H. ducreyi is highly susceptible to azithromycin (13), mass treatment with azithromycin in Papua New Guinea reduced the overall prevalence of CU by 90% but failed to reduce the proportion of ulcers that contained H. ducreyi DNA (14). The high prevalence of CU caused by H. ducreyi and the failure of mass treatment to eradicate H. ducreyi infection underscores the need to understand the factors that contribute to H. ducreyi pathogenicity.
To understand H. ducreyi pathogenesis, we developed an experimental infection model in which healthy adult volunteers are inoculated on the upper arm over the deltoid area with 1 to 100 CFU of the GU strain 35000HP via puncture wounds. Whole-genome sequencing shows that recently discovered CU strains are derived from the same lineage and are nearly genetically identical to 35000HP (15); thus, the model is relevant to both GU and CU. In the model, H. ducreyi initially causes papule formation at approximately 90% of the inoculated sites. All papules spontaneously resolve in approximately 30% of volunteers (resolvers), whereas 70% of the volunteers form one or more pustules (pustule formers) (16). Men are 2-fold more likely than women to form pustules, indicating that gender influences outcome (16). Gender-matched resolvers and pustule formers who are infected a second time tend to have the same outcome as their initial infection, suggesting that host factors in addition to gender influence outcome (17).
Within 24 h of experimental infection with H. ducreyi, macrophages and polymorphonuclear leukocytes (PMN) traffic on collagen and fibrin scaffolds deposited in the wounds, forming micropustules in the dermis and epidermis (18, 19). In pustule formers, PMN accumulate and form an abscess that eventually erodes the epidermis (18, 19). Below the abscess is a macrophage collar admixed with regulatory T cells (Treg) and a dermal infiltrate of memory and effector memory subsets of CD4 and CD8 T cells, activated natural killer (NK) cells, myeloid dendritic cells (DC), and Langerhans cells (19–25). DC likely phagocytose H. ducreyi, migrate to regional nodes, and sensitize naive T cells to become H. ducreyi-specific memory cells that eventually home to the lesions (23, 26). Throughout experimental infection and in natural ulcers, H. ducreyi associates with PMN and macrophages, which fail to ingest the organism (19, 25, 27). Thus, evasion of phagocytosis is a major mechanism of H. ducreyi survival. In resolvers, the mechanism of resolution is unknown but is presumably due to enhanced phagocytic clearance of the organism at the papular stage of disease and the subsequent termination of the inflammatory response.
Comparison of gene expression profiles of biopsy specimens obtained from resolvers and pustule formers who were reinfected indicated that the two groups share a core immune response but that pustule formers also express transcripts consistent with a hyperinflammatory, dysregulated state (28). When infected with H. ducreyi, myeloid DC derived from resolvers express transcripts consistent with promoting Th1 and Th17 responses, while DC from pustule formers express transcripts that should promote Th1 and Treg responses (28). A combined Th1 and Th17 response may result in a cytokine environment that promotes phagocytosis and disease resolution, whereas a combined Th1 and Treg response may result in a cytokine environment that inhibits phagocytosis and promotes abscess formation. Transcripts associated with M2 macrophage polarization are also upregulated in resolvers (28, 29). M2 cells are more phagocytic for and equally potent at killing H. ducreyi than M1 cells (29). Thus, M1 polarization may lead to hyperinflammation and bacterial persistence in pustule formers, whereas a switch from M1 to M2 may result in resolution of inflammation and bacterial clearance (29). Taken together, these data suggest that differential innate immune responses play a major role in the outcome of H. ducreyi infection (28, 29).
The presence of serum in the wounds and the recruitment of PMN, macrophages, dendritic cells, NK cells, and T cells lead to a local environment marked by activated complement, proinflammatory cytokines, antimicrobial peptides, and an increased concentration of reactive oxygen and nitrogen species. Although H. ducreyi has mechanisms that allow it to survive in an abscess and resist phagocytic uptake, complement-mediated killing, antimicrobial peptides, and reactive oxygen species (30–33), these nonspecific host defenses likely affect the resident microbiota. Inflammatory responses are known to change the composition of the microbiome (34), but whether the cutaneous immune response to an infectious agent influences the skin microbiome is unknown.
As the microbiome influences the outcome of certain infectious disease (35), we hypothesized that in addition to the quality of the innate immune response and gender, the structure of the skin microbiome may also play a role in the ability of H. ducreyi to progress to abscess formation in some hosts and resolve in others. Here we evaluated the skin microbiome before and after inoculation of human volunteers to investigate baseline differences in pustule formers and resolvers and changes that occur throughout experimental H. ducreyi infection. In accordance with the damage response framework of infectious diseases (36), “infection” is defined to include both the contributions of H. ducreyi and the differential inflammatory responses observed in pustule formers and resolvers. In an attempt to incorporate the microbiota into the damage response framework (35), we asked whether the microbiome present prior to infection was associated with host susceptibility to H. ducreyi infection and what, if any, changes in the microbiome occurred following infection with H. ducreyi. This is the first prospective study to assess associations among the skin microbiome, a bacterial pathogen, and differential immune responses in humans.
RESULTS
Infection outcomes in volunteers.
Four sets of dose-matched resolvers and pustule formers, who were inoculated with estimated delivered doses of 32 to 69 CFU of strain 35000HP, were included in the study (Table 1). In the model, infection with these doses typically results in the formation of a papule within 24 h (16). By day 1, papules developed at 22 of 24 inoculated sites (Table 1). Volunteers 396, 391A, 407, and 408 resolved all papules between days 2 and 13. Volunteers 395, 406, and 409 formed pustules at 2 of the 3 inoculated sites, while volunteer 398 formed pustules at all 3 sites (Table 1).
TABLE 1 .
Responses to inoculation with live H. ducreyi 35000HPa
| Volunteer no., gender |
No. of days infected |
Site no. |
Inoculum (CFU) |
Clinical endpoint |
Induration size and presence of pustule(s) on postinoculation dayb: |
|||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 5 | Endpoint | |||||
| 395, F | 7 | 1 | 32 | R | 6 × 6 (−) | 9 × 9 (−) | R | R |
| 2 | P | 6 × 6 (−) | 8 × 9 (−) | 4 × 4 (−) | 4 × 4 (+) | |||
| 3 | P | 5 × 5 (−) | 5 × 8 (−) | 4 × 4 (−) | 4 × 4 (+) | |||
| 396, F | 13 | 1 | 32 | R | 3 × 3 (−) | 4 × 4 (−) | 6 × 7 (−) | R |
| 2 | R | ND | ND | ND | ND | |||
| 3 | R | ND | ND | ND | ND | |||
| 398, M | 6 | 1 | 63 | P | 5 × 5 (−) | 5 × 6 (−) | 9 × 7 (+) | 10 × 15 (+) |
| 2 | P | 4 × 4 (−) | 5 × 5 (−) | 5 × 5 (+) | 6 × 6 (+) | |||
| 3 | P | 5 × 5 (−) | 5 × 5 (−) | 6 × 5 (+) | 5 × 6 (+) | |||
| 391A, M | 7 | 1 | 63 | R | 4 × 4 (−) | R | R | R |
| 2 | R | 2 × 2 (−) | 2 × 2 (−) | R | R | |||
| 3 | R | 3 × 3 (−) | 4 × 4 (−) | 3 × 2 (−) | R | |||
| 406, M | 6 | 1 | 69 | R | 2 × 2 (−) | 2 × 2 (−) | R | R |
| 2 | P | 4 × 4 (−) | 3 × 3 (−) | 4 × 4 (+) | 5 × 5 (+) | |||
| 3 | P | 4 × 4 (−) | 3 × 3 (−) | 15 × 7 (+) | 7 × 7 (+) | |||
| 407, M | 9 | 1 | 69 | R | 4 × 4 (−) | 5 × 5 (−) | Rc | R |
| 2 | R | 4 × 4 (−) | 5 × 5 (−) | R | R | |||
| 3 | R | 4 × 4 (−) | 5 × 5 (−) | R | R | |||
| 408, F | 8 | 1 | 35 | R | 2 × 2 (−) | 3 × 3 (−) | 3 × 3 (−) | R |
| 2 | R | 4 × 4 (−) | R | R | R | |||
| 3 | R | 3 × 3 (−) | R | R | R | |||
| 409, M | 7 | 1 | 35 | P | 3 × 3 (−) | 3 × 3 (−) | 8 × 8 (+) | 12 × 10 (+) |
| 2 | R | 3 × 3 (−) | 2 × 2 (−) | R | R | |||
| 3 | P | 4 × 4 (−) | 5 × 4 (−) | 5 × 5 (+) | 12 × 12 (+) | |||
Abbreviations: F, female; M, male; ND, no disease; R, resolved; P, pustule.
Induration dimensions are reported in millimeters; + and − symbols in parentheses indicate the presence or absence of pustules, respectively.
Subject 407 did not achieve the endpoint on day 5 because disease was present at sites injected with the mutant strain.
Sample processing and enrichment of skin-specific sequences.
We processed skin swabs collected prior to inoculation, at days 1, 2, and 5 postinoculation, at the clinical endpoint, and at the test-of-cure visit (TOC) (Fig. 1). Thus, each volunteer provided 18 clinical samples. Of the 144 swabs, 6 yielded insufficient DNA for 16S rRNA gene PCR (see Table S1 in the supplemental material). High-quality sequences were obtained from the remaining 138 samples.
FIG 1 .
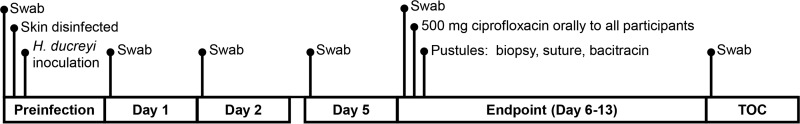
Schematic of times for swab sampling chosen for processing prior to and following experimental inoculation with H. ducreyi.
Because skin swabs yield relatively low bacterial numbers, environmental contamination can affect the analysis. To control for contamination, a swab that was dipped in the phosphate-buffered saline (PBS) used to moisten the skin swabs at each visit was processed in parallel to the skin swabs. Of 48 PBS control samples, sequences were obtained from 46. After bacterial OTUs were assigned, the OTUs that were less than 2-fold greater in the skin samples over the PBS controls were discarded as likely environmental contaminants. Initially 2,462 OTUs were formed at greater than 97% identity from 309,161 16S rRNA sequence reads. Applying the 2-fold cutoff removed 16% of the OTUs, resulting in 2,068 OTUs and 190,569 reads from the samples. The number of corrected reads obtained from each site is shown in Table S1 in the supplemental material; all subsequent analyses were performed on this corrected data set. The average corrected number of reads per sample was 1,347.
We defined the skin microbiome as the collective bacterial community identified by the V1-V3 region of the 16S rRNA gene. OTUs were grouped according to genus and phylum to determine the relative abundances at these taxonomic levels. Species designations were based on the best alignment of an OTU consensus sequence to a species in the RDP or NCBI database; two species are listed in the cases of identical hits.
Characteristics of the upper deltoid microbiome.
We examined the relative abundance and prevalence of major taxa in all swabs collected at preinfection, prior to cleansing and inoculation with H. ducreyi. As observed in other studies characterizing dry skin, the microbiome of the upper deltoid region was dominated by Actinobacteria (resolvers versus pustule formers, 50% versus 41%), Firmicutes (32% versus 18%), Proteobacteria (7% versus 34%) and, to a lesser extent, Bacteroidetes (6% versus 2%) (Fig. 2). Of the remaining taxa, approximately 2% belonged to 11 minor phyla and the remaining 2% were unclassified at the phylum level (Fig. 2).
FIG 2 .
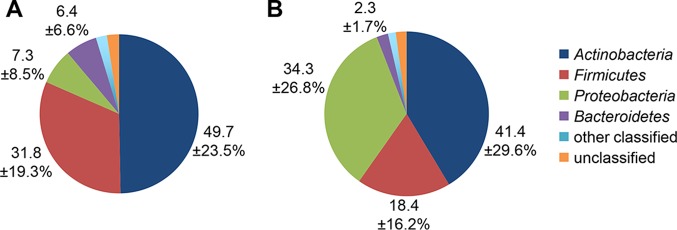
Distribution of the four dominant phyla in resolvers (A) and pustule formers (B) preinfection. Pie charts represent the percent proportions of each phylum across all days from the corrected sequence data, with averages and standard deviations shown for Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes.
The prevalence of the four major phyla differed between each volunteer and changed over the course of the study (see Fig. S1 in the supplemental material). In general, the microbiomes were similar at the 3 sampling sites from each volunteer, at both the phylum (see Fig. S1) and genus (Fig. 3) levels.
FIG 3 .

Distribution of the 20 most abundant genera, other classified genera, and unclassified genera in resolvers (A) and pustule formers (B). All samples for which sequence data were obtained are shown; no sequences were obtained for the missing samples.
The most abundant genera consisted primarily of taxa within one of the four dominant phyla. The distribution of the top 20 genera at each site on each day is shown in Fig. 3. Across all days, the most prevalent members of Actinobacteria were Propionibacterium (resolvers versus pustule formers, 33% versus 15%), Corynebacterium (11% versus 10%), and Micrococcus (0.8% versus 8%) (Table 2). The most abundant taxa at the OTU level were classified as Propionibacterium acnes, Corynebacterium tuberculostearicum, Corynebacterium accolens/Corynebacterium aurimucosum, Corynebacterium afermentans, Corynebacterium mucifaciens, and Micrococcus luteus. Within the Firmicutes, Staphylococcus (16% versus 14%), Streptococcus (7% versus 1%), and Finegoldia (1% versus 0.4%) were the most prevalent genera. The most abundant OTUs in these genera were Staphylococcus caprae/Staphylococcus capitis, Staphylococcus petrasii, and Streptococcus mitis. Members of the Proteobacteria, including Paracoccus (0.1% versus 18%) and Haematobacter (0.05% versus 7%), were detected primarily in pustule formers; these genera were dominated by Paracoccus aminovorans, Paracoccus versutus, and Haematobacter massiliensis. The most abundant genera of Bacteroidetes were Porphyromonas (1.7% versus 0.2%) and Chryseobacterium (0.2% versus 1.5%).
TABLE 2 .
Relative abundance and prevalence levels of major taxa at preinfectiona
| Major taxonomy group | % relative abundance (mean ± SD) |
No. of subjects colonizedb |
||
|---|---|---|---|---|
| R | P | R | P | |
| Actinobacteria | ||||
| Propionibacterium acnes | 33.5 ± 29.2 | 15.2 ± 25.3 | ●●●● | ●●●● |
| Corynebacterium spp. | 11.6 ± 14.5 | 10.6 ± 13.4 | ●●●● | ●●●● |
| C. tuberculostearicum | 2.3 ± 4.9 | 3.1 ± 3.6 | ●●●● | ●●●● |
| C. aurimucosum/accolens | 1.6 ± 2.6 | 2.3 ± 4.7 | ●●●● | ●●●● |
| C. afermentans | 4.0 ± 5.5 | 1.4 ± 2.3 | ●● | ●●● |
| C. mucifaciens | 1.4 ± 2.6 | 1.3 ± 2.6 | ●●● | ●● |
| Micrococcus luteus | 0.8 ± 1.5 | 8.4 ± 12.2 | ●●● | ●●●● |
| Actinomyces | 1.9 ± 2.6 | 0.08 ± 0.1 | ●●● | ●● |
| Microbacterium | 0.01 ± 0.3 | 1.3 ± 2.2 | ● | ●● |
| Kocuria | 0.39 ± 0.9 | 3.6 ± 5.0 | ●● | ●●● |
| K. palustris | 0.2 ± 0.7 | 2.9 ± 5.2 | ● | ●● |
| Firmicutes | ||||
| Staphylococcus spp. | 16.5 ± 11.3 | 14.1 ± 16.4 | ●●●● | ●●●● |
| S. capitis/S. caprae | 9.9 ± 6.1 | 11.4 ± 17 | ●●●● | ●●●● |
| S. petrasii | 5.4 ± 7 | 1.7 ± 2.5 | ●●● | ●●●● |
| Streptococcus spp. | 7.0 ± 16.7 | 1.2 ± 2.0 | ●●●● | ●●●● |
| S. mitis | 5.7 ± 13.6 | 0.3 ± 4 | ●●●● | ●●●● |
| Nosocomiicoccus | 1.3 ± 246 | 0 ± 0 | ● | |
| Finegoldia spp. | 1.1 ± 1.8 | 0.4 ± 0.8 | ●● | ●●● |
| F. magna | 1.0 ± 1.7 | 0.4 ± 0.8 | ●●●● | ●●●● |
| Aerococcus urinaeequi/A. viridans | 1.3 ± 2.5 | 0.13 ± 0.2 | ● | ●● |
| Proteobacteria | ||||
| Paracoccus spp. | 0.15 ± 0.3 | 17.8 ± 20.6 | ● | ●●● |
| P. aminovorans | 0.08 ± 0.17 | 12.2 ± 21.9 | ● | ●●● |
| P. versutus | 0.06 ± 0.2 | 4.1 ± 7.1 | ● | ● |
| Haematobacter massiliensis | 0.05 ± 0.2 | 7.2 ± 12.4 | ● | ●●● |
| Sphingomonas spp. | 0.5 ± 1.2 | 2.9 ± 4.6 | ●●●● | ●●●● |
| S. hankookensis | 0.3 ± 1.0 | 2.8 ± 4.8 | ● | ● |
| Mesorhizobium amorphae | 1.0 ± 2.8 | 0.5 ± 1.7 | ●●● | ●● |
| Bacteroidetes | ||||
| Porphyromonas | 1.7 ± 5.2 | 0.18 ± 0.4 | ●● | ●● |
| Chryseobacterium | 0.2 ± 0.5 | 1.5 ± 2.7 | ●● | ●● |
| Prevotella | 1.2 ± 1.7 | 0.4 ± 0.6 | ●●● | ●●●● |
| Flavobacterium | 0.8 ± 0.8 | 0.2 ± 0.4 | ●●● | ●●●● |
R, resolvers; P, pustule formers.
Each dot represents a volunteer in which the respective genus or species was detected.
The microbiome at preinfection differs between resolvers and pustule formers.
To understand if the microbiome present prior to inoculation influences disease susceptibility, we analyzed the samples collected at preinfection. We hypothesized that the microbiomes in resolvers would have different diversity levels than those of pustule formers; that is, the bacterial communities may harbor different amounts of unique taxa (richness) that are present in relatively different proportions (evenness). To assess this hypothesis, alpha diversity measurements including richness, evenness, and diversity were determined using the Chao1 and ACE, Pielou, and Shannon and Simpson algorithms, respectively. No significant differences were detected at preinfection for any of these parameters between those volunteers who eventually resolved infection versus those who formed pustules (data not shown).
To assess differences in bacterial community compositions, we determined the Bray-Curtis dissimilarity, which measures how similar two communities are based on taxa present and their relative abundances. Nonmetric multidimensional scaling (NMDS) using Bray-Curtis dissimilarities showed that the microbiomes of the sites clustered by host as well as by the clinical outcomes (Fig. 4). The permutational multivariate analysis of variance (PERMANOVA) test indicated that the difference between resolvers and pustule formers was statistically significant (P = 0.001). We also performed Unifrac analysis, which measures differences in communities based on phylogenetic relationships among bacterial taxa. Weighted and unweighted Unifrac analysis results corroborated the Bray-Curtis analysis; significant separation was observed between pustule formers and resolvers (data not shown). Thus, the microbiomes of resolvers and pustule formers are different from each other.
FIG 4 .

Nonmetric multidimensional scaling using Bray-Curtis dissimilarity of OTUs detected in preinfection samples from resolvers (R; n = 10) and pustule formers (P; n = 10) (P = 0.001, PERMANOVA). Samples that contained fewer than 450 sequence reads were excluded from the analysis. At preinfection, resolver sites and sites within volunteers clustered.
Pustule formers share common microbiomes at the endpoint.
We next asked if pustule formation or infection resolution correlated with changes in the microbiomes throughout the infection. We hypothesized that outgrowth of H. ducreyi in pustule formers would decrease the diversity, richness, and evenness of the microbiome. Samples collected at days 1, 2, and 5 and at the clinical endpoint were analyzed. Chao1 and ACE richness, Shannon and Simpson diversity indices, and Pielou evenness did not differ significantly between resolved and pustule sites (data not shown). Thus, we were not able to detect differences in the alpha diversity associated with H. ducreyi infections based on the current sample size.
The NMDS using Bray-Curtis dissimilarities of endpoint samples showed that pustule sites clustered together but away from the resolved sites (Fig. 5), and the separation of pustule formers and resolvers was statistically significant (P = 0.001, PERMANOVA). Sites from the same individual tended to cluster together; even the resolved sites from the pustule formers clustered with their adjacent pustule-forming sites, suggesting that the host effect remained a strong factor influencing the microbiome (Fig. 5). In contrast to the preinfection data, at the endpoint the resolver sites did not cluster together, while sites from the pustule formers appeared to cluster. Weighted and unweighted Unifrac analyses also showed statistically significant separation of pustule formers and resolvers (data not shown).
FIG 5 .

Nonmetric multidimensional scaling using Bray-Curtis dissimilarity of OTUs detected in endpoint samples from resolved sites (R; n = 11) and pustule sites (P; n = 9) (P = 0.001, PERMANOVA). At the endpoint, pustule sites and sites within volunteers clustered.
Dynamics of the microbiome during experimental infection.
In addition to differences at preinfection and the endpoint between resolvers and pustule formers, we were interested in how the microbiomes changed over the course of the experimental infection. To investigate kinetic changes in alpha diversity, we measured the changes in Chao1 and ACE richness, Shannon and Simpson diversity indices, and Pielou evenness on days 1, 2, 5, the endpoint, and TOC relative to preinfection (baseline). We expected to observe a difference in the community dynamic, for example, a relative loss in diversity as pustules developed. However, no significant differences in the kinetic alpha diversity measurements were observed throughout experimental infection (data not shown).
We also compared the Bray-Curtis dissimilarities on each day relative to those at preinfection. We hypothesized that as H. ducreyi grew in the abscess, a relative change in community composition would occur. Although the Bray-Curtis dissimilarities at preinfection and at endpoint were significantly different between pustule formers and resolvers (Fig. 4 and 5), the relative change in beta diversity on each day compared to preinfection was not significantly different between pustule formers and resolvers (data not shown).
Detection of H. ducreyi.
H. ducreyi 16S rRNA gene sequences were detected in 14 samples. Importantly, no H. ducreyi was detected in swabs collected at preinfection or at TOC, and no H. ducreyi was detected in the PBS controls. Among the resolvers, H. ducreyi was detected in 3 samples; one read of H. ducreyi was observed in each of the day 1 samples from volunteers 396 and 408 and in the endpoint sample from volunteer 408. H. ducreyi was identified in 11 samples obtained from pustule formers. Samples from all 3 sites of volunteer 398 contained H. ducreyi, and H. ducreyi 16S rRNA gene sequences comprised 15 and 33% of the total reads in two of the endpoint swabs for this volunteer. H. ducreyi positive surface cultures were obtained from the 3 samples of volunteer 398, who had the highest H. ducreyi reads. Additionally, H. ducreyi reads ranged from 1 to 12% of the total reads in 3 pustule sites among volunteers 406 (two positive sites) and 409 (one positive site).
Prevalence rates of specific taxa correlate with infection outcome.
To investigate whether specific bacteria were associated with infection resolution or progression, we compared the relative abundance of taxa in resolved and pustule-forming sites. Although the Bray-Curtis analysis (Fig. 4 and 5) and phylum- and genus-level distributions (Fig. 3; see also Fig. S1 in the supplemental material) showed that the microbiomes of these 3 resolved sites resembled their respective adjacent pustule-forming sites, we excluded these 3 sites so that we could compare resolved sites from resolvers to pustule-forming sites from pustule formers. We only analyzed taxa that were present in at least 25% of the sites and comprised 1% (at the phylum or genus level) or 0.1% (at the OTU level) of the microbiome. We analyzed these taxa using two statistical models, the negative binomial (NB) model and the zero-inflated negative binomial (ZINB) mixed model. The NB model accounted for the small sample size, and the ZINB model also factored in the large proportion of zero values observed for certain taxa in the microbiome samples (37). P values were corrected for multiple comparisons. Taxa that were significantly different (P < 0.05) between resolvers and pustule formers by either or both the NB and ZINB models were considered signature bacteria. We used the Akaike Information Criterion (AIC) value to measure the quality of the fit of the model for the data set. Analyses of the data via the two models generally produced the same signature taxa. We report the relative abundance of each signature phylum, genus, and species in Table 3. More detailed information regarding signature taxa is compiled in Tables S2, S3, and S4 in the supplemental material.
TABLE 3 .
Relative abundance of signature taxa in resolvers and pustule formersa
| Outcome and signature taxonomical group | Relative abundance (%) at: |
|||||
|---|---|---|---|---|---|---|
| Pre | Day 1 | Day 2 | Day 5 | End | TOC | |
| Organisms in resolvers | ||||||
| Propionibacterium spp. | 39.99b | 42.3 | 39.2b | |||
| P. acnes | 38.97b* | 42.02 | 38.3 | |||
| Actinobacteria | 58.3b | |||||
| Organisms in pustule formers | ||||||
| Proteobacteria | 37.4 | 14.4 | 18.2 | 16.4 | ||
| Kocuria | 1.9c | |||||
| Micrococcus spp. | 6.4b | 2.1c | 4.7 | 4.3c | 7.4b | |
| M. luteus | 5.9 | 4.3 | ||||
| Flavobacterium | 1.1c | 0.5 | ||||
| Bacteroidetes | 6.5 | 2.6 | ||||
| Corynebacterium spp. | 17.6 | 12.7 | 11.2 | |||
| C. tuberculostearicum | 4.00 | 5.9c | 4.95 | 5.9 | ||
| C. aurimucosum/C. accolens | 8.7 | 6.03 | 2.6 | |||
| Haemophilus | 10.2b | |||||
| Paracoccus | 3.3* | 12.4 | ||||
| Staphylococcus spp. | 41.4 | |||||
| S. capitis/S. caprae | 30.8b | 24.6 | ||||
| Firmicutes | 46.0b | 40.4b | ||||
| Unclassified | 1.0 | |||||
A taxonomical group was considered “signature” if it was detected in ≥25% of sites, comprised ≥1% (at the phylum or genus level) or 0.1% (at the OTU level) of the total microbiome of resolvers or pustule formers, and its relative abundance was significantly different (P < 0.05) between resolvers and pustule formers. Pre, preinfection; End, endpoint. *, trended towards significance.
The NB model fit the data better.
The ZINB model fit the data better.
At preinfection, the genus Propionibacterium was significantly overrepresented in the resolvers; the major Propionibacterium species, P. acnes, trended toward being significantly overrepresented in resolvers (Table 3). Propionibacterium and P. acnes remained associated with resolution on day 2 and at the endpoint. Likewise, the phylum Actinobacteria was higher in resolvers on day 2.
In contrast, the phylum Proteobacteria was significantly more abundant in pustule formers at preinfection, days 1 and 2, and at TOC. The genus Kocuria (family Micrococcacae) was also associated with pustule formation on day 1. The genus Micrococcus was significantly overrepresented in pustule formers on days 1, 2, and 5, at the endpoint, and at TOC. Likewise, the dominant micrococcal species, M. luteus, was associated with pustule formation on day 1 and at the endpoint.
Other bacteria associated with pustule formers included the phylum Bacteroidetes, which was significantly overrepresented on day 2 and at TOC. Similarly, Flavobacterium was higher in pustule formers on day 1, the endpoint, and TOC. The genus Corynebacterium and 2 dominant species, C. tuberculostearicum and C. aurimucosum/C. accolens, were associated with pustule formers on day 5 and at the endpoint and TOC.
The abundance of the genus Haemophilus was significantly higher in pustule formers at the endpoint. H. ducreyi was the only Haemophilus species detected. Another member of the Proteobacteria, Paracoccus, trended toward being significantly associated with pustule formation at the endpoint and was significantly different at TOC. The genus Staphylococcus was significantly overrepresented in pustule formers at the endpoint, and the OTU classified as S. capitis/S. caprae was higher at the endpoint and TOC.
Local and systemic treatments do not impact the microbiome.
We wondered whether any changes in the microbiome occurred due to systemic treatment with ciprofloxacin (resolved sites) or treatment with ciprofloxacin along with punch biopsy, suture, and topical bacitracin application (pustule sites). Because the procedures differed between resolvers and pustule formers, directly comparing the two groups was not appropriate. Rather, we compared the Bray-Curtis dissimilarity values between the endpoint and TOC samples to Bray-Curtis values between any other 2 days within each group. The differences between the endpoint and TOC were not significantly different from the dissimilarities between any other day, for either resolvers or pustule formers (data not shown). These results suggest that the treatment at the endpoint did not significantly impact changes in the microbiome. At TOC the microbiome of the resolved sites in the pustule formers, which were not biopsied, resembled their adjacent pustule-forming sites, suggesting that the biopsy and topical bacitracin did not significantly alter the microbiome (Fig. 3).
DISCUSSION
In this study, we examined the bacterial communities on the upper arms of 8 volunteers before and after inoculation with H. ducreyi. The microbiomes were similar to those of reported forearm microbiomes (1, 2, 5). At preinfection, we found the skin of the deltoid region to be largely dominated by Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes. Similarly, a recent study on two human volunteers who were asked not to bathe 3 days prior to sampling identified these 4 phyla and Cyanobacteria on the upper arm (38). In contrast, the same study found low levels of Propionibacterium and Staphylococcus on the upper arm, whereas these genera comprised a large proportion of our volunteers’ microbiomes. Both studies identified moderate levels of Corynebacterium on the upper arm, and we found moderate levels of Streptococcus, Paracoccus, and Micrococcus species. We did not ask our participants to refrain from bathing prior to infection; perhaps hygiene and person-to-person variations contributed to the differences in the studies.
In our longitudinal study, we tracked changes in the microbiome over a 1- to 2-week period, compared the bacterial communities in 3 adjacent sites on the upper deltoid region, and identified differences in the microbiome between resolvers and pustule formers. Although the skin was cleansed with Betadine and ethanol prior to experimental infection, the microbiome was reestablished within 24 h. In general, the bacterial community on each volunteer’s arm changed over the course of the 6 samplings. Furthermore, the changes were consistent across the 3 sites spaced 3 cm apart, suggesting that the intrinsic (temperature, pH, immune response) and extrinsic (environment, hygiene, etc.) factors influencing the microbiome were conserved across the deltoid region of each volunteer.
To understand the interaction between the microbiome and H. ducreyi infection, we compared the bacterial communities of resolvers and pustule formers. Community ecological theory predicts that resistance to invading species correlates with high species diversity (39). The effect of community diversity has also been addressed in psoriasis; there is no consensus regarding whether higher diversity (3) or lower diversity (40) is associated with psoriatic lesions compared to unaffected or healthy skin. We hypothesized that a more diverse bacterial community present at both preinfection and the endpoint would correlate with resolution of H. ducreyi infection. However, alpha diversity measurements between resolvers and pustule formers were not significantly different on any day. The data suggest that community diversity, richness, and evenness do not contribute to protection from or susceptibility to H. ducreyi.
Although we did not find differences in diversity, community composition differed between resolvers and pustule formers at both preinfection and the endpoint. At preinfection, the microbiomes on the arms of resolvers were more similar to each other and were significantly different than the microbiomes present in the pustule formers. Host factors also influenced the microbiome, as sites from the same volunteer tended to cluster together; this effect was observed in samples analyzed at both preinfection and the endpoint.
Infection outcome also played a role in community composition; the microbiomes differed between resolvers and pustule formers at the endpoint. In contrast to what was found at preinfection, at the endpoint the microbiomes in pustule sites clustered together. Furthermore, 10 signature bacteria were significantly overrepresented in pustule-forming sites at the endpoint. These data suggest that pustule formation, which coincides with an ineffective hyperinflammatory immune response, is associated with a shift toward a more similar community composition. Similarly, Horton and colleagues found that in patients with abscesses caused by S. aureus, the microbiomes of periabscess and contralateral skin were more similar to each other than to the microbiomes of control samples obtained from uninfected volunteers (41). Additionally, natural and experimental infection with the fungal pathogen Batrachochytrium dendrobatidis drives common changes in the skin bacterial communities of frogs (42). We also found that the resolved sites of pustule formers maintained bacterial communities similar to adjacent pustule sites, suggesting that host factors have a strong influence on the microbiome. Consistent with this host effect, the microbiome of the skin adjacent to S. aureus abscesses resembles the microbiome of the uninfected contralateral arm within the same person (41). Taken together, these results suggest that the community structure of the skin microbiome is associated with susceptibility to infection and that the skin microbiomes of infected hosts converge toward a similar composition, perhaps driven by the persistence of the host immune response that failed to clear the pathogen. These observations may have important implications regarding the host-microbiome interactions in our understanding the susceptibility and treatment of skin infections.
Relative to preinfection, we anticipated that shifts in richness, evenness, and community composition in pustule-forming sites would coincide with the failure of the immune system (day 2 or day 5, when pustules appear) and increased abundance of H. ducreyi (endpoint). However, the relative changes in alpha or beta diversity throughout the experimental infection did not differ significantly between resolvers and pustule formers. The lack of significant differences in temporal changes in alpha or beta diversity was likely due to the small sample size and the high variance between the four volunteers, which could mask any temporal changes. Perhaps sampling of larger cohorts would reveal day-to-day shifts in alpha and beta diversity indices that would correspond to the observed disease state.
H. ducreyi was detected in endpoint samples of pustule formers. We hypothesized that an increased proportion of H. ducreyi would drive down the diversity at the pustule sites; however, no difference in Shannon or Simpson diversity indices between pustule sites and resolved sites was observed at either the endpoint or on each day of experimental infection relative to the endpoint. Thus, although H. ducreyi can comprise a large proportion of the microbiome (up to 30%), the alpha diversity measurements suggest that H. ducreyi does not completely alter the microbiome. This observation is in contrast to sites of atopic dermatitis flares, where staphylococci dominate the microbiome and diversity concomitantly decreases (7).
Despite the high variance within the clinical groups, specific taxa were associated with disease outcome. Proteobacteria, Bacteroidetes, Micrococcus, Corynebacterium, Paracoccus, and Staphylococcus were significantly overrepresented in pustule-forming sites on at least 1 day throughout the infection. Members of these phyla and genera may present an advantage to H. ducreyi, represent a disrupted infection-susceptible community, or thrive in the humid, inflamed pustular environments. On the other hand, Actinobacteria and Propionibacterium were significantly associated with resolvers. These bacteria may protect against infection by outcompeting H. ducreyi, secreting factors that inhibit H. ducreyi growth or by stimulating the immune system in a protective manner. Alternatively, the presence of these taxa may reflect a bias of the innate immunity of the infected host.
Our samples were obtained by swabbing the skin at the inoculation sites. Grice and colleagues showed that skin samples obtained by swab, scrape, or punch biopsy were highly similar and sufficiently represented the bacterial community (43). Thus, our sampling method likely captured an accurate representation of the total microbiome, further supported by the detection of H. ducreyi in higher levels in the pustule sites than in resolved sites.
Changes in the microbiome during infection or a disease state may be due to direct interactions between the invading pathogen and the commensal bacteria, by the host immune response, or by a combination of the two. One study showed that chemically or genetically induced host-mediated inflammation perturbed the composition of the mouse intestinal microbiome, whereas both enteropathogenic infection and the host inflammatory response were necessary to significantly reduce the number of intestinal microbes (34). Grice and colleagues established a correlation between an increase in Staphylococcus spp. and a persistent inflammatory cutaneous immune response in chronic wounds of diabetic mice (44). Lastly, patients with primary immunodeficiencies characterized by STAT1/STAT3 mutations and Th17 defects showed increased levels of Gram-negative bacteria, such as Acinetobacter spp., and decreased levels of Corynebacterium spp. in the skin microbiome (45). Those authors demonstrated that this shift in the microbiota had the capacity to impair host defense responses against staphylococci and Candida spp., which are commonly responsible for disease flares in these patients (45). These studies supported the notion that the changes we observed in the skin microbiome are likely due to the cutaneous immune response mounted against H. ducreyi and that changes in the microbiota can in turn affect the immune response.
Susceptibility to H. ducreyi infection is influenced by gender; men are more likely than women to form pustules or ulcers in both experimental and natural infections (16, 46, 47). Gender influences on the skin microbiome (6) and differences in the skin microbiomes between men and women may offer clues to the gender disparity observed in H. ducreyi infection. Three of four pairs of our volunteers were gender matched, but our study was not designed to address gender effects on the skin microbiome or how this factor relates to H. ducreyi susceptibility.
Except for participant 391A, all of the volunteers included in this study had no previous exposure to H. ducreyi. In a previous mutant-versus-parent trial (48) and in this study, all infected sites in volunteer 391A resolved. This is consistent with the observation that after a second challenge, resolvers tend to resolve infection while pustule formers tend to form pustules (17, 28, 49). Thus, there is evidence for differential host susceptibility to H. ducreyi infection but no evidence for the development of protective immunity following challenge. All of the volunteers had not taken antibiotics 1 month prior to infection; volunteer 391A was treated with a single dose of ciprofloxacin 2 months prior to this challenge. We showed here that a single dose of ciprofloxacin has little effect on the skin microbiome. Thus, it is unlikely that prior exposure to the organism, protective immunity, or receipt of prior antibiotics affected the study results for 391A or the other participants.
In conclusion, we observed that the preinfection skin microbiome community structure is associated with susceptibility to H. ducreyi infection and that the community structure appears to change in response to pustule formation. Although H. ducreyi comprises a large proportion of the bacterial community in pustules, it does not cause total loss of community diversity. Host effects and the cutaneous immune response appear to have strong influences on microbiome composition, and several taxa were associated with pustule formation versus resolution. Further studies analyzing the transcriptomes of signature taxa in pustule formers and resolvers may shed light on functional differences between the microbiomes of susceptible and resistant hosts and on how the microbiome interacts with H. ducreyi. Additionally, analyzing the microbiomes from persons with CU and GU naturally infected with H. ducreyi may lend insight into the effect of H. ducreyi infection on the microbiome.
MATERIALS AND METHODS
Human inoculation experiments.
Fifteen consecutive healthy adult volunteers who were infected in mutant-versus-parent challenge trials (references 48 and 50 and unpublished data) and one volunteer (391A) who was infected with the parent alone donated daily swabs for microbiome analysis. The volunteers gave informed consent for participation and for HIV serology in compliance with the human experimentation guidelines of the U.S. Department of Health and Human Services and the Institutional Review Board of Indiana University.
In the mutant-versus-parent trials, volunteers were inoculated over the upper deltoid region on one arm at 3 sites with the parent strain, 35000HP, and at 3 sites on the other arm with an isogenic mutant derived from 35000HP. The sites on each arm were vertically spaced 3 cm apart. A resolver was defined as a participant who resolved at all 3 sites infected with 35000HP. A pustule former was defined as a person who formed pustules at 2 or 3 sites inoculated with 35000HP, regardless of the outcome of the sites infected with the mutant strain. We included samples from pairs of resolvers and pustule formers who were dose matched and inoculated with at least 30 CFU of 35000HP. Of the 16 volunteers, 2 were excluded because they were inoculated with doses of <30 CFU; 3 were excluded because they formed only 1 pustule at parent-inoculated sites; 2 resolvers and 1 pustule former were excluded because they lacked a dose-matched counterpart.
Samples from 4 pairs of dose-matched resolvers and pustule formers were available for microbiome analysis (Table 1). The resolver group consisted of two men and two women (mean age ± standard deviation [SD] of 35.5 ± 13.0 years), two of whom were African-American (AA) and two of whom were European American (EA). The pustule-forming group consisted of three men and one woman (mean age ± SD, 38.5 ± 15.9 years), three of whom were EA and one of whom was AA.
Specimen collection and processing.
On the day of H. ducreyi inoculation, swabs were obtained from the 6 sites that were to be inoculated with either the mutant or the parent prior to cleansing the skin with Betadine and ethanol as required by the study protocol. The preinfection swabs were processed and stored as described below except that they were not cultured. Volunteers were examined daily on weekdays and followed by phone on weekends for up to 14 days or until they achieved the clinical endpoint, defined as resolution of a papule or development of a painful pustule (16). At the endpoint, all volunteers were treated with a single 500-mg oral dose of ciprofloxacin. All of the pustule sites were biopsied using a 6-mm punch biopsy forceps. Before biopsy, the skin was disinfected with ethanol; after biopsy, the skin was sutured and treated with bacitracin. All volunteers returned 1 day after biopsy for a TOC visit.
For each visit, the volunteer was assigned a tube of sterile PBS. A sterile Dacron swab was moistened in the PBS and rolled 10 times over one of the six inoculation sites. The swab was then used to inoculate a chocolate agar plate containing 3 µg/ml vancomycin and was placed immediately in a tube containing a DNA-stabilizing reagent (Gene Lock molecular specimen transport and preservation system; Sierra Molecular). This procedure was repeated for each inoculated site; all six swabs were moistened in the same tube of PBS. The tubes containing the Dacron swabs and the tube containing the PBS were stored within 1 h at −20°C. After the code was broken, swabs obtained from the parent-inoculated sites and the PBS controls were processed for DNA isolation and microbiome analysis; swabs from mutant-inoculated sites were not analyzed.
DNA isolation, multiplex 16S rRNA PCR, and pyrosequencing.
Skin and control swab samples were thawed, suspended in 1 ml PBS, and vortexed for 1 min. The resultant suspensions were pelleted by centrifugation for 15 min at 4,000 × g at 4°C. Genomic DNA (gDNA) was isolated using a DNeasy blood and tissue extraction kit (Qiagen) according to the manufacturer’s instructions, eluted in nuclease-free water, and stored at 4°C until library construction. Reagent-only controls were processed in parallel to monitor for reagent contamination. The 16S rRNA gene V1-V3 region was amplified from 2 µl of gDNA using barcoded primers 27F and 534R as previously described (51). All 16S rRNA amplicons were quantified using the Quant-It high-sensitivity double-stranded DNA assay (Life Technologies), pooled in equimolar concentrations, and sequenced on the Roche/454 GS-FLX Titanium platform at the Indiana University Center for Genomics and Bioinformatics, Bloomington, IN.
Sequence processing.
The Mothur software (version 1.32.1) (52) was used to process the 454 sequence reads as described previously (53). Briefly, sequence reads were deconvoluted into individual samples based on perfect matches to the barcode sequences. Primers and barcodes were trimmed from each read, and trimmed sequences shorter than 200 bp were discarded. Low-quality and chimeric sequences were removed with default Mothur parameters. Using the built-in average-linkage clustering algorithm of Mothur, the cleaned high-quality sequences were clustered into species-level OTUs based on the commonly used 97% similarity cutoff. OTUs containing only one 16S rRNA sequence read were considered a potential sequencing error; 1,387 singletons were omitted from the data set. The OTU sequences were analyzed using BLAST (54) against the RDP 16S sequence database, release 11.1 (55), by first removing any RDP sequences derived from uncultured bacteria. Top BLAST hits were assigned tentative species-level annotations if the BLAST alignment achieved >95% coverage and >97% sequence identity. Taxonomic classification from phylum to genus levels of the sequence reads was performed by using the RDP Classifier (version 2.5) (56) with a default 0.8 confidence threshold.
To remove environmental contamination, we performed a data enrichment step. For each OTU, the number of 16S sequence reads obtained from the volunteers’ arm swabs were compared with the number of reads from the PBS-only control swabs. The number of reads was normalized to the number of samples obtained from each group (138 arm swabs, 46 PBS control swabs). OTUs comprised of at least 2-fold-more normalized sequence reads from the arm swabs than those from the PBS control swabs were retained; the rest were considered background and were omitted from the data set. We chose the 2-fold cutoff to remove environmental contaminants from the taxa list and to retain taxa that represent the skin microbiota. The remaining OTUs were analyzed and compiled into their respective genera and phyla for further analysis.
Sequence analysis and statistics.
To minimize unequal sampling effects, 500 16S sequences were randomly subsampled without replacement from each sample to achieve equal sequencing depth. The subsampling was repeated 10 times, and the average taxa counts were used for the subsequent data analysis. The data were then filtered to exclude samples with fewer than 450 reads. The cutoff of 450 reads retained 93% of the samples, allowing us to maximize power from our small cohort while excluding samples that had inadequate sequence coverage. Nine samples had fewer than 450 reads and were excluded from statistical analysis (see Table S1 in the supplemental material). The R statistical package was used for the statistical analyses. The R packages phyloseq (57) and vegan (58) were used to determine Bray-Curtis dissimilarities, Shannon and Simpson diversity indices, Chao1 and ACE richness estimates, and Pielou evenness. NMDS analysis of microbial communities was performed using Bray-Curtis dissimilarity values. The PERMANOVA was applied to test whether within-group distances were significantly different from the between-group distances. Weighted and unweighted Unifrac analyses were done to confirm the results of the Bray-Curtis analyses (59, 60). The negative binomial mixed model and zero-inflated negative binomial mixed model (glmmADBM of the R package) were applied to analyze whether any taxa were significantly different between pustule formers and resolvers, as recommended by Fang and colleagues (37). We focused on the phylum, genus, and OTU levels, based on precedents in the published literature (2, 3, 61). Briefly, a taxa count was used as the response variable; the predictor was the clinical outcome (i.e., a dichotomous categorical variable for either pustule forming or resolved). The model also considered age, gender, and ethnic group as confounding factors. The repeated measurements from the same subject were modeled as random effects. Using the R package glmPQL, a generalized linear mixed model was employed to test whether any alpha diversity indices (response variable) were different between pustule formers and resolvers (predictor) by also considering age, gender, and ethnic group as confounding factors. Multiple test corrections were performed with the Benjamini and Hochberg procedure (62). Corrected P values of less than 0.05 were considered significant.
Nucleotide sequence accession number.
All of the sequences and associated metadata were deposited with the NCBI Sequence Read Archive (63) under the accession number SRP056364.
SUPPLEMENTAL MATERIAL
Distribution of the four dominant phyla in resolvers (A) and pustule formers (B). Pie charts represent the percent proportion of each phylum. R, resolved site; P, pustule site. All samples for which sequence data were obtained are shown here; no sequences were obtained from the missing samples. Download
OTUs observed in samples and breakdown of sites used for statistical analysis.
Signature phyla.
Signature genera.
Signature species.
ACKNOWLEDGMENTS
The human challenge trials were supported by grants R01 AI27863, R01 AI27863S1, and U19 AI31494 (to S.M.S.) from NIAID and by a grant to the Indiana Clinical and Translational Sciences Institute and the Indiana Clinical Research Center (UL RR052761).
We have no relevant financial relationships to disclose.
We thank the volunteers who participated in the trial.
Footnotes
Citation van Rensburg JJ, Lin H, Gao X, Toh E, Fortney KR, Ellinger S, Zwickl B, Janowicz DM, Katz BP, Nelson DE, Dong Q, Spinola SM. 2015. The human skin microbiome associates with the outcome of and is influenced by bacterial infection. mBio 6(5):e01315-15. doi:10.1128/mBio.01315-15.
REFERENCES
- 1.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, NISC Comparative Sequencing Program, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. 2009. Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao Z, Tseng CH, Pei Z, Blaser MJ. 2007. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci U S A 104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. 2008. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 3:e2719. doi: 10.1371/journal.pone.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaser MJ, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Estrada I, Gao Z, Clemente JC, Costello EK, Knight R. 2013. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J 7:85–95. doi: 10.1038/ismej.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. 2009. Bacterial community variation in human body habitats across space and time. Science 326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenthal M, Goldberg D, Aiello A, Larson E, Foxman B. 2011. Skin microbiota: microbial community structure and its potential association with health and disease. Infect Genet Evol 11:839–848. doi: 10.1016/j.meegid.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Program NCS, Murray PR, Turner ML, Segre JA. 2012. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinola SM. 2008. Chancroid and Haemophilus ducreyi, p 689–699. In Holmes KK, Sparling PF, Stamm WE, Piot P, Wasserheit JN, Corey L, Cohen MS, Watts DH (ed), Sexually transmitted diseases, 4th ed. McGraw-Hill, New York, NY. [Google Scholar]
- 9.Lewis DA. 2014. Epidemiology, clinical features, diagnosis and treatment of Haemophilus ducreyi—a disappearing pathogen? Expert Rev Anti Infect Ther 12:687–696. doi: 10.1586/14787210.2014.892414. [DOI] [PubMed] [Google Scholar]
- 10.Mitjà O, Lukehart SA, Pokowas G, Moses P, Kapa A, Godornes C, Robson J, Cherian S, Houinei W, Kazadi W, Siba P, de Lazzari E, Bassat Q. 2014. Haemophilus ducreyi as a cause of skin ulcers in children from a yaws-endemic area of Papua New Guinea: a prospective cohort study. Lancet Glob Health 2:e235–e241. doi: 10.1016/S2214-109X(14)70019-1. [DOI] [PubMed] [Google Scholar]
- 11.Marks M, Chi KH, Vahi V, Pillay A, Sokana O, Pavluck A, Mabey DC, Chen CY, Solomon AW. 2014. Haemophilus ducreyi associated with skin ulcers among children, Solomon Islands. Emerg Infect Dis 20:1705–1707. doi: 10.3201/eid2010.140573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghinai R, El-Duah P, Chi KH, Pillay A, Solomon AW, Bailey RL, Agana N, Mabey DC, Chen CY, Adu-Sarkodie Y, Marks M. 2015. A cross-sectional study of “yaws” in districts of Ghana which have previously undertaken azithromycin mass drug administration for trachoma control. PLoS Negl Trop Dis 9:e0003496. doi: 10.1371/journal.pntd.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thornton AC, O’Mara EM Jr, Sorensen SJ, Hiltke TJ, Fortney K, Katz B, Shoup RE, Hood AF, Spinola SM. 1998. Prevention of experimental Haemophilus ducreyi infection: a randomized, controlled clinical trial. J Infect Dis 177:1608–1613. doi: 10.1086/515320. [DOI] [PubMed] [Google Scholar]
- 14.Mitjà O, Houinei W, Moses P, Kapa A, Paru R, Hays R, Lukehart S, Godornes C, Bieb SV, Grice T, Siba P, Mabey D, Sanz S, Alonso PL, Asiedu K, Bassat Q. 2015. Mass treatment with single-dose azithromycin for yaws. N Engl J Med 372:703–710. doi: 10.1056/NEJMoa1408586. [DOI] [PubMed] [Google Scholar]
- 15.Gangaiah D, Webb KM, Humphreys TL, Fortney KR, Toh E, Tai A, Katz SS, Pillay A, Chen CY, Roberts SA, Munson RS Jr, Spinola SM. 2015. Haemophilus ducreyi cutaneous ulcer strains are nearly identical to class I genital ulcer strains. PLoS Negl Trop 9:e0003918. doi: 10.1371/journal.pntd.0003918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janowicz DM, Ofner S, Katz BP, Spinola SM. 2009. Experimental infection of human volunteers with Haemophilus ducreyi: fifteen years of clinical data and experience. J Infect Dis 199:1671–1679. doi: 10.1086/598966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spinola SM, Bong CT, Faber AL, Fortney KR, Bennett SL, Townsend CA, Zwickl BE, Billings SD, Humphreys TL, Bauer ME, Katz BP. 2003. Differences in host susceptibility to disease progression in the human challenge model of Haemophilus ducreyi infection. Infect Immun 71:6658–6663. doi: 10.1128/IAI.71.11.6658-6663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer ME, Spinola SM. 2000. Localization of Haemophilus ducreyi at the pustular stage of disease in the human model of infection. Infect Immun 68:2309–2314. doi: 10.1128/IAI.68.4.2309-2314.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer ME, Goheen MP, Townsend CA, Spinola SM. 2001. Haemophilus ducreyi associates with phagocytes, collagen, and fibrin and remains extracellular throughout infection of human volunteers. Infect Immun 69:2549–2557. doi: 10.1128/IAI.69.4.2549-2557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer KL, Schnizlein-Bick CT, Orazi A, John K, Chen C-Y, Hood AF, Spinola SM. 1998. The immune response to Haemophilus ducreyi resembles a delayed-type hypersensitivity reaction throughout experimental infection of human subjects. J Infect Dis 178:1688–1697. doi: 10.1086/314489. [DOI] [PubMed] [Google Scholar]
- 21.Humphreys TL, Schnizlein-Bick CT, Katz BP, Baldridge LA, Hood AF, Hromas RA, Spinola SM. 2002. Evolution of the cutaneous immune response to experimental Haemophilus ducreyi infection and its relevance to HIV-1 acquisition. J Immunol 169:6316–6323. doi: 10.4049/jimmunol.169.11.6316. [DOI] [PubMed] [Google Scholar]
- 22.Humphreys TL, Baldridge LA, Billings SD, Campbell JJ, Spinola SM. 2005. Trafficking pathways and characterization of CD4 and CD8 cells recruited to the skin of humans experimentally infected with Haemophilus ducreyi. Infect Immun 73:3896–3902. doi: 10.1128/IAI.73.7.3896-3902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banks KE, Humphreys TL, Li W, Katz BP, Wilkes DS, Spinola SM. 2007. Haemophilus ducreyi partially activates human myeloid dendritic cells. Infect Immun 75:5678–5685. doi: 10.1128/IAI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Janowicz DM, Fortney KR, Katz BP, Spinola SM. 2009. Mechanism of human natural killer cell activation by Haemophilus ducreyi. J Infect Dis 200:590–598. doi: 10.1086/600123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Tenner-Racz K, Racz P, Janowicz DM, Fortney KR, Katz BP, Spinola SM. 2010. Role played by CD4+FOXP3+ regulatory T cells in suppression of host responses to Haemophilus ducreyi during experimental infection of human volunteers. J Infect Dis 201:1839–1848. doi: 10.1086/652781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelfanova V, Humphreys TL, Spinola SM. 2001. Characterization of Haemophilus ducreyi-specific T cell lines from lesions of experimentally infected human subjects. Infect Immun 69:4224–4231. doi: 10.1128/IAI.69.7.4224-4231.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer ME, Townsend CA, Ronald AR, Spinola SM. 2006. Localization of Haemophilus ducreyi in naturally acquired chancroidal ulcers. Microbes Infect 8:2465–2468. doi: 10.1016/j.micinf.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Humphreys TL, Li L, Li X, Janowicz DM, Fortney KR, Zhao Q, Li W, McClintick J, Katz BP, Wilkes DS, Edenberg HJ, Spinola SM. 2007. Dysregulated immune profiles for skin and dendritic cells are associated with increased host susceptibility to Haemophilus ducreyi infection in human volunteers. Infect Immun 75:5686–5697. doi: 10.1128/IAI.00777-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W, Katz BP, Spinola SM. 2012. Haemophilus ducreyi-induced IL-10 promotes a mixed M1 and M2 activation program in human macrophages. Infect Immun 80:4426–4434. doi: 10.1128/IAI.00912-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vakevainen M, Greenberg S, Hansen EJ. 2003. Inhibition of phagocytosis by Haemophilus ducreyi requires expression of the LspA1 and LspA2 proteins. Infect Immun 71:5994–6003. doi: 10.1128/IAI.71.10.5994-6003.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elkins C, Morrow KJ, Olsen B. 2000. Serum resistance in Haemophilus ducreyi requires outer membrane protein DsrA. Infect Immun 68:1608–1619. doi: 10.1128/IAI.68.3.1608-1619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mount KL, Townsend CA, Bauer ME. 2007. Haemophilus ducreyi is resistant to human antimicrobial peptides. Antimicrob Agents Chemother 51:3391–3393. doi: 10.1128/AAC.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holley CL, Zhang X, Fortney KR, Ellinger S, Johnson P, Baker B, Liu Y, Janowicz DM, Katz BP, Munson RS Jr, Spinola SM. 2015. DksA and (p)ppGpp have unique and overlapping contributions to Haemophilus ducreyi pathogenesis in humans. Infect Immun 83:3281–3292 doi: 10.1128/IAI.00692-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. 2007. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Casadevall A, Pirofski LA. 2015. What is a host? Incorporating the microbiota into the damage-response framework. Infect Immun 83:2–7. doi: 10.1128/IAI.02627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casadevall A, Pirofski LA. 1999. Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun 67:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang R, Wagner BD, Harris JK, Fillon SA. 2014. Application of zero-inflated negative binomial mixed model to human microbiota sequence data. Peer J PrePrints 2:e215v1. doi: 10.7287/peerj.preprints.215v1. [DOI] [Google Scholar]
- 38.Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, Berg-Lyon D, Ackermann G, Moeller Christensen GJ, Nakatsuji T, Zhang L, Borkowski AW, Meehan MJ, Dorrestein K, Gallo RL, Bandeira N, Knight R, Alexandrov T, Dorrestein PC. 2015. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci U S A 112:E2120–E2129. doi: 10.1073/pnas.1424409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shea K, Chesson P. 2002. Community ecology theory as a framework for biological invasions. Trends Ecol Evol 17:170–176. doi: 10.1016/S0169-5347(02)02495-3. [DOI] [Google Scholar]
- 40.Alekseyenko AV, Perez-Perez GI, De Souza A, Strober B, Gao Z, Bihan M, Li K, Methé BA, Blaser MJ. 2013. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 1:31. doi: 10.1186/2049-2618-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horton JM, Gao Z, Sullivan DM, Shopsin B, Perez-Perez GI, Blaser MJ. 2015. The cutaneous microbiome in outpatients presenting with acute skin abscesses. J Infect Dis 211:1895–1904 doi: 10.1093/infdis/jiv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jani AJ, Briggs CJ. 2014. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc Natl Acad Sci U S A 111:E5049–E5058. doi: 10.1073/pnas.1412752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grice EA, Kong HH, Renaud G, Young AC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA. 2008. A diversity profile of the human skin microbiota. Genome Res 18:1043–1050. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, Program NCS, Liechty KW, Segre JA. 2010. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A 107:14799–14804. doi: 10.1073/pnas.1004204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smeekens SP, Huttenhower C, Riza A, van de Veerdonk FL, Zeeuwen PL, Schalkwijk J, van der Meer JW, Xavier RJ, Netea MG, Gevers D. 2014. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. J Innate Immun 6:253–262. doi: 10.1159/000351912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morse SA. 1989. Chancroid and Haemophilus ducreyi. Clin Microbiol Rev 2:137–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bong CT, Harezlak J, Katz BP, Spinola SM. 2002. Men are more susceptible to pustule formation than women in the experimental model of Haemophilus ducreyi infection. Sex Transm Dis 29:114–118. doi: 10.1097/00007435-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Rinker SD, Gu X, Fortney KR, Zwickl BW, Katz BP, Janowicz DM, Spinola SM, Bauer ME. 2012. Permeases of the Sap transporter are required for cathelicidin resistance and virulence of Haemophilus ducreyi in humans. J Infect Dis 206:1407–1414. doi: 10.1093/infdis/jis525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Tawfiq JA, Palmer KL, Chen C-Y, Haley JC, Katz BP, Hood AF, Spinola SM. 1999. Experimental infection of human volunteers with Haemophilus ducreyi does not confer protection against subsequent challenge. J Infect Dis 179:1283–1287. doi: 10.1086/314732. [DOI] [PubMed] [Google Scholar]
- 50.Spinola SM, Li W, Fortney KR, Janowicz DM, Zwickl B, Katz BP, Munson RS. 2012. Sialylation of lipooligosaccharides is dispensable for the virulence of Haemophilus ducreyi in human. Infect Immun 80:679–687. doi: 10.1128/IAI.05826-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong Q, Nelson DE, Toh E, Diao L, Gao X, Fortenberry JD, Van der Pol B. 2011. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS One 6:e19709. doi: 10.1371/journal.pone.0019709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nienhouse V, Gao X, Dong Q, Nelson DE, Toh E, McKinley K, Schreckenberger P, Shibata N, Fok CS, Mueller ER, Brubaker L, Wolfe AJ, Radek KA. 2014. Interplay between bladder microbiota and urinary antimicrobial peptides: mechanisms for human urinary tract infection risk and symptom severity. PLoS One 9:e114185. doi: 10.1371/journal.pone.0114185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 55.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. 2014. Ribosomal Database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMurdie PJ, Holmes S. 2012. phyloseq: a Bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pac Symp Biocomput 17:235–246. doi: 10.1142/9789814366496_0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2015. vegan: the community ecology package. R package, version 2.2-1 The R Foundation, [Google Scholar]
- 59.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lozupone CA, Hamady M, Kelley ST, Knight R. 2007. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson RC, Ellis MW, Lanier JB, Schlett CD, Cui T, Merrell DS. 2015. Correlation between nasal microbiome composition and remote purulent skin and soft tissue infections. Infect Immun 83:802–811. doi: 10.1128/IAI.02664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- 63.Shumway M, Cochrane G, Sugawara H. 2010. Archiving next generation sequencing data. Nucleic Acids Res 38:D870–D871. doi: 10.1093/nar/gkp1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of the four dominant phyla in resolvers (A) and pustule formers (B). Pie charts represent the percent proportion of each phylum. R, resolved site; P, pustule site. All samples for which sequence data were obtained are shown here; no sequences were obtained from the missing samples. Download
OTUs observed in samples and breakdown of sites used for statistical analysis.
Signature phyla.
Signature genera.
Signature species.