

ABSTRACT
Blastomyces adhesin-1 (BAD-1) protein mediates the virulence of the yeast Blastomyces dermatitidis, in part by binding host lung tissue, the extracellular matrix, and cellular receptors via glycosaminoglycans (GAGs), such as heparan sulfate. The tandem repeats that make up over 90% of BAD-1 appear in their native state to be tightly folded into an inactive conformation, but recent work has shown that they become activated and adhesive upon reduction of a disulfide linkage. Here, atomic force microscopy (AFM) of a single BAD-1 molecule interacting with immobilized heparin revealed that binding is enhanced upon treatment with protein disulfide isomerase and dithiothreitol (PDI/DTT). PDI/DTT treatment of BAD-1 induced a plateau effect in atomic force signatures that was consistent with sequential rupture of tandem binding domains. Inhibition of PDI in murine macrophages blunted BAD-1 binding to heparin in vitro. Based on AFM, we found that a short Cardin-Weintraub sequence paired with a WxxWxxW sequence in the first, degenerate repeat at the N terminus of BAD-1 was sufficient to initiate heparin binding. Removal of half of the 41 BAD-1 tandem repeats led to weaker adhesion, illustrating their role in enhanced binding. Mass spectroscopy of the tandem repeat revealed that the PDI-induced interaction with heparin is characterized by ruptured disulfide bonds and that cysteine thiols remain reduced. Further binding studies showed direct involvement of thiols in heparin ligation. Thus, we propose that the N-terminal domain of BAD-1 governs the initial association with host GAGs and that proximity to GAG-associated host PDI catalyzes activation of additional binding motifs conserved within the tandem repeats, leading to enhanced avidity and availability of reduced thiols.
IMPORTANCE
Pathogenic fungi and other microbes must adhere to host tissue to initiate infection. Surface adhesins promote this event and may be required for disease pathogenesis. We studied a fungal adhesin essential for virulence (BAD-1; Blastomyces adhesin-1) and found that host products induce its structural reconfiguration and foster its optimal binding to tissue structures.
INTRODUCTION
The dimorphic fungus Blastomyces dermatitidis is the causative agent of blastomycosis, one of the major systemic mycoses of humans in areas where it is endemic. The seven dimorphic fungi that cause systemic fungal disease collectively account for several million new infections annually worldwide. The infections are acquired when mold spores in soil are inhaled into the lung and convert to pathogenic yeast in the tissue. Infections can lead to pneumonia, dissemination, and death. Blastomyces adhesin-1 (BAD-1) protein is required for pathogenicity of Blastomyces dermatitidis (1). It is a yeast-phase-specific protein and confers multiple functions for virulence: adherence to host lung tissue and matrix (1, 2), binding of CR3 receptors leading to “stealth” entry into phagocytes, suppression of tumor necrosis factor alpha in a manner that requires transforming growth factor β, and siderophore-like scavenging of divalent cations, including calcium (3–5).
The 120-kDa BAD-1 adhesin contains 3 domains, the smallest being an N-terminal domain just 18 amino acids long and the largest comprising 41 copies of a 25-amino-acid tandem repeat. Each tandem repeat contains two consistently conserved cysteines linked via disulfide bonds. Deletion of even half of the tandem repeats curtails virulence of the yeast, whereas deletion of the next-largest domain, the C-terminal domain, does not affect virulence (2). The primary sequence of the tandem repeats shows similarity to the type 1 repeat of thrombospondin-1 (TSP-1) and the malaria thrombospondin-related adhesive protein (TRAP). Like those proteins, BAD-1 mediates binding to glycosaminoglycans (GAGs), such as heparan sulfate (2). This may contribute to its capacity to mediate entry into host cells, as heparan sulfate is a known cell surface endocytosis receptor (6). BAD-1 binding of heparan sulfate-modified CD47 on T cells impairs activation and effector function, as does TSP-1, likely promoting immune evasion and progressive disease (2).
There are many examples of microbial adhesins with repeat domains in their primary sequence. Agglutination-like sequence (Als) proteins in Candida albicans (7), YadA in Yersinia enterolytica (8), and Cna in Staphylococcus aureus (9) are examples. It is plausible that the avidity of BAD-1 for GAGs is due to its numerous repeats, especially given their conservation of heparin-binding consensus sequences. In binding assays, however, peptides containing four tandem repeats held in the native conformation failed to bind immobilized heparin. It was only after the reduction of its disulfide linkages that this peptide approached the binding capability of full-length BAD-1 (2). This is not the first example of an adhesin regulated by its disulfide structure. In integrin αIIbβ3, failure to form disulfide bridges leaves its fibrinogen-binding site constitutively active (10), and in the malaria circumsporozoite protein, the disulfide arrangement can either increase or decrease its binding affinity (11).
Nuclear magnetic resonance (NMR) structural studies of BAD-1 have permitted new insights into the nature of its heparin-binding mechanism (2). In both TSP-1 and TRAP repeats, the heparin-binding cleft is formed as three tryptophans (WxxWxxW) on an α-helix stack alternating with two basic residues (BxB) that project from an antiparallel strand. In the native BAD-1 repeat, however, a conformation was resolved in which such intercalation was precluded, largely due to the constraint of the disulfide linkage (2) (Fig. 1A). This structure would account for the failure of the tandem repeat model peptide to bind heparin. The evolution of heparin-binding structures could thus be contingent upon a conformational switch, once scission of the disulfide bond alleviates this constraint. In this model, it follows that either a novel pattern of disulfide bonds would develop in repeats as heparin becomes engaged or that, alternatively, cysteines persist as free thiols (Fig. 1B). In the first scenario, novel disulfide bonds would be expected to afford the heparin-bound conformation additional stability, while in the latter scenario, free thiol groups could promote oligomerization of BAD-1 or cross-linking with host cell surface proteins.
FIG 1 .

The BAD-1 adhesin. (A) The N-terminal region is only 18 amino acids long and contains a Cardin-Weintraub domain (BBxB). Immediately adjacent to this region, the first tandem repeat is degenerate, lacking a span of nine residues typical of the consensus repeats. Residues with basic (positively charged) side chains are displayed in blue, and conserved tryptophans are shown in red. The degenerate repeat is followed by the consensus sequence of the other 40 tandem repeats. (B) Repeats in the native conformation (two repeats are depicted) are predicted to hold the elements of a heparin-binding motif apart (2). Blue and red residues are as noted for panel A. Upon reduction of disulfide bonds, these elements would be able to interact freely and thus create a TSP-1-like heparin-binding cleft (2). This motif could form in one of two possible ways: with cysteines joined in novel disulfides (above) or remaining free (below).
Herein, we use atomic force microscopy (AFM) (12, 13) to investigate the role of distinct BAD-1 domains in binding to heparin, and in particular, the role of disulfide bond reduction in heparin-specific binding by BAD-1. In addition to the tandem repeats, BAD-1 contains one additional predicted heparin-binding motif (as described by Cardin-Weintraub [2]) within its short N-terminal domain. We show here that (i) the Cardin-Weintraub (C-W) motif, when paired with an initial degenerate repeat, has the capacity to initiate a low-affinity association with host GAGs, (ii) that the treatment of BAD-1 with protein disulfide isomerase (PDI) enhances binding to heparin on solid surfaces, (iii) that inhibition of PDI impairs binding mediated by BAD-1 to heparin on the surface of macrophages, and (iv) that the conformation of heparin-bound BAD-1 repeats favors the maintenance of reduced cysteine thiols. Our findings suggest a model in which an initial low-affinity binding, mediated via the N-terminal domain with its C-W motif, triggers host cell PDI-mediated scission of disulfide bonds in BAD-1, thereby exposing multiple, neo-adhesive domains—ligand-induced binding sites (LIBS) (14)—that greatly augment the avidity of BAD-1 toward heparin on selected host cell surfaces. Exposure of unpaired thiols in this model may then cement stable adhesion with the host cell surface, as has been shown to be the case in integrin-dependent adhesion (15, 16).
RESULTS
BAD-1 binding to heparin by AFM.
To probe single BAD-1–heparin interaction forces, BAD-1 protein bearing a His tag at its C-terminal end was attached in an oriented manner to gold-coated AFM tips bearing Ni2+-nitrilotriacetate (NTA) groups, and biotinylated heparin molecules were immobilized on flat gold substrates by using a biotin-streptavidin sandwich system previously described (Fig. 2A) (17). Topographic imaging revealed that the heparin surface was homogeneous and stable upon repeated scanning, indicating good stability of the heparin film. Scanning a small area with strong applied forces revealed the thickness of the biomolecular layer (2.8 ± 0.9 nm [mean ± standard deviation]) (Fig. 2B). We recorded force-distance curves at a pulling speed of 1,000 nm s−1 between BAD-1 tips and heparin-coated surfaces. Many curves (61%; mean of 2,048 curves from 2 independent experiments) showed well-defined, single adhesion forces, with a magnitude ranging from 50 to 300 pN (Fig. 2C). The remaining 39% of the recorded force curves displayed no adhesive events, as represented by the first bin of the adhesion histogram of Fig. 2C. The observed adhesive signatures were attributed to the binding of BAD-1 to heparin because (i) most force peaks were well-fitted by the worm-like chain (WLC) model (using a persistence length of 0.4 nm) (Fig. 2C, red lines) (18, 19), suggesting they reflect the force-induced unfolding of individual proteins, and (ii) the adhesion frequency was significantly reduced (39%) upon injection of a 100-µg ml−1 heparin solution (Fig. 2D), confirming the specificity of the measured BAD-1–heparin interactions.
FIG 2 .

Strength and dynamics of the BAD-1–heparin interaction. (A) Schematics of the strategy for measuring the interaction between BAD-1 protein and heparin-coated surfaces. Histidine-tagged BAD-1 proteins were attached to the AFM tip terminated with Ni2+-NTA groups, and biotinylated heparin was bound to a gold substrate via streptavidin and biotinylated bovine serum albumin (BBSA) layers. (B) AFM height image (z = 20 nm; a vertical cross-section from the center of the image is shown beneath the image) was recorded with a silicon nitride tip, confirming the presence of a smooth, homogeneous heparin layer. (C) Adhesion-force histogram, with representative force curves, recorded in HEPES buffer between the heparin-coated surface and BAD-1-functionalized tip. The red lines on the force curves correspond to the fit using the WLC (worm-like chain) model with a persistence length of 0.4 nm. The first bin corresponds to nonadhesive events (n.a.). The data correspond to results from 2,048 force curves pooled from two independent experiments (performed with different tips, substrates, and batch of recombinant proteins). (D) Similar experiments after injection of a 100-µg ml−1 free heparin solution in the AFM setup. (E) Dependence of the adhesion force on the loading rate applied during retraction (means ± standard errors of 400 force curves for each data point; rates were measured with a contact time of 250 ms). (F) Dependence of the adhesion frequency on the interaction time, measured at a constant retraction speed of 1,000 nm s−1.
To assess the binding affinity, we explored the dynamics of the BAD-1–heparin interaction. Consistent with other receptor-ligand bonds (20, 21), the mean adhesion force (F) increased linearly with the logarithm of the loading rate [ln(r)] (Fig. 2E). The length scale of the energy barrier, xβ, was assessed from the slope fβ ([1.70 ± 0.15] × 10−11) of the plot of F versus ln(r) and found to be 0.24 nm, which is in the range of values typically measured by single-molecule AFM (20). Extrapolation to zero force yielded a kinetic off-rate constant of dissociation at zero force, koff (koff = rF=0 ⋅ xβ/kBT, where kB is the Boltzmann constant), of 0.66 s−1, indicating a rapid dissociation of BAD-1–heparin bonds. We then measured the adhesion frequency while increasing the contact time (Fig. 2F). The binding frequency increased exponentially and reached a plateau after about 2 s. From this plot, we found that the interaction time needed for half-maximal probability of binding, t0.5, was 0.38 s. We estimated the association constant (kon) by using the following equation: kon = t0.5−1 NA Veff = 1,260.8 M−1 s−1, where Veff is the effective volume explored by the tip-tethered BAD-1 (approximated here as a half-sphere of 10-nm radius) and NA is the Avogadro constant. This allowed us to estimate the equilibrium dissociation constant (KD) with the following equation: KD = koff/kon = 0.52 mM, a value suggesting that a single molecule of BAD-1 binds heparin with low affinity under these conditions.
Heparin-binding capacity is enhanced upon treatment with PDI/DTT.
NMR spectroscopy results reported previously pointed to a mechanism requiring redox catalysis to trigger specific interactions with the glycosaminoglycans that decorate host cell surfaces (2). We hypothesized that the high-frequency binding of BAD-1 to heparin would be initiated by the Cardin-Weintraub site located at the N-terminal end of the protein (Fig. 1A), in concert with the first degenerate repeat. Thereafter, reduction of the cysteine bonds in the next tandem repeat would lead to a domino effect, with the exposure of neo-adhesive domains in each repeat binding to heparin (Fig. 1B). Enhanced adhesion following protein reconfiguration has been reported for the Candida albicans adhesin Als5p, which is triggered by mechano-chemical stress (22), and for the neutrophil adhesin Mac1 through sulfhydryl cleavage by PDI (23). To test this hypothesis, we studied the effect of PDI/DTT on the specific binding forces. To ensure that BAD-1 linkage to the AFM tip remained stable under reducing conditions, we covalently grafted the BAD-1 adhesin via random N-hydroxysuccinimide (NHS) chemistry. To do so, AFM tips were coated with carboxylate-terminated thiols (COOH). The carboxylates were then allowed to react with NHS in the presence of carbodiimide (EDC), resulting in a semistable NHS-ester, which was then reacted with the free primary amines of the proteins to form amide cross-links (24).
Multiple force curves obtained between BAD-1 and heparin (Fig. 3A) revealed adhesion events similar to those measured with the His tag chemistry (Fig. 2C). However, the binding frequency (26%; mean from 6,144 curves from 6 independent experiments) (Fig. 3A) was lower, an effect that most likely arose from differences in the orientation of the attached proteins (oriented versus random orientation). Notably, injection of a PDI/DTT solution resulted in a substantial increase of the adhesion probability (from 26 to 45%) (Fig. 3B), indicating that reduction of the disulfide bonds plays a key role in binding. In addition, plateau force profiles were observed after reduction (Fig. 3B). These remarkable adhesion signatures likely represent the mechanical breakdown of a molecular zipper-like association between heparin and tandem repeat regions, i.e., the sequential rupture of tandem binding domains. Such zipper-like adhesion has previously been reported for Gram-positive bacteria pili (25) and microbial amyloids (26). Injection of free heparin into the reduced system decreased the adhesion frequency (from 45% to 26%; n = 6,144 curves) (Fig. 3C) and led to the disappearance of plateau signatures, indicating that these features are specific to heparin binding.
FIG 3 .

Mechanisms of BAD-1 binding to heparin via the tandem repeat region. (A) Adhesion-force histogram and representative force curves obtained in HEPES buffer between the heparin-coated surfaces and BAD-1 protein covalently attached via NHS-EDC chemistry. The first bin corresponds to nonadhesive events (n.a.). The histogram was created from 6,144 curves from 6 independent experiments using independent tips, substrates, and batches of recombinant proteins. (B) Force curves and adhesion histogram obtained from 6,144 curves obtained after replacing the HEPES buffer with phosphate buffer containing DTT and DPI. The increased adhesion frequency and presence of plateau force curves reveal that, in the presence of DPI, the protein binds to heparin through the reduction of the tandem repeat disulfide bonds, in a zipper-like mechanism. (C) Forces curves and adhesion-force histogram (n = 6,144) obtained after injection of free heparin (100 µg ml−1), confirming the specificity of the measured interaction by blocking of BAD-1 adhesion sites.
The short N-terminal sequence of BAD-1 is sufficient for low-affinity heparin binding, but tandem repeats confer avidity.
The Cardin-Weintraub BBxB motif has been shown to mediate binding to heparin (27). Because the sequence within the N-terminal domain of BAD-1 is HHPK, with conditionally protonated histidine residues rather than consistently protonated lysine and arginine residues (Fig. 4A), we ascertained the binding capacity of this motif. At biological pH, a peptide containing the HHPK motif bound to heparin-agarose beads only slightly better than the negative (Fluor-only) control (Fig. 4B). Likewise, neither the basic half of the tandem repeat motif (BxB peptide) nor the aromatic half (WxxWxxW peptide) mediated binding above control levels. In contrast, heparin binding was achieved when the basic and aromatic motifs were combined on one peptide to duplicate the complete N-terminal domain. Heparin binding by this peptide was similar to that mediated by full-length BAD-1.
FIG 4 .

Functional role of the different BAD-1 domains. (A) Synthetic peptides of the BAD-1 N terminus were tested for binding to heparin-agarose. The N terminus of BAD-1 contains a Cardin-Weintraub BBxB heparin-binding motif, in which two of the basic residues (B, shown in blue) are histidines and are adjacent to a truncated, degenerate first tandem repeat (containing only WxxWxxW [red]) that lacks the BxB basic residue motif present in the subsequent tandem repeats. Sequences of the four synthetic peptides used in the binding assay are shown. (B) Fluorescence assay of binding to heparin-agarose by labeled synthetic peptides, BAD-1, and Fluor alone. (C) Adhesion-force histogram and representative force curves obtained with HEPES buffer between the heparin-coated surfaces and a peptide made of the N-terminal C-W domain and the first degenerate tandem repeat domain of BAD-1 (“Complete N-term peptide”). The significant adhesion frequency revealed that the N-terminal part of the protein initiates binding to heparin. The first bin corresponds to nonadhesive events (n.a.). The histogram was created from results of 4 independent experiments (n = 4,096 force curves). (D) Forces curves and adhesion-force histogram (n = 4,096) obtained after injection of free heparin (100 µg ml−1) in the AFM setup. (E) Force curves and the adhesion histogram of the binding of BAD-1 protein from which 20 of the 42 tandem repeat regions were deleted (ΔTR20; n = 4,096 force curves). (F) Results for the same experiment as shown in panel E, except phosphate buffer containing DTT and PDI was used (n = 4,096 curves). The increased adhesion and presence of plateau force curves are mediated by the remaining tandem repeats after reduction of their disulfide bonds by PDI.
To further dissect the role played by the different domains of BAD-1 via AFM, we measured the binding forces of the short peptide bearing the Cardin-Weintraub sequence at the N terminus paired with the first adjacent tandem repeat of the protein (“complete N-term peptide”). Forces ranging from 50 to 300 pN were observed in 22% of the cases (mean from 4,096 curves from 4 independent experiments) (Fig. 4C), very similar to the forces of the full protein (26%) (Fig. 3A). Blocking with free heparin confirmed the specificity of the interaction (Fig. 4D). Thus, the N-terminal Cardin-Weintraub-containing domain can initiate association with heparin. Finally, to further assess the role of the tandem repeats, BAD-1 truncated by 20 of the 41 tandem repeats (ΔTR20) normally present in the protein structure was tested for adhesion. The frequency of initial adhesion was lower (17%, mean from 4,096 curves from 4 independent experiments) (Fig. 4E) than observed with the full protein (Fig. 3A), yet adhesion was enhanced upon PDI/DTT treatment (Fig. 4F). Hence, removal of half of the total 41 BAD-1 tandem repeats reduced the adhesive properties of BAD-1.
Roles of heparin and PDI in BAD-1-mediated adherence to macrophages.
To explore the biological relevance of the BAD-1–heparin interaction during adhesin-phagocyte interactions, we performed binding assays with BAD-1-coated polystyrene beads and RAW 264.7 macrophages. BAD-1-coated beads bound 2- to 5-fold more macrophages than did control beads coated with polyvinylpyrrolidone (PVP) (Fig. 5A and B). Up to 80 BAD-1-coated beads bound to 300 RAW 264.7 macrophages in a high-power field, compared to about 18 PVP-coated beads. The addition of soluble heparin to the binding assay mixture blocked the binding of BAD-1-coated beads to cells by up to 50% and did so in a concentration-dependent manner (Fig. 5A). To test the role of host PDI in this interaction, we preincubated cell monolayers with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), a known inhibitor of PDI. DTNB blocked binding of BAD-1-coated beads to macrophages in a concentration-dependent manner, inhibiting binding by up to 67%. In contrast, binding of PVP control beads was not altered by DTNB, nor was the binding of nonspecific protein-coated control beads (data not shown).
FIG 5 .
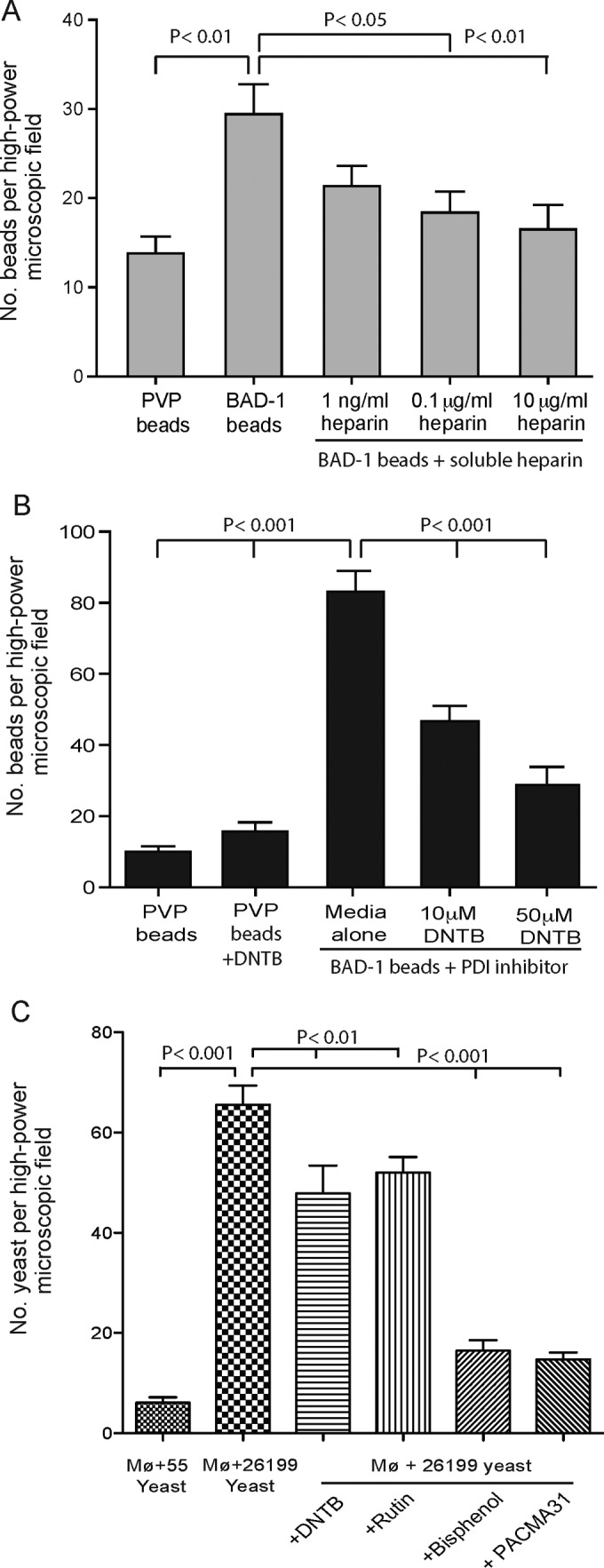
Interaction of BAD-1 coated beads with RAW macrophages. (A) Binding of 5-µm-diameter polystyrene beads coated with BAD-1 or PVP (control) after 20 min of incubation with RAW macrophages. The y axis shows the number of beads bound per high-power field, which contained ≈300 macrophages. Assay results shown are representative of three separate experiments. Soluble heparin was added during coincubation with beads. Binding in the presence of 10 µg ml−1 heparin was not significantly different from that for the negative control (PVP). (B) RAW macrophages were preincubated for 5 min with the PDI inhibitor DTNB. (C) RAW macrophages were preincubated for 5 min with the PDI inhibitors Rutin, DTNB, bisphenol, or PACMA31 before the addition of yeast. Strain 26199 is BAD-1 sufficient; strain 55 is BAD-1 deficient.
To further establish that the effect of PDI inhibitors was biologically relevant, we repeated macrophage-binding studies with BAD-1-sufficient and -deficient B. dermatitidis yeast cells (isogenic ATCC strains 26199 and 55, respectively). BAD-1-sufficient yeast bound RAW macrophages avidly, whereas BAD-1-deficient yeast bound poorly. Each of the four PDI inhibitors blunted yeast adherence to RAW macrophages significantly, albeit to varied degrees (Fig. 5C). Thus, BAD-1 mediates adherence to the surface of macrophages at least in part by interacting with heparin, and that interaction is augmented significantly by PDI.
MS analysis of the redox state of cysteines in a heparin-bound tandem repeat model peptide, TR4.
In our recently reported study of the BAD-1 tandem repeats, in which we used a model containing four tandem repeat sequences (TR4), reduction of the disulfide bonds was requisite for heparin association (2). Here, to ascertain the postbinding state of the cysteines released from these bonds, we incubated heparin-bound TR4 with iodoacetic acid (IAA) to carboxymethylate free sulfhydryls. After removal of this reagent, we released TR4 from heparin-agarose beads by boiling in SDS-PAGE buffer. TR4 was applied to a PAGE gel, and a solitary band visualized by using Coomassie blue stain was excised. After gel destaining, trypsin-digested peptides were reduced and treated with N-ethylmaleimide (NEM) to differentially label any cysteines that were paired via a novel disulfide bond in the heparin-bound conformation. Labeled peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Major peaks corresponding to the mass of each expected tryptic peptide were thus identified, but in every case peptides that included a cysteine residue were detected exclusively in the carboxymethyl-modified form (Fig. 6A). Controls verified that all disulfides were cleavable via DTT and that reduced thiols were accessible to modification. The thoroughness of carboxymethylation in the heparin-bound TR4 peptide was confirmed by comparison to a fully carboxymethylated control (data not shown). This result established that the heparin-binding motifs that evolve in response to disulfide reduction are not only stable in the absence of new disulfide linkages but also that they actually seem to stabilize the cysteine thiols in their reduced form.
FIG 6 .

Analysis of the redox state of cysteine thiols in tandem repeat peptide models bound to heparin. (A) LC-MS/MS was used to determine whether the thiols that are reduced to permit TR4 repeats to engage heparin remain free (reduced) in the binding conformation, or if this conformation favors the formation of novel disulfide pairings. In the bound conformation, free thiols were carboxymethylated with IAA. After PAGE separation, reduction, and tryptic digestion, all other cysteines were labeled with NEM. Detected tryptic peptides are shown in red and underlined (inset). Corresponding peptide masses appear in the boxes, and modified cysteines are shown in red. (B) Peptide binding to heparin-agarose. Wild-type TR2 synthetic peptide comprises two tandem repeats flanked by two respective pairs of cysteines, while the mutant (C → A) TR2 peptide is two tandem repeats in which the cysteines have been replaced with alanines. Peptides were treated with 10 mM DTT before analysis of binding to heparin-agarose.
To determine whether the free cysteine residues themselves contribute to heparin binding, we compared the binding capacity of a synthetic peptide containing two tandem repeats versus a mutant peptide in which the four cysteines residues were replaced by alanines (Fig. 6B). If the cysteines only constrain repeats to a nonbinding configuration, both peptides would be expected to bind equally well following DTT reduction. The mutant peptide bound heparin poorly, as did the unreduced form of the wild-type peptide. The wild-type peptide bound heparin significantly upon reduction of its cysteine residues by 10 mM DTT, whereas the mutant peptide failed to bind heparin under these conditions. An additional finding in these experiments was that DTT itself inhibited binding unless the reduced samples were diluted to <1 mM DTT. This inhibition by a dithiol may be supportive evidence that the reduced thiols of the tandem repeats are directly involved in adhesion to GAGs.
DISCUSSION
Herein, we report that the N-terminal region of BAD-1 is sufficient to initiate binding to immobilized heparin. We have ascertained that an N-terminal Cardin-Weintraub motif (BBXB), in which the first two basic residues are normally replaced with histidines, is not sufficient to mediate this binding, and neither is a cysteine-flanked WxxWxxW motif in the first repeat, but that together these motifs comprise a functional, low-affinity, heparin-binding domain. This may be ascribed to the strong affinity between histidines and tryptophans (28) and the capacity of these interactions to stabilize the protonated form of histidine (29). In contrast, similar motifs replicated within every repeat appear quiescent until activated by disulfide reduction (2). PDI-dependent activation fostered BAD-1 adherence to heparin and association with macrophages, and it may promote yeast entry into host cells during the early stages of infection.
It is not uncommon for disulfide bonds to frame and stabilize ligand-binding repeats in microbial adhesins (30), and reconfiguration of these bonds can be a prerequisite for adherence (30–32) and entry into host cells (33). Ideally, for a microbe such reactions would be contingent upon the proximity of the host ligand. In interactions between cell surfaces, cysteine-rich proteins are commonplace (34), but not as ubiquitous as mechano-chemical stress. With Candida Als5p, when a single adhesin molecule binds to its host ligand, it unfolds under the mechano-chemical stress of stretching, exposing neo-domains that promote protein clustering. Protein clustering stabilizes the extended conformation of adjacent adhesin molecules (22), potentially bringing more binding sites to bear. Thus, the forces to which adhesins are universally subject at the advent of target ligation may constitute a situational trigger, spurring reorganizations in the secondary structure that facilitate uptake. This could be through promotion of oligomerization, as with Als5p (22), catalysis of cross-linking, as with the circumsporozoite protein of P. falciparum (11), or elaboration of additional binding structures, as with the heparin-binding growth-associated protein (35).
Mechano-chemical stress is pertinent to disulfide bond chemistry in that it renders such bonds labile to reduction (36, 37). The notion that repeats under mechano-chemical stress may become reorganized structurally in the presence of reducing/isomerizing agents has been demonstrated by biophysicists but remains poorly appreciated as a mechanism of adhesive function in pathogenesis. A trigger mechanism requiring both stress and disulfide reorganization would derive significant tissue specificity from the fact that both GAGs and molecules mediating extracellular PDI activity are enriched on the surfaces of host immune cells (38, 39) and within damaged matrix (40). A mechanism in which heparin binding requires disulfide reduction may be relevant in the context of recent reports that B. dermatitidis can grow intracellularly after entry into host macrophages (41). Heparan sulfate GAGs and PDI are both exploited by numerous intracellular pathogens to effect macrophage-specific binding and promote their own phagocytic uptake (6, 42, 43). It is likely that various tissue-specific triggering mechanisms evolved to prevent deleterious off-target binding. If high-affinity GAG-binding motifs were constitutively active upon the surface of a pathogen, they might engender nonspecific adherence and/or enhance host immune response.
The structure of the BAD-1 adhesin appears to be rod-like, but not rigid (2), and anchored by its C-terminal domain to the surface of B. dermatitidis yeast (44). While its N-terminal domain is sufficient to initiate binding, the low affinity we calculated here based on AFM differs from the affinity estimated previously based on surface plasmon resonance (SPR) (2). This discrepancy may relate to the difficulty we had in achieving binding equilibria in our SPR work, which we attributed to the tendency of BAD-1 to self-associate. We have shown that BAD-1 coordinates calcium cations (~27 per monomer), is released in quantity from yeast cell surfaces by a lowered calcium concentration, and undergoes C-terminal domain-dependent calcium precipitation (3), suggesting a potential to aggregate under biological conditions. Whereas AFM measurements investigate interactions of a solitary molecule of the adhesin, SPR characterizes the binding behavior of many molecules as the mobile phase passes over a heparinized surface. If BAD-1 were to oligomerize in the SPR buffer, the resulting polyvalency of binding domains might explain both the elevation of the calculated affinity constant and the incapacity to achieve a stable plateau in binding versus time (45–47). A second possibility is that BAD-1 may oligomerize in response to ligand binding, as described for C. albicans Als5p (22). A last possibility is that the low affinity observed here reflects the absence of PDI, and thus represents only the binding of the short N-terminal domain. If the conditions used in SPR were to somehow permit disulfide reorganization (via contaminating PDI, reductive interactions with the gold surface, or an uncharacterized PDI capacity of CxxC and CxC motifs [40] present in the C-terminal domain of BAD-1), the higher affinity measured via SPR could be attributed to the development of additional binding motifs.
In addition to proven interactions between the BAD-1 repeats and calcium, heparin, and its own C-terminal domain, we have shown here that following PDI-catalyzed ligation to heparin, the cysteine thiols of TR4 become stabilized in their reduced form rather than forming a new pattern of disulfide bonds. Considering that there are 82 cysteines in the tandem repeat domain of BAD-1, this constitutes an exceptional number of thiols with potential to cross-link with host proteins, cement BAD-1 oligomers into extended, highly polyvalent complexes, or contribute directly to heparin ligation through coordination of surface-associated cations (like zinc [48, 49]). In addition to the assay and experimental design differences noted above, the capacity to form oligomers could also explain the differences between affinity values for truncated BAD-1 for heparin determined via AFM versus those via SPR (2).
In summary, we propose a model in which the BAD-1 adhesin mediates initial ligation of heparan sulfate on host cell surfaces, thus bringing it proximal to host PDI. This event, in concert with mechano-chemical stress, triggers the evolution of additional heparin-binding domains. These magnify the avidity of the BAD-1–heparin association and, in the process, expose reactive cysteine thiols capable of reacting with host thiols or cross-linking with other molecules of BAD-1. The resultant, high-avidity bond between yeast and host cells may promote retention in the lung and, possibly, uptake of yeast into the host cell itself, promoting the pathogenesis of infection.
MATERIALS AND METHODS
BAD-1 production and purification and growth of yeast.
American Type Culture Collection strain 26199 of B. dermatitidis and the BAD-1-null, isogenic strain 55, were propagated as yeast on 7H10 slants as described previously (1). BAD-1, BAD-1–6H (with His6 tag), and tandem repeat Δ20 (ΔTR20) proteins and TR4 peptide were expressed and purified as previously described (2, 3). Synthetic peptides were purchased from GenScript (Piscataway, NJ).
Heparin-agarose-binding experiments.
Heparin-agarose beads were obtained from Sigma-Aldrich (St. Louis, MO). Biotinylated peptides (100 µg ml−1) were allowed to interact with beads in a 96-well plate for 30 min at 25°C in binding buffer (20 mM Tricine [pH 7.6], 50 mM NaCl) and washed with binding buffer. Then, 1.0 µl of Efluor 605-streptavidin (eBioscience, San Diego, CA) was added to each well in binding buffer, incubated at 25°C for 20 min, and washed three times in binding buffer before fluorescence was determined on a FilterMax F5 multimode microplate reader (Molecular Devices, Sunnyvale, CA). In assays with unlabeled tandem repeat peptide, binding to heparin-agarose beads was quantified by measurement of the A280 as described elsewhere (2).
Preparation of heparin surfaces and BAD-1 tips.
For AFM, heparin-coated surfaces were prepared via a sandwich layer chemistry (17). Silicon wafers (Siltronix, Archamps, France) were coated using electron beam evaporation with a 5-nm-thick chromium layer followed by a 30-nm-thick gold layer. The gold-coated surfaces were cleaned for 15 min with UV-ozone, rinsed with ethanol, and dried with N2. The surfaces were then immersed overnight at room temperature in a 25-µg ml−1 solution of biotinylated bovine serum albumin (BBSA; Sigma) in HEPES buffer (40 mM HEPES, 100 mM NaCl; pH 7). After rinsing with buffer, the BBSA surfaces were exposed to a 10-µg ml−1 solution of streptavidin (Sigma) in HEPES for 2 h, followed by thorough rinsing with buffer. Then, the BBSA-streptavidin surfaces were immersed for 2 h in a buffer solution containing 10 µg ml−1 biotinylated heparin (Sigma), rinsed in buffer, and mounted to the AFM setup without dewetting.
His-tagged BAD-1 proteins were attached in an oriented manner onto Ni-NTA-terminated AFM tips. For this step, gold-coated cantilevers (Olympus; nominal spring constant, 0.02 N m−1) were cleaned for 10 min by UV-ozone treatment, rinsed with ethanol, dried with N2, and immersed overnight in ethanol containing 0.05 mM NTA-terminated (10% of mixture) and triethylene glycol-terminated (90% of mixture) alkane thiols. After rinsing with ethanol and drying, the tips coated with alkane thiols were immersed in a 40 mM aqueous solution of NiSO4 (pH 7.2) for 30 min, rinsed with buffer, immersed in protein solution at 0.2 mg ml−1 for 2 h, rinsed in buffer, and immediately used for measurements.
BAD-1 proteins and peptides were also covalently attached to tips via N-hydroxysuccinimide (NHS) surface chemistry. Gold-coated cantilevers were immersed overnight in ethanol solutions containing 1 mM 16-mercaptohexadecanoic acid (10% of mixture; Sigma) and 11-mercapto-1-undecanol (90% of mixture; Sigma) and then rinsed with ethanol. Cantilevers were immersed for 30 min in a solution containing 10 mg ml−1 NHS (Sigma) and 25 mg ml−1 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC; Sigma) and rinsed with water. The activated tips were incubated with protein solution at 0.2 mg ml−1 for 1 h, followed by rinsing with buffer.
AFM measurements.
AFM images and force-distance curves were obtained in HEPES buffer (40 mM HEPES, 100 mM NaCl; pH 7) at room temperature by using a Nanoscope VIII Multimode AFM instrument (Bruker, Santa Barbara, CA). To verify the homogeneity and quality of the heparin-coated surfaces, a small square (1 µm by 1 µm) was first scanned using a bare silicon nitride tip (MSCT cantilever; Bruker) under high applied force, followed by a larger image (5 µm by 5 µm) taken under low force. Bare tips were then exchanged with BAD-1-functionalized tips. Multiple (32 × 32) force-distance curves were recorded on areas of 5 µm by 5 µm, and the adhesion force was calculated for each curve. Adhesion force histograms were then constructed from the adhesion values of the rupture peaks using the Origin software (version 7.5). Unless otherwise stated, all force curves were obtained using a contact time of 1.1 s, a maximum applied force of 250 pN, and approach and retraction speeds both at 1,000 nm s−1. The loading rates (in pico-Newtons per second) were estimated by multiplying the tip retraction velocity by the spring constant of the cantilever. Reduction experiments were done by replacing HEPES buffer with 100 mM sodium phosphate buffer (pH 7.0) containing 1 mM sodium EDTA, 4 mM DTT, and 1 µg ml−1 PDI (Sigma) in the AFM setup, 30 min prior to the force measurements. Blocking experiments were carried out by injecting HEPES buffer containing 100 µg ml−1 of free heparin 30 min prior to force measurements.
Binding of BAD-1-coated beads to macrophages.
Five-micrometer-diameter fluorescent polystyrene beads were obtained from Phosphorex, Inc. (Hopkinton, MA). Beads were coated with purified BAD-1 by incubating 1 × 108/ml beads in 100 mM sodium carbonate buffer, pH 9.6, with 200 µg ml−1 BAD-1 for 1 h with agitation at 25°C and overnight at 4°C. Control beads were coated with 200 µg ml−1 polyvinyl pyrrolidone (PVP) in the same buffer. Beads were washed once with carbonate buffer and once with phosphate-buffered saline (PBS). RAW 264.7 macrophages were propagated in 3 ml Dulbecco’s modieifed Eagle’s medium (DMEM) plus 10% fetal bovine serum (FBS) on 6-well tissue culture plates (Celltreat Scientific Products, Shirley, MA). RAW macrophages were washed twice with 2 ml DMEM (no FBS) before addition of beads. Beads (5 × 106) were allowed to interact with macrophages for 20 min at 25°C before unattached beads were washed away with three 2-ml washes with DMEM. Microscope fields were evaluated on an Olympus IX50 inverted fluorescence microscope with a Zeiss LD Plan-NeoFluar 40× (no immersion) objective. The PDI inhibitor DTNB (Sigma) was dissolved in dimethyl sulfoxide (DMSO). DMSO alone was added to controls.
Adhesion of yeast to macrophages.
Yeast cells were washed once in PBS prior to introduction into wells with RAW 264.7 macrophages in DMEM (no FBS). Binding was allowed to proceed and was evaluated and quantified as described above for BAD-1-coated beads, except that prior to the last DMEM wash, yeast cells were briefly stained with Uvitex 3BSA (CIBA-Geigy, Basel, Switzerland) diluted 1:2,000 for contrast. The PDI inhibitors DTNB, Rutin, bisphenol, and PACMA31 were obtained from Sigma and dissolved at 100 mM in DMSO to make working stocks.
MS analysis of the heparin-binding conformation of BAD-1 tandem repeats.
The peptide TR4 contains four copies of the conserved consensus for the tandem repeats of BAD-1, and it fails to bind to heparin unless its disulfide bonds are first reduced (2). Upon reduction with 10 mM DTT and mild heating (55°C for 45 min), TR4 binds avidly to heparin-agarose, once the concentration of DTT is diluted to <1 mM (2). We assessed which, if any, cysteines were free in the heparin-binding conformation and whether any of them formed novel disulfide bonds. To accomplish this, we incubated reduced TR4 with heparin-agarose beads, removed the DTT, and incubated the bead mixture at 4°C overnight to allow favorable oxidation reactions to proceed. Heparin-associated peptides were treated with IAA, to label free cysteines with a carboxymethyl group. IAA was removed and TR4 was extracted from heparin-agarose beads by heating in SDS-PAGE loading buffer for 5 min at 95°C. TR4 was subjected to PAGE through a 15% acrylamide gel, visualized as a solitary band with Coomassie blue stain, and excised with a clean razor blade.
Enzymatic in-gel digestion.
In-gel digestion and mass spectrometric analysis were done at the Mass Spectrometry Facility (Biotechnology Center, University of Wisconsin—Madison). The digestion was performed as outlined on the Biotechnology Center website (http://www.biotech.wisc.edu/ServicesResearch/MassSpec). In short, Coomassie R-250-stained gel pieces were destained completely in 50/50 (vol/vol) methanol-H2O–100 mM NH4HCO3, dehydrated for 2 min in 50/50 (vol/vol) ACN-H2O–25 mM NH4HCO3, and then once more for 30 s in 100% ACN. The gel pieces were dried in a SpeedVac for 1 min, reduced in 25 mM DTT (in NH4HCO3) for 30 min at 56°C, alkylated with 20 mM N-ethylmaleimide in 25 mM Tris-HCl (pH 7.5) at room temperature for 30 min, washed once in H2O, dehydrated for 2 min in 50/50 (vol/vol) ACN-H2O–25 mM NH4HCO3, and then once more for 30 s in 100% ACN. The gel pieces were dried again, rehydrated with 20 µl of trypsin solution with 0.01% ProteaseMAX surfactant (10 ng/µl trypsin [Trypsin Gold; Promega Corp.]) in 25 mM NH4HCO3–0.01% (wt/vol) ProteaseMAX (Promega Corp.), and let stand for 2 min at room temperature. An additional 35 µl of overlay solution (25 mM NH4HCO3–0.01% [wt/vol] ProteaseMAX) was added to keep gel pieces immersed throughout the digestion. The digestion was conducted for 3 h at 42°C, peptides generated from digestion were transferred to a new Protein LoBind tube (~50-µl volume; Eppendorf), and digestion was terminated by acidification with 2.5% trifluoroacetic acid (TFA) to a 0.3% final concentration (7.5 µl added). Degraded ProteaseMAX was removed via centrifugation (at maximum speed, 10 min), and the peptide solid phase was extracted (ZipTip C18 pipette tips; Millipore, Billerica, MA). Peptides were eluted off the C18 column with 1 µl of 60/40 (vol/vol) acetonitrile-H2O–0.1% TFA, a 0.5-µl aliquot of eluent was deposited onto an Opti-TOF 384-well plate (Applied Biosystems, Foster City, CA), and proteins were recrystallized with 0.40 µl of matrix for matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) analysis (the matrix was 10-mg ml−1 60/40 [vol/vol] α-cyano-4-hydroxycinnamic acid in 60/40 [vol/vol] acetonitrile-H2O–0.2% TFA). The rest of the eluent was diluted to a 25-µl total volume with 0.1% formic acid, and 3 µl was loaded on an Orbitrap instrument for nano-LC-MS/MS.
MALDI-TOF MS analysis.
Peptide mass fingerprint analysis was performed on a 4800 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Foster City, CA). In short, the peptide fingerprint was generated by scanning the 700- to 4,000-Da mass range using 1,000 shots acquired from 20 randomized regions of the sample spot at a 3,200-eV intensity with an OptiBeam on-axis Nd:YAG laser with a 200-Hz firing rate and 3- to 7-ns pulse width in positive reflectron mode. Raw data were deconvoluted with GPS Explorer software.
Nano-LC-MS/MS.
Peptides were analyzed by nano-LC-MS/MS with the Agilent 1100 nanoflow system (Agilent, Palo Alto, CA) connected to a new-generation hybrid linear ion Trap-Orbitrap mass spectrometer (LTQ-Orbitrap Elite; Thermo, Fisher Scientific) equipped with an Easy-Spray electrospray source. Chromatography of peptides prior to mass spectral analysis was accomplished with a capillary emitter column (PepMap C18; 3 µM, 100 Å, 150 by 0.075 mm; Thermo, Fisher Scientific) onto which extracted peptides were automatically loaded. The nano-HPLC system delivered solvents A (0.1% [vol/vol] formic acid) and B (99.8% [vol/vol] acetonitrile, 0.2% [vol/vol] formic acid) at 0.60 µl/min to load the peptides and 0.3 µl/min to elute peptides directly into the nano-electrospray apparatus over a 35-min 0% (vol/vol) solvent B to 40% (vol/vol) solvent B gradient, followed by 5 min over a 40% (vol/vol) solvent B to 100% (vol/vol) solvent B gradient. As peptides eluted from the HPLC/electrospray source, survey MS scans were acquired in the Orbitrap with a resolution of 120,000 followed by MS2 fragmentation of the 20 most intense peptides detected in the MS1 scan from 300 to 2,000 m/z; redundancy was limited by dynamic exclusion. Raw MS/MS data were converted to an mgf format by using MSConvert (ProteoWizard open source software for rapid proteomics tools development). Resulting mgf files were used to search against the sequence database containing expressed TR4 protein sequences plus common lab contaminants, using an in-house Mascot search engine 2.2.07 (Matrix Science, London, United Kingdom) with cysteine glutathionation, carbamidomethylation, dithiothreitolation or NEM modification, methionine oxidation, and asparagine/glutamine deamidation as variable modifications. Peptide mass tolerance was set at 15 ppm and fragment mass was 0.6 Da.
Statistical analysis.
Statistical analysis of data was done with the graphing program Prism 5 (GraphPad Software). Tabulated data were subjected to a one-way analysis of variance with Dunnett’s multiple-comparison posttest.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI-035681 and AI-111024 (B.K.). Work at the Université catholique de Louvain was supported by the National Fund for Scientific Research (FNRS), the Federal Office for Scientific, Technical and Cultural Affairs (Interuniversity Poles of Attraction Programme), and the Research Department of the Communauté française de Belgique (Concerted Research Action). Y.F.D. is a Research Director at the FNRS.
We thank Greg Sabbat (University of Wisconsin—Madison, Biotechnology Center) for assistance with the mass spectrometry analysis and Robert Gordon (Department of Pediatrics, School of Medicine and Public Health, University of Wisconsin—Madison) for assistance with graphic illustrations.
Footnotes
Citation Beaussart A, Brandhorst T, Dufrêne YF, Klein BS. 2015. Blastomyces virulence adhesin-1 protein binding to glycosaminoglycans is enhanced by protein disulfide isomerase. mBio 6(5):e01403-15. doi:10.1128/mBio.01403-15.
REFERENCES
- 1.Brandhorst TT, Wüthrich M, Warner T, Klein B. 1999. Targeted gene disruption reveals an adhesin indispensable for pathogenicity of Blastomyces dermatitidis. J Exp Med 189:1207–1216. doi: 10.1084/jem.189.8.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandhorst TT, Roy R, Wüthrich M, Nanjappa S, Filutowicz H, Galles K, Tonelli M, McCaslin DR, Satyshur K, Klein B. 2013. Structure and function of a fungal adhesin that binds heparin and mimics thrombospondin-1 by blocking T cell activation and effector function. PLoS Pathog 9:e1003464. doi: 10.1371/journal.ppat.1003464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandhorst TT, Gauthier GM, Stein RA, Klein BS. 2005. Calcium binding by the essential virulence factor BAD-1 of Blastomyces dermatitidis. J Biol Chem 280:42156–42163. doi: 10.1074/jbc.M507188200. [DOI] [PubMed] [Google Scholar]
- 4.Brandhorst TT, Wüthrich M, Finkel-Jimenez B, Warner T, Klein BS. 2004. Exploiting type 3 complement receptor for TNF-alpha suppression, immune evasion, and progressive pulmonary fungal infection. J Immunol 173:7444–7453. doi: 10.4049/jimmunol.173.12.7444. [DOI] [PubMed] [Google Scholar]
- 5.Finkel-Jimenez B, Wüthrich M, Klein BS. 2002. BAD1, an essential virulence factor of Blastomyces dermatitidis, suppresses host TNF-alpha production through TGF-beta-dependent and -independent mechanisms. J Immunol 168:5746–5755. doi: 10.4049/jimmunol.168.11.5746. [DOI] [PubMed] [Google Scholar]
- 6.Christianson HC, Belting M. 2014. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol 35:51–55. doi: 10.1016/j.matbio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Frank AT, Ramsook CB, Otoo HN, Tan C, Soybelman G, Rauceo JM, Gaur NK, Klotz SA, Lipke PN. 2010. Structure and function of glycosylated tandem repeats from Candida albicans Als adhesins. Eukaryot Cell 9:405–414. doi: 10.1128/EC.00235-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoiczyk E, Roggenkamp A, Reichenbecher M, Lupas A, Heesemann J. 2000. Structure and sequence analysis of Yersinia YadA and Moraxella UspAs reveal a novel class of adhesins. EMBO J 19:5989–5999. doi: 10.1093/emboj/19.22.5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartford O, McDevitt D, Foster TJ. 1999. Matrix-binding proteins of Staphylococcus aureus: functional analysis of mutant and hybrid molecules. Microbiology 145:2497–2505. [DOI] [PubMed] [Google Scholar]
- 10.Butta N, Arias-Salgado EG, González-Manchón C, Ferrer M, Larrucea S, Ayuso MS, Parrilla R. 2003. Disruption of the β3 663-687 disulfide bridge confers constitutive activity to β3 integrins. Blood 102:2491–2497. doi: 10.1182/blood-2003-01-0213. [DOI] [PubMed] [Google Scholar]
- 11.Rathore D, McCutchan TF. 2000. Role of cysteines in Plasmodium falciparum circumsporozoite protein: interactions with heparin can rejuvenate inactive protein mutants. Proc Natl Acad Sci U S A 97:8530–8535. doi: 10.1073/pnas.140224597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alsteens D, Beaussart A, El-Kirat-Chatel S, Sullan RM, Dufrêne YF. 2013. Atomic force microscopy: a new look at pathogens. PLoS Pathog 9:e1003516. doi: 10.1371/journal.ppat.1003516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufrêne YF. 2014. Atomic force microscopy in microbiology: new structural and functional insights into the microbial cell surface. mBio 5(4):e01363-14. doi: 10.1128/mBio.01363-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frelinger AL III, Cohen I, Plow EF, Smith MA, Roberts J, Lam SC, Ginsberg MH. 1990. Selective inhibition of integrin function by antibodies specific for ligand-occupied receptor conformers. J Biol Chem 265:6346–6352. [PubMed] [Google Scholar]
- 15.Lahav J, Jurk K, Hess O, Barnes MJ, Farndale RW, Luboshitz J, Kehrel BE. 2002. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood 100:2472–2478. doi: 10.1182/blood-2001-12-0339. [DOI] [PubMed] [Google Scholar]
- 16.Lahav J, Gofer-Dadosh N, Luboshitz J, Hess O, Shaklai M. 2000. Protein disulfide isomerase mediates integrin-dependent adhesion. FEBS Lett 475:89–92. doi: 10.1016/S0014-5793(00)01630-6. [DOI] [PubMed] [Google Scholar]
- 17.Dupres V, Menozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, Bompard C, Raze D, Dufrêne YF. 2005. Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat Methods 2:515–520. doi: 10.1038/nmeth769. [DOI] [PubMed] [Google Scholar]
- 18.Bustamante C, Marko JF, Siggia ED, Smith S. 1994. Entropic elasticity of λ-phage DNA. Science 265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 19.Marko JF, Siggia ED. 1995. Stretching DNA. Macromolecules 28:8759–8770. doi: 10.1021/ma00130a008. [DOI] [Google Scholar]
- 20.Hinterdorfer P, Dufrêne YF. 2006. Detection and localization of single molecular recognition events using atomic force microscopy. Nat Methods 3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- 21.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 22.Alsteens D, Garcia MC, Lipke PN, Dufrêne YF. 2010. Force-induced formation and propagation of adhesion nanodomains in living fungal cells. Proc Natl Acad Sci U S A 107:20744–20749. doi: 10.1073/pnas.1013893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zarbock A. 2013. β2-integrin activity: the role of thiols. Blood 121:3779–3780. doi: 10.1182/blood-2013-03-489872. [DOI] [PubMed] [Google Scholar]
- 24.Sehgal D, Vijay IK. 1994. A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Anal Biochem 218:87–91. doi: 10.1006/abio.1994.1144. [DOI] [PubMed] [Google Scholar]
- 25.Tripathi P, Beaussart A, Alsteens D, Dupres V, Claes I, Von Ossowski I, De Vos WM, Palva A, Lebeer S, Vanderleyden J, Dufrêne YF. 2013. Adhesion and nanomechanics of pili from the probiotic Lactobacillus rhamnosus GG. ACS Nano 7:3685–3697. doi: 10.1021/nn400705u. [DOI] [PubMed] [Google Scholar]
- 26.Alsteens D, Ramsook CB, Lipke PN, Dufrêne YF. 2012. Unzipping a functional microbial amyloid. ACS Nano 6:7703–7711. doi: 10.1021/nn3025699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardin AD, Weintraub HJ. 1989. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9:21–32. doi: 10.1161/01.ATV.9.1.21. [DOI] [PubMed] [Google Scholar]
- 28.Fernández-Recio J, Vázquez A, Civera C, Sevilla P, Sancho J. 1997. The tryptophan/histidine interaction in alpha-helices. J Mol Biol 267:184–197. doi: 10.1006/jmbi.1996.0831. [DOI] [PubMed] [Google Scholar]
- 29.Loewenthal R, Sancho J, Fersht AR. 1992. Histidine-aromatic interactions in barnase. Elevation of histidine pKa and contribution to protein stability. J Mol Biol 224:759–770. doi: 10.1016/0022-2836(92)90560-7. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson LM, Yakovenko O, Tchesnokova V, Thomas WE, Schembri MA, Vogel V, Klemm P, Sokurenko EV. 2007. The cysteine bond in the Escherichia coli FimH adhesin is critical for adhesion under flow conditions. Mol Microbiol 65:1158–1169. doi: 10.1111/j.1365-2958.2007.05858.x. [DOI] [PubMed] [Google Scholar]
- 31.Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA. 2006. Importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry 45:15020–15028. doi: 10.1021/bi061566o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piatek R, Bruździak P, Wojciechowski M, Zalewska-Piatek B, Kur J. 2010. The noncanonical disulfide bond as the important stabilizing element of the immunoglobulin fold of the Dr fimbrial DraE subunit. Biochemistry 49:1460–1468. doi: 10.1021/bi901896b. [DOI] [PubMed] [Google Scholar]
- 33.Davis CH, Raulston JE, Wyrick PB. 2002. Protein disulfide isomerase, a component of the estrogen receptor complex, is associated with Chlamydia trachomatis serovar E attached to human endometrial epithelial cells. Infect Immun 70:3413–3418. doi: 10.1128/IAI.70.7.3413-3418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pathak DT, Wei X, Bucuvalas A, Haft DH, Gerloff DL, Wall D. 2012. Cell contact-dependent outer membrane exchange in myxobacteria: genetic determinants and mechanism. PLoS Genet 8:e1002626. doi: 10.1371/journal.pgen.1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilpelainen I, Kaksonen M, Kinnunen T, Avikainen H, Fath M, Linhardt RJ, Raulo E, Rauvala H. 2000. Heparin-binding growth-associated molecule contains two heparin-binding β-sheet domains that are homologous to the thrombospondin type I repeat. J Biol Chem 275:13564–13570. doi: 10.1074/jbc.275.18.13564. [DOI] [PubMed] [Google Scholar]
- 36.Iozzi MF, Helgaker T, Uggerud E. 2011. Influence of external force on properties and reactivity of disulfide bonds. J Phys Chem A 115:2308–2315. doi: 10.1021/jp109428g. [DOI] [PubMed] [Google Scholar]
- 37.Baldus IB, Gräter F. 2012. Mechanical force can fine-tune redox potentials of disulfide bonds. Biophys J 102:622–629. doi: 10.1016/j.bpj.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Täger M, Kröning H, Thiel U, Ansorge S. 1997. Membrane-bound protein disulfide isomerase (PDI) is involved in regulation of surface expression of thiols and drug sensitivity of B-CLL cells. Exp Hematol 25:601–607. [PubMed] [Google Scholar]
- 39.Hahm E, Li J, Kim K, Huh S, Rogelj S, Cho J. 2013. Extracellular protein disulfide isomerase regulates ligand-binding activity of αMβ2 integrin and neutrophil recruitment during vascular inflammation. Blood 121:3789–3800, S1–S15. doi: 10.1182/blood-2012-11-467985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langenbach KJ, Sottile J. 1999. Identification of protein-disulfide isomerase activity in fibronectin. J Biol Chem 274:7032–7038. doi: 10.1074/jbc.274.11.7032. [DOI] [PubMed] [Google Scholar]
- 41.Sterkel AK, Mettelman R, Wüthrich M, Klein BS. 2015. The unappreciated intracellular lifestyle of Blastomyces dermatitidis. J Immunol 194:1796–1805. doi: 10.4049/jimmunol.1303089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santos CX, Stolf BS, Takemoto PV, Amanso AM, Lopes LR, Souza EB, Goto H, Laurindo FR. 2009. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J Leukoc Biol 86:989–998. doi: 10.1189/jlb.0608354. [DOI] [PubMed] [Google Scholar]
- 43.Barbouche R, Lortat-Jacob H, Jones IM, Fenouillet E. 2005. Glycosaminoglycans and protein disulfide isomerase-mediated reduction of HIV Env. Mol Pharmacol 67:1111–1118. doi: 10.1124/mol.104.008276. [DOI] [PubMed] [Google Scholar]
- 44.Brandhorst T, Wüthrich M, Finkel-Jimenez B, Klein B. 2003. A C-terminal EGF-like domain governs BAD1 localization to the yeast surface and fungal adherence to phagocytes, but is dispensable in immune modulation and pathogenicity of Blastomyces dermatitidis. Mol Microbiol 48:53–65. doi: 10.1046/j.1365-2958.2003.03415.x. [DOI] [PubMed] [Google Scholar]
- 45.Mammen M, Choi S-K, Whitesides GM. 1998. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed 37:2754–2794. [DOI] [PubMed] [Google Scholar]
- 46.López C, Sánchez J, Hermida L, Zulueta A, Márquez G. 2004. Cysteine mediated multimerization of a recombinant dengue E fragment fused to the P64k protein following immobilized metal ion affinity chromatography. Protein Expr Purif 34:176–182. doi: 10.1016/j.pep.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 47.MacKenzie CR, Hirama T, Deng SJ, Bundle DR, Narang SA, Young NM. 1996. Analysis by surface plasmon resonance of the influence of valence on the ligand binding affinity and kinetics of an anti-carbohydrate antibody. J Biol Chem 271:1527–1533. doi: 10.1074/jbc.271.3.1527. [DOI] [PubMed] [Google Scholar]
- 48.Kluszynski BA, Kim C, Faulk WP. 1997. Zinc as a cofactor for heparin neutralization by histidine-rich glycoprotein. J Biol Chem 272:13541–13547. doi: 10.1074/jbc.272.21.13541. [DOI] [PubMed] [Google Scholar]
- 49.Woodhead NE, Long WF, Williamson FB. 1986. Binding of zinc ions to heparin. Analysis by equilibrium dialysis suggests the occurrence of two, entropy-driven, processes. Biochem J 237:281–284. doi: 10.1042/bj2370281. [DOI] [PMC free article] [PubMed] [Google Scholar]