

Introduction
Inflammation is an important contributor to the pathology of diseases implicated in skeletal muscle dysfunction. A number of disorders including inflammatory myopathies and chronic obstructive pulmonary disorder (COPD) are characterized by chronic inflammation or elevation of the inflammatory mediators. While these disease states exhibit different pathologies, all have in common the loss of skeletal muscle mass and a deregulated skeletal muscle physiology. Pro-inflammatory cytokines are key contributors to chronic inflammation found in many of these pathologies. This section of the review focuses on some of the known inflammatory disorders like COPD, Rheumatoid Arthritis (RA) and inflammatory myopathies that display skeletal muscle atrophy and also provides the reader an overview of the mediators of inflammation, their signaling pathways, and mechanisms of action.
Myogenic Regulatory Factors
Skeletal muscle arises from mesodermal precursor cells whose differentiation is controlled by four highly conserved basic loop helix (bHLH) proteins known as Myogenic Regulatory Factors (MRFs). These MRFs, namely MyoD, Myf5, MRF4, and myogenin have overlapping patterns of gene expression. However, each plays a distinct role in myogenesis1. Myogenin is the only MRF required for viability2; 3. Mice lacking myogenin die at birth and have severe muscle defects. Although the absence of Myf5, MRF4, and MyoD is not lethal, each mutant nevertheless exhibits a distinct phenotype4.
Signaling pathways involved in skeletal muscle development
In response to environmental cues, skeletal muscle activates a variety of signaling pathways to undergo remodeling and sustain a muscle performance. The Wnt pathway is required during embryonic muscle development as well as during muscle stem cell self renewal and differentiation in the adult5. Insulin-like growth factor (IGF-1) exerts a tremendous influence on skeletal muscle proliferation and myoblast differentiation. IGF-1 signaling also induces hypertrophy to skeletal muscle cells by stimulating the phosphatidylinositol-3 kinase (PI3K)/Akt pathway, which activates mTOR and other downstream targets that stimulate protein synthesis6; 7. Mice null for the IGF-1 receptor exhibit reduced skeletal muscle mass and growth retardation8; 9, whereas muscle specific overexpression of IGF-1 causes muscle hypertrophy and increases protein synthesis.10; 11; 12. Fibroblast growth factor (FGF) is another signaling effector that plays essential roles in skeletal muscle development, as loss of FGFR1 signaling leads to reduced skeletal muscle mass and perturbed myofiber organization13. While some pathways positively influence the development of skeletal muscle, others act as negative modulators. During the induction of muscle atrophy, distinct transcriptional pathways are activated, which catalyze increased protein turnover and degradation14; 15. One such pathway is the ubiquitin-proteasome system16. In multiple models of skeletal muscle atrophy, E3 ubiquitin ligase genes, MurF1 and MAFbx/Atrogin-1 are significantly elevated17; 18; 19; 20. The inhibition of MuRF1 and MAFbx/Atrogin-1 involves FoxO family of transcription factors, which are phosphorylated by Akt 21; 22. Upon dephosphorylation, FoxO transcription factors namely FoxO1 and FoxO3 translocate to the nucleus and upregulate MurF1 and MAFbx/Atrogin-121.
In addition, the nuclear factor kappa B (NF-κB) signaling pathway has also been implicated in regulating the atrophy of skeletal muscle. In cultured C2C12 myoblasts NF-κB is essential for TNF-α to mediate an inhibition of muscle differentiation23. Likewise, skeletal muscle specific over expression of the NF-κB pathway promotes severe atrophy via the regulation of MuRF1.
Regeneration of skeletal muscle post damage or injury
Skeletal muscle cells possess the remarkable ability to regenerate after injury. Whether the injury is inflicted on a day-to-day basis and involves normal wear and tear, or a direct physical trauma like extensive physical exercise, the process of muscle regeneration is divided into two main phases; a degenerative phase followed by a regenerative phase. The degenerative phase is characterized by extreme muscle necrosis and disruption of the muscular architecture. This early phase is also accompanied by accumulation of an inflammatory infiltrate and activation of quiescent, resident muscle stem cells called satellite cells, which are essential for efficient muscle regeneration24; 25. The signals generated from an injured muscle are thought to activate inflammatory cells residing within the muscle, which in turn provide chemotactic signals to other circulating inflammatory cells. Neutrophils promote revascularization in muscle cells and are amongst the first cells to arrive at the site of injury. Among the cells of the myeloid lineage, eosinophils and macrophages also positively influence muscle regeneration. Eosinophils promote muscle regeneration by removing cellular debris and activating fibroblastic/adipogenic mesenchymal progenitors (FAPs)26. Two distinct populations of macrophages, which are present at the site of injury at different times, play key roles in muscle regeneration27. The pro-inflammatory M1 subtype is present 1 or 2 days post-injury and coincides with the degenerative phase of muscle repair, marked by activation and proliferation of satellite cells. Conversely, the anti-inflammatory M2 subtype peaks at 4 to 5 days post-injury and is associated with the regenerative phase of muscle repair27. Targeted ablation of neutrophils, monocytes, and macrophages severely disrupts muscle regeneration demonstrating their importance in the repair process. Recent studies have highlighted the role of non satellite cells in muscle regeneration. These include mesoangioblasts, which are associated with blood vessels28; 29; 30 and interstitial cells that express PICs, PW1 interstitial cells (PW1)31. Furthermore, a permissive cellular environment that promotes interactions between FAPs and satellite cells helps regulate muscle homeostasis32; 33.
The regenerative phase of muscle repair is characterized by cellular proliferation of the activated satellite cells, which re-enter the cell cycle and expand. Activated satellite cells express the transcription factor Pax7 which is required for expansion and cell survival, and further express MyoD that commits cells to a myoblast fate34; 35; 36. The process of differentiation is largely driven by MyoD and other MRFs such as myogenin, which in part regulate the decline of Pax7, which if left intact, signals cells to self-renew to satellite cells for a subsequent round of regeneration 37. Committed myoblasts proceed through the differentiation program, characterized by fusion with neighboring myoblasts, to form terminally differentiated, multinucleated myotubes. A successfully regenerated, mature muscle fiber is almost indistinguishable from a non-injured, undamaged muscle fiber.
Mediators of inflammation
During injury to adult skeletal muscle there are a number of key inflammatory mediators that govern the repair process. Both physiologic and pathogenic activities have been attributed to a selective number of inflammatory cytokines described below.
Interferon gamma (IFN-γ)
IFN-γ belongs to the type II IFNs and is secreted by CD4+ T helper cells, CD8 cytotoxic T cells, and natural killer cells (NK) cells38; 39. Recent evidence suggests that macrophages, dendritic, B and professional antigen presenting cells (APCs) also secrete IFN-γ40; 41; 42; 43. Mice lacking IFN-γ are born normally, but are more susceptible to bacterial, viral, and parasitic infections44. IFN-γ acts as an antiviral factor and influences a myriad of cellular and physiological processes. In addition, IFN-γ provides cytotoxic immunity by upregulating the major histocompatibility complex (MHC) class I and class II antigens. IFN-γ and IL-12 are the main cytokines that direct the primary response to antigen towards Th1 differentiation, while IL-4 is responsible for directing the antigen response towards a Th2 differentiation. IFN-γ stimulates IL-12 production in phagocytes and inhibits IL-4 secretion45; 46. The cytokine also primes macrophages for a rapid and elevated response to lipopolysaccharide (LPS) and toll-like receptor (TLR) agonists45, and contributes to multiple M1 macrophage dependent activities that include enhanced pinocytosis, increased microbial killing activity, induction of the NADPH-dependent phagocyte oxidase (NADPH oxidase) system, and priming for NO (nitric oxide) production47.
IFN-γ primarily signals through the JAK (Janus kinase)-STAT1 (Signal transducer and activator of transcription) pathway. The IFN-γ receptor comprises of two signal transducing IFNGR2 chains, with associated signaling machinery, and two ligand binding IFNGR1 chains. Both the IFNGR1 and IFNGR2 belong to the class II cytokine signaling family48. When IFN-γ binds to its receptor, the receptor associated protein tyrosine kinases, JAK1 and JAK2 are activated49. This leads to the phosphorylation of STAT1, which then dimerizes and subsequently translocates to the nucleus, where it binds to its target promoters, including the pIV promoter of CIITA50, to activate gene expression. The JAK1-STAT1 pathway has been shown to play prominent roles in myogenesis51. JAK1 and STAT1 are required for myoblast proliferation and display a potent anti-differentiation effect, which appears specific to STAT1, as similar activities cannot be reproduced by family members, STAT2, 3, 5A, or 5B.
Numerous studies have also shown that IFN-γ influences skeletal muscle homeostasis and repair52. Transient administration of exogenous IFN-γ following injury has been shown to improve healing and limit fibrosis53. This response is consistent with the phenotype that IFN-γ null mice exhibit defective muscle regeneration and development of fibrosis52. During early stages of muscle regeneration, IFN-γ expression is upregulated in muscle itself 52 and its levels decline as the regeneration stage transitions from proliferation to differentiation. Mechanistically, IFN-γ improves muscle repair by regulating the migration of specific immune cells at the site of injury by upregulating chemokine and adhesion molecules that include chemokine C-C motif ligand 5 (Ccl-5, RANTES) and Ccl-254; 55; 56; 57 and intracellular adhesion molecule (ICAM).
Similar to inflammatory cytokines like TNF, where the effects on skeletal muscle differentiation appear to be dose dependent58, IFN-γ is also able to impede myogenesis when administered in high doses in vitro. In addition, IFN-γ expression is elevated in mdx mouse muscles, which is a mouse model for muscular dystrophy, at a time of macrophage-mediated muscle damage59. Ablation of IFN-γ in the mdx animals improves muscle function and promotes muscle strength60. These types of studies reinforce the dose dependent effects of IFN-γ and show that at chronic levels this cytokine exhibits anti-myogenic properties. Recently, the mechanism by which IFN-γ inhibits muscle differentiation was resolved. The cytokine induces the expression of the MHC class II transactivator, CIITA, which acts by directly binding to and inhibiting the function of myogenin61. The absence of myogenin function leads to a reduction in muscle specific gene expression and transcription factors that drive terminal differentiation61; 62; 63. However, in IFN-γ treated myotubes, myogenin expression is unaffected. CIITA mediates the anti-differentiation activity of IFN-γ activity by catalyzing the initial recruitment of a Jumonji family protein JARID2, followed by the subsequent recruitment of the polycomb repressive complex 2 (PRC2) to the promoters of muscle specific genes64. Studies have shown that the PRC2 complexes are silenced during muscle differentiation65. However, elevated levels of circulating IFN-γ maintain the expression of PRC2 which silences muscle specific genes by methylating the DNA associated histone mark, H3K2764.
Interleukin-17 (IL-17)
IL-17A and IL-17F belong to a six-member family of IL-17 cytokines66. Specialized T cells, known as Th17 cells, are the primary source of IL-17A and IL-17F in adaptive immunity67; 68. However, other sources such as lymphocytes and neutrophils also contribute to IL-17 production69. IL-17A, previously termed as CTLA8, signals via surface receptors (IL-17R) on target cells. IL-17RA was the first receptor to be identified, followed by the subsequent identification of IL-17RB, IL-17RC, IL-17RD, IL-17RE70; 71; 72.
IL-17 mainly mediates immune function by stimulating the production of pro-inflammatory cytokines TNF-α, IL-6, IL-1β, and chemokines C-X-C motif ligand 1 (CXCL1), C-C- motif ligand 2 (CCL2), CCL7, CCL20, as well as matrix metalloproteinase 3 and 9 (MMP3 and 9)73; 74. Albeit its importance in protecting the host from invasive pathogens, similar to IFN-γ, dysregulated IL-17 production can result in excessive cytokine production and chronic inflammation leading to tissue damage and autoimmunity. The IL-17 family has been implicated in several autoimmune diseases including multiple sclerosis (MS), RA and inflammatory bowel disease75; 76. Recent studies have shown that IL-17 mRNA is elevated in muscle biopsies from Duchenne muscular dystrophy (DMD) patients suggesting a possible pathogenic role77.
Interleukin-6 (IL-6)
IL-6 is a pleiotropic cytokine which controls and coordinates multiple immune responses78. IL-6, unlike other cytokines has a unique property of exerting both pro and anti-inflammatory effects depending on the local tissue mileu of the immune cells and the micro environment79. The IL-6 family of cytokines includes IL-11, IL-31, IL-27, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary inhibitory factor (CNTF), andcardiotropin-1 (CT-1)79. In classical IL-6 signaling, IL-6 exerts its signaling activities by binding to the membrane bound IL-6R receptor on the target cells. Subsequently, IL-6/IL-6R complex associates with a membrane glycoprotein receptor and a signal transduction subunit, gp130, which homodimerizes to allow signal initiation and activation of the JAK-STAT3, PI3K and ERK signaling pathways80; 81. The membrane bound IL-6 receptor, however, is expressed on selected cells such as neutrophils, macrophages, hepatocytes, and some T cells. IL-6 signaling can also occur in trans via gp130 and the soluble IL-6 receptor (sIL-6R). The trans signaling is critical for lymphocyte trafficking during inflammation, regulation of adhesion molecule expression on endothelial cells, and T cell proliferation during colon cancer82; 83; 84. Studies have shown that an upregulation in circulating levels of IL-6 enhances fat oxidation and improves glucose uptake.
Increasing evidence suggests that muscle cells are a source of IL-6. This cytokine is detected in a contracting skeletal muscle after 30 minutes of exercise85; 86; 87. In cultured C2C12 myoblasts, IL-6 mRNA knockdown reduces muscle specific gene expression88. IL-6 has also been identified as an essential regulator of muscle stem cell mediated hypertrophy89; 90. IL-6 deficient mice exhibit severe muscle atrophy and loss of IL-6 results in proliferation and migration defects in myoblasts possibly due to reduced activation of STAT389. In cases of muscle injury, IL-6 levels dramatically increase, but in control animals the levels return to normal unlike the age matched mdx mice, which exhibit persistently higher levels of IL-691. In its heightened state, IL-6 has been linked to muscle wasting and chronic inflammation in mdx mice. Although the underlying mechanism remains to be elucidated, chronic levels of IL-6 have been shown to decrease the pro-myogenic factor, IGF-1 by directly acting on the liver and muscle IGF-192. Elevated levels of IL-6 have also been associated with arthritis, Crohn’s disease, and other inflammatory diseases93; 94.
Interleukin-4 (IL-4)
IL-4 is produced by NK, activated T, mast, basophils, and eosinophil cells. This cytokine regulates a variety of immune functions including isotype switching in B cells and differentiation of T cells95; 96. IL-4 also induces the expression of MHC class II molecules and downregulates the expression of pro-inflammatory cytokines, TNF-α and IL-197; 98; 99. IL-4 signals through two distinct cell surface receptor complexes IL-4R type I, specific for IL-4 and IL-4R type 2, which is shared by IL-13100. Both IL-4 and IL-13 utilize the JAK-STAT signaling pathway for signal transduction. However, IL-4Rα associates with JAK1 and JAK3 while IL-13Ra associates with JAK2 and not JAK3. During signal initiation, IL-4 binds to its receptor, IL-4Rα, which gets autophosphorylated. This leads to the phosphorylation of JAK, which in turn phosphorylates and activates STAT6. Phosphorylated STAT6 dimerizes, migrates to the nucleus, and binds to the consensus sequences located in the promoters of IL-4 target genes101. A second signaling pathway that can be activated by IL-4 through the IRS family of proteins is the phosphatidylinositol 3-kinase (PI3K) pathway. Binding of IL-4 to IL-4Ra leads to autophosphorylation of IL-4Ra. IRS proteins get recruited to IL-4Ra and then get phosphorylated. Tyrosine phosphorylated IRS proteins in turn associate with cytoplasmic signaling molecules containing SH2 domains including the p85 subunit of PI3K leading to the activation of the catalytic subunit of PI3K, p110101; 102.
In muscle cells, induction of IL-4 by NFATc2 has been shown to promote myoblast fusion possibly by increasing the expression of cell adhesion103. In fact, IL-4 has been shown to regulate the expression of ICAM1 on myoblasts104 and vascular cell adhesion molecule 1(VCAM-1), which is required for myotube formation in vitro in smooth muscle cells60; 105. IL-4 is expressed during early stages of muscle injury and is a dominant regulator of alternative macrophage (M2) activation that increases during the later stages of the muscle injury and promotes efficient muscle regeneration106; 107. IL-4 also facilitates muscle regeneration by controlling the functions of FAPS26. Stimulation with IL-4 directs the FAPs to proliferate as fibroblasts and support myogenesis by clearing out necrotic debris. In the absence of IL-4, FAPs differentiate into adipocytes, resulting into fatty degeneration of skeletal muscle26. Muscle biopsies from patients suffering from idiopathic in ammatory myopathies show an upregulation in IL-4 gene and mRNA expression108; 109; 110.
Interleukin-10 (IL-10)
IL-10 also known as CSIF (cytokine synthesis inhibitory factor) was discovered through a screen for factors that inhibited cytokine production by Th1 cells111. IL-10 signals through IL-10R and IL-10R2 receptors, which belong to the interferon family112; 113. IL-10R1 (IL-10Rα) is expressed constitutively on most hemopoietic cells and exhibits an induced expression in non-hemopoietic cells114; 115; 116; 117. IL-10R2 (IL-10β) is expressed on most tissues113; 118. Like IFN-γ, IL-10 signals mainly through the JAK-STAT pathway with STAT3 being indispensible for IL-10 signaling in all IL-10 responsive cells119; 120.
IL-10 is primarily known for its function in inhibiting the pro-inflammatory cytokines such as IFN-γ, TNF-α and IL-6121. IL-10 also inhibits the production of CSF, IL-1α, IL-1β, IL-12,IL-18, G-CSF, M-CSF, GM-CSF, as well as C-C and C-X-C cytokines112. IL-10 converts the cytolytic M1 macrophages to the more regenerative M2c phenotype, which express markers like CD206, arginase, IL-4α, and CD163122; 123; 124; 125; 126. IL-10 has a direct effect on muscle cells. Muscle cells express IL-10 independent of the myeloid cell population that resides in the muscle127. The role of IL-10 in inhibiting the pro-inflammatory cytokines could be deemed protective as IL-10 rescues the block on myogenin by IGF-1, which is induced by TNF-α129. IL-10 also prevents TNF-α induced phosphorylation of JNK and prevents upregulation of IL-6 expression by TNF-α in myoblasts128; 129, all of which are considered anti-myogenic signals.
In an injured muscle, IL-10 is key in directing the switch between M1 to M2 macrophages130. A recent study from the Tidball lab demonstrated that the expression of IL-10 and its receptor are elevated in mdx mice at the onset of pathology as well as during the regeneration phase131. Therefore, unlike pro-inflammatory cytokines described above whose activities switch from pro to anti-myogenic in a dose dependent manner, persistent levels of IL-10, as shown in mdx mice, remain anti-inflammatory and pro-regenerative, a feature of this cytokine that might be exploited for therapy.
Transforming growth factor- beta (TGF-β)
The multifaceted TGF-β superfamily is crucial is regulating normal physiology and has also been described in a plethora of studies as a contributor to pathogenesis132,133 The TGF-β superfamily consists of various signaling molecules including isoforms of TGF-β (1 to 3), Bone morphogenic proteins (BMPs 1 to 20), growth and differentiation factors (GDFs), activins (A and B) and inhibins (A and B)133. TGF1-β is synthesized as a precursor molecule which eventually upon getting cleaved into a mature, but inactive form, complexes with a portion of the precursor peptide known as the latency associated peptide (LAP)134. This inactive TGF1-β-LAP complex associates with latent TGF binding proteins (LTBPs), which release TGF1-β from the ECM. For the initiation of signal transduction, TGF1-β binds to its receptor, TGF1-βR type II or ALK (activin like kinase receptor) 1 or ALK5, which leads to the phosphorylation of two receptor-associated Smads, Smad2 and Smad3. Phosphorylated Smad2 and Smad3 proteins then heterodimerize with a common mediator Smad, Smad4, which as a Smad2/3 -Smad4 complex, translocates to the nucleus to activate the transcription of its target genes by cooperatively associating with other transcriptional factors and coactivators135. Apart from this canonical signaling pathway, TGF-β also signals in a non-canonical manner, which is Smad independent. Induction of this pathway leads to Ras and TGF-β activated kinase 1(TAK1) activation, which subsequently stimulates the MAPK kinases, p38/JNK 136. Activation of the MAPK pathway however, can occur by both Smad dependent and independent fashion indicating that a possible cross talk exists between the TGF-β canonical and non-canonical signaling pathways136; 137. The TGF-β signaling is negatively regulated by the Smads, Smad6 and Smad7 or by a ubiquitin proteasomal degradation pathway mediated by Smad ubiquitin regulatory factors (SMURFs)138. In skeletal muscle, perhaps the most extensively studied ligand of TGF-β family is myostatin. Myostatin binds to the activin receptor type II A (ActR-IIA),ActR-IIB, or ALK 4 or 5. Both the pathways converge in the activation of Smad2 and 3 followed by the dimerization with Smad4139. Interestingly, Smad7, the inhibitor of the TGF-β/Myostatin signaling displays pro-myogenic functions through its interactions with MyoD1140.
Studies have shown that TGF-β inhibits skeletal muscle differentiation and also modulates proliferation of satellite cells141; 142; 143. Smad3, which is the key mediator of the inhibitory effects of TGF-β on myogenesis, physically interacts with MyoD1 to inhibit MyoD1 dependent transactivation141. Furthermore, TGF-β not only inhibits the transactivation properties of MyoD1, but also inhibits the transcription of MyoD1144. TGF-β has also been shown to block the transcriptional activity of myogenin and thereby inhibit muscle differentiation145. Studies also demonstrate that TGF-β is upregulated in the skeletal muscle post-injury or following exercise. TGF-β is thought to participate in the inflammatory response involved in muscle repair and plays a key role in promoting the transformation of myoblasts into fibrotic tissue. This role of TGF-β as a driver of fibrosis is repeated in numerous pathologies, such as in idiopathic pulmonary fibrosis. This disease is characterized by the accumulation of inflammatory infiltrate and increased collagen deposition resulting in loss of alveolar architecture. Lung biopsies from these patients show activated fibroblasts expressing collagen and fibronectin and alveolar macrophages expressing excessive levels of TGF-β protein and mRNA146.
GDF11 and myostatin are two highly related TGF-β family members, but have very distinct biological functions. GDF11 is more widely expressed and was recently identified for its ‘rejuvenating’ effects on skeletal muscle, suggesting that restoring systemic GDF11 levels may help prevent age related dysfunction in mice147. Myostatin however negatively regulates skeletal muscle mass during development. The myostatin gene is expressed in the heart, skeletal muscle, and adipose tissue. Mice homozygous null for myostatin exhibit hypermusculature due to increased muscle mass. Myostatin null animals also show decreased fat, increased muscle strength and change in fiber type distribution leaning more towards type IIb fibers148. In aged mice, short-term inhibition of myostatin enhances muscle regeneration and satellite cell activation. Not surprisingly, overexpression of myostatin leads to excessive muscle wasting, similarly to that observed in cancer cachexia149.
There is another distant and divergent member of the TGF-β family known as GDF15 (also known as macrophage inhibitory cytokine (MIC-1)) that plays a role during chronic inflammation150. Elevated circulatory levels of MIC-1 are found in chronic inflammatory diseases like atherosclerosis and RA indicating endothelial activation and vascular inflammation151; 152. In patients with acute myocardial infarction, enhanced levels of GDF15 have been reported which are correlated with inflammatory biomarkers153; 154. GDF15 deficiency inhibits the progression of atherosclerosis and regulates IL-6 and TGF-β dependent inflammatory responses155; 156. However, studies have revealed that GDF15 also has broad anti-inflammatory and immune suppressive properties157.
Tumor Necrosis Factor-α (TNF-α)
TNF-α, also known as cachectin is a prototypic ligand of the TNF super family. It plays central roles in inflammation, apoptosis and immune system development. TNF-α is produced by a wide variety of immune and epithelial cells158 and activates a number of signaling pathways that mediate cell type specific, pleotrophic responses. At least 3 major pathways are activated by TNF-α including activation of c-Jun terminal kinase (JNK) and activator protein-1 (AP1), stimulation of apoptosis via TNF-α receptor complex, and Fas associated protein with death domain (FADD) and activation of NF-κB, which is a primary mediator of transcriptional control and catabolic signaling. TNF-α signaling can be mediated by TNFR1 or TNFR2. Binding of TNF-α to its receptor, initiates a IKK-γ dependent signaling cascade that activates the inactive p50/p65 heterodimer and causes its translocation into the nucleus where it decreases the expression of the pro-myogenic transcription factor, MyoD159.
In skeletal muscle, TNF-α influences satellite cell proliferation and accelerates the G1 to S phase transition162. Administration of neutralizing antibodies against TNF-α to mdx mice increases the number of Pax7+ve cells and decreases the inflammation based activation of p38/MAPK signaling163. The observed increase in Pax7 expression is due to the inhibition of association of the repressive PRC2 complex subunits with Pax7 promoter163.
TNF-α stimulates the production of catabolic cytokines and induces anorexia. In dystrophic muscle, elevated levels of TNF-α inhibit the regenerative potential of satellite cells by epigenetically silencing Notch 1164. TNF-α has been attributed to a number of inflammatory diseases like COPD and is associated with loss of muscle mass in COPD patients165.
Tumor Necrosis Factor Like Weak Inducer of Apoptosis (TWEAK)
The cytokine tumor necrosis factor like weak inducer of apoptosis (TWEAK) is a member of the TNF superfamily. TWEAK is initially synthesized as a 249-amino-acid protein comprising of a C-terminal extracellular domain, a transmembrane domain, and a N-terminal intracellular domain, which gets proteolytically cleaved at its C terminal domain into an soluble form166. The soluble form trimerizes and functions as a homo trimer. While the specific conditions for the existence of both the forms of TWEAK have not been understood, TWEAK is fully functional both in its cell surface associated transmembrane form and its soluble form166. TWEAK has been detected as a membrane anchored protein in IFN-γ activated human monocytes167 and in human CD4+ cells168.
TWEAK binds to the fibroblast growth factor inducible 14 (Fn-14) receptor, which also belongs to the TNF superfamily of receptors and is characterized as a type Ia transmembrane receptor lacking a cytoplasmic death domain169; 170. The unprocessed TWEAKR/Fn-14 contains a 27-aa N terminal signal peptide sequence and a highly hydrophobic region which functions as a plasma membrane spanning domain. The mature form of TWEAKR/Fn-14, which is produced after proteolytic cleavage is predicted to be 102 aa in length, making it the smallest member of the TNF family of receptors169; 170. Both the human and murine forms Fn14 contain a highly conserved 29 aa cytoplasmic tail and a putative TRAF binding site171.
In cultured C2C12 myotubes, treatment with TWEAK leads to a reduction of MyHC, possibly through an upregulation of the muscle specific E3 ubiquitin ligases MuRF1 and MAFbx in a dose dependent manner172. In mice, the treatment with TWEAK results in reduction in body weight and fiber cross sectional area compared to the littermates172. Furthermore, transgenic overexpression of full-length TWEAK cDNA using a muscle creatine kinase promoter shows severe muscle wasting172.
The TWEAK-Fn14 axis regulates a number of physiological processes like apoptosis, proliferation, differentiation, cell survival and angiogenesis. In various cell types including skeletal muscle, TWEAK has been shown to activate NF-κB, p44/p42 MAPK, JNK, and AP-1. However, the TWEAK-Fn14 axis is also often linked to the pathogenesis of systemic lupus, neuro inflammation, cardiac dysfunction, RA, MS, and a number of cancers 166. Increased expression of TWEAK is also associated with the induction of fibrosis and a broad pro-inflammatory and cell death/tissue-damaging activity173. This could be through its direct action on fibroblasts and their progenitors, or the cooperation of TWEAK with other cytokines that become upregulated during various disease states171.
The TWEAK/Fn14 pathway is well known for its involvement in modulating inflammation in auto immune and chronic inflammatory disorders. TWEAK induced pro-inflammatory responses stimulate the expression of chemokines, cytokines, adhesion molecules and MMPs, from endothelial, epithelial, and other non hematopoetic cell types 174. TWEAK can also cooperate with other pro-inflammatory cytokines like TNF-α and IL-17, to name a few, to augment inflammatory response173; 175. In addition to TWEAK, a variety of Fn14 inducing stimuli like IFN-γ, TNF-α and IL-1β have been recently identified which could very well explain the diverse outcomes derived by the TWEAK-Fn14 pathway, alone or in combination with other cytokines166.
Inflammatory disorders leading to muscle loss
Chronic obstructive pulmonary disorder (COPD)
COPD is one of the leading causes of morbidity and mortality all around the world. Primarily, COPD is a respiratory disease and is diagnosed based on abnormal lung function and symptoms such as dyspnea and chronic cough production. However, along with the symptoms described above, COPD presents itself with a low-grade systemic inflammation, which results in skeletal muscle dysfunction.
The idea that skeletal muscle dysfunction could be an impairment in patients with COPD was first described in a study by Killian et. al176, in which the exercise capability in patients with COPD was tested. Approximately 40% of the COPD patients exhibited early termination of exercise due to symptoms of leg fatigue, which was far greater than their rating of shortness of breath at the end of the exercise study. In addition to contributing to a reduced ability to exercise, decreased health status and diminished muscle function, muscle wasting is a determinant to morbidity in COPD, independent of the pulmonary disorders. Muscle wasting in COPD has been demonstrated as the loss of fat free mass at the whole body level and also at the level of the extremities177. In addition to the depletion of muscle mass, fiber type switching from type I to type II occurs resulting in decreased muscle oxidative capacity. This switch not only reduces endurance,178 but also accelerates muscle atrophy179. Over the last two decades, research has focused on identifying the potential triggers of muscle wasting in COPD. Based on biochemical and immunohistochemical studies a number of factors have been identified as potential causes for muscle wasting in COPD. These include malnutrition, hypoxemia, disuse and inflammation. Low physical activity or a sedentary life style are common in COPD patients180. Inactivity and/or muscle disuse are well known triggers for muscle atrophy. Hypoxemia, i.e. reduced arterial oxygen tension is prevalent in COPD patients. Currently, most of the evidence implying that hypoxemia and subsequent tissue hypoxia can trigger muscle wasting is purely based on observations made from healthy patients and experimental models. Studies in mountaineering expeditions in which subjects are exposed to high altitudes and hypoxia are reported to have decreased muscle mass181 and reduced muscle fiber size despite of physical activity182. While being a potential trigger, the precise mechanisms by which hypoxemia induces muscle atrophy are still unknown. Malnutrition is reported in at least one third of the patients with COPD and the severity advances with the progression of the disease. Some data demonstrate that the positive effects of nutritional supplementation lead to preservation of fat free mass in COPD patients. However, for most patients increasing energy intake alone does not rescue muscle atrophy. The protein synthetic rate presumably goes down during starvation, which is supported by one study183, but follow up reports have argued against such a mechanism184; 185. While malnutrition, hypoxemia, and inactivity have all been linked to muscle wasting in COPD, more recent attention has shifted to the relation between cachexia and inflammation.
Findings show that COPD is characterized by the elevation of inflammatory factors such as IL-6, TNF-α, IL-8, and C-reactive protein. In addition, COPD patients also show evidence of elevated expression of adhesion molecules in plasma and bronchoalveolar fluids, as well as an increase in the generation of ROS186. Possibly, muscle wasting in COPD results from bursts of ROS in combination with inflammatory cytokines. How downstream factors such as NF-κB are involved in COPD-induced muscle loss remains controversial. The variable levels of NF-κB that have been reported in COPD patients may reflect the different stages of disease progression that involve both stable and severe muscle loss. Results from animal models of COPD appear more consistent that NF-κB is activated and associated with acute pulmonary inflammation where there is a connection with muscle atrophy, but further evidence is required to validate the role of inflammatory factors and NF-κB in this pathology.
Rheumatoid arthritis (RA)
Rheumatoid arthritis is a chronic, autoimmune, debilitating disease that generally occurs within the fourth and sixth decade of life. The disease is more common in men than women. RA is primarily characterized by joint pain, swelling, stiffness, and accompanied by skeletal muscle wasting. RA is also characterized by sustained inflammatory synovitis187. Persistent synovial inflammation results in bone erosion and cartilage damage, due to the loss of functionality in individuals affected by RA188. Although RA is classified as a multi-system disease, inflammatory cytokines are recognized as key mediators in its pathology.
The synovial membrane in patients with RA is characterized by hyperplasia, increased vascularity, and infiltration of inflammatory cells, primarily of CD4+ T cell origin188. Antigen activated CD4+ T cells stimulate monocytes, macrophages, and synovial fibroblasts to release pro-inflammatory cytokines, IL-1, IL-6, IL-18 and TNF-α188, which for the most part can be detected in the synovial fluid of RA patients 189. Furthermore TNF-α and IL-1 act as potential stimulators of mesenchymal cells that release MMPs to destroy tissue and at the same time inhibit the production of TIMPs, inhibitors of MMPs190. Transgenic mice over expressing TNF-α spontaneously develop inflammatory arthritis191. In vitro studies with synovial cultures from RA patients demonstrate that blocking TNF-α with antibodies drastically reduces the expression of pro-inflammatory cytokines192. This suggests that inhibition of TNF-α might have a more global effect in treatment of RA than neutralizing other cytokines.
IL-18 is another cytokine elevated in synovial fluids and synovial tissues of patients with RA. Within the RA joint, IL-18 contributes to the inflammatory process by stimulating leukocyte extravasation through the upregulation of endothelial adhesion molecules193; 194 and the release of chemokines195 from RA synovial fibroblasts through activation of NF-κB. Additionally, IL-18 acts synergistically with IL-12 to induce production of IFN-γ from T cells further aggravating joint inflammation and cartilage destruction. IL-32 is another inducer of pro-inflammatory cytokine produced from lymphocytes that infiltrate severely inflamed synovial tissues in patients with RA, and intensity of IL-32 staining correlates disease severity196. In mice models of inflammatory arthritis, studies have shown that recombinant IL-32 injections in naive mice result in joint swelling and infiltration of inflammatory infiltrates196. However, similar injections in TNF-α deficient mice did not show the same phenotype, suggesting that the ability of IL-32 to induce joint inflammation is in part dependent on TNF-α167. Due to its close relationship with TNF-α, IL-32 is being considered as a potential target for therapies against RA.
IL-6 has been regarded as a key player in promoting joint and systemic inflammation and inducing immunological abnormalities in RA. IL-6 promotes muscle wasting and joint destruction in RA by activating release of adhesion molecules and inducing the secretion of monocyte chemoattractant protein 1 (MCP-1) and IL-8197; 198. IL-6 and IL-1 can synergistically enhance the production of MMPs from synovial cells, which leads to joint and cartilage destruction199. Also, in synovial fibroblasts, IL-6 induces the secretion of vascular endothelial growth factor (VEGF), which leads to enhanced angiogenesis and vascular permeability of the synovial tissue200. The pathological effect of IL-6 has been well documented in animal models. Collagen induced arthritis is an established model for RA in which an injection of type II collagen in mice causes an immune response directed at connective tissue. In this model, activated T cells produce augmented amounts of Th1 and Th17 cytokines. Suppression of IL-6 through gene knockout experiments reduces cytokine production and ameliorates the symptoms of RA201; 202. Inhibiting IL-6 by antibody or gene deletion has yielded similar results in other models of RA203. Such results are consistent with findings documenting elevated levels of IL-6 in the serum and synovial fluid of RA patients204.
More than two thirds of the people with RA suffer from loss of skeletal muscle mass or ‘rheumatoid cachexia’, a term coined by James Paget in 1873. Unlike the general definition of cachexia, which includes wasting of skeletal muscle and adipose tissue, rheumatoid cachexia is defined as a loss of body cell mass, predominantly in the skeletal muscle and with no or little weight loss in the presence of increased or stable fat mass205. While precise mechanisms for the cause are still under investigation, it is believed that elevated levels of pro-inflammatory cytokines are one of the leading causes of rheumatoid cachexia. TNF-α and IL-1 likely act as central mediators of muscle wasting in RA206. Studies in rat models of adjuvant arthritis207 show that TNF-α blockade alone rescues the loss of skeletal muscle, suggesting that TNF-α functions as an important contributor of cachexia in RA, but is also likely not the sole mediator. In support of this notion, inhibition of both IL-1 and TNF-α is more effective in reducing muscle wasting in cachexia208 than individual blockage alone, thus reinforcing the concept that IL-1 and TNF-α act synergistically to promote cachexia in RA208.
TNF-α has also been shown to reduce the action of peripheral insulin, which might be another mechanism by which this cytokine contributes to cachexia209; 210. Another peculiar characteristic of patients with RA is that they exhibit elevated resting energy expenditure211. Generally, under normal conditions, there is a balance between the rate of protein degradation and the rate of protein synthesis212. This balance regulates important physiological functions and enables adaptation to physiological and environmental cues. In RA, chronic inflammation alters this balance towards net protein catabolism causing an increase in the resting energy expenditure211, a net efflux of amino acids from muscle to the liver, and an increase in the synthesis of acute phase proteins, fibrinogen and CRP, the sum of which is predicted to lead to cachexia.
Effective therapies for RA have concentrated on targeting the cytokines that mediate rheumatoid cachexia. For example, Tocilizumab, a humanized anti-IL-6 receptor antibody, is already in clinical trials213. Patients treated with Tocilizumab alone or in combination with methotrexate exhibit significant improvements214. TNF-α blocking antibodies like D2E7215 or Infliximab216 or the decoy receptor, Etanercept217; 218, all demonstrate some form of clinical improvement for RA. In addition, in a randomized, double blind, placebo controlled trial of patients with RA, treatment with recombinant human IL-1 receptor antagonist resulted in moderate clinical improvement and decreased progression of erosions as assessed by radiography219. A drawback of using an IL-1 receptor antagonist therapeutically is its short half-life (6 hours)220, which demands frequent injections and high concentrations. As opposed to the responses obtained with blocking IL-1, IL-6, and TNF, clinical trials undertaken to target IL-4 and IL-10188 have met with limited benefit.
Inflammatory myopathies (Myositis)
Idiopathic inflammatory myopathies are autoimmune muscle disorders that involve inflammation of the muscle or the surrounding tissues such as blood vessels that supply blood to the muscles. Another term used to describe inflammatory myopathies is myositis- ‘myo’ meaning muscle and ‘itis’ meaning inflammation. These myopathies are considered to be auto immune in origin, due to their predominance of T and B cells in the affected muscle, the over expression of MHC class I and II molecules by muscle cells, and the association with myositis specific auto antibodies. Nevertheless, the exact nature of the antigens mediating these myopathies remains to be defined. Inflammatory myopathies are classified into three main types, polymyositis, inclusion body myositis, and dermatomyositis. Although each subtype presents with their own distinct clinical features, there are some common symptoms shared among all three subtypes including progressive muscle weakness, muscle atrophy, and vasculature damage surrounding muscle fibers. Progressive muscle weakness leads to additional symptoms such as shortness of breath, difficulty in swallowing and speaking, heart arrhythmias, and fatigue.
Polymyositis (PM) and inclusion body myositis (IBM)
PM (‘inflammation of many muscles’) is generally regarded as a prototypic T cell mediated autoimmune myopathy whereas IBM on the other hand is classified by a more peculiar pattern of muscle wasting, longer clinical course, and a T cell dominant auto immune response in combination with myofiber degeneration. The degeneration aspect is characterized by the appearance of vacuoles in muscle cells, deposition of abnormal proteins, and filamentous inclusions, from which IBM derives its name. The existence of PM as a separate entity is controversial given the frequent coexistence of PM and IBM221; 222. The controversy relates to whether PM occurs as a muscle specific disease or an autoimmune disorder, given the similarities between IBM and PM and the more frequent occurrence of IBM223.
In both PM and IBM, CD8+ T cells are thought to be the primary effector cells causing muscle damage and weakness224. CD8+ T cells proliferate and differentiate locally in the muscle. Because muscle presents antigen, these cells are targeted by autoinvasive CD8+ T cells to induce their turnover 225; 226; 227. This is thought to occur through the secretion of MCP1 by the T cells to recruit monocytes228, which in turn express pro-inflammatory cytokines like TNF-α, IFN-γ and IL-1 to induce a toxic effect on skeletal muscle cells 229. Yet another pro-inflammatory cytokine, macrophage inhibitory factor (MIF), is elevated in PM and thought to contribute to the turnover of muscle230.
Dermatomyositis (DM)
DM is characterized by the presence of a typical DM rash on the face (heliotrophe rash), hand, elbows (Gottron’s papules), and torso231. DM can be classified into various subgroups based on its childhood or adult forms (Juvenile DM or adult DM), or based on whether it is associated with malignancy or a part of an overlapping syndrome (Cancer associated DM). DM can be further classified based on cutaneous manifestations and the severity of muscle weakness (DM with systemic manifestations or amyopathic DM)231; 232; 233. DM is thought to be initiated by the activation of the complement pathway leading to depletion of muscle fibers 229. However, how the complement pathway is activated in DM, is still unknown. One notion is that immunoglobins accumulate on intramuscular capillaries, which causes the activation of the complement cascade, and in turn triggers the production of pro-inflammatory cytokines and chemokines. These pro-inflammatory molecules, then upregulate adhesion molecules on endothelial cells that go on to stimulate B, T, and dendritic cells, leading to muscle necrosis229; 232. This inflammatory cascade within the perivascular and perimysial mileu is comprised of B and CD4+ T helper cells, IFN-γ producing Th1 cells, IL-17 secreting Th17 cells, and IFN-α producing dendritic cells234; 235. IL-17 is believed to be one of the factors responsible for upregulating MHC class I molecules in muscle cells as well as for facilitating the migration of mononuclear cells to muscle cells236.
In summary, while there are other causal factors to muscle wasting chronic inflammatory diseases, pro-inflammatory cytokines act as major contributors to muscle loss in these diseases (Fig. 1). While therapies targeting the cytokines are already in clinical trials, efforts are being focused on generating more efficient strategies to better target and reduce the deleterious effects of inflammation in these diseases.
Figure 1. The drivers of muscle wasting in chronic inflammatory diseases.
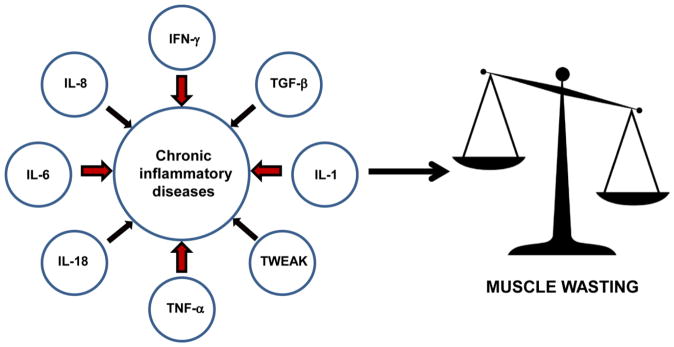
The figure depicts the inflammatory cytokines that contribute to muscle wasting. The degree of involvement of a particular cytokine is denoted by the width of individual arrows. Due to elevated expression of cytokines in these diseases, the balance is tipped towards hypercatabolism resulting in loss of muscle mass
The concept of skeletal muscle and myokines
The term ‘myokine’ was initially coined to strictly include proteins that were secreted by skeletal muscle cells. Nevertheless, the recent extended definition of a myokine includes proteins that are synthesized by skeletal muscle tissue and exert either paracrine or autocrine effects87. Chronic diseases like type 2 diabetes, cardiovascular diseases, colon cancer, breast cancer, to name a few, have highly different phenotypical presentations237. However, they share a few common pathogenic mechanisms such as physical inactivity. Chronic systemic inflammation goes hand in hand with physical inactivity, independent of obesity238. Evidence suggests that physical inactivity can lead to visceral fat, which can result in obesity and health consequences. Obesity coupled with lack of exercise subsequently results in activation of inflammatory pathways that lead to deleterious effects such as neuro degeneration, atherosclerosis, and development of insulin resistance. It has been long known that adipose tissue can function as an endocrine organ to release pro-inflammatory factors to promote obesity induced cardiovascular diseases and metabolic disorders. Recent studies have introduced the concept of ‘myokines’ which are released by skeletal muscle cells and predominantly function to counter the pro-inflammatory factors released by adipocytes239.
To date, IL-6 is perhaps the best recognized myokine. IL-6 was shown to be released in high amounts from contracting skeletal muscle following prolonged exercise, without exhibiting prominent muscle damage240. Besides IL-6, FGF2 can also be secreted from cultured C2C12 myoblasts241, but whether this factor qualifies as bonafide myofiber releasing myokine awaits further study. Although muscle derived IGF-1 is not detected in circulation, it is considered a myokine that functions in regulating muscle hypertrophy in an autocrine/paracrine manner, in response to exercise242; 243. Several activities of IGF-1 are regulated by muscle derived IGFBPs (IGF- binding proteins), which modulate IGF-1 availability and biological activity244. The cytokine, IL-15, also falls in the myokine category due to its anabolic activity on muscle cells and its possible role in reducing adipose tissue mass as part of a muscle, fat cross-talk245. Similar to IL-6, IL-15 is elevated in skeletal muscle cells post-exercise. Interestingly, administering IL-15 has also been found to improve glucose homeostasis and insulin resistance in obese mice246. Additional myokines include Fstl1 (also known as TSC36), which when secreted from skeletal muscle, exhibits an anti-apoptotic activity on endothelial cells247 through an Akt-eNOS signaling pathway247. This can be manifested under conditions of ischemic stress, where the addition of Fstl1 has been seen to accelerate revascularization247.
The myokine field is one that continues to emerge (reviewed in239; 248), as more recent candidates, such as IL-7, myonectin, and BDNF, have been proposed to be produced from skeletal muscle cells and act in a paracrine and autocrine fashions to maintain skeletal muscle homeostasis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kablar B, Rudnicki MA. Skeletal muscle development in the mouse embryo. Histol Histopathol. 2000;15:649–56. doi: 10.14670/HH-15.649. [DOI] [PubMed] [Google Scholar]
- 2.Nabeshima Y, Hanaoka K, Hayasaka M, Esumi E, Li S, Nonaka I, Nabeshima Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 1993;364:532–5. doi: 10.1038/364532a0. [DOI] [PubMed] [Google Scholar]
- 3.Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–6. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- 4.Parker MH, Seale P, Rudnicki MA. Looking back to the embryo: defining transcriptional networks in adult myogenesis. Nat Rev Genet. 2003;4:497–507. doi: 10.1038/nrg1109. [DOI] [PubMed] [Google Scholar]
- 5.von Maltzahn J, Chang NC, Bentzinger CF, Rudnicki MA. Wnt signaling in myogenesis. Trends Cell Biol. 2012;22:602–9. doi: 10.1016/j.tcb.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–9. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 7.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–13. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 8.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 9.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 10.Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A. 1998;95:15603–7. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman ME, DeMayo F, Yin KC, Lee HM, Geske R, Montgomery C, Schwartz RJ. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–16. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- 12.Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3′-kinase/Akt signaling pathway. J Biol Chem. 2000;275:35942–52. doi: 10.1074/jbc.M005832200. [DOI] [PubMed] [Google Scholar]
- 13.Flanagan-Steet H, Hannon K, McAvoy MJ, Hullinger R, Olwin BB. Loss of FGF receptor 1 signaling reduces skeletal muscle mass and disrupts myofiber organization in the developing limb. Dev Biol. 2000;218:21–37. doi: 10.1006/dbio.1999.9535. [DOI] [PubMed] [Google Scholar]
- 14.Haddad F, Roy RR, Zhong H, Edgerton VR, Baldwin KM. Atrophy responses to muscle inactivity. II. Molecular markers of protein deficits. J Appl Physiol (1985) 2003;95:791–802. doi: 10.1152/japplphysiol.01113.2002. [DOI] [PubMed] [Google Scholar]
- 15.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 16.Jagoe RT, Lecker SH, Gomes M, Goldberg AL. Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J. 2002;16:1697–712. doi: 10.1096/fj.02-0312com. [DOI] [PubMed] [Google Scholar]
- 17.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 18.Dehoux MJ, van Beneden RP, Fernandez-Celemin L, Lause PL, Thissen JP. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett. 2003;544:214–7. doi: 10.1016/s0014-5793(03)00505-2. [DOI] [PubMed] [Google Scholar]
- 19.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–5. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–71. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15:1537–45. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 23.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–6. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 24.Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–56. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 25.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–5. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–88. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tidball JG, Wehling-Henricks M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J Physiol. 2007;578:327–36. doi: 10.1113/jphysiol.2006.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Tonlorenzi, A Dellavalle, E Schnapp, G Cossu, M Sampaolesi. Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell Biol. 2007;Chapter 2(Unit 2B):1. doi: 10.1002/9780470151808.sc02b01s3. [DOI] [PubMed] [Google Scholar]
- 29.Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I, Perani L, Mantero S, Guttinger M, Pansarasa O, Rinaldi C, Cusella De Angelis MG, Torrente Y, Bordignon C, Bottinelli R, Cossu G. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–9. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- 30.Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP, Bottinelli R, Cossu G. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–92. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol. 2010;12:257–66. doi: 10.1038/ncb2025. [DOI] [PubMed] [Google Scholar]
- 32.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–63. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–52. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 34.Grounds MD, Garrett KL, Lai MC, Wright WE, Beilharz MW. Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 1992;267:99–104. doi: 10.1007/BF00318695. [DOI] [PubMed] [Google Scholar]
- 35.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–86. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 36.Yablonka-Reuveni Z, Rivera AJ. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev Biol. 1994;164:588–603. doi: 10.1006/dbio.1994.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cornelison DD, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol. 1997;191:270–83. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- 38.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 39.Young HA. Regulation of interferon-gamma gene expression. J Interferon Cytokine Res. 1996;16:563–8. doi: 10.1089/jir.1996.16.563. [DOI] [PubMed] [Google Scholar]
- 40.Frucht DM, Fukao T, Bogdan C, Schindler H, O’Shea JJ, Koyasu S. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol. 2001;22:556–60. doi: 10.1016/s1471-4906(01)02005-1. [DOI] [PubMed] [Google Scholar]
- 41.Flaishon L, Hershkoviz R, Lantner F, Lider O, Alon R, Levo Y, Flavell RA, Shachar I. Autocrine secretion of interferon gamma negatively regulates homing of immature B cells. J Exp Med. 2000;192:1381–8. doi: 10.1084/jem.192.9.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukao T, Matsuda S, Koyasu S. Synergistic effects of IL-4 and IL-18 on IL-12-dependent IFN-gamma production by dendritic cells. J Immunol. 2000;164:64–71. doi: 10.4049/jimmunol.164.1.64. [DOI] [PubMed] [Google Scholar]
- 43.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–8. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–5. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 45.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 46.Gajewski TF, Fitch FW. Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol. 1988;140:4245–52. [PubMed] [Google Scholar]
- 47.Decker T, Stockinger S, Karaghiosoff M, Muller M, Kovarik P. IFNs and STATs in innate immunity to microorganisms. J Clin Invest. 2002;109:1271–7. doi: 10.1172/JCI15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci U S A. 1990;87:6934–8. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 50.Morris AC, Beresford GW, Mooney MR, Boss JM. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol Cell Biol. 2002;22:4781–91. doi: 10.1128/MCB.22.13.4781-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun L, Ma K, Wang H, Xiao F, Gao Y, Zhang W, Wang K, Gao X, Ip N, Wu Z. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J Cell Biol. 2007;179:129–38. doi: 10.1083/jcb.200703184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng M, Nguyen MH, Fantuzzi G, Koh TJ. Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am J Physiol Cell Physiol. 2008;294:C1183–91. doi: 10.1152/ajpcell.00568.2007. [DOI] [PubMed] [Google Scholar]
- 53.Foster W, Li Y, Usas A, Somogyi G, Huard J. Gamma interferon as an antifibrosis agent in skeletal muscle. J Orthop Res. 2003;21:798–804. doi: 10.1016/S0736-0266(03)00059-7. [DOI] [PubMed] [Google Scholar]
- 54.Gasque P, Morgan BP, Legoedec J, Chan P, Fontaine M. Human skeletal myoblasts spontaneously activate allogeneic complement but are resistant to killing. J Immunol. 1996;156:3402–11. [PubMed] [Google Scholar]
- 55.Legoedec J, Gasque P, Jeanne JF, Scotte M, Fontaine M. Complement classical pathway expression by human skeletal myoblasts in vitro. Mol Immunol. 1997;34:735–41. doi: 10.1016/s0161-5890(97)00093-x. [DOI] [PubMed] [Google Scholar]
- 56.Mantegazza R, Hughes SM, Mitchell D, Travis M, Blau HM, Steinman L. Modulation of MHC class II antigen expression in human myoblasts after treatment with IFN-gamma. Neurology. 1991;41:1128–32. doi: 10.1212/wnl.41.7.1128. [DOI] [PubMed] [Google Scholar]
- 57.Reyes-Reyna SM, Krolick KA. Chemokine production by rat myocytes exposed to interferon-gamma. Clin Immunol. 2000;94:105–13. doi: 10.1006/clim.1999.4828. [DOI] [PubMed] [Google Scholar]
- 58.Chen SE, Jin B, Li YP. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Physiol Cell Physiol. 2007;292:C1660–71. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lagrota-Candido J, Vasconcellos R, Cavalcanti M, Bozza M, Savino W, Quirico-Santos T. Resolution of skeletal muscle inflammation in mdx dystrophic mouse is accompanied by increased immunoglobulin and interferon-gamma production. Int J Exp Pathol. 2002;83:121–32. doi: 10.1046/j.1365-2613.2002.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosen GD, Sanes JR, LaChance R, Cunningham JM, Roman J, Dean DC. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 1992;69:1107–19. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- 61.Londhe P, Davie JK. Gamma interferon modulates myogenesis through the major histocompatibility complex class II transactivator, CIITA. Mol Cell Biol. 2011;31:2854–66. doi: 10.1128/MCB.05397-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, Tidball JG. IFN-gamma promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J Immunol. 2011;187:5419–28. doi: 10.4049/jimmunol.1101267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalovidouris AE, Plotkin Z, Graesser D. Interferon-gamma inhibits proliferation, differentiation, and creatine kinase activity of cultured human muscle cells. II. A possible role in myositis. J Rheumatol. 1993;20:1718–23. [PubMed] [Google Scholar]
- 64.Londhe P, Davie JK. Interferon-gamma resets muscle cell fate by stimulating the sequential recruitment of JARID2 and PRC2 to promoters to repress myogenesis. Sci Signal. 2013;6:ra107. doi: 10.1126/scisignal.2004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18:2627–38. doi: 10.1101/gad.1241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, Ojeda SS, Kang HS, Schluns KS, Gui J, Jetten AM, Dong C. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity. 2012;36:23–31. doi: 10.1016/j.immuni.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 68.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–15. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 69.Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–12. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- 70.Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L, Jr, Singh H, Ouyang W, Pappu R. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12:1159–66. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- 71.Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. 2011;12:1151–8. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- 72.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 73.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–21. [PubMed] [Google Scholar]
- 75.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–14. [PubMed] [Google Scholar]
- 76.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.De Pasquale L, D’Amico A, Verardo M, Petrini S, Bertini E, De Benedetti F. Increased muscle expression of interleukin-17 in Duchenne muscular dystrophy. Neurology. 2012;78:1309–14. doi: 10.1212/WNL.0b013e3182518302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kishimoto T. Interleukin-6: from basic science to medicine--40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 79.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 80.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol. 2006;195:173–83. doi: 10.1007/s00430-006-0019-9. [DOI] [PubMed] [Google Scholar]
- 82.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–55. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 83.Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, Jones SA, Rose-John S, Scheller J. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111:1021–8. doi: 10.1182/blood-2007-07-102137. [DOI] [PubMed] [Google Scholar]
- 84.Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, Rose-John S, von Andrian UH, Baumann H, Evans SS. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol. 2006;7:1299–308. doi: 10.1038/ni1406. [DOI] [PubMed] [Google Scholar]
- 85.Bartoccioni E, Michaelis D, Hohlfeld R. Constitutive and cytokine-induced production of interleukin-6 by human myoblasts. Immunol Lett. 1994;42:135–8. doi: 10.1016/0165-2478(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 86.Fassone L, Gaidano G, Ariatti C, Vivenza D, Capello D, Gloghini A, Cilia AM, Buonaiuto D, Rossi D, Pastore C, Carbone A, Saglio G. The role of cytokines in the pathogenesis and management of AIDS-related lymphomas. Leuk Lymphoma. 2000;38:481–8. doi: 10.3109/10428190009059266. [DOI] [PubMed] [Google Scholar]
- 87.Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. 2008;88:1379–406. doi: 10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- 88.Baeza-Raja B, Munoz-Canoves P. p38 MAPK-induced nuclear factor-kappaB activity is required for skeletal muscle differentiation: role of interleukin-6. Mol Biol Cell. 2004;15:2013–26. doi: 10.1091/mbc.E03-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44. doi: 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 90.Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H, Monden M. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. 1996;97:244–9. doi: 10.1172/JCI118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurek JB, Nouri S, Kannourakis G, Murphy M, Austin L. Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve. 1996;19:1291–301. doi: 10.1002/(SICI)1097-4598(199610)19:10<1291::AID-MUS6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 92.Ahmed TA, Buzzelli MD, Lang CH, Capen JB, Shumate ML, Navaratnarajah M, Nagarajan M, Cooney RN. Interleukin-6 inhibits growth hormone-mediated gene expression in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1793–803. doi: 10.1152/ajpgi.00547.2006. [DOI] [PubMed] [Google Scholar]
- 93.Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, Cheung N, Williams B, Hazleman B, Price R, Yoshizaki K, Nishimoto N, Kishimoto T, Panayi GS. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002;46:3143–50. doi: 10.1002/art.10623. [DOI] [PubMed] [Google Scholar]
- 94.Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106:2627–32. doi: 10.1182/blood-2004-12-4602. [DOI] [PubMed] [Google Scholar]
- 95.Brown MA, Hural J. Functions of IL-4 and control of its expression. Crit Rev Immunol. 1997;17:1–32. doi: 10.1615/critrevimmunol.v17.i1.10. [DOI] [PubMed] [Google Scholar]
- 96.Choi P, Reiser H. IL-4: role in disease and regulation of production. Clin Exp Immunol. 1998;113:317–9. doi: 10.1046/j.1365-2249.1998.00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lombard-Platet S, Meyer V, Ceredig R. Both IFN-gamma and IL-4 induce MHC class II expression at the surface of mouse pro-B cells. Dev Immunol. 1997;5:115–20. doi: 10.1155/1997/76506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mijatovic T, Kruys V, Caput D, Defrance P, Huez G. Interleukin-4 and -13 inhibit tumor necrosis factor-alpha mRNA translational activation in lipopolysaccharide-induced mouse macrophages. J Biol Chem. 1997;272:14394–8. doi: 10.1074/jbc.272.22.14394. [DOI] [PubMed] [Google Scholar]
- 99.Manna SK, Aggarwal BB. Interleukin-4 down-regulates both forms of tumor necrosis factor receptor and receptor-mediated apoptosis, NF-kappaB, AP-1, and c-Jun N-terminal kinase. Comparison with interleukin-13. J Biol Chem. 1998;273:33333–41. doi: 10.1074/jbc.273.50.33333. [DOI] [PubMed] [Google Scholar]
- 100.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300:1527–8. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 101.Kelly-Welch A, Hanson EM, Keegan AD. Interleukin-4 (IL-4) pathway. Sci STKE. 2005:cm9. doi: 10.1126/stke.2932005cm9. [DOI] [PubMed] [Google Scholar]
- 102.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 103.Horsley V, Jansen KM, Mills ST, Pavlath GK. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell. 2003;113:483–94. doi: 10.1016/s0092-8674(03)00319-2. [DOI] [PubMed] [Google Scholar]
- 104.Marino M, Scuderi F, Mazzarelli P, Mannella F, Provenzano C, Bartoccioni E. Constitutive and cytokine-induced expression of MHC and intercellular adhesion molecule-1 (ICAM-1) on human myoblasts. J Neuroimmunol. 2001;116:94–101. doi: 10.1016/s0165-5728(01)00287-9. [DOI] [PubMed] [Google Scholar]
- 105.Barks JL, McQuillan JJ, Iademarco MF. TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol. 1997;159:4532–8. [PubMed] [Google Scholar]
- 106.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1173–87. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–97. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lundberg I, Brengman JM, Engel AG. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and non-weak controls. J Neuroimmunol. 1995;63:9–16. doi: 10.1016/0165-5728(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 109.Lepidi H, Frances V, Figarella-Branger D, Bartoli C, Machado-Baeta A, Pellissier JF. Local expression of cytokines in idiopathic inflammatory myopathies. Neuropathol Appl Neurobiol. 1998;24:73–9. doi: 10.1046/j.1365-2990.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 110.Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:865–74. doi: 10.1002/art.1780400514. [DOI] [PubMed] [Google Scholar]
- 111.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–95. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 113.Spencer SD, Di Marco F, Hooley J, Pitts-Meek S, Bauer M, Ryan AM, Sordat B, Gibbs VC, Aguet M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–8. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y, Wei SH, Ho AS, de Waal Malefyt R, Moore KW. Expression cloning and characterization of a human IL-10 receptor. J Immunol. 1994;152:1821–9. [PubMed] [Google Scholar]
- 115.Tan JC, Indelicato SR, Narula SK, Zavodny PJ, Chou CC. Characterization of interleukin-10 receptors on human and mouse cells. J Biol Chem. 1993;268:21053–9. [PubMed] [Google Scholar]
- 116.Carson WE, Lindemann MJ, Baiocchi R, Linett M, Tan JC, Chou CC, Narula S, Caligiuri MA. The functional characterization of interleukin-10 receptor expression on human natural killer cells. Blood. 1995;85:3577–85. [PubMed] [Google Scholar]
- 117.Weber-Nordt RM, Meraz MA, Schreiber RD. Lipopolysaccharide-dependent induction of IL-10 receptor expression on murine fibroblasts. J Immunol. 1994;153:3734–44. [PubMed] [Google Scholar]
- 118.Gibbs VC, Pennica D. CRF2-4: isolation of cDNA clones encoding the human and mouse proteins. Gene. 1997;186:97–101. doi: 10.1016/s0378-1119(96)00690-7. [DOI] [PubMed] [Google Scholar]
- 119.O’Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17:1006–18. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. 1999;274:16513–21. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 121.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–22. [PubMed] [Google Scholar]
- 122.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 123.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–11. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 124.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–63. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 125.Schaer DJ, Boretti FS, Hongegger A, Poehler D, Linnscheid P, Staege H, Muller C, Schoedon G, Schaffner A. Molecular cloning and characterization of the mouse CD163 homologue, a highly glucocorticoid-inducible member of the scavenger receptor cysteine-rich family. Immunogenetics. 2001;53:170–7. doi: 10.1007/s002510100304. [DOI] [PubMed] [Google Scholar]
- 126.Sulahian TH, Hogger P, Wahner AE, Wardwell K, Goulding NJ, Sorg C, Droste A, Stehling M, Wallace PK, Morganelli PM, Guyre PM. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12:1312–21. doi: 10.1006/cyto.2000.0720. [DOI] [PubMed] [Google Scholar]
- 127.Alvarez B, Quinn LS, Busquets S, Lopez-Soriano FJ, Argiles JM. TNF-alpha modulates cytokine and cytokine receptors in C2C12 myotubes. Cancer Lett. 2002;175:181–5. doi: 10.1016/s0304-3835(01)00717-0. [DOI] [PubMed] [Google Scholar]
- 128.Strle K, McCusker RH, Tran L, King A, Johnson RW, Freund GG, Dantzer R, Kelley KW. Novel activity of an anti-inflammatory cytokine: IL-10 prevents TNFalpha-induced resistance to IGF-I in myoblasts. J Neuroimmunol. 2007;188:48–55. doi: 10.1016/j.jneuroim.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Strle K, McCusker RH, Johnson RW, Zunich SM, Dantzer R, Kelley KW. Prototypical anti-inflammatory cytokine IL-10 prevents loss of IGF-I-induced myogenin protein expression caused by IL-1beta. Am J Physiol Endocrinol Metab. 2008;294:E709–18. doi: 10.1152/ajpendo.00662.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol. 2012;189:3669–80. doi: 10.4049/jimmunol.1103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum Mol Genet. 2011;20:790–805. doi: 10.1093/hmg/ddq523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 134.Barcellos-Hoff MH. Latency and activation in the control of TGF-beta. J Mammary Gland Biol Neoplasia. 1996;1:353–63. [PubMed] [Google Scholar]
- 135.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–40. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 136.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 137.Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–50. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 138.Attisano L, Silvestri C, Izzi L, Labbe E. The transcriptional role of Smads and FAST (FoxH1) in TGFbeta and activin signalling. Mol Cell Endocrinol. 2001;180:3–11. doi: 10.1016/s0303-7207(01)00524-x. [DOI] [PubMed] [Google Scholar]
- 139.Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–42. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kollias HD, Perry RL, Miyake T, Aziz A, McDermott JC. Smad7 promotes and enhances skeletal muscle differentiation. Mol Cell Biol. 2006;26:6248–60. doi: 10.1128/MCB.00384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–66. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol. 1989;138:311–5. doi: 10.1002/jcp.1041380213. [DOI] [PubMed] [Google Scholar]
- 143.Florini JR, Roberts AB, Ewton DZ, Falen SL, Flanders KC, Sporn MB. Transforming growth factor-beta. A very potent inhibitor of myoblast differentiation, identical to the differentiation inhibitor secreted by Buffalo rat liver cells. J Biol Chem. 1986;261:16509–13. [PubMed] [Google Scholar]
- 144.Vaidya TB, Rhodes SJ, Taparowsky EJ, Konieczny SF. Fibroblast growth factor and transforming growth factor beta repress transcription of the myogenic regulatory gene MyoD1. Mol Cell Biol. 1989;9:3576–9. doi: 10.1128/mcb.9.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brennan TJ, Edmondson DG, Li L, Olson EN. Transforming growth factor beta represses the actions of myogenin through a mechanism independent of DNA binding. Proc Natl Acad Sci U S A. 1991;88:3822–6. doi: 10.1073/pnas.88.9.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–76. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, Hirshman MF, Lebowitz J, Shadrach JL, Cerletti M, Kim MJ, Serwold T, Goodyear LJ, Rosner B, Lee RT, Wagers AJ. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–52. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mendias CL, Marcin JE, Calerdon DR, Faulkner JA. Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol (1985) 2006;101:898–905. doi: 10.1152/japplphysiol.00126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 150.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A. 1997;94:11514–9. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Eggers KM, Kempf T, Lind L, Sundstrom J, Wallentin L, Wollert KC, Siegbahn A. Relations of growth-differentiation factor-15 to biomarkers reflecting vascular pathologies in a population-based sample of elderly subjects. Scand J Clin Lab Invest. 2012;72:45–51. doi: 10.3109/00365513.2011.626072. [DOI] [PubMed] [Google Scholar]
- 152.Brown DA, Moore J, Johnen H, Smeets TJ, Bauskin AR, Kuffner T, Weedon H, Milliken ST, Tak PP, Smith MD, Breit SN. Serum macrophage inhibitory cytokine 1 in rheumatoid arthritis: a potential marker of erosive joint destruction. Arthritis Rheum. 2007;56:753–64. doi: 10.1002/art.22410. [DOI] [PubMed] [Google Scholar]
- 153.Wollert KC, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, Allhoff T, Peter T, Siegbahn A, Venge P, Drexler H, Wallentin L. Growth differentiation factor 15 for risk stratification and selection of an invasive treatment strategy in non ST-elevation acute coronary syndrome. Circulation. 2007;116:1540–8. doi: 10.1161/CIRCULATIONAHA.107.697714. [DOI] [PubMed] [Google Scholar]
- 154.Kempf T, Horn-Wichmann R, Brabant G, Peter T, Allhoff T, Klein G, Drexler H, Johnston N, Wallentin L, Wollert KC. Circulating concentrations of growth-differentiation factor 15 in apparently healthy elderly individuals and patients with chronic heart failure as assessed by a new immunoradiometric sandwich assay. Clin Chem. 2007;53:284–91. doi: 10.1373/clinchem.2006.076828. [DOI] [PubMed] [Google Scholar]
- 155.de Jager SC, Bermudez B, Bot I, Koenen RR, Bot M, Kavelaars A, de Waard V, Heijnen CJ, Muriana FJ, Weber C, van Berkel TJ, Kuiper J, Lee SJ, Abia R, Biessen EA. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med. 2011;208:217–25. doi: 10.1084/jem.20100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bonaterra GA, Zugel S, Thogersen J, Walter SA, Haberkorn U, Strelau J, Kinscherf R. Growth differentiation factor-15 deficiency inhibits atherosclerosis progression by regulating interleukin-6-dependent inflammatory response to vascular injury. J Am Heart Assoc. 2012;1:e002550. doi: 10.1161/JAHA.112.002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Breit SN, Johnen H, Cook AD, Tsai VW, Mohammad MG, Kuffner T, Zhang HP, Marquis CP, Jiang L, Lockwood G, Lee-Ng M, Husaini Y, Wu L, Hamilton JA, Brown DA. The TGF-beta superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors. 2011;29:187–95. doi: 10.3109/08977194.2011.607137. [DOI] [PubMed] [Google Scholar]
- 158.Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, Murakami T, Drutskaya LN, Forster I, Clausen BE, Tessarollo L, Ryffel B, Kuprash DV, Nedospasov SA. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity. 2005;22:93–104. doi: 10.1016/j.immuni.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 159.Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, Acharyya S, Rudnicki MA, Hollenbach AD, Guttridge DC. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–6. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 161.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 162.Li YP. TNF-alpha is a mitogen in skeletal muscle. Am J Physiol Cell Physiol. 2003;285:C370–6. doi: 10.1152/ajpcell.00453.2002. [DOI] [PubMed] [Google Scholar]
- 163.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–69. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010;5:e12479. doi: 10.1371/journal.pone.0012479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Farber MO, Mannix ET. Tissue wasting in patients with chronic obstructive pulmonary disease. Neurol Clin. 2000;18:245–62. doi: 10.1016/s0733-8619(05)70188-2. [DOI] [PubMed] [Google Scholar]
- 166.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7:411–25. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, Kominami E, Okumura K, Yagita H. Multiple pathways of TWEAK-induced cell death. J Immunol. 2002;168:734–43. doi: 10.4049/jimmunol.168.2.734. [DOI] [PubMed] [Google Scholar]
- 168.Kaplan MJ, Ray D, Mo RR, Yung RL, Richardson BC. TRAIL (Apo2 ligand) and TWEAK (Apo3 ligand) mediate CD4+ T cell killing of antigen-presenting macrophages. J Immunol. 2000;164:2897–904. doi: 10.4049/jimmunol.164.6.2897. [DOI] [PubMed] [Google Scholar]
- 169.Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ, Jenkins NA, Richards CM, Winkles JA. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–76. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- 170.Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–46. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- 171.Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003;14:241–9. doi: 10.1016/s1359-6101(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 172.Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007;21:1857–69. doi: 10.1096/fj.06-7537com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dohi T, Burkly LC. The TWEAK/Fn14 pathway as an aggravating and perpetuating factor in inflammatory diseases: focus on inflammatory bowel diseases. J Leukoc Biol. 2012;92:265–79. doi: 10.1189/jlb.0112042. [DOI] [PubMed] [Google Scholar]
- 174.Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine. 2007;40:1–16. doi: 10.1016/j.cyto.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 175.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–41. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 176.Killian KJ, Leblanc P, Martin DH, Summers E, Jones NL, Campbell EJ. Exercise capacity and ventilatory, circulatory, and symptom limitation in patients with chronic airflow limitation. Am Rev Respir Dis. 1992;146:935–40. doi: 10.1164/ajrccm/146.4.935. [DOI] [PubMed] [Google Scholar]
- 177.Bernard S, LeBlanc P, Whittom F, Carrier G, Jobin J, Belleau R, Maltais F. Peripheral muscle weakness in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:629–34. doi: 10.1164/ajrccm.158.2.9711023. [DOI] [PubMed] [Google Scholar]
- 178.Gosker HR, Langen RC, Bracke KR, Joos GF, Brusselle GG, Steele C, Ward KA, Wouters EF, Schols AM. Extrapulmonary manifestations of chronic obstructive pulmonary disease in a mouse model of chronic cigarette smoke exposure. Am J Respir Cell Mol Biol. 2009;40:710–6. doi: 10.1165/rcmb.2008-0312OC. [DOI] [PubMed] [Google Scholar]
- 179.Remels AH, Gosker HR, Langen RC, Schols AM. The mechanisms of cachexia underlying muscle dysfunction in COPD. J Appl Physiol (1985) 2013;114:1253–62. doi: 10.1152/japplphysiol.00790.2012. [DOI] [PubMed] [Google Scholar]
- 180.Vorrink SN, Kort HS, Troosters T, Lammers JW. Level of daily physical activity in individuals with COPD compared with healthy controls. Respir Res. 2011;12:33. doi: 10.1186/1465-9921-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR, Cerretelli P. Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med. 1990;11(Suppl 1):S3–9. doi: 10.1055/s-2007-1024846. [DOI] [PubMed] [Google Scholar]
- 182.Mizuno M, Savard GK, Areskog NH, Lundby C, Saltin B. Skeletal muscle adaptations to prolonged exposure to extreme altitude: a role of physical activity? High Alt Med Biol. 2008;9:311–7. doi: 10.1089/ham.2008.1009. [DOI] [PubMed] [Google Scholar]
- 183.Morrison WL, Gibson JN, Scrimgeour C, Rennie MJ. Muscle wasting in emphysema. Clin Sci (Lond) 1988;75:415–20. doi: 10.1042/cs0750415. [DOI] [PubMed] [Google Scholar]
- 184.Shah OJ, Anthony JC, Kimball SR, Jefferson LS. 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab. 2000;279:E715–29. doi: 10.1152/ajpendo.2000.279.4.E715. [DOI] [PubMed] [Google Scholar]
- 185.Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, Correa J, Pearce D, Binnie M, Batt J. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2010;42:461–71. doi: 10.1165/rcmb.2008-0382OC. [DOI] [PubMed] [Google Scholar]
- 186.Kirkham P, Rahman I. Oxidative stress in asthma and COPD: antioxidants as a therapeutic strategy. Pharmacol Ther. 2006;111:476–94. doi: 10.1016/j.pharmthera.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 187.Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study G. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594–602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- 188.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–16. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 189.Houssiau FA. Cytokines in rheumatoid arthritis. Clin Rheumatol. 1995;14(Suppl 2):10–3. doi: 10.1007/BF02215851. [DOI] [PubMed] [Google Scholar]
- 190.Shingu M, Nagai Y, Isayama T, Naono T, Nobunaga M, Nagai Y. The effects of cytokines on metalloproteinase inhibitors (TIMP) and collagenase production by human chondrocytes and TIMP production by synovial cells and endothelial cells. Clin Exp Immunol. 1993;94:145–9. doi: 10.1111/j.1365-2249.1993.tb05992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Butler DM, Maini RN, Feldmann M, Brennan FM. Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw. 1995;6:225–30. [PubMed] [Google Scholar]
- 193.Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin-18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem. 2001;276:37069–75. doi: 10.1074/jbc.M103574200. [DOI] [PubMed] [Google Scholar]
- 194.Morel JC, Park CC, Zhu K, Kumar P, Ruth JH, Koch AE. Signal transduction pathways involved in rheumatoid arthritis synovial fibroblast interleukin-18-induced vascular cell adhesion molecule-1 expression. J Biol Chem. 2002;277:34679–91. doi: 10.1074/jbc.M206337200. [DOI] [PubMed] [Google Scholar]
- 195.Morel JC, Park CC, Kumar P, Koch AE. Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation. Lab Invest. 2001;81:1371–83. doi: 10.1038/labinvest.3780351. [DOI] [PubMed] [Google Scholar]
- 196.Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2006;103:3298–303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Suzuki M, Hashizume M, Yoshida H, Mihara M. Anti-inflammatory mechanism of tocilizumab, a humanized anti-IL-6R antibody: effect on the expression of chemokine and adhesion molecule. Rheumatol Int. 2010;30:309–15. doi: 10.1007/s00296-009-0953-0. [DOI] [PubMed] [Google Scholar]
- 198.Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 2012;122:143–59. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- 199.Suzuki M, Hashizume M, Yoshida H, Shiina M, Mihara M. IL-6 and IL-1 synergistically enhanced the production of MMPs from synovial cells by up-regulating IL-6 production and IL-1 receptor I expression. Cytokine. 2010;51:178–83. doi: 10.1016/j.cyto.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 200.Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, Yoshizaki K, Nishimoto N. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48:1521–9. doi: 10.1002/art.11143. [DOI] [PubMed] [Google Scholar]
- 201.Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–8. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sasai M, Saeki Y, Ohshima S, Nishioka K, Mima T, Tanaka T, Katada Y, Yoshizaki K, Suemura M, Kishimoto T. Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum. 1999;42:1635–43. doi: 10.1002/1529-0131(199908)42:8<1635::AID-ANR11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 203.Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M, Kishimoto T. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci U S A. 1998;95:8222–6. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988;18:1797–801. doi: 10.1002/eji.1830181122. [DOI] [PubMed] [Google Scholar]
- 205.Walsmith J, Roubenoff R. Cachexia in rheumatoid arthritis. Int J Cardiol. 2002;85:89–99. doi: 10.1016/s0167-5273(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 206.Dinarello CA, Roubenoff R. George Clowes’ classic paper and the search for “muscle proteolysis factor”. Nutrition. 1996;12:67–8. doi: 10.1016/0899-9007(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 207.Roubenoff R, Freeman LM, Smith DE, Abad LW, Dinarello CA, Kehayias JJ. Adjuvant arthritis as a model of inflammatory cachexia. Arthritis Rheum. 1997;40:534–9. doi: 10.1002/art.1780400320. [DOI] [PubMed] [Google Scholar]
- 208.Dayer JM. Interleukin 1 or tumor necrosis factor-alpha: which is the real target in rheumatoid arthritis? J Rheumatol Suppl. 2002;65:10–5. [PubMed] [Google Scholar]
- 209.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A. 1994;91:4854–8. doi: 10.1073/pnas.91.11.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 211.Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson-Hughes B, Dinarello CA, Rosenberg IH. Rheumatoid cachexia: cytokine-driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest. 1994;93:2379–86. doi: 10.1172/JCI117244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 213.Nishimoto N, Yoshizaki K, Maeda K, Kuritani T, Deguchi H, Sato B, Imai N, Suemura M, Kakehi T, Takagi N, Kishimoto T. Toxicity, pharmacokinetics, and dose-finding study of repetitive treatment with the humanized anti-interleukin 6 receptor antibody MRA in rheumatoid arthritis. Phase I/II clinical study. J Rheumatol. 2003;30:1426–35. [PubMed] [Google Scholar]
- 214.Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, Thomson D, Kishimoto T, Group CS. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–29. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- 215.van de Putte LB, Rau R, Breedveld FC, Kalden JR, Malaise MG, van Riel PL, Schattenkirchner M, Emery P, Burmester GR, Zeidler H, Moutsopoulos HM, Beck K, Kupper H. Efficacy and safety of the fully human anti-tumour necrosis factor alpha monoclonal antibody adalimumab (D2E7) in DMARD refractory patients with rheumatoid arthritis: a 12 week, phase II study. Ann Rheum Dis. 2003;62:1168–77. doi: 10.1136/ard.2003.009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 217.Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Ruderman EM, Horwitz DA, Arkfeld DG, Garrison L, Burge DJ, Blosch CM, Lange ML, McDonnell ND, Weinblatt ME. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130:478–86. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- 218.Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151:1548–61. [PubMed] [Google Scholar]
- 219.Bresnihan B, Alvaro-Gracia JM, Cobby M, Doherty M, Domljan Z, Emery P, Nuki G, Pavelka K, Rau R, Rozman B, Watt I, Williams B, Aitchison R, McCabe D, Musikic P. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 220.Campion GV, Lebsack ME, Lookabaugh J, Gordon G, Catalano M. Dose-range and dose-frequency study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis. The IL-1Ra Arthritis Study Group. Arthritis Rheum. 1996;39:1092–101. doi: 10.1002/art.1780390704. [DOI] [PubMed] [Google Scholar]
- 221.van der Meulen MF, Bronner IM, Hoogendijk JE, Burger H, van Venrooij WJ, Voskuyl AE, Dinant HJ, Linssen WH, Wokke JH, de Visser M. Polymyositis: an overdiagnosed entity. Neurology. 2003;61:316–21. doi: 10.1212/wnl.61.3.316. [DOI] [PubMed] [Google Scholar]
- 222.Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology. 2003;61:288–9. doi: 10.1212/wnl.61.3.288. [DOI] [PubMed] [Google Scholar]
- 223.Carpenter S, Karpati G, Heller I, Eisen A. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy. Neurology. 1978;28:8–17. doi: 10.1212/wnl.28.1.8. [DOI] [PubMed] [Google Scholar]
- 224.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. V: Identification and quantitation of T8+ cytotoxic and T8+ suppressor cells. Ann Neurol. 1988;23:493–9. doi: 10.1002/ana.410230511. [DOI] [PubMed] [Google Scholar]
- 225.Wiendl H, Mitsdoerffer M, Schneider D, Melms A, Lochmuller H, Hohlfeld R, Weller M. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible co-stimulatory molecule, ICOSL: implications for the pathogenesis of inflammatory myopathies. Brain. 2003;126:1026–35. doi: 10.1093/brain/awg114. [DOI] [PubMed] [Google Scholar]
- 226.Murata K, Dalakas MC. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol. 1999;155:453–60. doi: 10.1016/s0002-9440(10)65141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–27. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- 228.De Bleecker JL, De Paepe B, Vanwalleghem IE, Schroder JM. Differential expression of chemokines in inflammatory myopathies. Neurology. 2002;58:1779–85. doi: 10.1212/wnl.58.12.1779. [DOI] [PubMed] [Google Scholar]
- 229.Dalakas MC. Pathogenesis and therapies of immune-mediated myopathies. Autoimmun Rev. 2012;11:203–6. doi: 10.1016/j.autrev.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 230.Reimann J, Schnell S, Schwartz S, Kappes-Horn K, Dodel R, Bacher M. Macrophage migration inhibitory factor in normal human skeletal muscle and inflammatory myopathies. J Neuropathol Exp Neurol. 2010;69:654–62. doi: 10.1097/NEN.0b013e3181e10925. [DOI] [PubMed] [Google Scholar]
- 231.Luo YB, Mastaglia FL. Dermatomyositis, polymyositis and immune-mediated necrotising myopathies. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 232.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82. doi: 10.1016/S0140-6736(03)14368-1. [DOI] [PubMed] [Google Scholar]
- 233.Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011;7:664–75. doi: 10.1038/nrrheum.2011.139. [DOI] [PubMed] [Google Scholar]
- 234.Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, Park P, Amato AA. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–78. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- 235.Page G, Chevrel G, Miossec P. Anatomic localization of immature and mature dendritic cell subsets in dermatomyositis and polymyositis: Interaction with chemokines and Th1 cytokine-producing cells. Arthritis Rheum. 2004;50:199–208. doi: 10.1002/art.11428. [DOI] [PubMed] [Google Scholar]
- 236.Tournadre A, Miossec P. Interleukin-17 in inflammatory myopathies. Curr Rheumatol Rep. 2012;14:252–6. doi: 10.1007/s11926-012-0242-x. [DOI] [PubMed] [Google Scholar]
- 237.Pedersen BK. The diseasome of physical inactivity--and the role of myokines in muscle--fat cross talk. J Physiol. 2009;587:5559–68. doi: 10.1113/jphysiol.2009.179515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fischer CP, Berntsen A, Perstrup LB, Eskildsen P, Pedersen BK. Plasma levels of interleukin-6 and C-reactive protein are associated with physical inactivity independent of obesity. Scand J Med Sci Sports. 2007;17:580–7. doi: 10.1111/j.1600-0838.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- 239.Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214:337–46. doi: 10.1242/jeb.048074. [DOI] [PubMed] [Google Scholar]
- 240.Steensberg A, van Hall G, Osada T, Sacchetti M, Saltin B, Klarlund Pedersen B. Production of interleukin-6 in contracting human skeletal muscles can account for the exercise-induced increase in plasma interleukin-6. J Physiol. 2000;529(Pt 1):237–42. doi: 10.1111/j.1469-7793.2000.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008;582:3805–10. doi: 10.1016/j.febslet.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hede MS, Salimova E, Piszczek A, Perlas E, Winn N, Nastasi T, Rosenthal N. E-peptides control bioavailability of IGF-1. PLoS One. 2012;7:e51152. doi: 10.1371/journal.pone.0051152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Vinciguerra M, Musaro A, Rosenthal N. Regulation of muscle atrophy in aging and disease. Adv Exp Med Biol. 2010;694:211–33. doi: 10.1007/978-1-4419-7002-2_15. [DOI] [PubMed] [Google Scholar]
- 244.Duan C, Ren H, Gao S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: roles in skeletal muscle growth and differentiation. Gen Comp Endocrinol. 2010;167:344–51. doi: 10.1016/j.ygcen.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 245.Carbo N, Lopez-Soriano J, Costelli P, Alvarez B, Busquets S, Baccino FM, Quinn LS, Lopez-Soriano FJ, Argiles JM. Interleukin-15 mediates reciprocal regulation of adipose and muscle mass: a potential role in body weight control. Biochim Biophys Acta. 2001;1526:17–24. doi: 10.1016/s0304-4165(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 246.Barra NG, Chew MV, Holloway AC, Ashkar AA. Interleukin-15 treatment improves glucose homeostasis and insulin sensitivity in obese mice. Diabetes Obes Metab. 2012;14:190–3. doi: 10.1111/j.1463-1326.2011.01495.x. [DOI] [PubMed] [Google Scholar]
- 247.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, Walsh K. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J Biol Chem. 2008;283:32802–11. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Raschke S, Eckel J. Adipo-myokines: two sides of the same coin--mediators of inflammation and mediators of exercise. Mediators Inflamm. 2013;2013:320724. doi: 10.1155/2013/320724. [DOI] [PMC free article] [PubMed] [Google Scholar]