

Abstract
While advances in technology and medicine have improved both longevity and quality of life in patients living with a spinal cord injury, restoration of full motor function is not often achieved. This is due to the failure of repair and regeneration of neuronal connections in the spinal cord after injury. In this review, the complicated nature of spinal cord injury is described, noting the numerous cellular and molecular events that occur in the central nervous system following a traumatic lesion. In short, postinjury tissue changes create a complex and dynamic environment that is highly inhibitory to the process of neural regeneration. Strategies for repair are outlined with a particular focus on the important role of biomaterials in designing a therapeutic treatment that can overcome this inhibitory environment. The importance of considering the inherent biological response of the central nervous system to both injury and subsequent therapeutic interventions is highlighted as a key consideration for all attempts at improving functional recovery.
1. Introduction
“One having a crushed vertebra in his neck; he is unconscious of his two arms (and) his two legs, (and) he is speechless. An ailment not to be treated” [1]. This excerpt from the Edwin Smith papyrus was the diagnosis of an ancient Egyptian physician, and some of the first ever medical observations regarding the limited ability of the central nervous system (CNS) to heal following a traumatic injury [1, 2]. This passage is also one of the first written accounts as to the grave nature of injuries to the CNS. While advances in modern medicine and technology have improved both lifespan and quality of life for victims of spinal cord injury (SCI), injury sustained to the spinal cord generally results in a permanent loss or impairment of motor function and sensation below the level of injury. This impairment presents victims of SCI with numerous financial, physical, emotional, and social burdens [3].
Current strategies to treat spinal cord injury have focused on restoring function via enhancement of neuronal survival after injury, regeneration of damaged axons, and neuroplasticity of spared axons. Ideally, a single treatment paradigm would be used to accomplish all of these tasks simultaneously. Unfortunately, however, research efforts have thus far demonstrated no single therapy or treatment that will reverse the damage after SCI. Such findings center on the fact that the spinal cord is a unique and complex environment, posing many challenges to the restoration of function. Given that combinations of pharmacologic and rehabilitative therapies may be necessary to address all of these challenges, researchers in this field need to consider the biological implications of each type of therapy in conjunction with the inherent response to spinal cord injury. Therefore, this paper is aimed at providing a comprehensive discussion of the challenges posed by the postinjury response of spinal cord, current strategies aimed at enhancing functional repair, and the potential use of biomaterials in aiding the recovery process.
2. Part I: The Complex Nature of Spinal Cord Injury
2.1. SCI: Etiology and Prognosis of Lesions
On average, there are approximately 12,500 newly reported cases of SCI in the United States each year, with a prevalence estimated to be approximately 276,000 persons [3]. Of the reported cases, a vast majority of SCIs (79%) occur in males and result from either a contusion or compression style injury [3]. During a contusion injury, forces are rapidly applied to and removed from the spinal cord. This causes a sudden and focal compression, with displacement of spinal tissue both rostrally and caudally, severing any axons within the affected region [4, 11, 12]. This style of injury is most commonly associated with blunt force trauma due to motor vehicle accidents (38%), falls (30%), and sporting injuries (9%) [3]. While compression injuries of the spinal cord occur as a result of a sustained force, crush injuries can result from slipped intervertebral discs, dislocation/fractures of the vertebrae, subluxation of the vertebrae during trauma, or spinal subdural hematomas and are known to produce larger and more diffuse areas of injury [3]. Although the area of injury can be quite extensive, trauma involving sharp penetrating injury or the complete dislocation of two adjacent vertebrae represents only a minority of SCI cases [3].
The human spinal cord, on average, is approximately 45 cm long in males and 42-43 cm long in females [13]. Trauma can occur anywhere along the length of the spinal cord. However, spinal injuries are largely localized to two anatomic regions: low cervical (C5-C7) and mid-thoracic (T9-T10, [3]). The anatomic level at which the injury occurs has a significant determination on the level and degree of impairment or paralysis that follows. Injuries to the cervical levels of the spinal cord most often result in some degree of quadriplegia, while those occurring at the thoracic region most often result in some degree of paraplegia [3].
While the anatomic level of the injury determines what regions and appendages of the body are affected, the completeness of the SCI determines the severity of loss in function and sensation. There are two categories into which SCIs can be classified: complete and incomplete. In a complete injury, the spinal cord is severed into two distinct stumps, axotomizing all of the ascending and descending axonal tracts. Incomplete injuries, on the other hand, axotomize some motor and sensory axonal tracts without separating the spinal cord into two distinct sections. The more complete the injury, the more severe the resulting impairment. In a complete lesion, the lack of tissue connectivity between the two cut ends results in physical retraction of the stumps, essentially negating any chance of translesion recovery. The spared rim of white matter in an incomplete lesion, however, holds the spinal cord together and provides a potential bridge for axonal regrowth, thus allowing for the possibility that some limited translesion recovery may occur. Unfortunately, even though a majority of SCI cases are the result of incomplete lesions [3], for which at least limited translesion recovery should be possible, the inherent biological response of the spinal cord to the injury often limits the success of the regenerative response. As such, characterizing the molecular features of the biological response, such that they may be addressed by therapeutic approaches, has largely been the target of SCI regenerative research.
2.2. Biological Response to Injury
2.2.1. Neuronal Response to SCI
The basic anatomic organization of the spinal cord places the white matter tracts on the outer periphery, making them susceptible to physical trauma [13]. In considering contusive or crush style injuries, the physical stretching forces applied to the spinal cord appear to be concentrated at the nodes of Ranvier, which, being devoid of a myelin sheath, is the weakest point of the axon [4, 14]. Following axonal rupture, exposure of the axoplasm to the extracellular environment allows for the rapid influx of extracellular calcium, in turn activating phospholipase A2 and triggering the cut end of the axon to reseal [15, 16]. More importantly, the calcium dependent events that occur at the cut end following injury also appear to play a role in determining if the damaged axonal tip develops into an end bulb or a functional growth cone [17]. As the severed distal end of the axon undergoes the process of Wallerian degeneration, the proximal part of the neuron also undergoes a chromatolytic reaction [18]. This reaction is characterized by the movement of the nucleus to a more lateral eccentric position within the neuron's cell body and the rough endoplasmic reticulum taking on a fragmented appearance [18, 19].
The postinjury changes that occur in neurons are thought to be a result of a disruption in sustained neurotrophic support. As neurons rely on neurotrophins for survival, any alterations in the availability of such molecules could result in irreparable damage. In general, neurotrophic support can be provided through autocrine or paracrine sources or from axonal connections with a neuronal target [20]. The majority of neurotrophins arise from innervated targets of the neuron, at least during development [20, 21]. Following SCI, the termination of the link between the neuron and its target disrupts the continued supply of neurotrophic molecules, causing the neuron to atrophy and further decreasing its ability to mount a regenerative response [4, 19].
It is well known that supraspinal neurons lack a strong intrinsic regenerative response following an axotomy in the spinal cord. This class of neurons, which includes the corticospinal tract neurons (CST), vestibulospinal tract (VST) neurons, and rubrospinal tract (RuST) neurons, has, therefore, become the most frequent targets of neural regeneration research. Although the observed lack of a regenerative response fostered the belief that CNS neurons were incapable of undergoing any type of regeneration, some studies have demonstrated the contrary. It has been noted that CNS neurons, particularly those that were axotomized near the neuronal cell body, are able to grow axons within a peripheral nerve graft [22–24]. Similarly, propriospinal (PS) neurons, another class of CNS neuron intrinsic to the spinal cord, have also been demonstrated to grow into peripheral nerve grafts [25]. These studies suggest that if presented with the appropriate triggers and environmental conditions, CNS neurons can mount a regenerative response. However, it is also clear that the postinjury environment plays a significant role in quashing the regenerative capacity of CNS neurons.
2.2.2. Glial Scar Formation
The complex orchestration of molecular changes in the spinal cord following any trauma creates an environment that is well documented as hostile to the regenerative processes [4, 5, 26–31]. The physical forces applied to the spinal cord during a contusion injury result in the destruction of blood vessels, leading to a massive inflammatory response (Figure 1) and the creation of a hypoxic postinjury environment [6, 32–35]. This inflammatory reaction triggers the process of reactive astrogliosis, which leads to the production of a chemophysical barrier that inhibits regenerative activity [4, 5, 26–28, 36–38]. More specifically, immediately following an SCI, astrocytes located within the zone of injury become hypertrophic, extending their processes, proliferating, and organizing into a dense astrocytic rich border at the lesion site [5, 26, 29]. Overall, this process functions to produce a glial scar surrounding the lesioned area.
Figure 1.
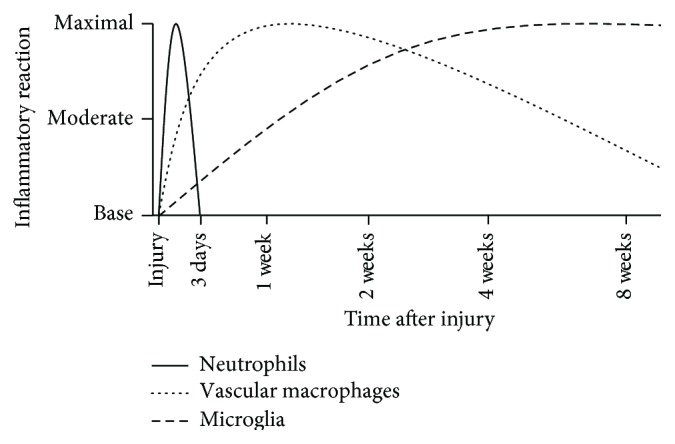
Immune response following SCI. Trauma to the spinal cord elicits an immune response, which begins almost immediately after injury. Neutrophils are the first immune cells to respond to the lesion site, arriving within the first few hours after injury, and remaining for up to 3 days after injury. Vascular macrophages are the second class of immune cell to arrive at the lesion, arriving after the initial infiltration of neutrophils. Activation and infiltration of vascular macrophages subsequently activate and recruit microglial cells, which can persist in the lesion site for months after injury [4–7].
The glial scar has been widely accepted as a primary reason for the lack of a maintained regenerative response following SCI. However recent evidence is beginning to cast a new light on the glial scar as an important protective barrier preventing further secondary tissue damage [28, 39–41]. These studies also suggest that the formation of the glial scar may be beneficial for the initiation of the axonal sprouting response. For example, Faulkner and colleagues [39] demonstrated that ablation of reactive astrocytes following a stab or crush style injury to the mouse spinal cord effectively prevented formation of the glial scar. This process resulted in increased demyelination and degeneration of neurons but decreased number of oligodendrocytes and functional ability. Overall, reactive astrocytes have been discussed as benevolent in nature, at least to some degree, due to their ability to reduce the excitotoxic levels of glutamate in the extracellular environment, produce molecules that prevent oxidative damage and toxicity, allow for reformation of the blood brain barrier, and regulate the fluid and ion balance of the extracellular space [28].
2.2.3. Chondroitin Sulfate Proteoglycans (CSPGs) as Barriers to Repair
Reactive astrogliosis produces an upregulation of CSPGs in the tissue surrounding the lesion site (reviewed by [5, 37, 38, 42–45]). Immediately following injury, astrocytes upregulate and synthesize the CSPGs brevican, neurocan, and phosphacan, while infiltrating vascular macrophages and microglia increase their expression of the proteoglycan NG2 (Figure 2, [8, 43, 46]). Demyelination triggers the recruitment of oligodendrocyte progenitor cells (OPCs) to the lesion site, which also causes the increase in expression of the proteoglycans NG2 and versican [8, 43, 46]. High levels of CSPGs in the postlesion environment have a significant role in inhibiting the regenerative capabilities of the CNS. For example, in the presence of CSPGs, OPCs fail to undergo differentiation into myelin forming oligodendrocytes [47–49]. The interaction of neurons with CSPGs activates the Rho-ROCK and/or protein kinase C (PKC) inhibitory signaling cascades, which have been demonstrated to negatively regulate axonal outgrowth and extension by inducing growth cone collapse [50, 51]. This leads to growth cone retraction and an overall abortion of the regenerative process [4, 5, 36]. Blocking the Rho-Rock and/or PKC pathways has been noted to reverse the inhibitory effects of CSPGs on axonal regeneration and OPC process outgrowth [45, 47, 52, 53], adding additional confirmation for the effects of these signaling cascades.
Figure 2.
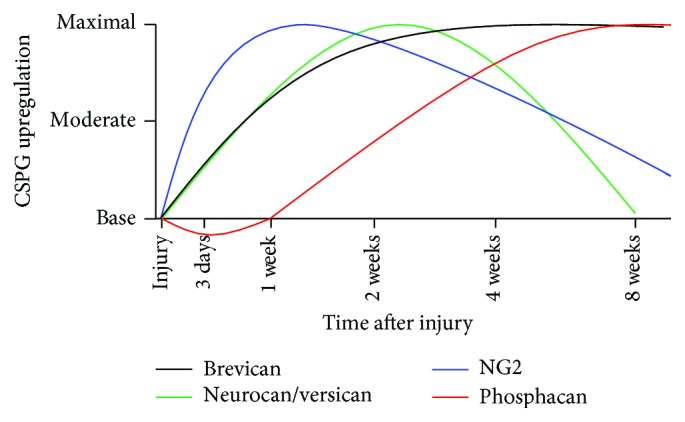
Upregulation and expression of CSPGs. Almost immediately following an SCI, astrocytes located within the area of trauma begin to undergo hypertrophy, synthesizing and secreting CSPGs, including neurocan, phosphacan, and brevican. Additionally, the infiltration of vascular macrophages, activated microglial cells, and OPCs results in the increase in the proteoglycans NG2 and versican. The temporal expression of these proteoglycans is important to factor into any treatment, as they have differential effects on the regenerative process. Neurocan and versican are upregulated quickly following injury, with maximal expression observed 2 weeks after injury. Their expression begins to wane at longer times, approaching base levels by 8 weeks after injury. Brevican is also upregulated after injury, reaching maximal expression 2 weeks after injury. However unlike neurocan and versican, brevican expression remains elevated over time. Phosphacan is initially downregulated following SCI, with significantly reduced levels 1 week after injury. The expression begins to increase at longer times after injury and peaks around 8 weeks after injury. NG2 expression can be generally correlated to the infiltration of vascular macrophages, activated microglia, and OPCs, with maximal expression being found 1 week after injury. This differential expression pattern of CSPGs plays a large role in governing the regenerative response as many CSPGs are inhibitory to both process of atonal regeneration and remyelination (adapted from [8]).
While CSPGs, overall, exert a largely inhibitory influence to the regenerative process, the specific inhibitory nature varies among the different proteoglycans. In vitro, purified brevican, neurocan, and phosphacan have all been identified as inhibitory to axonal attachment and growth [5, 42]. The ability of neurocan and phosphacan to interact with neural cell adhesion molecules (N-CAM) on neurons is thought to be the mechanism underlying their inhibitory effects [5, 42]. Versican, however, is not inhibitory to either axonal regrowth or adhesion. This is evidenced by the in vitro finding that axons not only are able to grow through deposits of versican but also show no signs of inhibition in the presence of the purified proteoglycan [54, 55]. Some in vitro studies have demonstrated that neural/glial antigen 2 (NG2) is inhibitory to the process of axonal outgrowth, although the effects of NG2 in vivo remain undetermined [8, 56, 57]. The most recently characterized brain-derived proteoglycan, Te38, has been found to be highly inhibitory to axonal regeneration [58] and is readily present within the lesion site following SCI [59]. While Te38 is able to be detected for up to 4 weeks after injury, the exact expression pattern for this proteoglycan has yet to be determined [59].
Neurocan and phosphacan are also both highly inhibitory to OPC process outgrowth and differentiation [47]. Importantly, studies have shown that CSPGs interact with adhesion molecules expressed on various cell types, mediating their inhibitory effects via the surface receptor protein tyrosine phosphatase sigma (PTPσ) that is found on both neurons and OPCs [49, 60–63]. This is an important finding since, to date, all other CSPG receptors, including Nogo-66 Receptor 1 (NgR1), Nogo-66 Receptor 3 (NgR3), and leukocyte common antigen receptor (LAR), have only been found on neurons [64, 65].
Overall, the inhibitory influence of CSPGs on the postinjury environment is a major barrier to the regenerative process. Further complicating this matter is the temporal expression of these molecules. While the induction of the CSPG synthesis begins immediately after injury, the upregulation of specific CSPGs happens at different intervals. Brevican, neurocan, and versican expression is found to be maximal at two weeks after injury, while NG2 achieves peak expression one-week after injury (Figure 2, [8]). Interestingly, however, the expression of phosphacan is initially downregulated and then begins to be expressed, with peak levels found approximately eight weeks after injury [8]. The continual upregulation of different CSPGs makes the postinjury environment of the spinal cord inhibitory to the regenerative process for many months.
The glial scar appears to be a paradoxical structure, identified as highly inhibitory to axonal regeneration, while also protecting and isolating the damaged tissue. The dual nature of the glial scar suggests that while it will need to be modified in order to create a permissive environment, the scar is necessary to prevent additional tissue damage. Additionally, the fact that reactive astrogliosis is a response that is graded to the nature of the CNS insult [29] indicates that treatments targeting this process will bring the most benefit to individuals suffering from severe injury.
2.2.4. Myelin Degradation
Both direct physical destruction and indirect damage due to inflammatory activity result in the death of oligodendrocytes, the myelin producing cell of the CNS. Oligodendrocytes are particularly sensitive to SCI [66] with tissue damage after an SCI resulting in the death of oligodendrocytes at the lesion site and, over time, even at a distance from the initial lesion [5]. Oligodendrocyte death occurs in two stages, with the initial loss being due to physical damage during the injury process and a delayed secondary loss resulting from ongoing pathology [4, 5, 7]. While the majority of early oligodendrocyte death is necrotic [7], oligodendrocyte apoptosis can be observed, both locally and in segments at a distance from the site of original lesion, for weeks following injury [67–74]. It has been demonstrated that a compression injury inflicted at the T8-9 level can lead to oligodendrocyte apoptosis at spinal levels as far away as T1-L2 [68]. This secondary process occurs, in part, due to SCI induced glutamate release, which reaches levels that are toxic to oligodendrocytes (550 mM ± 80 mM, [75]). Other events that induce apoptosis are the formation of free radicals in the lesioned tissue [7] and p75 neurotrophic receptor (p75NTR) mediated cell death. The latter results from p75NTR upregulation following pathologic stress to the oligodendrocytes [76, 77]. Specifically, trauma to the spinal cord leads to increases in the synthesis and production of nerve growth factor (NGF) by astrocytes, activated microglia, and vascular macrophages [78]. The immature form of NGF (proNGF) interacts with p75NTR on the surface of oligodendrocytes, resulting in apoptosis [79].
Loss of oligodendrocytes creates an excess of myelin breakdown products in the lesion. This myelin debris contains variety of myelin proteins, including Myelin Associated Glycoprotein (MAG), Myelin Oligodendrocyte Glycoprotein (MOG), Nogo-66, and Nogo-A, all of which have been demonstrated to be highly inhibitory to regenerating neurons [63, 80–90]. These molecules interact with a variety of surface receptors, such as Nogo receptor (NgR aka NgR1), p75NTR, and TROY (aka TAJ), and are documented to provide repulsive axonal guidance cues, by collapsing or causing retraction of the axonal growth cone [5, 44, 84, 89, 91, 92]. Due to the slow phagocytic nature of the central nervous systems macrophages and microglia, myelin proteins are able to remain in the postinjury environment for several months [5, 32, 93]. These proteins, however, are not the only inhibitory elements found at the lesion site. The regenerative process is also inhibited by the presence of glycoprotein CD44, tenascins, and semaphorins at the site of injury (reviewed by [4, 5, 26]).
2.2.5. Cavitation and Cyst Formation
As the macrophages and microglia clear cellular debris, the lesion cavity will eventually become nothing more than a fluid filled cyst called a syrinx [4, 7, 94]. While the pathophysiology underlying the formation of the posttraumatic syrinx is not fully understood, the process of postinjury cavitation is observed in both human and rodent cases of SCI [94, 95]. In the rat model of spinal cord injury, cavity formation is usually observed 15 days after injury [95]. Mouse models of SCI, interestingly, do not typically demonstrate such postinjury cavitation [95]. Syrinx formation in humans after SCI can take up to several months or years to manifest [94]. These cysts, once formed, run the risk of enlarging, producing a degenerative condition known as syringomyelia, which has the potential to cause further deterioration of sensory or motor function [94]. This fluid filled cavity also presents the surviving neurons, yet another challenge to overcome in the regenerative process, by eliminating the availability of an extracellular matrix on which their axons can grow.
Given the substantial role that cellular and molecular responses play in the regenerative ability of the spinal cord, it is critical that these aspects are considered when attempting to successfully approach the development and use of methods aimed at neurorestoration following SCI. Therefore, the remainder of review will be devoted to discussion of common research strategies for enhancing repair as well as the potential role of biomaterials in promoting more substantial neurorestorative effects.
3. Part II: Current Research Strategies to Enhance Repair
Given the complexity of the biological response to SCI, a number of different therapeutic approaches have been developed to target one or more of the issues preventing functional recovery. In general, spinal cord injury research focuses on a few broad topics: neutralization of inhibitory elements within the postinjury environment; promotion of neuronal survival (neuroprotection); stimulation of axonal regeneration and/or plasticity (neuroregeneration); and remyelination of denuded axons. Research in each one of these areas has yielded important insight into the ability for neurorestoration of the functional spinal cord as a result of postinjury environment manipulation.
3.1. Neutralization of Inhibitory Factors
Neutralization of inhibitory factors in the postinjury environment is one promising approach for enhancing the regenerative response following an SCI. While there are many different inhibitory elements that can be targeted within the postinjury environment, the most progress has been made on developing agents to neutralize the inhibitory influence of either CSPGs or myelin debris.
CSPGs expressed in and around the glial scar are widely accepted as a primary reason for the lack of axonal regeneration and/or remyelination following an SCI. However, the inhibitory nature of the CSPGs can actually be neutralized using the enzyme chondroitinase ABC (cABC). Chondroitinase is an enzyme produced by the bacteria Proteus vulgaris, which catalyzes the removal of the glycosaminoglycan side chains from the central core protein [96]. Many studies have shown that by treating a CNS lesion site with the enzyme cABC both axonal sprouting and axonal growth into and around the lesion are significantly increased [97–103]. The use of this agent has been shown to effectively reverse CSPG inhibition and promote axonal sprouting and outgrowth [97–102, 104]. Use of cABC also enables the migration and differentiation of endogenous OPCs [47–49, 105].
One of the major limitations of cABC as a treatment for SCI is the mode of administration. Chondroitinase is a very labile enzyme that when reconstituted does not retain its biological activity for very long, due to its thermal instability. When incubated at 37°C, the enzymatic activity of cABC, in solution, is gone by 7–10 days [106]. While the therapeutic ability of cABC is very promising, experimental stabilization of the enzyme is needed for it to be a more effective therapeutic agent. Stabilization of cABC has been attempted in several ways including alteration of its structure [107], incorporation in viral vectors for constant in vivo expression [103, 108, 109], or incorporation into biomaterial delivery systems [110].
While the neutralization of CSPGs has demonstrated promise in reversing their inhibitory effects, a similar result has also been noted via modulation of the activity of the PTPσ receptor. The PTPσ receptor has been identified as a CSPG interacting receptor that is expressed on neurons [49, 60–63]. In general, PTP receptors are a group of surface receptors that contain two catalytic domains, D1 and D2. The D1 domain is the primary catalytic site, while D2 serves regulatory functions. One way in which the activity of these receptors is modulated is through a wedge shaped sequence that is located between the membrane and the proximal region of the D1 catalytic domain [111]. Therefore, the activity of the PTPσ receptor can be inhibited using a generated membrane-permeable peptide that mimics the PTPσ wedge sequence. The binding of this peptide to the PTPσ receptor has been noted to result in a significant increase in axonal growth after injury [112]. As a result, research has determined that systemic delivery of this peptide over time allows for both the enhancement of serotonergic innervation of the spinal cord below the level of injury and the facilitation of recovery of motor function and micturition in treated animals [112].
The myelin debris released into the lesion environment presents additional inhibition to the regenerative ability of the injured axons, which becomes further compounded by the slow phagocytic clearance of the debris [5, 113, 114]. One myelin associated protein known to be a potent inhibitor of axonal regeneration, Nogo-A [115], is a specific target in the quest to neutralize inhibitory factors. Just as the inhibitory CSPGs can be neutralized using cABC or by modulating CSPG receptors with a blocking peptide, the negative influence of Nogo-A can be ameliorated using antibodies against it.
Significant increases in both the number of axons regenerating and the overall length of the regenerating axons have been found following infusion or other systemic deliveries of the Nogo-A antibody [85–88, 90]. Even after a long interval of time following a stroke injury, treatment with anti-Nogo-A can still produce not only a sprouting response from the damaged axons but also an improvement in subsequent functional recovery [116]. While the effects of Nogo-A on axonal regeneration and sprouting have largely been studied utilizing the stroke model of injury, its use in SCI models also demonstrates axonal sprouting and increases in the length of axonal arbors [87]. The observed neurite outgrowth, attained by using antibodies to Nogo-A, is thought to be accomplished through the inhibition of intracellular pathways that are activated by the Nogo receptor, such as the Rho/Rock pathway [52, 117].
Similar effects on axonal regeneration have also been noted following administration of an antibody that is specific to the potent inhibitory domain of Nogo-A, IN-1 [118, 119]. Notably, neuroanatomical evidence demonstrating regenerative axonal growth in combination with marked improvements in the recovery of function has been found when the IN-1 antibody is delivered to the site of a cerebral cortical transection or stroke injury in mice [118, 119]. More importantly, when IN-1 is administered into the CNS of nonhuman primates following a thoracic SCI, significant increases in axonal sprouting and regenerative growth are also found [120].
Taken together, these studies collectively demonstrate that the use of cABC, Nogo-A, or IN-1 to neutralize the inhibitory elements found within the postinjury environment has potential to aid the neuroregenerative response following SCI.
3.2. Stimulation of Axonal Regeneration
CSPGs interfere with axonal regeneration by inducing collapse of axonal growth cones, producing premature abortion of the normal regenerative response. Axonal collapse is thought to be a result of molecular signaling events activated within the axon itself. Exposure of the damaged axonal tip to the CSPGs and myelin debris found within the lesion results in the activation of inhibitory signaling pathways, such as RhoA/Rock. This then triggers the breakdown of actin filaments and results in the cessation of axon growth [121]. As stability of the axonal growth cone is dependent on microtubule polymerization, which is regulated by microtubule-actin interactions, a recent avenue of research has focused on promoting axon regeneration via microtubule stabilization and/or the modification of axonal pathway signaling.
Microtubule stabilizing anticancer drugs, which achieve their anticancer properties by interfering with cellular division, have recently shown promise in the field of axonal regeneration. Two such drugs are paclitaxel (Taxol) and Epothilone B [121–123]. Taxol has been found, both in vitro and in vivo, to prevent the formation of retraction bulbs after injury, stabilize the cytoskeleton of the reactive growth cone, and promote the regeneration of axons in an injured optic nerve model [121, 123]. Epothilone B, when given systemically following an SCI in rodents, has been found to decrease glial scarring and increase microtubule polymerization in the tip of the axon. In short, induction of microtubule polarization in the growth cone appears to drive growth of the axon at the site of lesion [122]. Importantly, not only do both Taxol and Epothilone B have the potential to enhance axonal growth following injury but also both of these drugs are currently FDA approved for cancer therapy. Thus, some evidence related to a degree of safety for use of such drugs in humans has been previously established in the cancer literature.
Axonal regeneration may be stimulated after injury through direct modulation of signaling pathways within the axons. While the molecular signaling events that occur within the axon are complex and numerous, there are a few that warrant discussion due to their ability to facilitate axonal regrowth. One such molecular signaling target is Phosphate and Tensin homologue (PTEN), which is a negative regulator of the mammalian target of rapamycin (mTOR). Recent studies have demonstrated that silencing this molecule results in significant axonal growth [124–126]. The disruption of PTEN, via mouse knockout models, has also been noted to produce robust axonal regeneration following a crush injury to the optic nerve [124, 125]. Even injecting shRNA against PTEN prior to injury appears to allow for protection of the regenerative response. Injections of shRNA against PTEN into the CST neuronal cell bodies of neonatal mice have been found to produce significantly higher levels of postinjury axonal regeneration, as compared to controls, when spinal cord crush injury had occurred 7 weeks after administration of the injection [126]. The regeneration that occurred following the shRNA injections was even present across areas rich in GFAP.
In addition to PTEN, suppressor of cytokine signaling 3 (SOCS3), which is a negative regulator of Janus kinase/signal transducers and activators of transcription (JAK/STAT), has been described as inhibitory to axonal regeneration. For example, conditionally knocking out SOCS3 in mice results in a significant increase in the number of axons that cross a crush injury to the optic nerve [127]. Further, when both SOCS3 and PTEN are knocked out, the amount of axonal regeneration observed following an optic nerve crush injury is significantly greater than what is achieved by knocking out only PTEN or SOCS3 alone [125].
Finally, another target for axonal regeneration therapies is the Krüppel-like factors (KLF) family of transcription factors. This family of transcription factors plays a large and important role in the regulation of neural growth and regeneration by either suppressing or enhancing axonal growth abilities. Interestingly, KLF family members known to be inhibitory to axonal growth (KLF 4 and 9) have been found to be upregulated postnatally, while those that are growth promoting (KLF 6 and 7) are downregulated at this time [128, 129]. Although this may sound counterintuitive to axon growth, which occurs at a high rate during development, knowledge of the levels of expression during times of high development provides another avenue for research into mechanisms for targeting regenerative potential.
These studies, when considered collectively, indicate that both strategies that target growth inhibitory signaling elements and therapies that stabilize the growth cone may be necessary in order to achieve the long distance growth needed for functional recovery following SCI.
3.3. Neurotrophic Factor Supplementation
The inhibitory nature of the postinjury environment is well described, and the physiologic and metabolic stresses experienced by the neuron are extensive. While it is clear that the neutralization of inhibitory elements found within the post-SCI environment has beneficial effects on axonal sprouting/growth, the overall health of neurons following injury still needs to be maintained. If the neuron dies, then any hope of a regenerative response is lost. Therefore, another active research area in SCI regeneration has focused specifically on the neuron, identifying ways to promote neuronal survival, axonal regeneration, and axonal plasticity through the use of neurotrophic (NT) agents and other growth promoting molecules.
Neurotrophic molecules consist of a family of proteins which are structurally similar and bind to one of three tyrosine kinase (Trk) surface receptors or the p75 neurotrophic receptor (p75NTR). Members of the NT family include Brain-Derived Neurotrophic Factor (BDNF) and neurotrophin-4 (NT4/5) which preferentially bind to TrkB, NGF which binds TrkA, neurotrophic factor-3 (NT-3), and its receptor TrkC [130–132]. While NTs bind to a specific Trk receptor, all of the NT molecules can bind to p75NTR, which has important physiological implications on neurons. A NT binding to p75NTR only without the expression of the appropriate Trk receptors can be harmful. For example, when sympathetic neurons expressing p75 and TrkA receptors were exposed to BDNF, the binding of BDNF to p75NTR without the presence of TrkB resulted in p75NTR induced apoptosis of the neurons [131–133].
Another family of growth promoting molecules is the glial derived neurotrophic factors, which require two surface receptor components. The GDNF family of molecules directly binds to one of four GDNF family receptor alphas (GFRα), which then complex with the Ret receptor tyrosine kinase. Members of this family include glial derived neurotrophic factor (GDNF) which binds GFRα1, neurturin which binds GFRα2, artemin which binds GFRα3, and persephin which binds to GFRα4 [132, 134].
The NT and GDNF family of molecules represents only a small sample of the plethora of neurotrophic substances and growth factors that may contribute to the regenerative quality of the CNS. Additional neurotrophic agents, which have also been shown to be potent in enhancing neuronal survival or axonal regeneration, include leukemia inhibitory factor (LIF) and ciliary neurotrophic factor (CNTF) [135–138]. Importantly, each of these additional agents binds to specific surface receptors, whose location and binding affinity must be considered when investigating their potential to assist in the regenerative process. Through the use of in situ hybridization, immunofluorescence, and genetic screening techniques, Trk, Ret, and GFRα receptors have been localized in several classes of afferent neurons, efferent neurons, and interneurons (e.g., [139–143]). If a specific neuronal population expresses a certain class of neurotrophin receptors, the neurons are considered to be responsive to that neurotrophin. Thus, BDNF, GDNF, NGF, NT-3, and NT-4/5 are commonly utilized in attempts to prevent injured neurons from undergoing apoptosis. Moreover, these factors have been used in order to coax such neurons into a regenerative response.
Studies have demonstrated that classes of efferent neurons such as CST, RuST, and coerulospinal and reticulospinal neurons are receptive to the NT agents, BDNF, GDNF, NT-3, and NT-4/5, with varying responses of neuronal survival, axonal sprouting, and even axonal growth [141, 144–148]. BDNF and NT-4/5 treatment has been shown to prevent the atrophy of RuST neurons, stimulate an upregulation of genes known to be associated with axonal regeneration, and even promote the regeneration of RuST axons [146, 147]. CST neurons have shown resistance to postinjury apoptosis following BDNF treatment [20] and have also been shown to be protected from cell death by the neurotrophic factors GDNF and NT-3 [141, 145, 148]. NT-4/5 promotes the growth of reticulospinal, coerulospinal, and PS axons, while appearing to have no significant effect on other efferent classes of neurons, such as the CST neurons [149]. While BDNF, NT-4/5, and GDNF are effective in coaxing classes of efferent neurons to undergo some degree of post-SCI sprouting, afferent classes of neurons, such as the dorsal root ganglion cells, are responsive to NT-3 and NGF [150, 151]. When considering or utilizing NT agents as a therapeutic strategy, there are three major issues that need to be carefully examined: the location of NT administration, the timing after injury in which the NT agent should be administered, and finally the number or combination of NT agents that need to be or can be administered at one time.
Overall, in vitro studies have shown that supplying neurotrophins, at the cell body and axon terminals combined or simply at the axon terminals alone, can maintain the neuronal cell body and induce axon growth [152]. In some cases where neurotrophic treatment is only applied to the cell body, axons are found to retract despite survival of the neuron [152]. However, in vivo studies of CST neurons demonstrate that BDNF treatment of the CST neuron at the cell body following axotomy saves the neuron from cell death and promotes sprouting of the injured axon [20, 153, 154], while damaged CST axons show no signs of regenerative growth when BDNF is applied at the lesion site in the spinal cord [20]. Treatment of RuST neurons with the neurotrophic factors at the level of the brain not only prevents their atrophy but also has been found to promote their axonal regeneration [146, 147]. Deciding on which location to apply the NT treatment is complicated, while the damaged end of the axons in cases of SCI is easily accessible, and the cell bodies of the major efferent neuronal classes are located in the brain and brainstem. This raises the question of whether or not performing invasive brain surgery to gain access to the neuroanatomical locations of the efferent neurons and potentially causing tissue damage via the administration of a NT agent are worth the risk posed to the patient.
Timing of NT delivery is another critical aspect when it comes to the use of NT agents to promote repair in the lesioned spinal cord. The lesion site and surrounding tissue present a very dynamic environment, with a multitude of events occurring concurrently (Figure 3). This complex orchestration of events (see review [9]) shifts from an acute phase of inflammation and tissue necrosis, resulting in an immediate loss of neurons and myelin, to a chronic phase of secondary injury, where the CNS structures distal to the site of injury undergo neuronal atrophy, CSPG upregulation, and demyelination (Figure 3). Neuronal response to NTs can be significantly impacted by the time of administration. In rodent models of SCI, BDNF and/or NT-3 delivered either immediately or 7 days after transection has been found to produce differential effects on axonal regeneration and functional recovery. Specifically, significantly greater amounts of axonal regeneration and behavioral recovery were observed in animals that received the delayed NT treatment, as compared to those having received treatment immediately following the spinal transection [155]. In contrast, immediate treatment of injured RuST neurons with BDNF results in robust growth of damaged axons in the spinal white matter [156]. However, when BDNF is provided at the lesion site several days following an injury, it appears to have no effect on RuST neurons [142]. The response of RuST neurons to BDNF illustrates another critical aspect of the issue of timing; traumatic injury to CNS neurons can cause a differential and transient expression of surface receptors that bind to specific neurotrophins. Examples of this postinjury shift in expression were observed in TrkB surface receptor on RuST neurons [142]. While TrkB is expressed along the entire axon and cell body of uninjured RuST neurons, the axonal expression of TrkB following a traumatic injury was found to diminish as the interval after injury increased [142, 147]. At 1 and 2 months after injury, TrkB receptor expression is found to be localized only to the RuST cell body with no expression on the reactive ending of the injured axon [142, 147]. This finding offers an explanation as to why BDNF treatment at the RuST cell body is successful in promoting growth and survival as well as axonal sprouting, while treatment at the damaged reactive ending appears to have no effect on the RuST neurons [142, 147].
Figure 3.
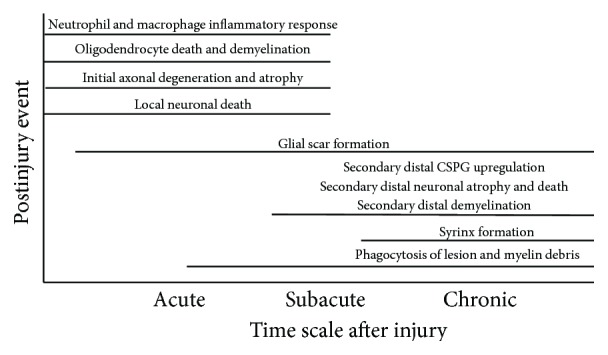
Chronology of postinjury events. The lesion site in an injured spinal cord is also a very dynamic environment that undergoes many different changes, as the lesion changes from an acute injury to a chronic injury. In addition to the inhibitory environment established after SCI, the ever-changing nature of these postinjury events needs to be factored into the design of any therapeutic treatment [4, 5, 7, 9, 10].
Microarray studies examining the postinjury response of specific classes of CNS neurons have also demonstrated how critical the issue of timing is in regard to the regenerative response. In a study examining the response of short thoracic propriospinal (TPS) neurons to axotomy, a strong upregulation in the genes for the receptors of GDNF and LIF was observed 3 days after injury [143]. Even more interesting was the upregulation in the NT receptor genes that occurred concurrently with the upregulation in several genes commonly associated with axonal regeneration. Following the 3 days after injury, gene expression level for both NT receptors and regeneration-associated genes began to decrease [143]. Therefore, the timing of NT administration may need to be specifically tailored to the postinjury expression curve of the NT receptors for individual populations of neurons in order to maximize the regenerative potential of these cells. Further, another important aspect of NT treatment is the timeframe in which the NT agents will be needed in the postinjury environment. Axonal growth proceeds at a very slow rate. Therefore, NT therapy will need to be administered in a manner that will allow the agent to be present in the lesion site for many months after injury.
Given the sheer number NT and growth factor receptors that are expressed on neurons and the differential expression of these receptors in efferent and afferent neuron populations, it is likely that different combinations of these NT molecules will be needed in order to elicit full regenerative potential after injury. To this end, studies have shown enhanced regenerative responses in retinal ganglion cells following application of a combination of BDNF, CNTF, fibroblast growth factor (FGF2), and NT-3, as compared with use of each factor independently [157–159]. In the study examining the postinjury effects of TPS neurons, examination of PS neurons after injury showed an upregulation in the receptors for GDNF and LIF, with no change in expression for the BDNF, NT-4/5, and NT-3 receptors [143]. The expression of many different NT receptors in TPS neurons strongly suggests that multiple agents (BDNF, NT-3, NT4/5, GDNF, and LIF) may be necessary for a strong and sustained regenerative response.
Administering neurotrophic agents after SCI results in an increase in the percentage of neurons spared from atrophy and apoptosis, as well as an enhancement of axonal sprouting or regeneration, when compared to control groups [20, 147, 156, 160]. It is clear however that many questions and problems with NT supplementation still exist. While considering which NT agents to give and how best to provide them simultaneously, the response of the other cell populations, glial cells, and immune cells, to each individual NT administered will have to be addressed. In addition to the critical issues of location and timing noted above, another important limitation that will need to be overcome is the formation of the “sink” or “honeypot” effect [161]. This occurs when the injection or infusion site has such a high concentration of NT agent and the sprouting/regenerating axons or migrating OPCs do not move beyond or outside of borders of this location [159, 161]. While NT administration will definitely have to be part of any postinjury regenerative therapy, there are still many issues that need to be resolved.
3.4. Remyelination
An additional avenue that can enhance functional recovery after SCI is the process of remyelination. As previously discussed, the survival of myelin producing oligodendrocytes can be limited by both direct and indirect factors following SCI. While the exact axonal cues that mediate oligodendrocyte survival have not been fully elucidated, both in vivo and in vitro experiments have demonstrated that the degeneration of axons subsequently results in the death and degeneration of oligodendrocytes (see review [162]). Further, when oligodendrocytes undergo apoptosis, all axons that are wrapped by that particular oligodendrocyte undergo the process of demyelination. Without myelin, the saltatory conduction of action potentials across the demyelinated portions of the intact axons can be severely impaired, exacerbating the postinjury deterioration of function [4, 5]. This process may be a critical component of the limited potential for functional recovery, given that one oligodendrocyte can be responsible for myelinating up to 60 different axons [163].
Remyelination of axons depends upon the health and availability of OPCs. Upon the completion of initial axon myelination, populations of adult OPCs remain throughout the brain and spinal cord. In order for OPCs to be successful in remyelinating axons, they must be able to proliferate, migrate towards the site of demyelination, make contact with an axon, and then mature into myelin forming oligodendrocytes [5]. These remaining progenitor cells are highly responsive to a demyelinating lesion (see reviews [164–166]), with those located within about 2 mm of the lesion site appearing to migrate towards the site of demyelination [167]. Unfortunately, however, the postinjury environment created following an SCI is highly inhibitory to the OPC, preventing both migration and development of these cells [47–49]. Additionally, the infiltration of OPCs into the lesion site and subsequent remyelination of denuded axons are minimal to nonexistent as the OPCs only accumulate at the border of the lesion [105, 165, 166]. CSPGs expressed in the glial scar are also highly inhibitory to the process outgrowth and differentiation of OPCs [5, 47–49, 63, 165, 168]. While this inhibitory influence is thought to be a major reason for failure of spared axon remyelination, an alternative theory posits that mature and damaged axons are no longer able to undergo the process of myelination.
The hypothesis that adult axons are no longer capable of myelination has been addressed in a series of different studies. In the adult retina, the nerve fiber layer contains naturally unmyelinated axons, as OPCs are unable to migrate out of the optic nerve and myelinate these axons. However, when OPCs are transplanted into the nerve fiber layer and then the layer is examined 4 weeks after OPC transplantation, axons have been found to undergo myelination [169]. The ability of demyelinated axons to remyelinate has also been demonstrated utilizing the cuprizone model of demyelination. Although cuprizone, when added to the diet of lab animals, results in demyelination, it has been demonstrated that spontaneous remyelination occurs quickly after removal of this drug from the animal's diet [170–173]. Collectively, these studies argue against the hypothesis that adult or demyelinated axons are no longer capable of undergoing (re)myelination and that the one likely cause of remyelination failure after CNS injury is the formation of the glial scar.
Interestingly, experimental therapies commonly used to stimulate axonal sprouting and regeneration after SCI have also demonstrated effects on the biology of OPCs. Supplementing the lesion site with various neurotrophic factors such as BDNF, NT-3 [174], or other growth promoting agents, that is, apotransferrin [175] resulted in enhanced levels of remyelination after injury. It has also been demonstrated in vitro that when BDNF, NT-3, and GDNF are supplied to OPCs grown in the presence concentrations of CSPGs, the OPCs are able to overcome the CSPG mediated inhibition, undergoing bipolar process outgrowth and differentiation [176].
In addition to NT treatment, another method for promoting remyelination of spared axons is through the use of antibodies to block the protein Leucine Rich Repeat and Ig Domain Containing 1 (LINGO-1). LINGO-1 is highly inhibitory to the myelination process and is selectively expressed in both oligodendrocytes and neurons. The expression of this protein is developmentally controlled, is known to be upregulated following CNS disease or injury, and inhibits the differentiation and maturation of OPCs via the activation of RhoA pathway [177, 178]. Studies have demonstrated in animal models of demyelination (autoimmune encephalomyelitis or lysolecithin-induced) that utilization of LINGO-1 knockout animals or administration of anti-LINGO-1 antibodies results in significantly increased levels of remyelination [178–180]. With respect to human populations, the use of anti-LINGO-1 as a method of medical treatment for multiple sclerosis (MS) cleared phase I clinical trials in April 2012 and has since moved into phase II [181–183]. While the findings and clinical trials for anti-LINGO-1 antibodies revolve around demyelinating conditions such as MS, anti-LINGO-1 does present another potential therapeutic opportunity for the treatment of SCI.
Promotion of remyelination has also been attempted via cellular transplantation. Transplanting cells, such as OPCs [184–186], Schwann cells (SCs) [187, 188], olfactory ensheathing cells (OECs) [186, 187, 189], or stem cells [186, 190–193], into the site of a demyelinating lesion, has been found to enhance CNS remyelination and subsequent recovery of function. While the results from these studies suggest that cell implantation may be an effective method for remyelinating spared axons, there are some technical difficulties and biological incompatibilities [186, 194] that need to be considered. One such issue is the potential incompatibility between the implanted cell and the endogenous environment of the lesioned CNS. This phenomenon has been noted following transplantation of SCs, which, capable of remyelinating denuded CNS axons, are unable to migrate within CNS tissue or integrate with astrocytes. This unfavorable interaction between SCs and the CNS environment has been documented both in vitro, with the failure of SCs to integrate with astrocytes [195], and in vivo, with implanted SCs failing to migrate beyond the lesion border [196]. OECs, on the other hand, do integrate with astrocytes [195] but still do not migrate within the damaged spinal cord following injury [197, 198]. Given that transplanted cells are unable to migrate and integrate appropriately, it is not surprising that other problems related to the implantation of stem cells include the possibility of tumorigenicity and the inability to ensure that the stem cells will differentiate into the desired myelin forming cell, as opposed to another phenotype dictated by local environmental influences [185, 194].
Remyelination of spared axons is an enticing avenue of research, given that it may explain an apparent disconnect within the reported findings of many axonal regeneration studies, which have demonstrated paradoxical functional recovery without full anatomical regeneration. While most studies can show evidence of increased axonal sprouting into the spinal cord lesion site, very few studies show that these axons grow beyond the lesion [199]. Thus, functional recovery must be due to some mechanism other than axonal reconnection. This leaves open the possibility that observed recovery in function could be due to the remyelination of spared axons.
4. Part III: Use of Biomaterials to Promote Repair
The complex nature of the spinal cord injury dictates that multiple agents will be needed to maximize repair (reviewed by [200]). The lesion itself is usually an irregular size, more often being a partial injury, as opposed to a complete transection. The cellular response, as described in detail above, creates an environment that is not very conductive to repair. Astrogliosis produces a gliotic scar expressing high levels of CSPGs that inhibit axonal regeneration. Neurons that are not connected to their target cells will attempt to regenerate axons and reconnect but are most often unsuccessful. These neurons can survive for several months, but the cell bodies themselves will atrophy and eventually die if connections are not restored [147, 201, 202]. In order to achieve complete restoration of motor function, it is clear that a combination of NTs, to maintain neuronal survival and stimulate axonal regrowth, as well as agents such as cABC, to neutralize the inhibitory effects of the scar, will be required.
One major limitation of many promising treatment strategies for spinal cord injuries is the method of delivery. Most of the therapeutic agents described above have to be delivered via an injection, series of injections, implantation of a pump or intrathecal catheter, use of a viral vector, or implantation of fibroblasts or other cells genetically engineered to produce a given NT or cABC [100, 142, 147, 156, 174, 203–206]. These delivery methods are highly invasive, which could trigger further astrogliotic scarring and inflammation, potentially causing additional neurological damage. Furthermore, the use of viral vectors and implanted genetically engineered cells that deliver a NT or cABC treatment presents an uncontrolled method of delivery and a potential tumor hazard. The use of minipumps and intrathecal catheters provide a nonspecific method of treatment prone to clogging or infection [207]. Thus, utilizing biomaterials that can assist with delivery and control of these agents is a promising area in the field of spinal cord repair.
4.1. Critical Issues in the Design of Biomaterials
At the present time, there is no agreement on the optimal characteristics for biomaterials used to repair of the damaged spinal cord [208]. While many different polymers and molecules have been utilized to treat SCI, there are clearly important considerations that cannot be ignored in the development and engineering phase of such biomaterials. First there is the biocompatibility of the material with the host tissue. The developed materials should not elicit an immune response nor be toxic to cells over long periods of time. If the material is biodegradable, the degradation products also should not be toxic to the surrounding tissue [209, 210]. Second, any biomaterial device should be easily introduced into the spinal cord without producing further damage. This can be challenging, as the natural response to spinal injury is the generation of a glial scar. Third, the device must be able to remain in place over long periods of time, in order to allow for nerve growth. This is particularly important for nanoparticles, which are often used for drug delivery. However this consideration is equally necessary for scaffolds and other types of implants. The final critical concern is the ability of the material to bind growth promoting molecules like NT, peptides, and cells, all of which would be delivered in bioactive forms to the injury site to stimulate tissue repair. Scaffolds alone will not be sufficient to enable the maximal repair of the damaged lesions.
The use of biomaterials in the spinal cord generally falls into one of three classes: guidance channels and scaffolds, hydrogels, and nanoparticles. Each one of these can be produced with different chemical compositions, uses, and biological compatibilities. The bioengineering and design of these materials is a very active research field and have been extensively described in the literature [211–214]. Thus, the remainder of this review will be focused on promising applications of biomaterials in the treatment of spinal cord injury.
4.2. Guidance Channels and Scaffolds
Guidance channels have been proposed as far back as the late 1800s, with the thought that demineralized bone tubes could be used to fix nerve gaps [215]. The idea behind guidance channels or conduits is to seal the two severed ends of the nerve in a hollow tube, which will direct new axonal regrowth towards the distal nerve stump. Nerve conduits have been successful in repairing peripheral nerve damage, since it is relatively easy to isolate the two individual nerve endings. This is particularly true of larger nerves. However, when considered for use in a spinal cord injury, guidance channels may be better for the transected spinal cord rather than the contusion injury. Unfortunately, the majority of SCIs are incomplete contusion injuries [3], which do not leave discrete nerve stumps and are unable to be sealed effectively using guidance channels. The lesion site in such injuries is irregular and ultimately becomes a cyst-like structure. Therefore, methods of either filling in or crossing these irregularly shaped gaps with a growth promoting substrate are needed. As such, scaffolds that can be inserted into the lesion site are more often being utilized for the contused spinal cord lesion.
In the case of peripheral nerve injury, there are several nerve conduits that are FDA approved for the repair of peripheral nerve gaps that are 30 mm or less (reviewed by [216, 217]). Almost all are made from biodegradable materials that include natural materials like collagen I or synthetic polymers such as polyglycolic acid (PGA) and poly-DL-caprolactone. While they all degrade at slow rates to allow time for the nerve to regenerate, it is imperative that total degradation occurs, ensuring that remaining fragments do not trigger scarring. The current conduits are permeable to nutrients and oxygen and flexible but strong enough to support the nerve and maintain its position during the repair process. While this form of treatment is being utilized in the clinic, there is a clear need for more clinical studies to evaluate the relative efficacy in promoting peripheral nerve repair.
Various forms of scaffolds have been designed to be placed into the spinal cord lesion in order to provide a bridge through the cavitations formed following injury (reviewed by [210]). These provide a permissive and growth promoting environment that allows axons to grow through the lesion unimpeded. Similar to guidance channels, these can be made out of natural materials such a collagen I, agarose, or fibronectin, as well as synthetic polymers like polylactic acid (PLA), PGA, or poly(2-hydroxyethyl methacrylate) (pHEMA). The structure of these scaffolds can vary greatly: they can be cylindrical or rectangular, resemble a multichannel guidance channel, or are sponge-like, with numerous scattered pores. There are also some designs with complex, defined paths which are intended to direct axon growth from specific nerve tracts [218, 219].
Scaffolds and guidance channels can incorporate both growth promoting molecules and a variety of cells that may assist in speeding axon growth through the lesion. The growth factors can be incorporated or attached to the scaffold itself [204, 220]. More often, the scaffold is seeded with cells that produce neurotrophic factors. Schwann cells, genetically altered fibroblasts, or neural stem cells have often been included in various scaffold matrices [221–226]. All scaffold designs are porous structures that are being optimized for long term survival of transplanted cells, while allowing for the infusion of nutrients, oxygen, and formation of new vasculature.
One important feature of any scaffold is that it should not elicit a host reaction to the implant. It was noted early that any implant into the spinal cord that was not biodegradable would activate a tissue response that ultimately resulted in the implant being encapsulated in reactive cells and separated from the host tissue [227]. Activation of immune cells such as macrophages and microglia may alter the effectiveness of any implant. Thus, most scaffolds are biodegradable over time, with their surfaces being modified to manage the host tissue response. This has been accomplished by using materials that encourage the attachment and even infiltration of endogenous cells such as fibroblasts, immune cells, and OPCs that are found close to the lesion site.
The advantage of scaffolds is that they bridge an area of the lesion that is inhospitable with axon regeneration. There are numerous studies that can demonstrate enhanced axonal growth and even some motor improvement, when such techniques are utilized in an experimental model of SCI (reviewed by [210, 228]). Moreover, scaffolds can be designed to guide the direction of new axonal growth through the use of microchannels or other tracts patterned into the scaffold. The disadvantage with this technique is that scaffolds have to be surgically implanted directly into the lesion. Since the dimensions of an injury site tend to be quite irregular, it may be difficult to find an optimal design that will work within each instance of spinal lesion.
4.3. Hydrogels
Hydrogels are water saturated polymers that can be developed to mimic the three-dimensional physical properties of the host environment (reviewed by [229, 230]). These polymers can be used in the creation of implantable scaffolds which, as described above, provide a bridge across irregular lesion sites. However, one important quality that makes hydrogels especially appealing for use in SCI repair is that many are able to be injected directly into the lesion site, where they can polymerize in vivo. Such polymers are extremely flexible and can fill irregularly shaped lesion cavities by absorbing water, expanding, and forming a flexible three-dimensional structure that closely resembles the extracellular matrix (ECM). Additionally, peptides can be designed to undergo triggered self-assembly, allowing for the formation of hydrogel scaffolds in response to specific changes in the physiological environment (reviewed by [230, 231]). Such hydrogels can also be loaded with growth promoting molecules such as NT or cells that can stimulate axon growth and tissue repair and can even be utilized with a defined scaffold design to maximize growth potential.
Hydrogels can be classified into two general categories: natural and synthetic, referring to the origin of the molecules being used. Mammalian ECM-based natural polymers such as collagen, fibronectin, hyaluronic acid, or combinations are often used in hydrogel creations because of their biocompatibility and the fact that they are part of the naturally occurring ECM. Such substances can be used as a cell-delivery vehicle to promote neurite outgrowth while also providing structural support to the regenerating tissues [206, 232]. Other naturally occurring polysaccharides such as chitosan, agarose, alginate, xyloglucan, gellan gum, and methylcellulose have also shown promise in treating SCI. Often these are used in various combinations, in an attempt to optimize growth promoting properties. All are slowly biodegradable over time, but the rates of degradation are set based on the properties of the molecules and cannot be readily altered.
Synthetic polymers are being developed which can be optimized for maximal protein, cell binding, and rates of degradation. Some of the most common synthetics used for CNS repair have been developed from poly(hydroxyethyl methacrylate) (pHEMA) and derivatives, poly-ethylene-glycol (PEG)/poly-ethylene oxide (PEO), poly(vinyl alcohol) (PVA), and poly(alpha-hydroxyacids). Being synthetic, these substances have some inherent advantages over the natural molecules. They can be manufactured easily, and the properties of the polymers can be customized, to maximize the desired capabilities. For example, the surface of the polymer gel can be optimized for cell attachment. Control of degradation rate can protect or release cells that are transplanted in the hydrogel, depending on the need and the role of these cells in the repair process. Since they are not derived from animals, the potential for allergic reactions to the hydrogel is also minimal [229]. Like scaffolds, both natural and synthetic polymers can be modified to deliver a range of agents, from NT and other growth factors to antagonists for axonal growth inhibitors like Nogo-66 [233–235].
While hydrogels show promise as a potential strategy to maximize repair of the spinal cord, there is no obvious polymer or combination of polymers that are optimal for this application. Therefore the identification of such agents is an active field of research. There are concerns that the mechanical strength of the hydrogel is not sufficient to sustain the lesion and that hydrogels have a shorter durability than fabricated scaffolds because they degrade quickly. More importantly, there is no directionality of the microchannels formed after polymerization in vivo. If the hydrogel is rich in growth promoting molecules, regenerating processes could extend into the hydrogel and remain there, mimicking the “honey pot” effect [161]. Overall, specific patterning may be required to actually direct the growth of regenerating axons through the lesion.
4.4. Nanoparticles
An alternative approach to treatment of the damaged spinal cord is the use of nanoparticles, which can be used to administer growth factors, NT, and antagonists to inhibitory substances in the lesion. Nanoparticles and microspheres are polymer derived particles that can degrade over time to release any encapsulated agents. These are being widely tested in a myriad of drug delivery applications in multiple tissues, from alleviating tissue rejections from allografts, to targeting cancer cells [236, 237]. These are an attractive delivery system as they are injectable, they provide localized drug delivery without systemic effects, the dose can be titrated easily, and they can target a specific cell type by modification of the cell surface properties. Based on these features, nanoparticles are a major focus of drug delivery methods.
Nanosphere delivery of growth factors and other agents can be successfully used to treat the spinal cord lesion. Drug delivery after an SCI is difficult because of the loss of vascularization and the instability of some of the more promising agents. Chondroitinase ABC is one such agent: it can degrade the glycan residues attached to CSPGs that inhibit axonal growth, neutralizing the effects of these CSPGs. However, it is highly unstable in solution, losing most of its activity within days [106]. It shows great promise in experimental models of spinal cord injury, allowing for substantial growth through a spinal lesion and improved functional recovery [98–100, 103, 238–242]. However, for translation to a human patient population, new delivery methods need to be developed. Nanospheres containing cABC have been developed and utilized in rat spinal contusion models [110]. These nanoparticles can release a sustained supply of active enzyme over the course of three weeks minimum and generate substantial axonal growth through the lesion site. They are nontoxic and do not elicit an inflammatory reaction in the spinal cord. Modulation of the surface charge ensures that they remain in the injury site. They can also be effective in the digestion of CSPGs at chronic times after injury, when the glial scar is fully established.
Nanoparticles and nanospheres that deliver GDNF, BDNF, and NT3 are being developed for several applications in the CNS, including SCI (reviewed by [218, 243, 244]). Other drugs are also being delivered to the spinal cord by nanospheres, including methylprednisolone and estrogen, which are anti-inflammatory agents that can have untoward side effects if administered systemically [245, 246]. Many nanoparticles are fabricated from synthetic polymers such as PLGA and PGA, which can be titrated to regulate the release kinetics. However, they can also be produced using natural polymers such as chitosan. At present, nanoparticles have to be injected into the spinal cord directly; however, recent research efforts are focused on surface modifications that can allow for the nanoparticles to cross the blood-brain-barrier and enter the brain and spinal cord without a direct injection into the tissue. Nonetheless, a direct injection of nanoparticles is much less invasive than the introduction of a scaffold at the lesion.
Nanosphere delivery of therapeutic molecules is attractive for treatment of SCI for many reasons: they are minimally invasive and provide sustained local drug release, which results in a higher dose locally without systemic side effects. Moreover, administration of multiple agents of growth factors could be accomplished by an injection of a mix of nanospheres containing growth factors, cABC, and Nogo antagonists. However, there are many questions that need to be answered prior to use of nanoparticles in a clinical setting. Some of the more critical questions concern the release rates and dose of the agents released from nanoparticles. For example, how much of a particular agent will be required before therapeutic effects occur? Moreover, how long will such agents need to be released into the post-lesion site? cABC and methylprednisolone may be needed acutely, but NT and other molecules may be needed at later stages following injury. A formulation that is released for months has not yet been manufactured in any experimental condition. Nanoparticles could be included in hydrogels to extend release times at the lesion site, if needed [242]. Details on optimal doses and release times in the context of an SCI will need to be determined in order to optimize the use of nanoparticles as a drug delivery system.
5. Discussion and Future Directions
Spinal cord injuries are complex and difficult to repair. Research efforts thus far have characterized many molecular events that occur at the injury site, allowing for the identification of several avenues for therapeutic intervention. In short, interventions aimed at promoting functional recovery following spinal cord injury may be targeted towards the neutralization of inhibitory proteoglycans, support of neuronal survival, and stimulation of axonal regeneration and remyelination. The clinical consensus is that there is no single therapeutic agent that can effectively address these issues and that maximal restoration of motor function will most likely be achieved with a mix of agents, each optimized to target a specific aspect of the postinjury response. The other significant problem that persists in developing treatments for SCI relates to the delivery of therapeutic agents to the spinal cord at the appropriate time to facilitate repair. Axonal regeneration is a slow process which, depending on the size of the lesion, could take many months or years in humans. Unfortunately, at present, little is known about how long various therapeutic agents will be needed, their biological half-life and bioactivity in vivo, or exactly when they should be introduced into the damaged spinal cord in order to obtain maximal therapeutic effects.
The use of biomaterials provides a promising avenue for addressing the above-mentioned concerns regarding spinal cord repair. Such materials can be utilized not only to deliver therapeutic agents but also to provide physical support for the damaged tissue. Additionally, both natural and synthetic polymers can be used to fabricate several types of structures that can release therapeutic agents with customizable release kinetics. While extensive literature on scaffolds, hydrogels, and nanoparticles exists, there is currently no consensus as to which material is most optimal in repairing the lesioned spinal cord. Ultimately, as new biological requirements of damaged spinal tissue are discovered, the development of biomaterials specialized for the treatment of SCI will need to consider how each of these requirements plays a role in the natural injury response process and potential for functional recovery.
Conflict of Interests
All contributing authors have no conflict of interests.
References
- 1.Hughes J. T. The Edwin Smith Surgical Papyrus: an analysis of the first case reports of spinal cord injuries. Paraplegia. 1988;26(2):71–82. doi: 10.1038/sc.1988.15. [DOI] [PubMed] [Google Scholar]
- 2.Elatorai I. M. History of spinal cord medicine. In: Lin V. W., editor. Spinal Cord Medicine: Principles & Practice. New York, NY, USA: Demos Medical; 2003. pp. 3–14. [Google Scholar]
- 3.SCI Information Network. University of Alabama at Birmingham, 2014, https://www.nscisc.uab.edu/
- 4.Profyris C., Cheema S. S., Zang D., Azari M. F., Boyle K., Petratos S. Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiology of Disease. 2004;15(3):415–436. doi: 10.1016/j.nbd.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Fawcett J. W., Asher R. A. The glial scar and central nervous system repair. Brain Research Bulletin. 1999;49(6):377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- 6.Popovich P. G., Wei P., Stokes B. T. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. Journal of Comparative Neurology. 1997;377(3):443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 7.Norenberg M. D., Smith J., Marcillo A. The pathology of human spinal cord injury: defining the problems. Journal of Neurotrauma. 2004;21(4):429–440. doi: 10.1089/089771504323004575. [DOI] [PubMed] [Google Scholar]
- 8.Jones L. L., Margolis R. U., Tuszynski M. H. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Experimental Neurology. 2003;182(2):399–411. doi: 10.1016/S0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- 9.Oyinbo C. A. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiologiae Experimentalis. 2011;71(2):281–299. doi: 10.55782/ane-2011-1848. [DOI] [PubMed] [Google Scholar]
- 10.Andrews E. M., Richards R. J., Yin F. Q., Viapiano M. S., Jakeman L. B. Alterations in chondroitin sulfate proteoglycan expression occur both at and far from the site of spinal contusion injury. Experimental Neurology. 2012;235(1):174–187. doi: 10.1016/j.expneurol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyor W. R., Avgeropoulos N., Ohlandt G., Hogan E. L. Treatment of spinal cord impact injury in the rat with transforming growth factor-β . Journal of the Neurological Sciences. 2002;200(1-2):33–41. doi: 10.1016/s0022-510x(02)00113-2. [DOI] [PubMed] [Google Scholar]
- 12.Young W. Spinal cord contusion models. Progress in Brain Research. 2002;137:231–255. doi: 10.1016/s0079-6123(02)37019-5. [DOI] [PubMed] [Google Scholar]
- 13.Watson C., Kayalioglu G. The organization of the spinal cord. In: Watson C., Paxinos G., Kayalioglu G., editors. The Spinal Cord: A Christopher and Dana Reeve Foundation Text and Atlas. London, UK: Elsevier; 2008. pp. 1–6. [Google Scholar]
- 14.Maxwell W. L. Histopathological changes at central nodes of Ranvier after stretch-injury. Microscopy Research & Technique. 1996;34(6):522–535. doi: 10.1002/(sici)1097-0029(19960815)34:660;522::aid-jemt462;3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 15.Yawo H., Kuno M. Calcium dependence of membrane sealing at the cut end of the cockroach giant axon. The Journal of Neuroscience. 1985;5(6):1626–1632. doi: 10.1523/JNEUROSCI.05-06-01626.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geddis M. S., Rehder V. Initial stages of neural regeneration in Helisoma trivolvis are dependent upon PLA2 activity. Journal of Neurobiology. 2003;54(4):555–565. doi: 10.1002/neu.10183. [DOI] [PubMed] [Google Scholar]
- 17.Kamber D., Erez H., Spira M. E. Local calcium-dependent mechanisms determine whether a cut axonal end assembles a retarded endbulb or competent growth cone. Experimental Neurology. 2009;219(1):112–125. doi: 10.1016/j.expneurol.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Kandel E. R., Schwartz J. H., Jessell T. M. Principles of Neuroscience. New York, NY, USA: McGraw-Hill; 1991. (edited by: E. R. Kandel, J. H. Schwartz, T. M. Jessell). [Google Scholar]
- 19.Dahlin L. B. The biology of nerve injury and repair. Journal of the American Society for Surgery of the Hand. 2004;4(3):143–155. doi: 10.1016/j.jassh.2004.06.006. [DOI] [Google Scholar]
- 20.Lu P., Blesch A., Tuszynski M. H. Neurotrophism without neurotropism: BDNF promotes survival but not growth of lesioned corticospinal neurons. Journal of Comparative Neurology. 2001;436(4):456–470. doi: 10.1002/cne.1080. [DOI] [PubMed] [Google Scholar]
- 21.Bregman B. S., McAtee M., Dai H. N., Kuhn P. L. Neurotrophic factors increase axonal growth after spinal cord injury and transplantation in the adult rat. Experimental Neurology. 1997;148(2):475–494. doi: 10.1006/exnr.1997.6705. [DOI] [PubMed] [Google Scholar]
- 22.David S., Aguayo A. J. Axonal elongation into peripheral nervous system ‘bridges’ after central nervous system injury in adult rats. Science. 1981;214(4523):931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- 23.Benfey M., Aguayo A. J. Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature. 1982;296(5853):150–152. doi: 10.1038/296150a0. [DOI] [PubMed] [Google Scholar]
- 24.Richardson P. M., Issa V. M. K., Aguayo A. J. Regeneration of long spinal axons in the rat. Journal of Neurocytology. 1984;13(1):165–182. doi: 10.1007/bf01148324. [DOI] [PubMed] [Google Scholar]
- 25.Chung K., Coggeshall R. E. Propriospinal fibers in the rat. Journal of Comparative Neurology. 1983;217(1):47–53. doi: 10.1002/cne.902170105. [DOI] [PubMed] [Google Scholar]
- 26.Silver J., Miller J. H. Regeneration beyond the glial scar. Nature Reviews Neuroscience. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 27.Fitch M. T., Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Experimental Neurology. 2008;209(2):294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sofroniew M. V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sofroniew M. V., Vinters H. V. Astrocytes: biology and pathology. Acta Neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raposo C., Schwartz M. Glial scar and immune cell involvement in tissue remodeling and repair following acute CNS injuries. Glia. 2014;62(11):1895–1904. doi: 10.1002/glia.22676. [DOI] [PubMed] [Google Scholar]
- 31.Beller J. A., Snow D. M. Proteoglycans: road signs for neurite outgrowth. Neural Regeneration Research. 2014;9(4):343–355. doi: 10.4103/1673-5374.128235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoll G., Trapp B. D., Griffin J. W. Macrophage function during Wallerian degeneration of rat optic nerve: clearance of degenerating myelin and Ia expression. The Journal of Neuroscience. 1989;9(7):2327–2335. doi: 10.1523/JNEUROSCI.09-07-02327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry V. H., Brown M. C., Gordon S. The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. The Journal of Experimental Medicine. 1987;165(4):1218–1223. doi: 10.1084/jem.165.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleming J. C., Norenberg M. D., Ramsay D. A., et al. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129(12):3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- 35.Kigerl K. A., Gensel J. C., Ankeny D. P., Alexander J. K., Donnelly D. J., Popovich P. G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of Neuroscience. 2009;29(43):13435–13444. doi: 10.1523/jneurosci.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laabs T., Carulli D., Geller H. M., Fawcett J. W. Chondroitin sulfate proteoglycans in neural development and regeneration. Current Opinion in Neurobiology. 2005;15(1):116–120. doi: 10.1016/j.conb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Galtrey C. M., Fawcett J. W. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Research Reviews. 2007;54(1):1–18. doi: 10.1016/j.brainresrev.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Siebert J. R., Steencken A. C., Osterhout D. J. Chondroitin sulfate proteoglycans in the nervous system: inhibitors to repair. BioMed Research International. 2014;2014:15. doi: 10.1155/2014/845323.845323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faulkner J. R., Herrmann J. E., Woo M. J., Tansey K. E., Doan N. B., Sofroniew M. V. Reactive astrocytes protect tissue and preserve function after spinal cord injury. The Journal of Neuroscience. 2004;24(9):2143–2155. doi: 10.1523/jneurosci.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myer D. J., Gurkoff G. G., Lee S. M., Hovda D. A., Sofroniew M. V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129(10):2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 41.Rolls A., Shechter R., Schwartz M. The bright side of the glial scar in CNS repair. Nature Reviews Neuroscience. 2009;10(3):235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- 42.Dow K. E., Wang W. Cell biology of astrocyte proteoglycans. Cellular and Molecular Life Sciences. 1998;54(6):567–581. doi: 10.1007/s000180050185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viapiano M. S., Matthews R. T. From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends in Molecular Medicine. 2006;12(10):488–496. doi: 10.1016/j.molmed.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Yiu G., He Z. Glial inhibition of CNS axon regeneration. Nature Reviews Neuroscience. 2006;7(8):617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Busch S. A., Silver J. The role of extracellular matrix in CNS regeneration. Current Opinion in Neurobiology. 2007;17(1):120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Asher R. A., Morgenstern D. A., Shearer M. C., Adcock K. H., Pesheva P., Fawcett J. W. Versican is up-regulated in CNS injury and is a product of oligodendrocyte lineage cells. The Journal of Neuroscience. 2002;22(6):2225–2236. doi: 10.1523/JNEUROSCI.22-06-02225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siebert J. R., Osterhout D. J. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. Journal of Neurochemistry. 2011;119(1):176–188. doi: 10.1111/j.1471-4159.2011.07370.x. [DOI] [PubMed] [Google Scholar]
- 48.Lau L. W., Keough M. B., Haylock-Jacobs S., et al. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Annals of Neurology. 2012;72(3):419–432. doi: 10.1002/ana.23599. [DOI] [PubMed] [Google Scholar]
- 49.Pendleton J. C., Shamblott M. J., Gary D. S., et al. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPσ . Experimental Neurology. 2013;247:113–121. doi: 10.1016/j.expneurol.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Hall A., Lalli G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harbor Perspectives in Biology. 2010;2(2) doi: 10.1101/cshperspect.a001818.a001818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujita Y., Yamashita T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Frontiers in Neuroscience. 2014;8, article 338 doi: 10.3389/fnins.2014.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubo T., Yamaguchi A., Iwata N., Yamashita T. The therapeutic effects of Rho-ROCK inhibitors on CNS disorders. Therapeutics and Clinical Risk Management. 2008;4(3):605–615. doi: 10.2147/tcrm.s2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu H., Yu S. P., Gutekunst C.-A., Gross R. E., Wei L. Inhibition of the Rho signaling pathway improves neurite outgrowth and neuronal differentiation of mouse neural stem cells. International Journal of Physiology, Pathophysiology and Pharmacology. 2013;5(1):11–20. [PMC free article] [PubMed] [Google Scholar]
- 54.Braunewell K.-H., Pesheva P., McCarthy J. B., Furcht L. T., Schmitz B., Schachner M. Functional involvement of sciatic nerve-derived versican- and decorin-like molecules and other chondroitin sulfate proteoglycans in ECM-mediated cell adhesion and neurite outgrowth. European Journal of Neuroscience. 1995;7(4):805–814. doi: 10.1111/j.1460-9568.1995.tb00683.x. [DOI] [PubMed] [Google Scholar]
- 55.Fidler P. S., Schuette K., Asher R. A., et al. Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. The Journal of Neuroscience. 1999;19(20):8778–8788. doi: 10.1523/JNEUROSCI.19-20-08778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan A. M., Zhang W., Levine J. M. NG2: a component of the glial scar that inhibits axon growth. Journal of Anatomy. 2005;207(6):717–725. doi: 10.1111/j.1469-7580.2005.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McTigue D. M., Tripathi R., Wei P. NG2 colocalizes with axons and is expressed by a mixed cell population in spinal cord lesions. Journal of Neuropathology and Experimental Neurology. 2006;65(4):406–420. doi: 10.1097/01.jnen.0000218447.32320.52. [DOI] [PubMed] [Google Scholar]
- 58.Henke-Fahle S., Wild K., Sierra A., Monnier P. P. Characterization of a new brain-derived proteoglycan inhibiting retinal ganglion cell axon outgrowth. Molecular and Cellular Neuroscience. 2001;18(5):541–556. doi: 10.1006/mcne.2001.1034. [DOI] [PubMed] [Google Scholar]
- 59.Monnier P. P., Sierra A., Schwab J. M., Henke-Fahle S., Mueller B. K. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Molecular and Cellular Neuroscience. 2003;22(3):319–330. doi: 10.1016/S1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- 60.Friedlander D. R., Milev P., Karthikeyan L., Margolis R. K., Margolis R. U., Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. Journal of Cell Biology. 1994;125(3):669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen Y., Tenney A. P., Busch S. A., et al. PTPσ Is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326(5952):592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bugiani M., Postma N., Polder E., et al. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain. 2013;136(1):209–222. doi: 10.1093/brain/aws320. [DOI] [PubMed] [Google Scholar]
- 63.Harlow D. E., Macklin W. B. Inhibitors of myelination: ECM changes, CSPGs and PTPs. Experimental Neurology. 2014;251:39–46. doi: 10.1016/j.expneurol.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fisher D., Xing B., Dill J., et al. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. Journal of Neuroscience. 2011;31(40):14051–14066. doi: 10.1523/JNEUROSCI.1737-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dickendesher T. L., Baldwin K. T., Mironova Y. A., et al. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nature Neuroscience. 2012;15(5):703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bunge R. P., Puckett W. R., Becerra J. L., Marcillo A., Quencer R. M. Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Advances in Neurology. 1993;59:75–89. [PubMed] [Google Scholar]
- 67.Li G. L., Brodin G., Farooque M., et al. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. Journal of Neuropathology and Experimental Neurology. 1996;55(3):280–289. doi: 10.1097/00005072-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Li G. L., Farooque M., Holtz A., Olsson Y. Apoptosis of oligodendrocytes occurs for long distances away from the primary injury after compression trauma to rat spinal cord. Acta Neuropathologica. 1999;98(5):473–480. doi: 10.1007/s004010051112. [DOI] [PubMed] [Google Scholar]
- 69.Crowe M. J., Bresnahan J. C., Shuman S. L., Masters J. N., Beattie M. S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nature Medicine. 1997;3(1):73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 70.Shuman S. L., Bresnahan J. C., Beattie M. S. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. Journal of Neuroscience Research. 1997;50(5):798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 71.Grossman S. D., Rosenberg L. J., Wrathall J. R. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Experimental Neurology. 2001;168(2):273–282. doi: 10.1006/exnr.2001.7628. [DOI] [PubMed] [Google Scholar]
- 72.Casha S., Yu W. R., Fehlings M. G. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103(1):203–218. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- 73.Kim D. H., Vaccaro A. R., Henderson F. C., Benzel E. C. Molecular biology of cervical myelopathy and spinal cord injury: role of oligodendrocyte apoptosis. The Spine Journal. 2003;3(6):510–519. doi: 10.1016/s1529-9430(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 74.Totoiu M. O., Keirstead H. S. Spinal cord injury is accompanied by chronic progressive demyelination. Journal of Comparative Neurology. 2005;486(4):373–383. doi: 10.1002/cne.20517. [DOI] [PubMed] [Google Scholar]
- 75.Xu G.-Y., Hughes M. G., Ye Z., Hulsebosch C. E., McAdoo D. J. Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Experimental Neurology. 2004;187(2):329–336. doi: 10.1016/j.expneurol.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 76.Dowling P., Ming X., Raval S., et al. Up-regulated p75(NTR) neurotrophin receptor on glial cells in MS plaques. Neurology. 1999;53(8):1676–1682. doi: 10.1212/WNL.53.8.1676. [DOI] [PubMed] [Google Scholar]
- 77.Chang A., Nishiyama A., Peterson J., Prineas J., Trapp B. D. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. The Journal of Neuroscience. 2000;20(17):6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krenz N. R., Weaver L. C. Nerve growth factor in glia and inflammatory cells of the injured rat spinal cord. Journal of Neurochemistry. 2000;74(2):730–739. doi: 10.1046/j.1471-4159.2000.740730.x. [DOI] [PubMed] [Google Scholar]
- 79.Beattie M. S., Harrington A. W., Lee R., et al. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36(3):375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filbin M. T. Myelin-associated glycoprotein: a role in myelination and in the inhibition of axonal regeneration? Current Opinion in Neurobiology. 1995;5(5):588–595. doi: 10.1016/0959-4388(95)80063-8. [DOI] [PubMed] [Google Scholar]
- 81.DeBellard M.-E., Tang S., Mukhopadhyay G., Shen Y.-J., Filbin M. T. Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Molecular and Cellular Neurosciences. 1996;7(2):89–101. doi: 10.1006/mcne.1996.0007. [DOI] [PubMed] [Google Scholar]
- 82.Fournier A. E., GrandPre T., Strittmatter S. M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409(6818):341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- 83.Wang K. C., Koprivica V., Kim J. A., et al. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002;417(6892):941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- 84.Filbin M. T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nature Reviews Neuroscience. 2003;4(9):703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 85.Liebscher T., Schnell D., Scholl J., et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Annals of Neurology. 2005;58(5):706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- 86.Freund P., Schmidlin E., Wannier T., et al. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nature Medicine. 2006;12(7):790–792. doi: 10.1038/nm1436. [DOI] [PubMed] [Google Scholar]
- 87.Freund P., Wannier T., Schmidlin E., et al. Anti-Nogo-A antibody treatment enhances sprouting of corticospinal axons rostral to a unilateral cervical spinal cord lesion in adult macaque monkey. Journal of Comparative Neurology. 2007;502(4):644–659. doi: 10.1002/cne.21321. [DOI] [PubMed] [Google Scholar]
- 88.Tsai S.-Y., Markus T. M., Andrews E. M., et al. Intrathecal treatment with anti-Nogo-A antibody improves functional recovery in adult rats after stroke. Experimental Brain Research. 2007;182(2):261–266. doi: 10.1007/s00221-007-1067-0. [DOI] [PubMed] [Google Scholar]
- 89.Xie F., Zheng B. White matter inhibitors in CNS axon regeneration failure. Experimental Neurology. 2008;209(2):302–312. doi: 10.1016/j.expneurol.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maier I. C., Ichiyama R. M., Courtine G., et al. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain. 2009;132(6):1426–1440. doi: 10.1093/brain/awp085. [DOI] [PubMed] [Google Scholar]
- 91.Bandtlow C. E., Schmidt M. F., Hassinger T. D., Schwab M. E., Kater S. B. Role of intracellular calcium in NI-35-evoked collapse of neuronal growth cones. Science. 1993;259(5091):80–83. doi: 10.1126/science.8418499. [DOI] [PubMed] [Google Scholar]
- 92.Zhou X.-F., Li H.-Y. Roles of glial p75NTR in axonal regeneration. Journal of Neuroscience Research. 2007;85(8):1601–1605. doi: 10.1002/jnr.21220. [DOI] [PubMed] [Google Scholar]
- 93.Vargas M. E., Barres B. A. Why is Wallerian degeneration in the CNS so slow? Annual Review of Neuroscience. 2007;30:153–179. doi: 10.1146/annurev.neuro.30.051606.094354. [DOI] [PubMed] [Google Scholar]
- 94.Goetz L. L., Mummaneni R. Posttraumatic syringomyelia. Medscape. 2014 http://emedicine.medscape.com/article/322348-overview. [Google Scholar]
- 95.Surey S., Berry M., Logan A., Bicknell R., Ahmed Z. Differential cavitation, angiogenesis and wound-healing responses in injured mouse and rat spinal cords. Neuroscience. 2014;275:62–80. doi: 10.1016/j.neuroscience.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 96.Crespo D., Asher R. A., Lin R., Rhodes K. E., Fawcett J. W. How does chondroitinase promote functional recovery in the damaged CNS? Experimental Neurology. 2007;206(2):159–171. doi: 10.1016/j.expneurol.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 97.Zuo J., Neubauer D., Dyess K., Ferguson T. A., Muir D. Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Experimental Neurology. 1998;154(2):654–662. doi: 10.1006/exnr.1998.6951. [DOI] [PubMed] [Google Scholar]
- 98.Yick L.-W., Wu W., So K.-F., Yip H. K., Shum D. K.-Y. Chondroitinase ABC promotes axonal regeneration of Clarke's neurons after spinal cord injury. NeuroReport. 2000;11(5):1063–1067. doi: 10.1097/00001756-200004070-00032. [DOI] [PubMed] [Google Scholar]
- 99.Bradbury E. J., Moon L. D. F., Popat R. J., et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416(6881):636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 100.Yick L.-W., Cheung P.-T., So K.-F., Wu W. Axonal regeneration of Clarke's neurons beyond the spinal cord injury scar after treatment with chondroitinase ABC. Experimental Neurology. 2003;182(1):160–168. doi: 10.1016/s0014-4886(02)00052-3. [DOI] [PubMed] [Google Scholar]
- 101.Barritt A. W., Davies M., Marchand F., et al. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. The Journal of Neuroscience. 2006;26(42):10856–10867. doi: 10.1523/jneurosci.2980-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tom V. J., Kadakia R., Santi L., Houlé J. D. Administration of chondroitinase ABC rostral or caudal to a spinal cord injury site promotes anatomical but not functional plasticity. Journal of Neurotrauma. 2009;26(12):2323–2333. doi: 10.1089/neu.2009.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bartus K., James N. D., Didangelos A., et al. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. Journal of Neuroscience. 2014;34(14):4822–4836. doi: 10.1523/jneurosci.4369-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tester N. J., Howland D. R. Chondroitinase ABC improves basic and skilled locomotion in spinal cord injured cats. Experimental Neurology. 2008;209(2):483–496. doi: 10.1016/j.expneurol.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Siebert J. R., Stelzner D. J., Osterhout D. J. Chondroitinase treatment following spinal contusion injury increases migration of oligodendrocyte progenitor cells. Experimental Neurology. 2011;231(1):19–29. doi: 10.1016/j.expneurol.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 106.Tester N. J., Plaas A. H., Howland D. R. Effect of body temperature on chondroitinase ABC's ability to cleave chondroitin sulfate glycosaminoglycans. Journal of Neuroscience Research. 2007;85(5):1110–1118. doi: 10.1002/jnr.21199. [DOI] [PubMed] [Google Scholar]
- 107.Nazari-Robati M., Khajeh K., Aminian M., Mollania N., Golestani A. Enhancement of thermal stability of chondroitinase ABC i by site-directed mutagenesis: an insight from Ramachandran plot. Biochimica et Biophysica Acta—Proteins and Proteomics. 2013;1834(2):479–486. doi: 10.1016/j.bbapap.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 108.Jin Y., Ketschek A., Jiang Z., Smith G., Fischer I. Chondroitinase activity can be transduced by a lentiviral vector in vitro and in vivo. Journal of Neuroscience Methods. 2011;199(2):203–213. doi: 10.1016/j.jneumeth.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao R.-R., Muir E. M., Alves J. N., et al. Lentiviral vectors express chondroitinase ABC in cortical projections and promote sprouting of injured corticospinal axons. Journal of Neuroscience Methods. 2011;201(1):228–238. doi: 10.1016/j.jneumeth.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Osterhout D. J., Garma P. D., Au A., et al. Nanosphere delivery of chondroitinase produces digestion of CSPGs with enhanced axonal sprouting after spinal cord injury. Submitted. [Google Scholar]
- 111.Xie Y., Massa S. M., Ensslen-Craig S. E., et al. Protein-tyrosine phosphatase (PTP) wedge domain peptides: a novel approach for inhibition of PTP function and augmentation of protein-tyrosine kinase function. The Journal of Biological Chemistry. 2006;281(24):16482–16492. doi: 10.1074/jbc.m603131200. [DOI] [PubMed] [Google Scholar]
- 112.Lang B. T., Cregg J. M., DePaul M. A., et al. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. 2014;518(7539):404–408. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schwab M. E. Nogo and axon regeneration. Current Opinion in Neurobiology. 2004;14(1):118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 114.Pernet V., Schwab M. E. The role of Nogo-A in axonal plasticity, regrowth and repair. Cell and Tissue Research. 2012;349(1):97–104. doi: 10.1007/s00441-012-1432-6. [DOI] [PubMed] [Google Scholar]
- 115.Wang T., Xiong J.-Q., Ren X.-B., Sun W. The role of Nogo-A in neuroregeneration: a review. Brain Research Bulletin. 2012;87(6):499–503. doi: 10.1016/j.brainresbull.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 116.Tsai S.-Y., Papadopoulos C. M., Schwab M. E., Kartje G. L. Delayed anti-Nogo-A therapy improves function after chronic stroke in adult rats. Stroke. 2011;42(1):186–190. doi: 10.1161/strokeaha.110.590083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lingor P., Teusch N., Schwarz K., et al. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. Journal of Neurochemistry. 2007;103(1):181–189. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 118.Schnell L., Schwab M. E. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343(6255):269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 119.Seymour A. B., Andrews E. M., Tsai S.-Y., et al. Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. Journal of Cerebral Blood Flow & Metabolism. 2005;25(10):1366–1375. doi: 10.1038/sj.jcbfm.9600134. [DOI] [PubMed] [Google Scholar]
- 120.Fouad K., Klusman I., Schwab M. E. Regenerating corticospinal fibers in the Marmoset (Callitrix jacchus) after spinal cord lesion and treatment with the anti-Nogo-A antibody IN-1. European Journal of Neuroscience. 2004;20(9):2479–2482. doi: 10.1111/j.1460-9568.2004.03716.x. [DOI] [PubMed] [Google Scholar]
- 121.Sengottuvel V., Fischer D. Facilitating axon regeneration in the injured CNS by microtubules stabilization. Communicative and Integrative Biology. 2011;4(4):391–393. doi: 10.4161/cib.15552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ruschel J., Hellal F., Flynn K. C., et al. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science. 2015;348(6232):347–352. doi: 10.1126/science.aaa2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sengottuvel V., Leibinger M., Pfreimer M., Andreadaki A., Fischer D. Taxol facilitates axon regeneration in the mature CNS. The Journal of Neuroscience. 2011;31(7):2688–2699. doi: 10.1523/jneurosci.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Park K. K., Liu K., Hu Y., et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322(5903):963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sun F., Park K. K., Belin S., et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480(7377):372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zukor K., Belin S., Wang C., Keelan N., Wang X., He Z. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. The Journal of Neuroscience. 2013;33(39):15350–15361. doi: 10.1523/jneurosci.2510-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith P. D., Sun F., Park K. K., et al. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64(5):617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moore D. L., Blackmore M. G., Hu Y., et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326(5950):298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Apara A., Goldberg J. L. Molecular mechanisms of the suppression of axon regeneration by KLF transcription factors. Neural Regeneration Research. 2014;9(15):1418–1421. doi: 10.4103/1673-5374.139454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reynolds A. J., Bartlett S. E., Hendry I. A. Molecular mechanisms regulating the retrograde axonal transport of neurotrophins. Brain Research Reviews. 2000;33(2-3):169–178. doi: 10.1016/s0165-0173(00)00028-x. [DOI] [PubMed] [Google Scholar]
- 131.Twiss J. L., Chang J. H., Schanen N. C. Pathophysiological mechanisms for actions of the neurotrophins. Brain Pathology. 2006;16(4):320–332. doi: 10.1111/j.1750-3639.2006.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Friedman W. Growth factor. In: Brady S., Siegel G., Albers R. W., Price D. L., editors. Basic Neurochemistry Principles of Molecular, Cellular, and Medical Neurobiology. 8th. New York, NY, USA: Academic Press; 2012. [Google Scholar]
- 133.Roux P. P., Barker P. A. Neurotrophin signaling through the p75 neurotrophin receptor. Progress in Neurobiology. 2002;67(3):203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 134.Airaksinen M. S., Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature Reviews Neuroscience. 2002;3(5):383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- 135.Blesch A., Uy H. S., Grill R. J., Cheng J.-G., Patterson P. H., Tuszynski M. H. Leukemia inhibitory factor augments neurotrophin expression and corticospinal axon growth after adult CNS injury. Journal of Neuroscience. 1999;19(9):3556–3566. doi: 10.1523/JNEUROSCI.19-09-03556.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cafferty W. B. J., Gardiner N. J., Gavazzi I., et al. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. Journal of Neuroscience. 2001;21(18):7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gregg C., Weiss S. CNTF/LIF/gp130 receptor complex signaling maintains a VZ precursor differentiation gradient in the developing ventral forebrain. Development. 2005;132(3):565–578. doi: 10.1242/dev.01592. [DOI] [PubMed] [Google Scholar]
- 138.Leibinger M., Müller A., Andreadaki A., Hauk T. G., Kirsch M., Fischer D. Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. The Journal of Neuroscience. 2009;29(45):14334–14341. doi: 10.1523/jneurosci.2770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klein R., Martin-Zanca D., Barbacid M., Parada L. F. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109(4):845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- 140.Vazquez M. E., Ebendal T. Messenger RNAs for trk and the low-affinity NGF receptor in rat basal forebrain. NeuroReport. 1991;2(10):593–596. doi: 10.1097/00001756-199110000-00010. [DOI] [PubMed] [Google Scholar]
- 141.Giehl K. M., Schütte A., Mestres P., Yan Q. The survival-promoting effect of glial cell line-derived neurotrophic factor on axotomized corticospinal neurons in vivo is mediated by an endogenous brain-derived neurotrophic factor mechanism. The Journal of Neuroscience. 1998;18(18):7351–7360. doi: 10.1523/JNEUROSCI.18-18-07351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kwon B. K., Liu J., Oschipok L., Teh J., Liu Z. W., Tetzlaff W. Rubrospinal neurons fail to respond to brain-derived neurotrophic factor applied to the spinal cord injury site 2 months after cervical axotomy. Experimental Neurology. 2004;189(1):45–57. doi: 10.1016/j.expneurol.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 143.Siebert J. R., Middelton F. A., Stelzner D. J. Intrinsic response of thoracic propriospinal neurons to axotomy. BMC Neuroscience. 2010;11, article 69 doi: 10.1186/1471-2202-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grill R., Murai K., Blesch A., Gage F. H., Tuszynski M. H. Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth and partial functional recovery after spinal cord injury. The Journal of Neuroscience. 1997;17(14):5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Giehl K. M., Schacht C. M., Yan Q., Mestres P. GDNF is a trophic factor for adult rat corticospinal neurons and promotes their long-term survival after axotomy in vivo . European Journal of Neuroscience. 1997;9(11):2479–2489. doi: 10.1111/j.1460-9568.1997.tb01665.x. [DOI] [PubMed] [Google Scholar]
- 146.Kobayashi N. R., Fan D.-P., Giehl K. M., Bedard A. M., Wiegand S. J., Tetzlaff W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Tα1-tubulin mRNA expression, and promote axonal regeneration. The Journal of Neuroscience. 1997;17(24):9583–9595. doi: 10.1523/JNEUROSCI.17-24-09583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kwon B. K., Liu J., Messerer C., et al. Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):3246–3251. doi: 10.1073/pnas.052308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhou L., Shine H. D. Neurotrophic factors expressed in both cortex and spinal cord induce axonal plasticity after spinal cord injury. Journal of Neuroscience Research. 2003;74(2):221–226. doi: 10.1002/jnr.10718. [DOI] [PubMed] [Google Scholar]
- 149.Blesch A., Yang H., Weidner N., Hoang A., Otero D. Axonal responses to cellularly delivered NT-4/5 after spinal cord injury. Molecular and Cellular Neuroscience. 2004;27(2):190–201. doi: 10.1016/j.mcn.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 150.Romero M. I., Rangappa N., Garry M. G., Smith G. M. Functional regeneration of chronically injured sensory afferents into adult spinal cord after neurotrophin gene therapy. The Journal of Neuroscience. 2001;21(21):8408–8416. doi: 10.1523/JNEUROSCI.21-21-08408.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ramer M. S., Bishop T., Dockery P., et al. Neurotrophin-3-mediated regeneration and recovery of proprioception following dorsal rhizotomy. Molecular and Cellular Neuroscience. 2002;19(2):239–249. doi: 10.1006/mcne.2001.1067. [DOI] [PubMed] [Google Scholar]
- 152.Goldberg J. L. How does an axon grow? Genes & Development. 2003;17(8):941–958. doi: 10.1101/gad.1062303. [DOI] [PubMed] [Google Scholar]
- 153.Hiebert G. W., Khodarahmi K., McGraw J., Steeves J. D., Tetzlaff W. Brain-derived neurotrophic factor applied to the motor cortex promotes sprouting of corticospinal fibers but not regeneration into a peripheral nerve transplant. The Journal of Neuroscience Research. 2002;69(2):160–168. doi: 10.1002/jnr.10275. [DOI] [PubMed] [Google Scholar]
- 154.Vavrek R., Girgis J., Tetzlaff W., Hiebert G. W., Fouad K. BDNF promotes connections of corticospinal neurons onto spared descending interneurons in spinal cord injured rats. Brain. 2006;129(6):1534–1545. doi: 10.1093/brain/awl087. [DOI] [PubMed] [Google Scholar]
- 155.Coumans J. V., Lin T. T.-S., MacArthur L., McAtee M., Nash C., Bregman B. S. Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. The Journal of Neuroscience. 2001;21(23):9334–9344. doi: 10.1523/JNEUROSCI.21-23-09334.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu Y., Kim D., Himes B. T., et al. Transplants of fibroblasts genetically modified to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. The Journal of Neuroscience. 1999;19(11):4370–4387. doi: 10.1523/JNEUROSCI.19-11-04370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lindsay R. M., Thoenen H., Barde Y.-A. Placode and neural crest-derived sensory neurons are responsive at early developmental stages to brain-derived neurotrophic factor. Developmental Biology. 1985;112(2):319–328. doi: 10.1016/0012-1606(85)90402-6. [DOI] [PubMed] [Google Scholar]
- 158.Goldberg J. L., Espinosa J. S., Xu Y., Davidson N., Kovacs G. T. A., Barres B. A. Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron. 2002;33(5):689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- 159.Logan A., Ahmed Z., Baird A., Gonzalez A. M., Berry M. Neurotrophic factor synergy is required for neuronal survival and disinhibited axon regeneration after CNS injury. Brain. 2006;129(2):490–502. doi: 10.1093/brain/awh706. [DOI] [PubMed] [Google Scholar]
- 160.Hammond E. N. L., Tetzlaff W., Mestres P., Giehl K. M. BDNF, but not NT-3, promotes long-term survival of axotimized adult rat corticospinal neurons in vivo . NeuroReport. 1999;10(12):2671–2675. doi: 10.1097/00001756-199908200-00043. [DOI] [PubMed] [Google Scholar]
- 161.Fawcett J. W. Bridging spinal cord injuries. Journal of Biology. 2008;7(7, article 25) doi: 10.1186/jbiol89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barres B. A., Raff M. C. Axonal control of oligodendrocyte development. Journal of Cell Biology. 1999;147(6):1123–1128. doi: 10.1083/jcb.147.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Remahl S., Hilderbrand C. Relations between axons and oligodendroglial cells during initial myelination. II. The individual axon. Journal of Neurocytology. 1990;19(6):883–898. doi: 10.1007/BF01186817. [DOI] [PubMed] [Google Scholar]
- 164.Baumann N., Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiological Reviews. 2001;81(2):871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 165.Levine J. M., Reynolds R., Fawcett J. W. The oligodendrocyte precursor cell in health and disease. Trends in Neurosciences. 2001;24(1):39–47. doi: 10.1016/S0166-2236(00)01691-X. [DOI] [PubMed] [Google Scholar]
- 166.McTigue D. M., Tripathi R. B. The life, death, and replacement of oligodendrocytes in the adult CNS. Journal of Neurochemistry. 2008;107(1):1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- 167.Franklin R. J. M., Gilson J. M., Blakemore W. F. Local recruitment of remyelinating cells in the repair of demyelination in the central nervous system. Journal of Neuroscience Research. 1997;50(2):337–344. doi: 10.1002/(sici)1097-4547(19971015)50:2lt;337::aid-jnr21>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 168.Fok-Seang J., Mathews G. A., Ffrench-Constant C., Trotter J., Fawcett J. W. Migration of oligodendrocyte precursors on astrocytes and meningeal cells. Developmental Biology. 1995;171(1):1–15. doi: 10.1006/dbio.1995.1255. [DOI] [PubMed] [Google Scholar]
- 169.Setzu A., Ffrench-Constant C., Franklin R. J. M. CNS axons retain their competence for myelination throughout life. Glia. 2004;45(3):307–311. doi: 10.1002/glia.10321. [DOI] [PubMed] [Google Scholar]
- 170.Blakemore W. F. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. Nature Neuroscience. 1973;4(1):1116–1122. doi: 10.1016/0022-510x(73)90118-4. [DOI] [PubMed] [Google Scholar]
- 171.Matsushima G. K., Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathology. 2001;11(1):107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Armstrong R. C., Le T. Q., Frost E. E., Borke R. C., Vana A. C. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. The Journal of Neuroscience. 2002;22(19):8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stidworthy M. F., Genoud S., Suter U., Mantei N., Franklin R. J. M. Quantifying the early stages of remyelination following cuprizone-induced demyelination. Brain Pathology. 2003;13(3):329–339. doi: 10.1111/j.1750-3639.2003.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McTigue D. M., Horner P. J., Stokes B. T., Gage F. H. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. The Journal of Neuroscience. 1998;18(14):5354–5365. doi: 10.1523/JNEUROSCI.18-14-05354.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Adamo A. M., Paez P. M., Cabrera O. E. E., et al. Remyelination after cuprizone-induced demyelination in the rat is stimulated by apotransferrin. Experimental Neurology. 2006;198(2):519–529. doi: 10.1016/j.expneurol.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 176.Siebert J. R., Osterhout D. J. Strategies to reverse the effects of chondrotin sulfate proteoglycans on oligodendrocytes. doi: 10.1111/j.1471-4159.2011.07370.x. in press. [DOI] [PubMed] [Google Scholar]
- 177.Mi S., Miller R. H., Lee X., et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nature Neuroscience. 2005;8(6):745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 178.Mi S., Blake Pepinsky R., Cadavid D. Blocking LINGO-1 as a therapy to promote CNS repair: from concept to the clinic. CNS Drugs. 2013;27(7):493–503. doi: 10.1007/s40263-013-0068-8. [DOI] [PubMed] [Google Scholar]
- 179.Mi S., Hu B., Hahm K., et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nature Medicine. 2007;13(10):1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- 180.Zhang Y., Zhang Y. P., Pepinsky B., et al. Inhibition of LINGO-1 promotes functional recovery after experimental spinal cord demyelination. Experimental Neurology. 2015;266:68–73. doi: 10.1016/j.expneurol.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 181.Münzel E. J., Williams A. Promoting remyelination in multiple sclerosis-recent advances. Drugs. 2013;73(18):2017–2029. doi: 10.1007/s40265-013-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cadavid D., Phillips G., Dong-Si T., Tran J., Xy L. Efficacy and safety of anti LINGO-1 for the treatment of relapsing forms of multiple sclerosis: design of the phase 2 SYNERGY trial. Neurology. 2014;82(10, supplement P3.154) [Google Scholar]
- 183.Pepinsky R. B., Arndt J. W., Quan C., et al. Structure of the LINGO-1-anti-LINGO-1 Li81 antibody complex provides insights into the biology of LINGO-1 and the mechanism of action of the antibody therapy. Journal of Pharmacology and Experimental Therapeutics. 2014;350(1):110–123. doi: 10.1124/jpet.113.211771. [DOI] [PubMed] [Google Scholar]
- 184.Bambakidis N. C., Miller R. H. Transplantation of oligodendrocytes precursors and sonic hedgehog results in improved function and white matter sparing in the cords of adult rats after contusion. The Spine Journal. 2004;4(1):16–26. doi: 10.1016/j.spinee.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 185.Keirstead H. S., Nistor G., Bernal G., et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. The Journal of Neuroscience. 2005;25(19):4694–4705. doi: 10.1523/jneurosci.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wu B., Ren X. Promoting axonal myelination for improving neurological recovery in spinal cord injury. Journal of Neurotrauma. 2009;26(10):1847–1856. doi: 10.1089/neu.2008.0551. [DOI] [PubMed] [Google Scholar]
- 187.Akiyama Y., Lankford K., Radtke C., Greer C. A., Kocsis J. D. Remyelination of spinal cord axons by olfactory ensheathing cells and Schwann cells derived from a transgenic rat expressing alkaline phosphatase marker gene. Neuron Glia Biology. 2004;1(1) doi: 10.1017/s1740925x04000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Woodhoo A., Sahni V., Gilson J., et al. Schwann cell precursors: a favourable cell for myelin repair in the Central Nervous System. Brain. 2007;130(8):2175–2185. doi: 10.1093/brain/awm125. [DOI] [PubMed] [Google Scholar]
- 189.Radtke C., Akiyama Y., Brokaw J., et al. Remyelination of the nonhuman primate spinal cord by transplantation of H-transferase transgenic adult pig olfactory ensheathing cells. The FASEB Journal. 2004;18(2):335–337. doi: 10.1096/fj.03-0214fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Akiyama Y., Radtke C., Kocsis J. D. Remyelination of the rat spinal cord by transplantation of identified bone marrow stromal cells. Journal of Neuroscience. 2002;22(15):6623–6630. doi: 10.1523/JNEUROSCI.22-15-06623.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Akiyama Y., Radtke C., Honmou O., Kocsis J. D. Remyelination of the spinal cord following intravenous delivery of bone marrow cells. GLIA. 2002;39(3):229–236. doi: 10.1002/glia.10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.McKenzie I. A., Biernaskie J., Toma J. G., Midha R., Miller F. D. Skin-derived precursors generate myelinating Schwann cells for the injured and dysmyelinated nervous system. The Journal of Neuroscience. 2006;26(24):6651–6660. doi: 10.1523/jneurosci.1007-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dasari V. R., Spomar D. G., Gondi C. S., et al. Axonal remyelination by cord blood stem cells after spinal cord injury. Journal of Neurotrauma. 2007;24(2):391–410. doi: 10.1089/neu.2006.0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Franklin R. J. M. Remyelination of the demyelinated CNS: the case for and against transplantation of central, peripheral and olfactory glia. Brain Research Bulletin. 2002;57(6):827–832. doi: 10.1016/s0361-9230(01)00765-1. [DOI] [PubMed] [Google Scholar]
- 195.Lakatos A., Franklin R. J. M., Barnett S. C. Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia. 2000;32(3):214–225. doi: 10.1002/1098-1136(200012)32:3lt;214::aid-glia20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 196.Iwashita Y., Fawcett J. W., Crang A. J., Franklin R. J. M., Blakemore W. F. Schwann cells transplanted into normal and X-irradiated adult white matter do not migrate extensively and show poor long-term survival. Experimental Neurology. 2000;164(2):292–302. doi: 10.1006/exnr.2000.7440. [DOI] [PubMed] [Google Scholar]
- 197.Andrews M. R., Stelzner D. J. Modification of the regenerative response of dorsal column axons by olfactory ensheathing cells or peripheral axotomy in adult rat. Experimental Neurology. 2004;190(2):311–327. doi: 10.1016/j.expneurol.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 198.Andrews M. R., Stelzner D. J. Evaluation of olfactory ensheathing and Schwann cells after implantation into a dorsal injury of adult rat spinal cord. Journal of Neurotrauma. 2007;24(11):1773–1792. doi: 10.1089/neu.2007.0353. [DOI] [PubMed] [Google Scholar]
- 199.Bradbury E. J., McMahon S. B. Spinal cord repair strategies: why do they work? Nature Reviews Neuroscience. 2006;7(8):644–653. doi: 10.1038/nrn1964. [DOI] [PubMed] [Google Scholar]
- 200.Silva N. A., Sousa N., Reis R. L., Salgado A. J. From basics to clinical: a comprehensive review on spinal cord injury. Progress in Neurobiology. 2014;114:25–57. doi: 10.1016/j.pneurobio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 201.Conta Steencken A. C., Stelzner D. J. Loss of propriospinal neurons after spinal contusion injury as assessed by retrograde labeling. Neuroscience. 2010;170(3):971–980. doi: 10.1016/j.neuroscience.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 202.Hains B. C., Black J. A., Waxman S. G. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. Journal of Comparative Neurology. 2003;462(3):328–341. doi: 10.1002/cne.10733. [DOI] [PubMed] [Google Scholar]
- 203.Walthers C., Seidlits S. K. Gene delivery strategies to promote spinal cord repair. Biomarker Insights. 2015;10(supplement 1):11–29. doi: 10.4137/bmi.s20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Taylor L., Jones L., Tuszynski M. H., Blesch A. Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord. The Journal of Neuroscience. 2006;26(38):9713–9721. doi: 10.1523/jneurosci.0734-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu Y. L., Himes B. T., Murray M., Tessler A., Fischer I. Grafts of BDNF-producing fibroblasts rescue axotomized rubrospinal neurons and prevent their atrophy. Experimental Neurology. 2002;178(2):150–164. doi: 10.1006/exnr.2002.7977. [DOI] [PubMed] [Google Scholar]
- 206.Taylor S. J., Sakiyama-Elbert S. E. Effect of controlled delivery of neurotrophin-3 from fibrin on spinal cord injury in a long term model. Journal of Controlled Release. 2006;116(2):204–210. doi: 10.1016/j.jconrel.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jones L. L., Tuszynski M. H. Chronic intrathecal infusions after spinal cord injury cause scarring and compression. Microscopy Research and Technique. 2001;54(5):317–324. doi: 10.1002/jemt.1144. [DOI] [PubMed] [Google Scholar]
- 208.Haggerty A. E., Oudega M. Biomaterials for spinal cord repair. Neuroscience Bulletin. 2013;29(4):445–459. doi: 10.1007/s12264-013-1362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wang M., Zhai P., Chen X., Schreyer D. J., Sun X., Cui F. Bioengineered scaffolds for spinal cord repair. Tissue Engineering—Part B: Reviews. 2011;17(3):177–194. doi: 10.1089/ten.teb.2010.0648. [DOI] [PubMed] [Google Scholar]
- 210.Sakiyama-Elbert S., Johnson P. J., Hodgetts S. I., Plant G. W., Harvey A. R. Handbook of Clinical Neurology: Spinal Cord Injury. chapter 36. 2012. Scaffold to promote spinal cord regeneration; p. p. 109. [DOI] [PubMed] [Google Scholar]
- 211.Mitragotri S., Lahann J. Physical approaches to biomaterial design. Nature Materials. 2009;8(1):15–23. doi: 10.1038/nmat2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Danhier F., Ansorena E., Silva J. M., Coco R., Le Breton A., Préat V. PLGA-based nanoparticles: an overview of biomedical applications. Journal of Controlled Release. 2012;161(2):505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 213.Makadia H. K., Siegel S. J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Jiang Y., Chen J., Deng C., Suuronen E. J., Zhong Z. Click hydrogels, microgels and nanogels: emerging platforms for drug delivery and tissue engineering. Biomaterials. 2014;35(18):4969–4985. doi: 10.1016/j.biomaterials.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 215.Deal D. N., Griffin J. W., Hogan M. V. Nerve conduits for nerve repair or reconstruction. Journal of the American Academy of Orthopaedic Surgeons. 2012;20(2):63–68. doi: 10.5435/jaaos-20-02-063. [DOI] [PubMed] [Google Scholar]
- 216.Pabari A., Lloyd-Hughes H., Seifalian A. M., Mosahebi A. Nerve conduits for peripheral nerve surgery. Plastic and Reconstructive Surgery. 2014;133(6):1420–1430. doi: 10.1097/prs.0000000000000226. [DOI] [PubMed] [Google Scholar]
- 217.Daly W., Yao L., Zeugolis D., Windebank A., Pandit A. A biomaterials approach to peripheral nerve regeneration: bridging the peripheral nerve gap and enhancing functional recovery. Journal of the Royal Society Interface. 2012;9(67):202–221. doi: 10.1098/rsif.2011.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wong D. Y., Leveque J.-C., Brumblay H., Krebsbach P. H., Hollister S. J., LaMarca F. Macro-architectures in spinal cord scaffold implants influence regeneration. Journal of Neurotrauma. 2008;25(8):1027–1037. doi: 10.1089/neu.2007.0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Madigan N. N., McMahon S., O'Brien T., Yaszemski M. J., Windebank A. J. Current tissue engineering and novel therapeutic approaches to axonal regeneration following spinal cord injury using polymer scaffolds. Respiratory Physiology and Neurobiology. 2009;169(2):183–199. doi: 10.1016/j.resp.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Houweling D. A., Lankhorst A. J., Gispen W. H., Bär P. R., Joosten E. A. J. Collagen containing neurotrophin-3 (NT-3) attracts regrowing injured corticospinal axons in the adult rat spinal cord and promotes partial functional recovery. Experimental Neurology. 1998;153(1):49–59. doi: 10.1006/exnr.1998.6867. [DOI] [PubMed] [Google Scholar]
- 221.Xu X. M., Guenard V., Kleitman N., Bunge M. Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. Journal of Comparative Neurology. 1995;351(1):145–160. doi: 10.1002/cne.903510113. [DOI] [PubMed] [Google Scholar]
- 222.Loh N. K., Woerly S., Bunt S. M., Wilton S. D., Harvey A. R. The regrowth of axons within tissue defects in the cns is promoted by implanted hydrogel matrices that contain BDNF and CNTF producing fibroblasts. Experimental Neurology. 2001;170(1):72–84. doi: 10.1006/exnr.2001.7692. [DOI] [PubMed] [Google Scholar]
- 223.Tobias C. A., Dhoot N. O., Wheatley M. A., Tessler A., Murray M., Fischer I. Grafting of encapsulated BDNF-producing fibroblasts into the injured spinal cord without immune suppression in adult rats. Journal of Neurotrauma. 2001;18(3):287–301. doi: 10.1089/08977150151070937. [DOI] [PubMed] [Google Scholar]
- 224.Teng Y. D., Lavik E. B., Qu X., et al. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proceedings of the National Academy of Science of the United States of America. 2002;99(5):3024–3029. doi: 10.1073/pnas.052678899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tobias C. A., Han S. S. W., Shumsky J. S., et al. Alginate encapsulated BDNF-producing fibroblast grafts permit recovery of function after spinal cord injury in the absence of immune suppression. Journal of Neurotrauma. 2005;22(1):138–156. doi: 10.1089/neu.2005.22.138. [DOI] [PubMed] [Google Scholar]
- 226.Johnson P. J., Tatara A., Shiu A., Sakiyama-Elbert S. E. Controlled release of neurotrophin-3 and platelet-derived growth factor from fibrin scaffolds containing neural progenitor cells enhances survival and differentiation into neurons in a subacute model of SCI. Cell Transplantation. 2010;19(1):89–101. doi: 10.3727/096368909x477273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Stensaas S. S., Stensaas L. J. Histopathological evaluation of materials implanted in the cerebral cortex. Acta Neuropathologica. 1978;41(2):145–155. doi: 10.1007/bf00689766. [DOI] [PubMed] [Google Scholar]
- 228.Straley K. S., Foo C. W. P., Heilshorn S. C. Biomaterial design strategies for the treatment of spinal cord injuries. Journal of Neurotrauma. 2010;27(1):1–19. doi: 10.1089/neu.2009.0948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Perale G., Rossi F., Sundstrom E., et al. Hydrogels in spinal cord injury repair strategies. ACS Chemical Neuroscience. 2011;2(7):336–345. doi: 10.1021/cn200030w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Assunção-Silva R. C., Gomes E. D., Sousa N., Silva N. A., Salgado A. J. Hydrogels and cell based therapies in spinal cord injury regeneration. Stem Cells International. 2015;2015:24. doi: 10.1155/2015/948040.948040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chockalingam K., Blenner M., Banta S. Design and application of stimulus-responsive peptide systems. Protein Engineering, Design & Selection. 2007;20(4):155–161. doi: 10.1093/protein/gzm008. [DOI] [PubMed] [Google Scholar]
- 232.Nisbet D. R., Crompton K. E., Horne M. K., Finkelstein D. I., Forsythe J. S. Neural tissue engineering of the CNS using hydrogels. Journal of Biomedical Materials Research—Part B: Applied Biomaterials. 2008;87(1):251–263. doi: 10.1002/jbm.b.31000. [DOI] [PubMed] [Google Scholar]
- 233.Piantino J., Burdick J. A., Goldberg D., Langer R., Benowitz L. I. An injectable, biodegradable hydrogel for trophic factor delivery enhances axonal rewiring and improves performance after spinal cord injury. Experimental Neurology. 2006;201(2):359–367. doi: 10.1016/j.expneurol.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 234.Tsai E. C., Dalton P. D., Shoichet M. S., Tator C. H. Matrix inclusion within synthetic hydrogel guidance channels improves specific supraspinal and local axonal regeneration after complete spinal cord transection. Biomaterials. 2006;27(3):519–533. doi: 10.1016/j.biomaterials.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 235.Ma J., Tian W.-M., Hou S.-P., Xu Q.-Y., Spector M., Cui F.-Z. An experimental test of stroke recovery by implanting a hyaluronic acid hydrogel carrying a Nogo receptor antibody in a rat model. Biomedical Materials. 2007;2(4):233–240. doi: 10.1088/1748-6041/2/4/005. [DOI] [PubMed] [Google Scholar]
- 236.Fisher J. D., Acharya A. P., Little S. R. Micro and nanoparticle drug delivery systems for preventing allotransplant rejection. Clinical Immunology. 2015 doi: 10.1016/j.clim.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Oberoi H. S., Nukolova N. V., Kabanov A. V., Bronich T. K. Nanocarriers for delivery of platinum anticancer drugs. Advanced Drug Delivery Reviews. 2013;65(13-14):1667–1685. doi: 10.1016/j.addr.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Moon L. D. F., Asher R. A., Rhodes K. E., Fawcett J. W. Regeneration of CNS axons back to their target following treatment of adult rat brain with chondroitinase ABC. Nature Neuroscience. 2001;4(5):465–466. doi: 10.1038/87415. [DOI] [PubMed] [Google Scholar]
- 239.Zuo J., Neubauer D., Graham J., Krekoski C. A., Ferguson T. A., Muir D. Regeneration of axons after nerve transection repair is enhanced by degradation of chondroitin sulfate proteoglycan. Experimental Neurology. 2002;176(1):221–228. doi: 10.1006/exnr.2002.7922. [DOI] [PubMed] [Google Scholar]
- 240.Chau C. H., Shum D. K. Y., Li H., et al. Chondroitinase ABC enhances axonal regrowth through Schwann cell-seeded guidance channels after spinal cord injury. The FASEB Journal. 2004;18(1):194–196. doi: 10.1096/fj.03-0196fje. [DOI] [PubMed] [Google Scholar]
- 241.Caggiano A. O., Zimber M. P., Ganguly A., Blight A. R., Gruskin E. A. Chondroitinase ABCI improves locomotion and bladder function following contusion injury of the rat spinal cord. Journal of Neurotrauma. 2005;22(2):226–239. doi: 10.1089/neu.2005.22.226. [DOI] [PubMed] [Google Scholar]
- 242.Bradbury E. J., Carter L. M. Manipulating the glial scar: chondroitinase ABC as a therapy for spinal cord injury. Brain Research Bulletin. 2011;84(4-5):306–316. doi: 10.1016/j.brainresbull.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 243.Mohtaram N. K., Montgomery A., Willerth S. M. Biomaterial-based drug delivery systems for the controlled release of neurotrophic factors. Biomedical Materials. 2013;8(2) doi: 10.1088/1748-6041/8/2/022001.022001 [DOI] [PubMed] [Google Scholar]
- 244.Pilakka-Kanthikeel S., Atluri V. S. R., Sagar V., Saxena S. K., Nair M. Targeted brain derived neurotropic factors (BDNF) delivery across the blood-brain barrier for neuro-protection using magnetic nano carriers: an in-vitro study. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0062241.e62241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cox A. A., Varma A., Vertegel A., Barry J., Banik N. Nanoparticle estrogen in rat spinal cord injury elicits rapid anti-inflammatory effects in plasma, CSF and tissue. Journal of Neurotrauma. 2015 doi: 10.1089/neu.2014.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Saxena T., Loomis K. H., Pai S. B., et al. Nanocarrier-mediated inhibition of macrophage migration inhibitory factor attenuates secondary injury after spinal cord injury. ACS Nano. 2015;9(2):1492–1505. doi: 10.1021/nn505980z. [DOI] [PubMed] [Google Scholar]