

Abstract
The mitochondrial permeability transition (PT) is a permeability increase of the inner mitochondrial membrane mediated by a channel, the permeability transition pore (PTP). After a brief historical introduction, we cover the key regulatory features of the PTP and provide a critical assessment of putative protein components that have been tested by genetic analysis. The discovery that under conditions of oxidative stress the F-ATP synthases of mammals, yeast, and Drosophila can be turned into Ca2+-dependent channels, whose electrophysiological properties match those of the corresponding PTPs, opens new perspectives to the field. We discuss structural and functional features of F-ATP synthases that may provide clues to its transition from an energy-conserving into an energy-dissipating device as well as recent advances on signal transduction to the PTP and on its role in cellular pathophysiology.
I. INTRODUCTION
This review follows a previous Physiological Reviews article on the mitochondrial transport of cations and on the permeability transition (PT) (41). Progress in the field over the last 15 years has been astonishing, with the molecular identification of the Na+/Ca2+ exchanger NCLX (447), of the essential component of the mitochondrial Ca2+ uniporter MICU1 (466), of the Ca2+ uniporter MCU itself (36, 152), and of the channel function of F-ATP synthases and their role in the PT (218). Channel formation by F-ATP synthase has been demonstrated in mammals (9, 218), yeast (96), and Drosophila melanogaster (611) and appears to be a novel property of the eukaryotic complex. The features of the F-ATP synthase channel in mammals (activation by Ca2+ and thiol oxidants, inhibition by Mg2+/ADP, conductance properties) perfectly match those of the mitochondrial megachannel (MMC) (317, 475), which is the electrophysiological equivalent of the permeability transition pore (PTP) (569, 572, 573). While these findings leave little doubt that the PTP forms from the F-ATP synthase under conditions of oxidative stress (51), the mechanism of pore formation remains an open question (9, 218), as is the role of outer mitochondrial membrane (OMM) proteins in PTP modulation (51). In this review we cover regulation of both the PTP and of the F-ATP synthase and point to potential mechanisms that could turn the key enzyme of energy conservation into an energy-dissipating device. In addition, we discuss signaling pathways and posttranslational modifications that may affect pore formation and provide an update on the PTP in pathophysiology. It is our hope that the review will stimulate experiments eventually leading to a structural understanding of PTP formation.
A. Brief Historical Account
Increased permeability of the inner mitochondrial membrane (IMM) to solutes can be easily observed in isolated mammalian mitochondria, usually based on the onset of swelling (485). Stimulation by Ca2+ and coincidence of the permeability increase with loss of the ability to synthesize ATP has been recognized and studied very early (24, 26, 74, 100, 279, 354, 355, 390, 477, 485, 486, 582, 607, 636, 656). The term permeability transition was coined by Haworth and Hunter, who proposed that the permeabilization process was dependent on the opening of a regulated channel (the PTP) and that the process was regulated and potentially reversible (263, 276–278). This idea did not gain much consensus, also because of the acceptance of the chemiosmotic hypothesis, which had just been recognized with the award of the Nobel Prize to Peter Mitchell in 1978 (414). It was widely felt that the presence of a large pore within the IMM would contradict the basic principles of chemiosmosis because its opening would disrupt the proton gradient and prevent the synthesis of ATP. Furthermore, the estimated pore radius of 1.4 nm (390), which is large enough to allow diffusion of ions and solutes with molecular masses up to about 1,500 Da, suggested that the permeability pathway was unspecific and could not be mediated by a protein (41). These concerns substantially contributed to the widespread view that the PT was an in vitro artifact rather than a process of relevance to pathophysiology (52).
This attitude was to change radically toward the end of last century following the key discovery that the PT is inhibited by cyclosporin (Cs) A (76, 134, 144, 194). CsA binds matrix cyclophilin (CyP) D, a peptidyl-prolyl cis-trans isomerase whose enzymatic activity is inhibited by CsA with a matching inhibition of the pore (121, 122, 432, 638). These discoveries gave great impulse to the field because they provided a protein regulator to the PTP and a drug to test its occurrence in cells and living organisms. Through the use of CsA, the occurrence of PTP opening in cell death (133) could be tested in a series of pioneering studies in hepatocytes subjected to oxidative stress (75, 285), anoxia (459), or treatment with ATP (662) as well as in cardiomyocytes (168) and isolated hearts (232) exposed to ischemia followed by reperfusion. The discovery that release of cytochrome c and of additional proteins of the intermembrane space is a key determinant for activation of the intrinsic pathway to apoptosis (165, 172, 266, 362, 373, 565) rapidly made the PT one of the most popular areas of mitochondrial research.
A second key finding was the demonstration that mitochondrial ion channels can be studied by electrophysiology (540) and that the IMM is endowed with the MMC, a high-conductance channel (317, 475) possessing all the basic regulatory features of the PTP (49, 569, 572, 573). Electrophysiology has greatly contributed to our understanding of the PTP (149, 150, 475, 569, 572, 573, 575) and to the recent demonstration that the PTP forms from F-ATP synthase (9, 96, 218). The reader is referred to a recent Physiological Reviews article for an update on the tremendous advances made in the field of mitochondrial ion channels (575).
A third important observation was that both the membrane potential and matrix pH modulate the probability of PTP opening (40, 433, 468), as confirmed at the single-channel level (664). Control by the proton electrochemical gradient provided a conceptual framework to reconcile the PTP with chemiosmosis (41). Since the threshold voltage for PTP opening is affected by many effectors (469, 470), this finding also allowed accommodation of many individual agents affecting the PT (44, 238, 239). Regulation of the PTP and its role in cell death have been the subject of intense and fruitful research over the last 30 years, as testified by nearly 5,000 publications. We refer the reader to a number of reviews for primary references that could not be cited here (41, 47, 48, 52, 112, 130, 131, 167, 217, 226, 233, 239, 240, 247, 252, 254, 313, 318, 357, 436, 438, 476, 534, 549, 558, 665, 667, 668). On the other hand, the structure of the PTP has long remained a mystery (51). Before discussing facts and hypotheses about its molecular nature, we present an overview of the key features of PTP regulation. This overview is mostly based on the features of the pore in mammalian mitochondria, but we will also mention relevant data on the PTP of Drosophila and yeast mitochondria with the important proviso that multiple conductance pathways may exist in yeast (see Refs. 381, 483, 484, 506, 595).
B. Consequences and Regulation of the Permeability Transition
The consequences of PTP opening depend on the open time of individual pores and on the number of open pores per mitochondrion at any given time. The complex relationship between channel kinetics and population dynamics has been discussed in detail (41) and will only be briefly summarized here. The PTP flickers between closed and open states both at the single channel and at the organelle level (317, 471, 475, 569, 572), but occurrence of transient, reversible openings can go undetected in a population of mitochondria unless openings are synchronized or individual mitochondria are monitored. Measurements of the membrane potential resolved in individual mitochondria indeed demonstrated transient and asynchronous cycles of depolarization-repolarization, which over time tended to become long-lasting and were accompanied by permeabilization to calcein (281). Both reversible and long-lasting depolarizations could be traced to PTP opening (281). Conditions have been described under which the mammalian PTP undergoes synchronized cycles of opening-closure and is permeable to ions but not sucrose, suggesting a lower conductance state (284). Importantly, the Drosophila PTP is permeable to H+ and Ca2+ but not to sucrose (610), consistent with its lower conductance of 53 pS (611).
In isolated mitochondria PTP opening is affected by a large variety of compounds that act either as inducers (like thiol oxidants) or inhibitors (like Mg2+, adenine nucleotides and acidic matrix pH; see Ref. 239 for an exhaustive list). “Induction” must be intended in the sense that the lag phase between accumulation of Ca2+ (which is an essential permissive agent for pore opening) and onset of permeabilization decreases, with a matching increase of the rate of spreading of the process through a mitochondrial population. Conversely, “inhibition” means that the lag phase between Ca2+ accumulation and onset of permeabilization increases, with a matching decrease of the rate of spreading of the process through a mitochondrial population (41). Since no true blockers of the PTP are known, the effect of inhibitors is best described as desensitization, and that of inducers as sensitization (52).
When the PTP open probability increases, depolarization can be measured both in intact mitochondria and in situ (473). Collapse of the proton gradient prevents ATP synthesis as long as the pore is open, and ATP hydrolysis by the F-ATP synthase worsens ATP depletion. The consequences of the PT on respiration in vivo, and the related production of reactive oxygen species (ROS), depend on the extent of pyridine nucleotide depletion (52). In mammalian mitochondria, increased probability of PTP opening is also followed by equilibration of ionic gradients and solutes, which may cause swelling, cristae unfolding, and eventually OMM rupture (48).
PTP opening may also contribute to selective release of cytochrome c with an intact OMM. Two pools of cytochrome c can be identified in isolated rat liver mitochondria (42). About 15% of total cytochrome c can be reduced by added NADH (42) through the OMM rotenone-insensitive NADH-cytochrome b5 reductase (543), suggesting that this pool resides in the intermembrane space; while 85% can only be reduced by electrons fed by the IMM electron transfer chain, suggesting that most cytochrome c resides within the intracristal compartments (196). After proapoptotic stimulation, PTP-dependent, CsA-sensitive cristae remodeling occurs with widening of cristae junctions and increase of the fraction of cytochrome c that can be released through OMM Bax/Bak channels (524). Thus the Bax/Bak-dependent and PTP-dependent pathways for cytochrome c release are synergistic rather than mutually exclusive (47).
1. Matrix effectors
Divalent cations are key to PTP regulation. As already mentioned, the PT requires matrix Ca2+; although Ca2+ alone may not be sufficient to trigger a PT, it is an essential “permissive” factor (41). The threshold Ca2+ load required for PTP opening varies with the experimental conditions, in particular the presence of Pi (see below). Matrix Mg2+ desensitizes the PTP, and the effect is synergistic with that of adenine nucleotides. The desensitizing effect is also seen with other divalent cations (such as Sr2+ and Mn2+) that are transported by the MCU and therefore are taken up in energized mitochondria. It appears likely that Ca2+, Mg2+, Sr2+, and Mn2+ compete for the same binding site(s) (49). A second binding site for divalent cations is accessible from the intermembrane space (i.e., it does not require cation uptake) and mediates PTP desensitization with a Ki of ∼0.2 mM with all divalent cations tested (49).
The PTP is extremely sensitive to oxidation-reduction events. Pore opening is promoted by oxidation of matrix pyridine nucleotides and dithiols, and by treatment with dithiol reagents like phenylarsine oxide and arsenite (470). The inducing effects can be individually reversed with appropriate reducing agents (124) and can be blocked by 1-chloro-2,4-dinitrobenzene, suggesting involvement of matrix glutathione (107). It appears likely that oxidation of critical PTP thiols mediates the inducing effects of peroxides and redox-cycling agents, which are indeed prevented by low concentrations (Ki ∼5 μM) of N-ethylmaleimide (NEM) and monobromobimane (123, 470). PTP modulation by these redox-sensitive sites could also be the basis for the inducing effects of p66Shc, which oxidizes intermembrane cytochrome c producing superoxide anion (215, 590).
The PTP open probability increases with electron flux within complex I (191), and this finding led to the discovery that the PT is regulated by quinones, possibly through a specific binding site (615). Ubiquinone 0 or decylubiquinone prevent pore opening with all tested inducing agents, and their inhibitory effects (unlike those of CsA) can be relieved by pore-inactive quinones (615). It appears likely that PTP modulation by complex I is mediated, in part at least, by production of ROS. Indeed, oxidation of succinate, which induces reverse electron flow at complex I (548), greatly favors ROS production and PTP opening, both events being prevented by rotenone (361).
The PT is modulated by matrix pH. In de-energized mitochondria, the optimum for opening was at matrix pH 7.4, with a decrease both below and above this value (433). Inhibition by acidic pH occurs through reversible protonation of His residues that can be blocked by diethylpyrocarbonate (433, 525), while the basis for inhibition above pH 7.4 remains unknown. His126 of CyPA plays an important role both in ligand binding and catalysis (652), but PTP modulation by matrix pH was not affected by genetic ablation of CyPD (i.e., in Ppif−/− mice), demonstrating that the PTP-regulatory His are not located on CyPD (34). It should be mentioned that the PTP can also be affected by pH indirectly through compounds whose accumulation is affected by pH. For example, in energized mitochondria, an acidic external pH can promote rather than desensitize the PTP because it increases the rate of Pi uptake, worsening PTP opening and tissue damage in ischemic and postischemic acidosis (337). PTP regulation by matrix CyPD and CsA will be discussed specifically in an upcoming section.
Pi is the most puzzling PTP effector. In spite of the fact that increasing concentrations of Pi decrease matrix free [Ca2+] (661), in mammalian mitochondria Pi acts as an inducer (239). Interestingly, Pi instead desensitizes the PTP of yeast and Drosophila melanogaster, species where the PTP is insensitive to CsA (96, 236, 297, 507, 508, 610, 647). We found that Pi can inhibit the PTP in CyPD null mouse liver mitochondria (35), suggesting that the inducing effects of Pi may depend in part at least on CyPD binding, a mechanism that is supported by recent data on the Pi-dependent interaction of CyPD with the F-ATP synthase subunit oligomycin-sensitivity conferring protein (OSCP) (216, 218). It is possible that some of the in situ effects of Pi are mediated by formation of polyphosphate, which has been shown to be a potent activator of the PTP (3, 463, 530).
2. CsA and CyPD
As mentioned above, the PTP is modulated by CsA (76, 194, 253), a powerful immunosuppressive agent that targets CyPs, a class of ubiquitous proteins endowed with peptidyl prolyl cis-trans isomerase activity (189, 464, 579). Sixteen isoforms of CyPs have been found in humans; the most abundant is cytosolic CyPA (619). The enzymatic activity of all CyPs is inhibited by CsA (66), and the CsA/CyPA complex inhibits the cytosolic phosphatase calcineurin (371). As a result, NFAT is no longer dephosphorylated, an event that prevents its nuclear translocation causing immunosuppression (117, 614). It should also be mentioned that calcineurin inhibition prevents translocation of the pro-fission protein Drp-1 to mitochondria, an effect that does not relate to that of CsA on the PTP (98). Mammals possess a unique mitochondrial species called CyPD, which in the mouse is encoded by the Ppif gene (see Ref. 217 for a review). CyPD is the mitochondrial target for CsA and modulates the PTP by decreasing the Ca2+ load required for pore opening, an effect that is prevented in the presence of CsA (253). We, and others, successfully created Ppif−/− mice and examined the properties of the PTP in mitochondria lacking this protein (27, 34, 426, 518). CyPD-null mice showed no overt phenotype and no obvious changes in mitochondrial function. As expected, mitochondria from these mice lacked CyPD protein and were desensitized to Ca2+, as opening of the PTP required about twice the Ca2+ load necessary in strain-matched, wild-type mitochondria (27, 34, 426, 518). In other words, the PTP in CyPD-null mitochondria is desensitized, and its opening requires higher Ca2+ levels that match those of wild-type mitochondria treated with CsA. As would be predicted, mitochondria lacking CyPD were insensitive to CsA (27, 34, 426, 518). Other than for the requirement of higher Ca2+ loads, the PTP response to a variety of modulators was similar in mitochondria from wild-type and CyPD null mice, thereby demonstrating that CyPD has all the key aspects of a PTP modulator but is not an essential structural component of the PTP (27, 34, 426, 518). Consistently, the electrophysiological features of the PTP from CyPD-null mitochondria are indistinguishable from those of wild-type individuals (149). Importantly, largely through the use of CyPD-null mice, a wide variety of murine models of human degenerative diseases were shown to have their basis in mitochondrial pathogenesis and misregulation of the PTP (27, 166, 193, 200, 383, 410, 426, 445, 454, 518, 623).
CP3 is the Saccharomyces cerevisiae mitochondrial CyP isoform, but its genetic ablation does not affect the PT, indicating that it does not regulate the PTP in this species (96). The Drosophila melanogaster genome encodes 14 different CyPs (464). Of these, CyP1 has an NH2-terminal sequence that according to Mitoprot (115) confers high probability of mitochondrial import. Yet, full sequence analysis led to the conclusion that no mitochondrial CyP is present in this species (464). Consistent with this conclusion and the above data on yeast mitochondria, yeast and Drosophila PTPs are insensitive to CsA (297, 610).
3. Inner membrane
The inside-negative membrane potential tends to stabilize the PTP in the closed conformation, while depolarization favors pore opening (40). We have proposed that changes of both the transmembrane voltage and of the surface potential are decoded into changes of the PTP open probability by a voltage sensor (469) that may comprise critical Arg residues, as suggested by modulation of the PTP voltage dependence by Arg-selective reagents (175, 289, 368). This mechanism can explain pore opening by H+ and K+ currents (40, 527) and the effects of membrane-perturbing agents (77, 469) like amphipathic anions (which favor the PT) (465, 469, 526), polycations (343), amphipathic cations (77), and positively charged peptides (500) (which inhibit pore opening) (see Ref. 44 for review). We note that modulation by the surface potential could also account for the effects of atractylate (80) and bongkrekate (268), both ligands of the IMM adenine nucleotide translocase (ANT). Both compounds inhibit the ANT yet have opposite effects on pore opening, which is favored by atractylate and inhibited by bongkrekate (276). This set of observations was one of the bases for the proposal that the PTP forms from the ANT (637), an issue on which we shall return later in the review. Here we would like to observe that transition of the very abundant ANT from the “m” (bongkrekate-bound) to the “c” (atractylate-bound) conformation is accompanied by major structural rearrangements (378) that significantly perturb the surface potential (505, 516), which in turn may affect the PTP open-closed transition through the voltage sensor (see Ref. 44 for discussion).
4. Outer membrane
There is no doubt that the PT is primarily an IMM event, as it also occurs in mitoplasts, i.e., mitochondria stripped of the OMM (536). Yet, the OMM plays a role in pore modulation, as indicated by two sets of observations. The first is that PTP induction by rather high concentrations (0.5–1.0 mM) of NEM (478) and other substituted maleimides requires an intact OMM (347). The effect of NEM is due to secondary oxidation of thiol groups that can also be triggered by copper-ortho-phenantroline (125). We have fully confirmed that PTP opening by NEM is abolished in mitoplasts, and thus that its inducing effect depends on OMM proteins (535). We have demonstrated the “sensitizing” role of the OMM in a second paradigm, where the PTP is modulated by dicarboxylic porphyrins plus irradiation with visible light, a treatment that leads to the production of singlet oxygen (499). Low doses of light inactivate the PTP through degradation of His residues, which in turn prevents matrix Cys oxidation (511). In contrast, higher doses of light activate the PTP through the direct oxidation of Cys residues in the OMM (474). The inducing effect of hematoporphyrin plus high light doses was completely lost in mitoplasts, indicating that it requires an intact OMM (536). Largely based on the effect of its ligands on the PTP, we, as well as others, had proposed that the protein responsible for PTP sensitization was the peripheral benzodiazepine (Bz) receptor, an OMM protein today called TSPO (101, 181, 319, 334, 399, 458, 536), but based on recent experiments on TSPO-null mice, this conclusion turned out to be incorrect (535). The target of NEM and of photooxidative stress thus remains unknown, but a potential candidate is Abcb6, an ATP binding cassette transporter of the OMM involved in heme and porphyrin homeostasis (335).
5. Models for pore formation
In 1993, Kinnally et al. (319) reported that nanomolar concentrations of several ligands of TSPO affected the channel properties of the MMC. These ligands included Ro5-4864, PK11195, and protoporphyrin IX, one of the most powerful inducers of the PTP (458). Independent work from the Snyder laboratory had shown that TSPO copurified with the ANT and the OMM voltage-dependent anion channel (VDAC) in protocols based on detergent extraction followed by hydroxylapatite chromatography. In these studies, radiolabeled high-affinity ligands were recovered in fractions where TSPO could be detected together with VDAC and ANT (399). These data suggested that formation of the PTP could involve the OMM proteins TSPO and VDAC, and the IMM protein ANT.
A few years later, this suggestion was strengthened by the Brdiczka laboratory, who were characterizing OMM and IMM “contact sites,” specialized structures where the two membranes form close contacts mediated by protein-protein interactions (329). These dynamic sites would include hexokinase (HK) bound to the cytosolic surface of the OMM, VDAC within the OMM, creatine kinase and nucleoside diphosphate kinase in the intermembrane space, and ANT in the IMM. Contact sites were proposed to mediate channeling of adenine nucleotides to and from mitochondria, thus limiting the need for its diffusion (7, 83, 329). HK-enriched fractions from low-detergent extracts of mitochondria also formed channels with the conductance expected of the PTP (55). These fractions were not enriched in VDAC and/or ANT (55) and contained an extremely large number of proteins including members of the Bcl-2 family (387), which makes assignment of the channel activity to a specific species quite problematic. Furthermore, and unlike the case of PTP, currents were inhibited rather than induced by atractylate (55). This set of observations led to a model where the PTP would be a multiprotein complex spanning both mitochondrial membranes and comprising ANT, VDAC, TSPO, CyPD, as well as HK and Bcl-2 proteins (653).
As we will review more in detail below, this model did not stand the test of genetics as a CsA-sensitive PT could be easily detected in the absence of ANT (325), VDAC (28, 333), as well as of TSPO (535). Furthermore, mitochondria from the brine shrimp Artemia franciscana do not undergo a PT despite the presence of ANT, VDAC, and CyPD (406). An alternative model has been proposed where the PTP is formed by the Pi carrier (PiC) following its interaction with CyPD and ANT (359). However, results obtained by patch-clamp analysis of the reconstituted PiC do not match the electrophysiological PTP features (269), and genetic deletion of PiC does not support the idea that this protein is essential for PTP formation (339).
He and Lemasters (265) have proposed a model where the PTP would originate from misfolded membrane proteins that have been damaged by various forms of stress, which is similar to an earlier suggestion that the PT is due to oxidative damage of membrane proteins rather than a consequence of the opening of a preformed pore (330). Conductance through misfolded protein clusters would be normally blocked by chaperone-like proteins like CyPD. When the number of these protein clusters exceeds available chaperones opening of “unregulated” pores would occur, which would no longer be sensitive to CsA (265). The model accounts for both CsA-sensitive and -insensitive pores, as well as for the lack of selectivity of the permeability pathway. On the other hand, it is hard to see how a permeability increase mediated by denatured proteins would be regulated by voltage and matrix pH.
Ca2+-dependent, PT-like activities have also been observed in reconstituted systems with both fatty acids (413, 560) and 3-hydroxybutyrate/polyphosphate (3, 463, 530), suggesting that channels can also form in the absence of specific proteins. Lipids may participate in PTP formation and regulation as shown by earlier work from the Pfeiffer laboratory, who suggested that fatty acids produced by activation of Ca2+-dependent phospholipases profoundly affect the PTP and its sensitivity to CsA (77, 478).
A novel mechanism for PTP formation was recently proposed by our laboratories and first presented at the 17th European Bioenergetics Conference in 2012 (45). To identify the binding partners of CyPD, we monitored its associations to mitochondrial proteins after low-detergent extraction followed by protein separation by blue native gel electrophoresis. We found that CyPD comigrates with the F-ATP synthase, and the interaction was confirmed by immunoprecipitation of complex V followed by Western blotting (216). Binding of CyPD to the F-ATP synthase required Pi and caused a decrease of the enzyme's catalytic activity. This decrease was counteracted by CsA, which displaced CyPD and increased the catalytic activity (216). Cross-linking experiments with bifunctional reagents indicated that CyPD interacts with the lateral stalk subunits b, d, and OSCP (216). We could then immunoprecipitate each subunit individually and demonstrate that CyPD interacts with the OSCP subunit, from which it can be displaced by Bz-423 (218). Bz-423 was originally characterized as an apoptosis-inducing agent acting through mitochondria (59). OSCP was identified as its target through the unbiased screening of a human phage display library, and it was shown that Bz-423 is an inhibitor of the F-ATP synthase (290, 291, 551), an activity that is shared by several ligands of TSPO including PK-11195 (116). The demonstration that Bz-423 induces the PTP, and that its binding site on helices 3 and 4 of OSCP may coincide with that of CyPD (218) paved the way to the demonstration that F-ATP synthase can form channels with the features of the PTP-MMC.
Gel-purified dimers of F-ATP synthase from bovine heart were incorporated into lipid bilayers. Following treatment with Ca2+, Bz-243 and phenylarsine oxide, a vicinal dithiol cross linker that is a powerful PTP inducer (49, 358), opening of channels with a unit conductance of ∼500 pS was observed, which is compatible with that of the bona fide mammalian PTP-MMC (218). Monomers of F-ATP synthase, which have the same subunit composition as the dimers (588), were instead devoid of channel activity (218). The MMC has been consistently described as a multiconductance channel exhibiting many substates and a typical maximal (fully open) conductance of 0.9–1.0 nS, although a value of ∼1.3 nS in symmetrical 150 mM KCl can also be observed (475, 572). Electrophysiological studies have produced clear evidence for a binary structure of the pore, which displays a prominent half-conductance level (374, 569, 574). A channel of 0.4–0.5 nS can also be observed whose frequency of appearence is inversely related to that of the higher “full conductance” MMC, and which may correspond to one-half of the MMC (151, 663). Based on these data, we suspect that the 500-pS current observed in our study with purified F-ATP synthase (218) is mediated by dimers and that the full conductance in the native IMM originates from F-ATP synthase tetramers.
Channels were inhibited by Mg2+/ADP and by the F-ATP synthase inhibitor γ-imino ATP (a nonhydrolyzable ATP analog). Like the MMC of Ppif−/− mitochondria (149), and in keeping with the lack of CyPD in the preparations, channels were insensitive to CsA. Consistent with the absence of ANT, channel openings could not be induced by atractyloside and were still observed in the presence of bongkrekic acid (218). The channel-forming property is shared by purified F-ATP synthase dimers of mitochondria from Saccharomyces cerevisiae, which displayed Ca2+-dependent currents of ∼300 pS (96), and from Drosophila melanogaster, in which the conductance is 53 pS (611) in keeping with earlier results on solute permeation (610). Channel formation by F-ATP synthase has been confirmed in human cells (9) and is supported by a study where the c subunit was downregulated by siRNA in HeLa cells, which resulted in PTP inactivation (64). Potential mechanisms for PTP formation from F-ATP synthase is discussed in section III, while Table 1 summarizes the key features of the PTP of Saccharomyces cerevisiae, Drosophila melanogaster, and mammals.
Table 1.
Properties of the F-ATP synthase channel across species
| Conductance, pS | Matrix Ca2+ | Matrix Mg2+/ADP | Pi | CsA | Matrix CyP | |
|---|---|---|---|---|---|---|
| S. cerevisiae | 300 | Activates | Inhibits | Inhibits | No effect | Yes |
| D. melanogaster | 53 | Activates | Inhibits | Inhibits | No effect | No |
| Mammals | 500 | Activates | Inhibits | Activates | Inhibits | Yes |
The conductance of F-ATP synthase channels and the effects of some PTP effectors in the indicated organisms are summarized. CsA, cyclosporin A; CyP, cyclophilin.
C. Genetic Analysis of Putative Pore Components
As already mentioned, the hypothesis that the PTP is composed by ANT (and/or PiC) in the IMM and VDAC in the OMM, plus a variety of modulators including TSPO (653), has been put to a rigorous test by genetic inactivation of its putative components. The rationale is simple: inactivation of the gene(s) encoding key structural element(s) should eliminate PTP activity while genetic inactivation of a modulator should allow the formation of the pore, albeit with altered functional characteristics that may suggest a specific role for each modulator. As discussed below, the results leave little doubt that ANT, PiC, VDAC, and TSPO are not essential for PTP formation. Genetic analysis of F-ATP synthase subunits will be discussed after description of its structure and function.
1. Adenine nucleotide translocator
In respiring mitochondria, the ANT, the most abundant IMM protein, catalyzes efflux of ATP from the mitochondrial matrix and uptake of ADP into the matrix. Binding of the ANT to a CyPD affinity matrix has been reported (135, 637), but the relevance of this observation to PTP regulation remains unclear because CyPD bound equally well the ANT purified from rat liver or yeast (637) in spite of the fact that in yeast mitochondria the PT is not regulated by CyP or inhibited by CsA (96, 297). ANT from bovine heart mitochondria reconstituted in liposomes does exhibit high-conductance channel activity that is stimulated by Ca2+ and insensitive to CsA (81). Like the MMC, the channel displays prominent voltage-gating effects (81) with conductance ranging between 50 and 700 pS in 100 mM KCl (81) and low probability of current fluctuation at voltages lower than 150 mV. Unlike the MMC, however, currents could only be inhibited by ADP and bongkrekate together, while ADP alone had a marginal effect (73, 81, 82).
The Wallace laboratory conditionally inactivated the liver Ant2 gene in an Ant1−/− background, which generated mice with ANT1/ANT2-deficient liver mitochondria where respiration could not be stimulated by ADP (325). These mitochondria underwent CsA-sensitive PTP opening, but required a higher matrix Ca2+ load. The PT was inhibited by CsA and stimulated by H2O2 and diamide, indicating that the ANT is not the site of action of these oxidants or the relevant partner for CyPD binding; of note, the PTP was resistant to atractylate and ADP (325).
The survival of animals lacking all ANT isoforms is unexpected (249), and the main conclusion of this study has been challenged (65, 250, 254). Sequence analysis has indeed shown that, besides the genes encoding for ANT1 and -2, in most mammals two additional isoforms are present, SLC256A (ANT3) and SLC25A31 (ANT4). On these grounds, most recent reviews have called into question the idea that mitochondria prepared from ANT1/ANT2-null mitochondria were devoid of any ANT isoform, justifying its continued inclusion as a key structural element of the PTP (e.g., Refs. 65, 250). However, analysis of the mouse genome database (e.g., http://www.informatics.jax.org/) demonstrates that mice (and all rodents) lack a gene for ANT3 and hence can only express ANT1, ANT2, and ANT4. Moreover, ANT4 appears to be expressed specifically in germ cells and may be primarily compartmentalized to the sperm flagellum in all mammals (314, 364). One possible explanation would be the compensatory (mis)targeting of alternate isoforms normally directed to restricted locations (250, 251). While the liver may not normally express appreciable levels of ANT4, in this unique situation the viability of mice missing ANT1 and ANT2 may depend on higher levels of ANT4 in the liver (203). Even in this scenario, it is very difficult to see 1) how these putative ANT molecules would not transport adenine nucleotides yet promote a CsA-sensitive PT (325), and 2) why these putative pore-forming ANT4 molecules would not respond to atractylate and ADP (325), given that in wild-type mitochondria ANT4 is fully sensitive to both carboxyatractylate and bonkgrekate (163). Thus all available evidence points to the fact that ANT can modulate the PTP, possibly through its effects on the surface potential (44), but is not a core and required structural component.
2. Phosphate carrier
It has been pointed out that PTP formation in mitochondria missing major ANT isoforms could be due to its compensation by other members of the mitochondrial carrier family, PiC in particular (250). The PiC of the IMM is encoded by a single gene in mammals and is critical for ATP synthesis by serving as the primary means for mitochondrial Pi transport across the IMM. While long suspected as playing a role in PTP formation (e.g., Ref. 332), interest in the PiC was renewed following the realization that antibodies critical for the earlier studies on the ANT outlined above were, in fact, primarily directed toward the PiC (360). Additional biochemical studies demonstrated the CsA-sensitive binding of CyPD to the PiC and its modification by chemical reagents (e.g., by phenylarsine oxide and NEM) correlated with PTP opening and inhibition. These and a suite of additional biochemical studies led to the idea that PiC may be a key component of the PTP that undergoes a Ca2+-induced change in conformation to induce pore formation in cooperation with the ANT (250, 360). Consequently, in addition to the ANT, recent models of the PTP also include PiC (e.g., Refs. 65, 228, 254). Genetic tests of the role of PiC in PTP formation initially involved the application of siRNA techniques in either mammalian cells or transgenic animals (242, 601). In both situations, mitochondria lacking up to 60% PiC expression showed no alteration in PTP function, and no effects on PTP activity were observed after overexpression of PiC (242, 601). Furthermore, in mitochondria prepared from cells where the gene encoding PiC had been genetically inactivated (resulting in over 90% reduction in PiC levels), the PTP could still form and displayed marginally reduced sensitivity to activators (339). Finally, patch-clamp experiments with the reconstituted, functionally active PiC revealed an anion channel function with a mean conductance as low as 40 pS which was further decreased to 25 pS by Ca2+ and Mg2+, inhibited by Pi, and unaffected by ADP (269). Taken together, these experiments indicate that PiC, like the ANT, cannot constitute a key structural element of the PTP.
3. Voltage-dependent anion channels
VDAC is a major OMM protein which functions as a general diffusion pore for small hydrophilic molecules; in mammals, distinct genes encode three variants of the protein, VDAC1, -2, and -3 (487). VDAC1 displays channel activity that is similar to that of the PTP (571, 574). As already mentioned, because of its presumed preferential location at sites of interaction between the OMM and IMM (132) and of its copurification with TSPO and ANT, VDAC has been included in traditional models of the PTP (65, 254). The putative role of VDACs as a structural element or regulator of the PTP has been tested by genetic analysis. A thorough study of Vdac1−/− mitochondria lacking the major isoform VDAC1 demonstrated that the properties of the PTP (response to Ca2+ and Pi, sensitivity to oxidants and NEM, inhibitory profile with CsA and quinones) were identical to those obtained from wild-type mice of matched genetic background, indicating that VDAC1 is fully dispensable for PTP formation (333). Similar results were obtained with VDAC1/3 null mitochondria, while whether VDAC2 could compensate for the absence of VDAC1 and/or 3 was more complex to assess because ablation of Vdac2 results in embryonic lethality (106). This result is likely the consequence of the fact that VDAC2, while serving as a diffusion pore in the OMM like all VDACs, is also a potent inhibitor of the pro-apoptotic effects of OMM Bak (106). As a result, in the absence of VDAC2, Bak is unrestricted in its ability to induce cell death, likely leading to early embryonic lethality. To assess the role of VDAC2 in PTP formation, MEFs prepared from Vdac1−/−/Vdac3−/− mice were treated with VDAC2 siRNAs, which did lead to a decrease of VDAC2 protein levels to 2% of normal, but to no alteration in PTP onset and properties (28). As a result, there is currently no evidence, either biochemical or genetic, that supports a role for VDAC either as a structural element of the PTP or as a key modulator. Consequently, models that imply a role for VDAC in PTP formation must be called into question (65, 254). Regulation of the PTP through binding of HK to OMM will be discussed in section IV.
4. TSPO
TSPO was initially identified as an OMM protein that bound a series of Bz analogs that do not target Bz receptors in the central nervous system. Hence, it was initially referred to as the peripheral Bz receptor (451, 510). Early studies appeared to demonstrate that TSPO is intimately involved in two separate functions, mitochondrial transport of cholesterol and protoporphyrin IX, and PTP regulation. In cells that produce steroid hormones (e.g., in the adrenal cortex), TSPO was thought to promote the transport of cholesterol into the mitochondrial matrix, the rate-limiting step in steroid synthesis (411). However, the ubiquitous expression of TSPO suggested that it could serve more general functions, one of which was long thought to be the regulation of PTP activity. TSPO was initially linked to PTP function based on its association with other proteins thought to be required in traditional models of the PTP (i.e., VDAC and ANT) (319, 399, 458). In addition, experiments with Bz specifically targeting TSPO suggested that its ligands promoted the opening of the PTP, a view extended by recent studies (53, 101). However, important aspects of these studies remained confusing (for details, see Ref. 535). Ensuing studies have employed natural and synthetic ligands to assess the role of TSPO function in a number of natural and pathological circumstances. Largely through the use of these compounds and biochemical associations, TSPO was proposed to play a role in the PTP activity associated with cell death in many human pathological conditions. Since initial studies suggested that nonconditional inactivation of the nuclear gene encoding TSPO resulted in embryonic lethality (450), we assessed the role of TSPO in PTP function through the generation of mice in which the Tspo gene had been conditionally eliminated (535). These studies demonstrated that TSPO plays no role in the regulation or structure of the PTP (535). Consequently, we suspect that endogenous and synthetic ligands of TSPO regulate PTP activity because of their direct effects on the F-ATP synthase (116), as demonstrated for Bz-423 (218). Consistent with this conclusion, hearts lacking TSPO are as sensitive to damage caused by PTP opening following ischemia-reperfusion injury (see below) as are hearts from wild-type mice, in contrast to mice missing CyPD (27, 426). Consequently, OMM regulation of PTP activity must occur though a mechanism that does not require TSPO and is based on proteins that have yet to be identified. These results call into question a wide variety of studies implicating TSPO in a number of pathological processes through its actions on the PTP, and the validity of placing TSPO in recent representations of the structure of the PTP (65). Interestingly, in a separate study of conditional Tspo mice, the role of TSPO role in cholesterol metabolism has also been questioned (423, 591); consequently, the precise OMM function of TSPO has yet to be established.
5. Bcl-2 family members
Apoptosis regulators of the Bcl-2 family are evolutionarily related molecules that govern mitochondrial OMM permeabilization and can be either pro-apoptotic (e.g., Bax, Bad and Bak) or anti-apoptotic (e.g., Bcl-2 proper and Bcl-xL) (578). Genetic tests of the requirement of members of this family in the formation and regulation of the PTP have been carried out with pro-apoptotic Bax, Bak, and Bad. In mitochondria from MEFs in which genes encoding Bax and Bak have been genetically inactivated, OMM permeability decreased without significantly altering PTP function in the IMM (306). Indeed, physiological analysis of IMMs and biochemical studies showed no difference between wild-type and Bax/Bak-null mitochondria while the PTP was still inhibited by CsA. Thus Bax and Bak do not directly regulate IMM aspects of the PTP. In addition, studies on MEFs missing the BH3-only family member Bad have indicated that the basic characteristics of the PTP are no different in Bad-null cells, but they appear sensitized to various stress factors such as ceramide (537). These stress factors are proposed to lead to a complex cascade of kinase/phosphatase reactions that enable productive association, and inactivation, with the anti-apoptotic protein Bcl-xL. In sum, critical genetic studies provide no evidence for Bcl-2 family members in the direct regulation, or formation, of the PTP, while Bax and Bak are required for PTP-dependent OMM permeabilization, given that in their absence organelle rupture and cell death are prevented (306).
II. STRUCTURE AND FUNCTION OF F-ATP SYNTHASES
Given that PTP formation critically depends on the F-ATP synthase, a review of its structure and function may be useful to direct future research in the field. In the following discussion, we will also highlight issues related to the PTP that may inspire new experiments and foster progress in this developing area of research.
F-ATP synthase is responsible for the synthesis of most of ATP in living cells, a task achieved by coupling the chemical reaction ADP + Pi = ATP to transmembrane proton translocation from the intermembrane space to the matrix. This reaction is reversible, and in the absence of a H+ gradient, glycolytic ATP is hydrolyzed with coupled proton extrusion to the intermembrane space (for reviews, see Refs. 70, 187, 202, 298, 553, 628). In addition to the IMM, the F-ATP synthase is found in the thylakoid membrane of chloroplasts and in the plasma membrane of bacteria. Recent evidence supports its location also in the plasma membrane of mammalian cells, where orientation is opposite to what found in bacterial plasma membrane (385, 600).
A. Structure
The F-ATP synthase has a multi-subunit architecture whose overall structure and arrangement is extremely well conserved in spite of the early divergence of bacteria, plants, and animals. The complex is composed by a membrane-embedded FO subcomplex, through which the protons flow, as well as by a soluble catalytic F1 subcomplex linked to FO (and thus to the IMM) by central and peripheral stalks, which are clearly resolved by cryotomography of single particles (29, 345, 509). The overall arrangement of mitochondrial F-ATP synthase in its physiological dimeric form (see also sect. IIB) is reported in Figure 1.
Figure 1.
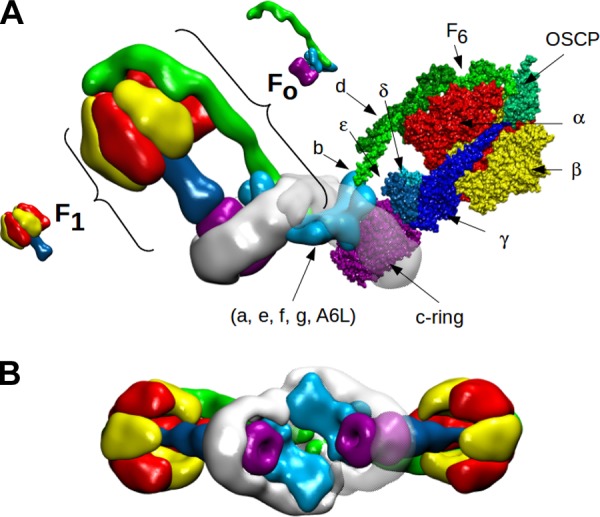
Model of F-ATP synthase dimer viewed from the lateral side (A) and from the intermembrane space (B). A: left monomer, the F1 and FO sectors are highlighted. Right monomer, the F1 and FO subunits are shown. In the F1 sector, the front α and β subunits have been removed to reveal the central stalk. The F1 α and β subunits are colored in red and yellow, respectively. The F1 γ, δ, and ε subunits are colored in shades of blue, the peripheral stalk subunits b, d, F6 and OSCP in shades of green, and the c-ring in purple. The remaining FO subunits a, e, f, g, and A6L are colored in light blue. The intramembrane FO is surrounded by detergent, shown in white. The image has been built starting from the yeast dimer molecular model (146) (PDB id. 4b2q) to which the cryoelectron microscopy (cryo-EM) map of bovine F-ATP synthase (29) (EMD id. EMD-2091) has been superimposed. The fit of molecular models to cryo-EM map was performed using the program ADP_EM (208). The molecular model for bovine F-ATP synthase was obtained by superimposing the 3D structure of the bovine F1-c-ring complex (PDB id. 2xnd) onto each corresponding monomer of the yeast dimer. The superposition was performed using the Swiss pdb viewer routine Iterative magic fit (237). The lateral stalk was taken from the yeast dimer (PDB id. 4b2q) which has been modeled using the bovine subunits. B: cryo-EM maps are rotated 180° to be viewed from the intermembrane space.
The F1 subcomplex always consists of three copies of subunits α and β and one copy of subunits γ, δ, and ε. The homologous α and β subunits carry the nucleotide binding sites and alternate to form a pseudo-hexameric ring around the coiled-coil structure of the γ subunit, which constitutes the central stalk along with subunit ε in bacteria, and with subunits ε and δ in chloroplasts and mitochondria [bacterial subunit ε is homologous to mitochondrial subunit δ (606)]. The steady-state catalytic mechanism for ATP synthesis/hydrolysis requires activity of all the catalytic sites, which are located in the β subunits at each α-β interface and interact in a highly cooperative manner. ATP (and sometimes ADP) molecules can also bind the α subunits at the α-β interfaces, but these nucleotides are not rapidly exchanged during catalysis; therefore, these sites, whose role is still unknown, are considered as noncatalytic (320).
The subunit composition of the FO subcomplex is more variable. The simplest form is present in bacteria, where it consists of 1 copy of subunit a, 2 copies of subunit b, and between 10 and 15 copies of subunit c depending on the species (202), which implies a H+/ATP stoichiometry between 3.3 and 5, respectively (626). The c subunits form a ring structure connected to F1 by the central stalk, where the central region is probably occupied by phospholipids (626). The a subunit associates with the c-ring peripherally, and at the interface, two half transmembrane water channels form through which H+ are transported via conserved carboxylic residues present in each c subunit. This residue is Asp61 in Escherichia coli and almost invariably Glu58 in other species, including mammals (202). Subunit a also associates with the lateral stalk, which is formed by a homodimer of subunit b and by δ subunit located at the top of F1. Single molecule approaches demonstrated that the catalytic and transport mechanisms are coupled by rotational catalysis at the rotor (constituted by the γ-ε-c-ring subcomplex) which is transmitted to the stator (formed by the α, β, δab subcomplex) (202). The lateral stalk of the stator is essential to prevent corotation of the α-β subunits with γ, which would curtail the catalytic activity.
The FO sector of mitochondria has a more complex structure than that of bacteria. Subunits a, b, and c share high homology with the bacterial subunits, with the important difference that the c-ring has 10 copies of the c subunit in yeast and only 8 copies in all vertebrates and most if not all invertebrates, which decreases the H+/ATP stoichiometry to 2.7 thus maximizing the ATP yield (626). Seven additional subunits have been identified in mitochondria, i.e., subunits d, e, f, g, F6, and A6L, which are unique to the mitochondrial complex, and OSCP, which is similar to the bacterial δ subunit (606). The membrane-embedded part of FO comprises subunits e, f, g, and A6L and is connected to F1 by a complex peripheral stalk (160, 495), which is composed by one copy each of subunits b, d, F6, and OSCP, the latter being located on top of F1 as is the δ subunit in bacteria. When phospholipids have not been extracted, the hydrophobic proteins MLQ/6.8-kDa proteolipid (105, 408) and AGP/DAPIT (441) can also be detected in the FO of mammals including humans (441), and their sequences are conserved in the genomes of vertebrates and metazoans (351). Conversely, the yeast enzyme additionally contains subunits i and k (19, 596) and a recently characterized subunit l, a homolog of subunit k (372). When all subunits are considered together, the mitochondrial FO membrane domain is constituted by ∼30 transmembrane α-helices (97), and the molecular mass of the whole complex varies between 540 and 585 kDa depending on the source.
The mitochondrial complex can also bind inhibitory factor 1 (IF1), a protein conserved from yeast to mammals. IF1 is a unidirectional inhibitor of ATP hydrolysis (31) that reversibly binds F1 with a 1:1 stoichiometry, resulting in full inhibition of enzyme activity (94, 227, 259). Along with IF1, in yeast mitochondria the enzyme can be regulated by two additional proteins, stabilizing factor 1 and 2 (90, 605). The subunit composition of the F-ATP synthase from prokaryotes and eukaryotes, along with the subunit homology and the corresponding nomenclature is reported in Table 2. These subunits are encoded by both nuclear and mitochondrial genes, and their assembly is a very complex process that is still under investigation (267). In yeast, the three FO core proteins 6, 8, and 9 are encoded by mtDNA, while in mammals only subunits a and A6L are encoded by the mitochondrial genome (267).
Table 2.
Equivalence of subunits of F-ATP synthase from different sources based on sequence homology
| Mitochondria |
||
|---|---|---|
| Bovine | Yeast | E. coli |
| α | α | α |
| β | β | β |
| γ | γ | γ |
| δ | δ | ε |
| ε | ε | |
| OSCP | OSCP | δ |
| b | 4 or b | b* |
| A6L | 8 | |
| F6 | h | |
| a | 6 or a | a |
| c | 9 or c | c |
| d | d | |
| e | e | |
| f | f | |
| g | g | |
| i/j | ||
| k/l | ||
| MLQ/6–8 kDa | ||
| AGP/DAPIT | ||
F-ATP synthase from Escherichia coli has two copies of the b subunit.
B. Supramolecular Organization: Clues to Pore Formation
Supramolecular organization of F-ATP synthase in situ was first reported by Allen (12), who observed that in Paramecium multimicronucatum F-ATP synthase complexes form a paired row around the outer curve of helical tubules, and noted that the complexes are closely and uniformly associated together within this helical band. Within the yeast, mammal, and plant IMM, F-ATP synthase is organized in dimers associated to form long rows of oligomers located at the cristae ridges, which are essential to maintain a high local curvature and normal cristae morphology (29, 147, 169, 246, 555). F-ATP synthase dimers have also been observed in chloroplasts of algae, where they appear to be susceptible to environmental effects like the Pi concentration (529), but not in bacteria. Dimers interact within the IMM through the FO subunits (29, 147, 169, 246, 555) with the peripheral stalks turned away from each other (Figure 1). Additional proposed roles of F-ATP synthase oligomers are higher efficiency and higher stability (57, 555, 586).
Studies in yeast established that preferential interactions in dimers occur through subunits 6 (635), 4 (546), e (20, 179), and g (86), which formed 6/6, 4/4, and e/g associations, and also through subunit h and the yeast-specific subunit i (460). In keeping with their general presence, subunits a (632), e (58, 246), and g (246) were shown to play a role in F-ATP synthase dimerization also in mammals. A stabilizing effect on mammalian F-ATP synthase dimers has also been reported for the inhibitor protein IF1 (56, 93, 206) and for the matrix metalloprotein Factor B (or subunit s), which interacts with e and g subunits of FO as well as with the ADP/ATP carrier (294, 352). A second interface (the oligomerization interface) stabilized through e/e and g/g interactions allowing oligomer formation has been characterized in yeast (246) (Figure 2). However, its existence is still debated because the distance between dimers appears variable in electron cryotomography of yeast mitochondria, making direct protein contacts difficult (146, 147). Furthermore, these latter studies showed fixed angles of >70° between two F1 in all species examined (146, 147), rather than the angles ranging between 40° (127, 412) and 70–90° (169, 586) observed in previous single-particle electron microscopy images of dimers.
Figure 2.

Model of F-ATP synthase dimer viewed from the matrix (A) and from the intermembrane space (B). A: the model (top view) was built and fitted to cryo-EM maps as described for Figure 1 to illustrate the region where the PTP could form between paired monomers. A second dimer is also depicted to illustrate another possible region for PTP formation in the area defined by two paired dimers. Left monomer: reversible binding of CyPD to F-ATP synthase is shown. Right monomer: the position of human Cys residues on the specified subunits (dots) is mapped onto the 3D structure of the bovine F1-c-ring complex and of the bovine lateral stalk (fC7 and A6LC59 are missing in the bovine complex). B: the same model shown in A is rotated 180° (view from the intermembrane space). Left monomer: the subunits involved in Ca2+-dependent interactions are highlighted, i.e., subunits α and β, which interact with the matrix protein S100A1 (62) (not shown in the picture), and the Fo region containing subunit e, which may interact with a hypothetical tropomyosin-like protein localized in the intermembrane space (17). Right monomer: Ca2+-regulatory sites located in the c-ring (23, 402) and the residues (T163, R189, E192, D256) of β subunits interacting with the catalytic metal ions are mapped onto the 3D structure of the bovine F1-c-ring complex. Numbering does not include the import sequences.
Both in yeast (246, 635) and mammals (56, 245, 632), the stabilizing contribution of the different subunits seems to be additive. Mutants lacking one or more of the above-mentioned subunits, such as ρ0 cells (632) and yeast and human cells totally or partially depleted of e and g subunits (245, 461), still possess lower amounts of F-ATP synthase dimers and oligomers as detected by native gel electrophoresis. It is striking that downregulation of e and g subunits in HEK and HeLa cells not only affected F-ATP synthase dimerization/oligomerization, but also overall oxidative phosphorylation and structure of the mitochondrial network (245).
The PT has been observed in ρ0 cells (389), while it was markedly inhibited in yeast mutants lacking subunits e and/or g (ΔTIM11, ΔATP20 and ΔTIM11;ΔATP20 strains) (96). These data support the hypothesis that dimerization of F-ATP synthase is necessary for PTP formation, but also suggest a different contribution of the FO subunits in forming the PTP. Based on the observation that PTP is conserved from human to yeast and Drosophila (25), it seems reasonable to suggest that subunits e and g of the FO subcomplex, which are involved in the dimerization and oligomerization interfaces, may also favor PTP formation possibly also through other FO/peripheral stalk subunits, such as subunits 4, h, and i which stabilize the dimers independently of the e and g subunits (199). F-ATP synthase contains several Cys residues, and consistently, treatment of mitochondria with CuCl2 stabilized preexisting dimers by formation of disulfide bridges between adjacent monomers in ΔTIM11, ΔATP20, and ΔTIM11;ΔATP20 yeast strains (96).
Numerous atomic structures have been resolved of the following: 1) various conformations of the bovine (1, 2, 33, 67, 68, 72, 214, 221, 301, 405, 442, 496, 598) and yeast F1 sector (299, 300); 2) the soluble part of the peripheral stalk (bdF6sol) (160) and bovine F1 in complex with full-length or truncated subunits of the stator (OSCP, F6; bT and dT) (495); and 3) the bovine F1 c8 (626) and yeast F1 c10 (143, 219, 554). A recent study of dimeric F-ATP synthase from the green alga Polytomella at 6.2 Å resolution shows that subunit a is arranged in two membrane-intrinsic hairpins at an angle of ∼70° relative to the c-ring helices (11). It remains to be established whether this arrangement is conserved in other F-ATP synthases (382). The structure and arrangement of most of the remaining membrane subunits has not been defined yet, and the available data derive from cross-linking experiments (20, 38, 86, 462, 544, 547, 552, 596) and from the cryo-electron tomography structures of monomeric bovine (29, 509) and yeast F-ATP synthase (345).
Yeast e and g “nonessential” subunits are small hydrophobic proteins exclusively associated with dimers of the enzyme (along with the species-specific subunit k) (19). In dimers, the presence of the e subunit is essential for preserving the g subunit, while the e subunit is not affected by deletion of the g subunit (461). They are located at the periphery of FO in the cryo-electron tomography difference maps (345), and their stoichiometry has been estimated to 2 subunits e and g per dimer (19). Subunit g could be cross-linked to subunit i (462) and to the membrane-embedded part of subunit 4 (544), which is anchored to the IMM by two transmembrane segments and contacts subunits f, 6, and 8. Both e and g subunits have one putative transmembrane domain inserted with the same orientation, and a soluble domain localized on opposite sides of the membrane with the COOH terminus of subunit e exposed to the intermembrane space. Both harbour a Cys residue (eCys28 and gCys75) and form e/g interactions in the dimerization interface through GXXXG motifs (20). As already mentioned, they also participate in the putative oligomerization interface though e/e and g/g interactions (246), as established in a yeast gCys75Ser/Leu109Cys mutant, which formed eCys28-eCys28 and gLeu109Cys-gLeu109Cys cross-links (20, 86). It is tempting to speculate that Ca2+ and dithiol reagents might induce conformational changes in e and g subunits, which could then contribute to PTP formation from F-ATP synthase dimers/tetramers. This may occur with or without association with the intramembrane parts of the two b subunits, whose proximity at the short hydrophilic loops linking their transmembrane domains has been demonstrated in the enzyme dimers (547). The involvement of other subunits, such as the species-specific subunit i (596) that is in contact with subunit g, cannot be excluded. On the other hand, subunit i and subunit f, which have one putative transmembrane domain, are in close proximity with subunits 6 (462) and 8 (552), respectively, whose absence did not preclude PTP opening (389), thus suggesting that i- and f-subunits do not contribute to channel formation.
A similar arrangement of e and g subunits in the IMM of bovine heart has been hypothesized based on cross-linking experiments (38), although, at variance from yeast, bovine e and g subunits remain associated to the enzyme monomers (118). Moreover, recent cryo-electron tomography data of intact bovine F-ATP synthase at 18 Å resolution (29) confirmed that these subunits extend from the a subunit density distal to the c8-ring. Most importantly, this map highlighted that e and g subunits deviate from the expected plane of the lipid bilayer with a bend of ∼43°, suggesting that two monomers placed in contact with their e and g subunits would bend the lipid bilayer at ∼90°, consistent with the electron tomography of intact mitochondria. The same map revealed that the b subunit spans the membrane without contacting the c8-ring in the enzyme monomer, suggesting to the authors that two subunits b are in close proximity in the dimer (29). We think that these observations support the hypothesis that the conserved subunits e, g, and b might contribute to PTP formation also in mammals.
It has been proposed that a latent H+-translocating pathway exists within FO, which would be formed by subunits e, f, g, and A6L, as well as the ANT. This pathway would be occluded in the matrix by the NH2 terminus of factor B, which is conserved in animal mitochondria (37). Consequently, the conductance of this pathway would be markedly increased upon displacement of factor B from FO subsequent to oxidation of Cys residues to the corresponding disulfide leading to mitochondrial uncoupling, as observed following treatment of IMM preparations with thiol modifiers (293). Although fascinating, this mechanism probably cannot account for PTP formation because 1) the proposed latent H+ pathway is sensitive to oligomycin, which binds to FO subunit c and fully inhibits enzyme catalysis (567), while the PTP is not inhibited by oligomycin; and 2) factor B is not present in yeast and Drosophila melanogaster, where the PTP is modulated by SH reagents, as in animal mitochondria (96, 610).
Interestingly, in F-ATP synthase from bovine heart mitochondria, another set of mono- and dithiols located in FO has been described, whose environment was altered by membrane energization and whose oxidation resulted in complete and reversible uncoupling (645, 646). Based on the observation that uncoupling was not inhibited by oligomycin, the authors proposed that the permeability pathway is located on the cytosolic side of the oligomycin inhibitory site (645). This position would suggest involvement of the unique Cys residue of subunit c, which is located near the Glu58 residue essential for proton translocation (626). It is tempting to speculate that oxidation of this Cys residue may favor PTP formation in another part of FO. This does not exclude the involvement of species-specific Cys residues located in other subunits, such as subunit b in bovine heart, whose modification also affects enzyme function (140, 370, 654). Oxidation of αCys294 and γCys103, which results in the formation of an intersubunit inhibitory disulfide bridge, has been observed in canine heart failure (620) and could be involved in PTP modulation. However, these residues are located far away from one another in the assembled complex (Figure 2), suggesting that the disulfide may form only in misfolded/aggregated enzymes (620). All together, the FO subunits form ∼30 transmembrane α helices (97), which might potentially contribute to PTP. Further genetic approaches are needed to test their potential role in PTP formation. A comparison of the Cys residues of human, Drosophila, and yeast F-ATP synthase is presented in Table 3.
Table 3.
Cys residues in human, Drosophila, and yeast F-ATP synthases
| Position |
|||
|---|---|---|---|
| Subunit | Human | Drosophila | Yeast |
| OSCP | 141 | 137 | 117 |
| α | 244,294 | 243,293,489 | 238 |
| β | |||
| γ | 103 | 105,131,197,246 | 117 |
| δ | 106 | ||
| ε | 19 | ||
| a | 112,144 | 33 | |
| b | 239 | 78,193,229 | |
| c | 125 | 123 | 65 |
| d | 101 | 48 | |
| e | 28 | ||
| f | 72 | ||
| g | 88 | 75 | |
| A6L (yeast 8) | 59 | 24 | |
| F6 (yeast h) | |||
| k (yeast) | |||
| j (yeast) | |||
Numbering includes the import sequences. Cys residues within import sequences are not included.
C. Catalytic Mechanism
Rotational catalysis as the coupling mechanism of F-ATP synthases was suggested at the end of the 1970s by Paul Boyer (69), who in 1993 proposed the binding change mechanism for ATP synthesis. His proposal was strongly supported by the X-ray structure of bovine heart F1 resolved in 1994 by the group of Sir John Walker (2), and by visualization of the ATP-driven rotation of bacterial F1 immobilized onto a glass surface (6, 434) made possible through a fluorescent actin filament or gold beads attached to the γ subunit (202). Rotary subunit movements within the whole bacterial H+-ATP synthase were also monitored in real time at subnanometer resolution through single-molecule fluorescence resonance energy transfer, which uses a double-labeled enzyme incorporated in liposomes driving either ATP hydrolysis or ATP synthesis (162, 177). It is now widely acknowledged that during ATP synthesis, the c-ring rotates clockwise (viewed from the matrix side of the membrane) powered by H+ translocation to the mitochondrial matrix. H+ translocation takes place through half channels located at the interface of the a and c subunits, and drives rotation of the γ subunit within the α3β3 F1 subcomplex at a rate of ∼100 revolutions/s. This rotation takes each of the three catalytic sites through at least three major functional states denoted as βE, βDP, and βTP, thereby synthesizing 3 ATP molecules from ADP and Pi per each 360° rotation. When the enzyme works in the direction of ATP hydrolysis, the transition between βE, βTP, and βDP functional states drives the counterclockwise rotation of the γ subunit and of the c-ring, thereby causing H+ translocation to the intermembrane space. Based on single-molecule measurements of bacterial F1, it has been established that rotation of the γ subunit is not continuous, but rather proceeds in 120° steps, each step being driven by hydrolysis of one ATP molecule. The 120° steps can be resolved into substeps of 80° and 40°, which are driven by ATP binding and by release of ADP or Pi, respectively (6). However, recent rotational studies show that in mammals the catalytic cycle can differ significantly from the 80° to 40° substeps seen in bacteria, in that with human F1 the substeps are rather ∼65°, 25°, and 30°, corresponding to ATP binding, Pi release, and catalytic dwell, respectively (566). The remaining challenge is now to understand how the chemical energy of ATP is converted into mechanical work, i.e., the cooperativity of the three β subunits in driving the rotation of γ (388, 428).
1. Me2+, Pi, and adenine nucleotides
Mg2+ is essential for catalysis, which requires the binding of the nucleotide as a complex with metal (195). Together with the γ subunit, Mg2+ contributes to determine the asymmetry of the catalytic sites necessary for the binding-change mechanism (69). X-ray crystallography clearly established that in the absence of Mg2+ and nucleotide, the α,β complex of thermophilic Bacillus PS3 shows a threefold symmetry (532) and that in yeast F1 the adenine-binding pocket of βTP is disrupted (300). Moreover, kinetic studies with Escherichia coli F1 mutants containing Trp replacements at the catalytic sites showed that in the absence of Mg2+, ATP binds the three catalytic sites with the same low affinity (627), highlighting the key role of Mg2+ in shaping the high-affinity catalytic sites of the enzyme. X-ray crystallography of bovine heart F1 loaded with Mg2+ and nucleotides demonstrated the presence of six metal binding sites, each coordinated to the bound nucleotides in the three α and β subunits (2). In the βTP precatalytic state and βDP catalytic state of ATP hydrolysis Mg2+ is hexa-coordinated by βThr163 (bovine numbering), by the COOH-terminal residue of the P-loop, by the oxygen atoms βO2 and γO2 of γ-imino ATP, and by three ordered water molecules (hydrogen-bonded to βArg189, βGlu192, and βAsp256) (496).
Uncoupled ATP hydrolysis can be induced by membrane de-energization (185, 190), by incubation with the already mentioned modifiers of SH groups located in the FO sector (645), by F-ATP synthase inhibitors (377, 421), by Ca2+-ATP (429, 452), by Mg2+-nucleotide complexes different from ATP (260), or when F1 contains two empty catalytic sites (139). Interestingly, uncoupled hydrolysis caused by Mg2+-nucleotide complexes different from ATP is inhibited much more by Mg2+-ADP than coupled hydrolysis (260). Indeed, mitochondrial matrix proteins able to revert ATP synthase uncoupling have been identified, such as the already-mentioned factor B (294, 352). Mutagenesis studies suggest that coupling between catalytic activity and H+ translocation (419) is due to the interaction between the DELSEED loop of β in a closed conformation with γ subunit at the bottom of the α,β cavity. In particular, the rigidity of the DELSEED loop, which is in an “up” or “down” position in β closed and β empty, respectively, seems important to transfer the torque from the nucleotide binding domain to the γ subunit (581). Nevertheless, how the above-mentioned proteins may restore enzyme coupling remains to be defined.
Mg2+ can be replaced by other divalent cations, including Ca2+, and the ionic radius is the chief determinant of the ability to activate F-ATP synthase (531). Intriguingly, Ca2+ ions, at variance from other divalent cations with similar ionic radii, support ATP hydrolysis but not H+ translocation both in bacteria (429) and in mammals (452). However, Ca2+-ATP was as effective as Mg2+-ATP at powering rotation of the γ subunit attached to an actin filament, as demonstrated in a highly active hybrid F1 consisting of α,β subunits from Rhodospirillum rubrum and γ subunit from spinach chloroplasts (592). Replacement of the equivalent βThr163 (bovine numbering) with Ser in Rhodospirillum rubrum F-ATP synthase produced a mutant unable to support the proton-decoupled, Ca2+-dependent ATP hydrolysis while maintaining proton-coupled ATP synthesis as well as Mg2+- and Mn2+-dependent ATP hydrolysis (429). Interestingly, this Thr residue is also involved in release of the inhibitory Mg2+-ADP (288), which remains entrapped during uncoupled ATP hydrolysis (659). Conversely, substitution of the conserved βPhe174 for a Ser in an Escherichia coli mutant caused 90% loss of Mg2+-dependent ATPase activity, while maintaining the Ca2+-dependent ATPase activity (303, 437). These results strongly suggest that the catalytic site has a different conformation state when it is occupied by Ca2+ and that this conformational state is unable to couple the chemical catalysis to H+ translocation. Our working hypothesis is that this specific coordination chemistry of Ca2+ ions in the catalytic site is able to induce PTP formation from F-ATP synthase dimers by long-range conformational changes in the FO sector that remain to be defined (218). Equivalent mutants in mammals will represent a unique test of our hypothesis, which could be (dis)proved by their propensity to form a PTP. It is interesting that Sr2+, which cannot substitute for Ca2+ as a trigger for PTP opening (49, 569), is unable to support ATP hydrolysis by soluble F1 (531), suggesting an explanation for the quite distinct effects of these two cations on the PTP.
Consistent with the involvement of F-ATP synthase in PTP formation, the latter is inhibited by Mg2+-ADP, which is also a strong inhibitor especially of uncoupled ATP hydrolysis (260). Because the inhibitory Mg2+-ADP is promptly expelled at the onset of ATP synthesis, but not hydrolysis, its binding to a catalytic site might be responsible for the higher sensitivity of PTP to Ca2+ during ATP synthesis compared with ATP hydrolysis that has been observed in intact mitochondria (218). In mammalian mitochondria PTP opening is favored by Pi, as well as by arsenate and vanadate, which revert the Mg2+-ADP inhibited form of F-ATP synthase (54, 85, 425, 659). In addition to this effect, Pi increases the binding of CyPD to the OSCP subunit (216). A “coupling” effect of F-ATP synthase activity by Pi at fairly low concentrations (in presence of very low concentration of ADP) has been described in membrane fragments obtained from Rhodobacter capsulatus and Escherichia coli by evaluating the efficiency of H+ transport coupled to ATP hydrolysis. A model has been proposed where this high-affinity site could coincide with a transition state of the hydrolysis/synthesis reaction (139). It is tempting to speculate that if such a mechanism was present in all F-ATP synthases, it could be responsible for PTP inhibition by low Pi, which has been selectively observed in the absence of CyPD (35). Interestingly, based on the atomic structure of bovine F1 inhibited by the Pi analog thiophosphate (32) and by use of molecular dynamics simulations, it has been proposed that during the 360° rotation cycle, Pi remains bound to βE after ADP release, where it blocks γ rotation (428). Finally, it should be recalled that the inducing effect of high concentrations of Pi is not seen in yeast and Drosophila melanogaster mitochondria, which rather inhibit the PTP. In summary, at present, it is not easy to sort the effects of Pi on the F-ATP synthase from those that may depend on the Pi-dependent decrease of the Ca2+ and Mg2+ concentrations.
In analogy with its binding partner CyPD, OSCP strongly influences the threshold Ca2+ required for PTP opening, which decreases in mitochondria with decreased OSCP levels (218). The role of OSCP in enzyme catalysis has been established by numerous studies (155) and matches its potential role in modulating accessibility to the catalytic sites of Ca2+ ions necessary for PT induction. Fluorescence resonance energy transfer measurements have indeed documented that stress develops between F1 and OSCP during ATP hydrolysis (210). Moreover, yeast OSCP mutants Gly166Asn (a highly conserved residue) showed partially uncoupled F-ATP synthase complexes that were more susceptible to dissociation than complexes containing native OSCP (71). In keeping with its regulatory role, OSCP has recently been recognized as the binding target of a variety of compounds and proteins. These include 17β-estradiol, whose binding promotes an intrinsically uncoupled state of F-ATP synthase (421); Bz-423, an apoptosis-inducing agent which inhibits both synthesis and hydrolysis of ATP (290, 551); sirtuin3, which mediates deacetylation of α and OSCP subunits in a nutrient-sensitive manner (603, 640); and the transcription factor p53, which has been proposed to take part in the assembly or stabilization of the mature FOF1 complexes (39). We found that CyPD affects both the synthetic and hydrolytic activity of F-ATP synthase (216) and possibly shares a common binding site with Bz-423 on OSCP (218) (Figure 2). In a striking analogy, both Bz-423 and CyPD decreased the threshold matrix Ca2+ required for PTP opening (218).
Although PTP formation is influenced by modulators of F-ATP synthase catalysis, in electrophysiology experiments we observed PTP opening using preparations of bovine (218), yeast (96), and Drosophila (611) F-ATP synthase dimers that presumably contained very low levels of endogenous nucleotides. Addition of Ca2+ was essential for PTP opening, which was also favored by the dithiol reagent phenylarsine oxide, while it was inhibited by Mg2+ and ADP (96, 218). ATP and ADP are equally effective at inhibiting the PTP in intact mitochondria, and phenylarsine oxide does not inhibit ATP hydrolysis catalyzed by F-ATP synthase (645). These findings suggest that Ca2+-ATP hydrolysis is not necessary to induce PTP opening. Furthermore, the PTP is not inhibited by oligomycin, while Ca2+-ATP hydrolysis has been reported to be, at least partially, inhibited by oligomycin (452). The propensity of the PTP to open is affected by pH, with a maximum at pH 7.4, while opening is strongly inhibited as pH decreases to pH 6.4 through reversible protonation of still unidentified His residues, as well as when pH increases to pH 8.0 (432, 433). This is quite different from the pH profile of ATP hydrolysis (at least in the presence of Mg2+), which has a maximum at about pH 8.0 (260). Such discrepancy suggests that the two events, ATP hydrolysis cycle and PTP opening, are not strictly related although, to the best of our knowledge, the pH profile of Ca2+-ATP hydrolysis has not been defined yet. As already mentioned, pore inhibition by low pH has been initially ascribed to unbinding of CyPD from the PTP (432), but the observation that in mitochondria devoid of CyPD the PTP response to pH was maintained suggests that the PTP-modulating His residues (which still remain to be identified) are located within F-ATP synthase (34).
2. Inhibitory factor 1
In principle, the low open probability of PTP at low pH might be due to the binding of the inhibitor protein IF1 to F1 (94, 227, 259), which is strongly favored by low pH when the inhibitory dimeric IF1 is formed (89, 90). This endogenous inhibitor could be responsible for the beneficial effects of F-ATP synthase inhibition during ischemia both in in vitro experimental models as well as in vivo (159, 192). Unexpectedly, mice with genetic ablation of IF1 have no overt phenotype (427). The mechanism of inhibition by IF1 is quite complex, and fundamental insights have been recently obtained by point mutations and X-ray crystallography of the bovine (31, 33) and yeast species (503). Considering that in mammalian mitochondria Mg2+-ADP is a PTP inhibitor, it might be hypothesized that the final inhibited IF1-F1 complex, which contains two Mg2+-ADP in the catalytic sites of βDP and βTP (31), is unable to properly bind Ca2+, necessary for its switch to the PTP. Intriguingly, the His reagent diethylpyrocarbonate, whose addition to mammalian mitochondria restores the ability to induce PTP opening at low pH (432, 433), also prevents IF1 binding to the inner membrane at acidic pH when preincubated with the isolated bovine heart IF1 (608). This result suggests that the His residues of IF1 involved in the pH modulation of mammalian IF1 activity (89) might be involved in PTP inhibition at low pH as well. On the other hand, the inhibited IF1-F1 complex does not contain Pi (31, 503), which is a PTP inhibitor at low concentrations, suggesting that IF1 binding might mask an inhibitory site favoring, rather than preventing, PTP opening. Modification of isolated β subunit from Rhodospirillum rubrum with diethylpyrocarbonate fully blocked Pi binding (310), suggesting that IF1 and diethylpyrocarbonate might act in a similar way.
IF1 has been also described to favor formation of F-ATP synthase dimers, although IF1 is not essential for dimerization either in yeast (161) or mammals (588). However, how IF1 binding improves the stability of the dimer structure remains to be clarified. In fact, overexpression of IF1 in HeLa cells promoted dimer/oligomer stabilization and increased ATP synthesis, but also inhibited ATP hydrolysis at low pH and was protective against ischemic injury (93). Conversely, in cardiomyoblasts undergoing differentiation, IF1 binding promoted dimer/oligomer stabilization and increased both ATP synthesis and ATP hydrolysis (56). These apparently conflicting data suggest that IF1 can associate with F-ATP synthase dimers in different conformations. This hypothesis would be consistent with the intrinsically disordered structure of IF1, which makes it potentially able to recognize, and interact with, more than one partner (31). Intriguingly, a pH-independent interaction of IF1 with OSCP has been described (655). Such additional interactions might favor PTP formation, but the recently reported protecting effect of IF1 overexpression against apoptotic cell death, which appeared related to F-ATP synthase dimerization, may rather suggest that IF1-mediated dimer stabilization counteracts PTP opening (180). We note, however, that in this study the amount of IF1 bound to F-ATP synthase dimers was not quantified, and that the pH profile for PTP opening was not addressed. In conclusion, PTP modulation by IF1 remains an intriguing possibility that should be further explored.
3. Ca2+ in regulation of F-ATP synthase
It has long been known that F-ATP synthase is activated by Ca2+ in normal heart and skeletal muscle (259), in parallel with the Ca2+-sensitive dehydrogenases of the Krebs cycle and pyruvate dehydrogenase (154, 248, 256, 397). Moreover, the response to Ca2+ is rapid enough to support steep changes in myocardial work load (585). Although an allosteric mechanisms for activation has been postulated already in the 1990s (259), how Ca2+ affects the F-ATP synthase remains unclear, yet these putative regulatory interactions are of obvious interest to the mechanism of PTP control.
Several mechanisms have been proposed to explain F-ATP synthase activation by Ca2+ (Figure 2). In cardiomyocytes a Ca2+-dependent interaction of the F1 subcomplex with the S100A1 protein, which is expressed predominantly in cardiac muscle, reportedly leads to increased ATP production (62). In rat liver and enamel cells, the β (but not the α) subunit displayed low-affinity binding of up to 7–18 mol Ca2+/mol in sites different from the nucleotide-binding sites, including the acidic sequence DELSEED (275), which is not present in the α subunit. Intriguingly, a Bacillus PS3 mutant where all the negative charges of the DELSEED sequence had been removed showed significantly higher ATP synthesis activity (419). However, no measurements have been performed in rat liver and enamel cells to confirm that Ca2+ binding induces enzyme activation.
FO subunit e has also been hypothesized to be a Ca2+-dependent activating region of F-ATP synthase, based on the homology of its residues 34–56 to the Ca2+-dependent tropomyosin-binding region for troponin T, and on the ability to mediate an increase of F-ATP synthase activity upon interaction with a specific antibody (17). Interestingly, these residues are exposed at the C-site of FO, potentially ensuring the rapid decoding of changes of cytosolic Ca2+. Evidence that this Ca2+-dependent regulatory region of subunit e is involved in Ca2+-dependent F-ATP synthase regulation, and that tropomyosin-like molecule(s) exist in the mitochondrial intermembrane space, is still lacking. Moreover, as for subunit β, subunit e seems to be a common rather than an activation mechanism preferentially occurring in heart and skeletal muscle, although its differently regulated stoichiometry among species and tissues (17, 58) might suggest different responses to Ca2+ ions. Due to exposure of the Ca2+-binding site to the mitochondrial intermembrane space, subunit e may be involved in Ca2+-dependent inhibition of PTP opening (49). Subunit e is important for F-ATP synthase dimer formation both in mammals and in yeast (58, 246, 461), suggesting that Ca2+ regulation could target the dimer structure/function, which is essential for PTP formation (218) as recently confirmed in yeast (96).
The NH2 terminus of FO subunit c, which is also exposed to the intermembrane space (568, 626), also contains a conserved Ca2+ binding site formed by two Asp residues, which could represent another candidate for PTP inhibition by Ca2+ in the intermembrane space. Purified subunit c from neuronal plasma membrane is able to form a voltage-sensitive pore that carries monovalent cation currents regulated by Ca2+ and cGMP (402), suggesting that Ca2+ could alter the c ring conformation (402). In bacteria and chloroplasts, Ca2+ binding to subunit c at the periplasmic/luminal site has been reported to block H+ translocation in both directions (599). However, while the Ca2+-binding capacity of subunit c from bacteria and chloroplasts is well established, that of the mammalian subunit c is debated (23, 599).
III. F-ATP SYNTHASE AND THE PERMEABILITY TRANSITION
A. Genetic Manipulation of F-ATP Synthase Subunits
In mammalian cells, genetic manipulations addressing the PTP-forming features of F-ATP synthase have been performed with siRNAs for OSCP (218) and c subunits (9, 64), and in ρ0 cells lacking subunits 6 and A6L (389). In Saccharomyces cerevisiae, the role of genetic inactivation of the “dimerization” subunits e (TIM11) and g (ATP20) on PTP formation has also been assessed (19).
Decreased expression of OSCP after treatment with specific siRNAs in human cells led to a parallel decrease of CyPD binding to F-ATP synthase, and to a matching decrease of the threshold matrix Ca2+ required for PTP opening in the face of a conserved catalytic activity and of a normal buildup of the proton gradient (218, 290). Accessibility of the metal binding sites of the catalytic F1 sector could be influenced by OSCP, which would then affect the probability of replacing Mg2+ with Ca2+ and thus to cause PTP opening (218). According to this hypothesis, OSCP would be a “negative” modulator whose effects can be counteracted by binding of the “positive” effector CyPD, which does increase the apparent Ca2+ affinity of the PTP. Under this scenario, CyPD binding to OSCP, or ablation of the latter, would induce equivalent effects causing increased probability of PTP opening (218).
Treatment with siRNA against the c subunit decreased PTP opening in response to various death stimuli (9, 64), while overexpression of Myc-tagged c subunit enhanced PTP opening (64). As also noted by Halestrap (254), while consistent with a role of the F-ATP synthase in PTP regulation, these experiments cannot determine whether the effects are linked to a direct involvement of the c subunit, are an indirect consequence of decreased ATP production by the knockdown (9, 64) or by a misfolded protein response by overexpression (64). Also, it is not clear whether and to what extent biogenesis of the F-ATP synthase was actually suppressed in this study, which only relied on an antibody against the uncleaved immature form of the c subunit, and where expression of the F1 subunits was not investigated (64). The proposal by Jonas and co-workers that the PTP channel may form within the c-ring (9) will be discussed in section IIIB.
ρ0 cells lack mtDNA (315) and therefore assemble an F-ATP synthase that lacks mitochondrially encoded subunits a and A6L (632). The F-ATP synthase of ρ0 cells does not pump protons but is catalytically active as an ATP hydrolase. This activity is essential to maintain a gradient of ATP (almost entirely produced by glycolysis) between the cytosol and the matrix, allowing the electrogenic exchange of extramitochondrial ATP for matrix ADP which generates a Δψm (84) of about −67 mV (16). Wittig et al. (632) have shown that dimers of F-ATP synthase do form in ρ0 cells. In these studies, the F-ATP synthase is almost fully assembled in spite of the absence of subunits a and A6L but results in the apparent lower stability of the complexes after digitonin treatment. While the levels of F-ATP synthase complexes were smaller, dimeric and even tetrameric forms of the large assembly intermediate were preserved, suggesting that the F-ATP synthase associated further into higher order structures in the mitochondrial membrane (632). ρ0 cells almost entirely depend on glycolysis for ATP production and upregulate HK II, which is largely bound to the OMM (389). Occurrence of the PT has been demonstrated in intact ρ0 cells based on mitochondrial depolarization following displacement of mitochondrial HK II (389) with clotrimazole or with an HK II-HIV-1 TAT fusion peptide (109). Since the assay for PTP detection was based on in situ depolarization (389), it is not known whether the properties of the PTP formed in the absence of the a and A6L subunits (in particular its conductance) are the same as those of wild-type F-ATP synthase.
Given that dimeric, but not monomeric, F-ATP synthase forms channels in mammals (218), yeast (96), and Drosophila (611), we have explored the role of the yeast dimerization subunits e and g in PTP formation by their genetic inactivation. Strains lacking subunits e (220) or g (146) display abnormal morphology, with balloon-shaped cristae and F-ATP synthase monomers distributed randomly in the membrane, but develop a normal membrane potential (96). In turn, this allowed measurements of their propensity to open the PTP following Ca2+ uptake in the presence of the ionophore ETH129, which catalyzes electrophoretic Ca2+ uptake in energized yeast mitochondria (297) when the concentration of Pi is optimized (647). Yeast strains lacking the e and/or g subunit showed a remarkable resistance to PTP opening, which suggests that dimer formation is important for pore formation in situ (96). In these experiments, onset of PTP opening is measured as the spontaneous Ca2+ release following Ca2+ uptake, which was observed also in the e and/or g subunit-null mitochondria at higher Ca2+ loads (96). One possible explanation is that dimers can transiently form even in the absence of e and g subunits, as shown in intact cells (211) and after treatment of mitochondria with copper, which promotes dimerization through formation of disulfide bridges between adjacent cysteine residues of the monomers (199, 246, 604). A second explanation should also be kept in mind, however. Yeast mitochondria lack the MCU (95), and therefore, the ionophore ETH129 must be added to allow Ca2+ uptake and determination of the Ca2+ sensitivity of the PTP (96, 297, 647). Onset of Ca2+ release can be due to PTP opening but also to increased H+ permeability, which would be followed by Ca2+ release via ETH129 itself. In other words, it cannot be excluded that the Ca2+ release occurring at higher Ca2+ loads in the yeast e and g null mutants (96) may not be due to the PTP, and thus that pore formation may strictly require the presence of the e and/or g subunits.
B. Mechanism of Channel Formation
The mechanism for channel formation within the F-ATP synthase is an open question. One recent proposal is that the actual channel forms within FO (9), while we have suggested that the pore forms in F-ATP synthase dimers (or higher order structures) at the interface between monomers (96, 218, 611).
1. FO and the c-ring
The first evidence that the FO sector can form a high-conductance voltage-dependent channel has been obtained in 1989 (541), but its functional role remained elusive. Subsequent patch-clamp studies of highly purified c subunit, which self-assembles into annular structures (18, 404), demonstrated the presence of channels inhibited by Ca2+ and activated by cGMP (400–402). Attention to subunit c as a potential regulator of the PTP was drawn by the observations that the PT could be induced by a phosphorylated peptide derived from the c subunit and by subunit c itself (22, 23).
Recently, Jonas and co-workers reconstituted the c subunit or the purified F-ATP synthase in liposomes, and measured Ca2+-activated channels (9) with properties similar to those described for purified dimers (218). They suggested that the channel of the PTP forms within the c-ring itself after Ca2+-dependent extrusion of F1, i.e., of the γ/δ/ε subunits (9). The “FO channel” could not be closed by subunits γ, δ, or ε, while it was blocked by subunit β, suggesting that this is the mechanism through which pore closure occurs in situ (9).
Displacement of F1 from FO requires very drastic conditions, such as treatment with 2 M urea (119, 302), yet a functional FOF1 complex can be easily reconstituted (178, 370), indicating that the γ/δ/ε subunits do reinsert into FO. It is hard to envision a plausible mechanism through which matrix Ca2+ would cause release of F1, and then create a channel within FO that cannot be closed by subunits γ/δ/ε (9). The reported closure of the “FO channel” by the β subunit is of some concern, because subunit β does not interact with the c-ring (Figure 1) (2) and the source of free β subunit is not obvious given that the F1 subcomplex is extremely resistant to denaturation. Furthermore, if PTP opening depends on expulsion of F1, yet F1 itself cannot reintegrate within FO through subunits γ/δ/ε, each event of pore opening would denature the corresponding F-ATP synthase, while the PTP flickers between open and closed states (280, 317, 475, 575) and is readily and fully reversible in mitoplasts (569), intact mitochondria (472), as well as in reconstituted dimers of F-ATP synthase (218). Of note, after PTP-dependent swelling, pore closure is followed by shrinkage and full recovery of mitochondrial structure and function, provided that the experiment is performed in K+-based media to allow K+ extrusion by the K+/H+ exchanger (41) and that cytochrome c, which is lost during swelling, is added back (472).
In the protocols of Alavian et al. (9), channel openings strictly required CyPD, were inhibited by CsA after the addition of Ca2+, and could also be detected with F-ATP synthase preparations. Like the MMC in the native IMM (149), the mammalian F-ATP synthase does not require CyPD for channel formation (218). Furthermore, neither the yeast nor the Drosophila PTPs are regulated by CyPs, yet their F-ATP synthases do form Ca2+ channels (96, 611). Thus the structural bases for the CyPD requirement and CsA sensitivity of “FO channels” remain obscure.
Mammals possess three isoforms (P1, P2, and P3) of subunit c of F-ATP synthase, which differ only in their mitochondrial targeting peptides, so that the mature c subunits are identical (170, 648). The targeting sequences are not redundant, however, and may have specific functions in maintenance of the respiratory chain (609). During F-ATP synthase biogenesis, the membrane-embedded c-ring attaches to F1 at an early stage (267), i.e., before the subunits of the peripheral stalk (b, d, F6, and OSCP), the membranous subcomplex (e, f, g, and other species-specific subunits), and the mtDNA-encoded subunits a and A6L are incorporated (632). The level of this complex (P1 isoform) appears crucial for defining the final content of F-ATP synthase, at least in mammals (273, 331). When present in “excess,” subunit c forms hydrophobic aggregates in the IMM as well as in lysosome-derived organelles that can be observed in Batten disease (401, 446). Importantly, these hydrophobic aggregates in the IMM do not increase membrane permeability, as recently observed after silencing of the ε subunit in HEK293 cells, which blocked the biogenesis of F-ATP synthase with accumulation of subunit c (262). Mitochondria in these cells were more coupled (262), which is unexpected if the c-ring can form high-conductance membrane channels.
A final point is our recent finding that the Drosophila melanogaster F-ATP synthase forms channels with a unit conductance of 53 pS (611). As already mentioned, vertebrates and most invertebrates have 8 c subunits per F-ATP synthase monomer, while the number of c subunits varies between 10 and 15 in prokaryotes, chloroplasts, yeasts, and fungi (626). In all c8-ring members, 1) Ala residues are conserved at positions 13, 19, and 23 (bovine numbering) of the NH2-terminal α-helix, which is essential to avoid clashes of the side chains (626), and 2) the COOH-terminal α-helix invariably has a Lys residue at position 43, which binds cardiolipin and allows tight packing of the c subunits that is essential for activity and stability (171, 222). These critical residues are conserved in Drosophila (626), which is also affected by loss of cardiolipin (4), indicating that F-ATP synthase of Drosophila melanogaster belongs to the c8-ring set. It is quite difficult to see how c-rings of the same size can form 500-pS channels in mammals and 53-pS channels in Drosophila. The lack of correlation between conductance of the channel and composition of the c-ring is also supported by measurements in yeast, where the c-ring has 10 subunits and the conductance is 300 pS (626). In summary, the ensemble of these findings is not consistent with the idea that the PTP channel forms within the c-ring, whose actual conductance and Ca2+ dependence remains controversial (9, 400–402).
2. F-ATP synthase dimers
We have observed channel formation after incorporation of gel-purified F-ATP synthase dimers of mitochondria from bovine heart, Saccharomyces cerevisiae, and Drosophila melanogaster in asolectin bilayers, while channel formation was not observed when monomers were used (96, 218, 611). Although dimers and monomers are functionally distinct in the native membrane (57), it is not known whether this difference is maintained after incorporation into an artificial membrane. Furthermore, it is not known whether dimers would maintain the dimeric structure, dissociate into monomers, or rather form higher-order structures like tetramers, a process that may also depend on the conditions used to obtain the F-ATP synthase preparations. Jonas and co-workers (9) have observed channel formation after incorporation of F-ATP synthase monomers into liposomes, but it is difficult to exclude that higher-order structures were formed, possibly because of the membrane curvature that may favor dimer and/or oligomer reassociation, which could be harder to obtain in planar bilayers. As already discussed, we think that the dimer/tetramer hypothesis is also strongly supported by the effects of genetic ablation of the e and/or g subunits on the yeast PTP (96) and by the maximal conductance of the reconstitued dimer, which corresponds to half the maximal conductance of the MMC in the native inner membrane (575).
We have proposed that the PTP channel forms at the interface of two monomers (associated into dimers) also because we were inspired by an atomic force microscopy study of native IMM (88). In this study, Buzhynskyy et al. (88) demonstrated the existence of two classes of dimers characterized by a stalk-to-stalk distance of 15 and 10 nm, respectively, with the latter presumably allowing binding of IF1. The authors concluded that the torque generated during ATP synthesis stabilizes the interfaces within each F-ATP synthase, within dimers and oligomers, and as a consequence stabilizes cristae morphology with a stalk-to-stalk distance of 15 nm. In contrast, ATP hydrolysis would destabilize dimers by pulling the stators apart, thus allowing the monomers to get closer to one another at a stalk-to-stalk distance of 10 nm, favoring IF1 binding (88). Since the sense of rotation of F-ATP synthase (synthesis vs. hydrolysis) strongly affects the threshold Ca2+ required for PTP opening in intact mitochondria (218, 611), the different conformation of dimers may affect the probability of replacing Mg2+ with Ca2+ at the F-ATP synthase catalytic site, triggering the conformational change that results in PTP opening (218). We would like to stress that the “dimer hypothesis” is only a working model that sets pore formation at a membrane-protein or protein-protein interface that does not directly involve the c-ring. This is consistent with the fact that the catalytic activity of F-ATP synthase can be inhibited (e.g., with oligomycin) without affecting PTP formation. Conversely, PTP formation can be inhibited (e.g., by CsA) without affecting, or even stimulating, activity of F-ATP synthase (216).
Structural work on the F-ATP synthase is fully consistent with the idea that substituting Ca2+ for Mg2+ at the catalytic site has a major conformational effect on the complex structure. We have already mentioned that, together with subunit γ, Mg2+ contributes to create the asymmetry of the catalytic sites that is essential for ATP synthesis (69, 195). In the absence of Mg2+ (and nucleotide), the α,β complex shows a threefold symmetry (532), while the adenine-binding pocket of βTP is disrupted (300). When Ca2+ replaces Mg2+, ATP hydrolysis takes place, but this is apparently not coupled to H+ translocation (429, 452), indicating that Ca2+ occupancy of the catalytic site causes a conformational change that decouples chemical catalysis from H+ translocation. If this conformational change is related to PTP opening, the lack of apparent H+ translocation with Ca2+-ATP hydrolysis may be rather due to backflow of H+ through the open pore. Our working model is that at the prevailing matrix Me2+ concentrations (millimolar Mg2+, micromolar Ca2+), the chances that Ca2+ replaces Mg2+ at the catalytic sites are minimal, and a PT does not occur. In mitochondria where the pore is modulated by CyPD, its binding to OSCP, which is favored by Pi (216), would cause a conformational change affecting accessibility of the Me2+ binding sites, but not necessarily causing the PT if matrix [Ca2+] remains low. Accessibility of the Me2+ binding site would be independently increased by thiol oxidation and counteracted by thiol reduction, making replacement of Mg2+ with Ca2+ more likely irrespective of whether the PTP is modulated by mitochondrial CyP. Once the PT occurs, ion/solute permeation would occur at the FO interfaces within dimers or higher order structures like tetramers. Note that this model predicts that the PT can occur at individual F-ATP synthase units, accounting for the fact that PTP-dependent depolarization can occur asynchronously in a mitochondrial population (281) (Figure 2).
Some recent PTP models have tried to incorporate the F-ATP synthase into the old two-membrane picture of the PTP, which requires an alteration of relative protein dimensions and of their topology (65, 479). We think that such cartoons do less to illuminate than serve as points of confusion as to which molecules play key structural roles, which are modulators and which have no function at all. This also applies to a recent review according to which the PTP would be related to the “ATP synthasome” (228). We note that the “ATP synthasome” (i.e., the physical association between F-ATP synthase, PiC, and ANT) has been seen after detergent extraction under conditions where only the monomeric form of F-ATP synthase is present (103). Moreover, recent studies have demonstrated that the ANT migrates as a monomer, and that higher molecular weight species are only observed under less favorable solubilization conditions, consistent with aggregation of the protein (129). Finally, these complexes are not apparent in cryo-electron tomography studies of mitochondria from various sources, where the density maps perfectly fit F-ATP synthase dimers (29, 146, 147, 509, 555).
IV. SIGNAL TRANSDUCTION TO THE PERMEABILITY TRANSITION PORE
Mitochondria dynamically integrate a variety of signals to adjust their metabolic activities to the changing cellular needs and conditions. Over the last few years, it has become clear that all complexes of oxidative phosphorylation (OXPHOS), including F-ATP synthase, undergo posttranslational modifications (PTMs) that may guarantee fine tuning of their activity, for instance, during cell differentiation or along the cell cycle (128, 283). Similarly, the PTP can be regulated by proteins that undergo complex PTMs by molecular cascades of signal transduction (493). Thus, for the first time, identification of F-ATP synthase as the key to PTP formation allows us to draw a tentative unifying picture connecting available data on PTMs of pore components and F-ATP synthase.
A. Posttranslational Modification of Pore Regulators
Given the lack of information on PTP structure, efforts had so far focused on PTP modulation by signals converging on pore regulators. This field of investigation is extremely complex for several reasons: crucial components of PTMs depend on the frequency, duration, and intensity of the signals, which in turn depend on the cell type and metabolic conditions. In addition, the subcellular localization of regulatory proteins and of multimeric enzyme complexes can further modulate PTMs of the PTP. Furthermore, most signal transducing enzymes (e.g., kinases and phosphatases) are located in multiple subcellular compartments, which demands a careful purification of mitochondria under conditions that preserve PTMs. Despite these difficulties, a complex regulatory network composed by phosphorylation, acetylation, and nitrosylation events that impact on PTP opening is emerging.
One key connection between the PTP and the metabolic status of the cell is provided by HK, the first enzyme of glucose metabolism. HK isoforms I and II have high affinity for glucose and possess hydrophobic NH2 termini that allow their docking to the OMM (394, 457, 502). In particular, HK II is the sole isoenzyme endowed with two catalytic domains, which maximize the rate of glucose metabolism to glucose-6-phosphate. This arrangement is strictly required in tissues such as brain and heart, insulin-sensitive muscle or fat depots, and in rapidly growing cancer cells (395). HK II expression is induced in most malignancies, where it crucially contributes to the Warburg effect, i.e., to the uncoupling between increased glycolysis and oxygen availability required for tumor cell survival during primary growth, when blood supply is scarce (274, 624). We have found that in different cell models with a high rate of glucose utilization, including neoplastic cells, cardiomyocytes, and cells lacking mtDNA, detachment of HK II from the mitochondrial external surface by the use of a cell-permeant, selective displacing peptide prompts PTP induction, which rapidly leads to cell demise (109, 389, 449).
HK II binding to mitochondria is controlled by kinase signaling. The Ser/Thr kinase AKT, a central transduction node that delivers anabolic and survival signals downstream to several growth factors, and whose dysregulation contributes to metabolic diseases and tumors (380), promotes HK II binding to mitochondria by phosphorylating HK II itself (418) or its upstream regulator GSK3 (394, 502). The exact mechanisms through which GSK3, a constitutively active Ser/Thr protein kinase that is inhibited by all major anabolic signaling cascades (292), controls the mitochondrial localization of HK II remain unclear (discussed in Ref. 493) as it remains to be established how HK II, which is externally associated to mitochondria, might regulate the PTP on the IMM. As a first step in elucidating this point, we have recently reported that HK II forms a multimeric complex with the myotonic dystrophy protein kinase and the active form of the Tyr kinase Src on the OMM (449). In these experiments, mitochondrial binding of HK II protects the PTP from opening independently of its enzymatic activity, and its detachment amplifies the oxidative damage caused by placing cells in conditions of serum and glucose depletion (449). Therefore, mitochondrial HK II could play an important antioxidant function (see also Ref. 564) possibly following interaction with another glycolytic enzyme, the fructose-2,6-bisphosphatase TIGAR (108), thus protecting cells from ROS-induced PTP opening. Taken together, these observations indicate that mitochondrial HK II might be an important “porekeeper,” which would act as a key survival device in conditions of adequate nutrition controlled by growth factor stimulation, or when kinase pathways are aberrantly induced by neoplastic transformation. Independently of the mode of PTP regulation by HK II, these data directly link the pore to crucial signal transduction pathways, providing an important example of how information on the status of the cell can be conveyed to the PTP.
Furthermore, PTMs serve as PTP regulators through mitochondria-restricted kinases, among which GSK3 plays a central role (111). Indeed, mitochondrial GSK3 (mGSK3) is inhibited by the survival kinases AKT and ERK1/2, PKCε, PKG, and p70s6K (261) and acts as the final effector of these transduction pathways in the regulation of PTP opening (295). A large body of information highlights the importance of PTP regulation by mGSK3 in pathological conditions, such as ischemia-reperfusion of the heart (see below). Notably, maintenance of cardiomyocyte viability during ischemic pre- and postconditioning requires phosphorylation-dependent inhibition of mGSK3, which in turn inhibits the PTP in response to ROS or Ca2+ overload (296, 415, 417). mGSK3 activation is dramatically increased during heart ischemia (416). Accordingly, administration of a GSK3 inhibitor together with CsA reduces infarct size in mice (99), and expression of a constitutively active GSK3 could not rescue myocytes from oxidative insults, whereas transfection with an inactive enzyme was protective (296, 440). Moreover, ROS-dependent activation of mGSK3 enhances cell death in neurons, probably following PTP induction (316, 467). Taken together, these observations suggest that mGSK3 coordinates diverse signaling pathways to connect PTP opening with stress and survival signals by changing the ROS and/or Ca2+ threshold of PTP opening. At present, it is unknown whether the pool of mGSK3 can directly phosphorylate any F-ATP synthase components, but we and others have reported that CyPD is a target of GSK3 phosphorylation in tumor cells (492, 589) and in cells lacking mtDNA (389).
By in silico analysis, we identified Ser/Thr residues on CyPD that are possible GSK targets, and we showed that recombinant GSK3 can phosphorylate CyPD in vitro (492). When moving to in-cell experiments, we found that mGSK3 could be inhibited by a mitochondrial-restricted fraction of the Ser/Thr kinase ERK (492). ERK activation by the protooncogene Ras delivers crucial signals that govern cell growth, proliferation, motility, and survival (398); Ras dysregulation is mandatory in a number of diseases, notably in the process of neoplastic transformation (156, 520). Our data demonstrate that in several cancer cell models mitochondrial ERK is constitutively active, and it confers resistance to PTP inducers through GSK3 inhibition, which in turn leads to the negative regulation of CyPD phosphorylation. Accordingly, PTP inhibition could be ablated by inducing GSK3-dependent phosphorylation of CyPD through treatment of cells with ERK inhibitors. This enhancement of PTP opening was increased after CyPD overexpression and absent in CyPD knockout cells; conversely, pharmacological inhibition of GSK3 protected cells from PTP opening (492). Therefore, inhibition of mGSK3 by active mitochondrial ERK prevents CyPD phosphorylation and desensitizes PTP opening. These data provide a mechanistic link between the ability to escape death, a hallmark of cancer cells (255), and PTP regulation by kinase signaling (discussed in Refs. 493, 494). Further evidence supports the importance of pore regulation by PTMs acting on CyPD: 1) the signal transducer and activator of transcription 3 (STAT3), which is regulated by several kinase pathways, interacts with CyPD and inhibits PTP opening (61); 2) PTP sensitization by acetylation is prevented by AMPK activation of the sirtuin-3 deacetylase, which targets CyPD (533); and 3) nitrosylation targeting a specific Cys residue of CyPD inhibits PTP opening (324, 431) (Figure 3).
Figure 3.
Regulation of PTP opening by posttranslational modifications of CyPD. CyPD is a molecular terminal of many signaling axes that regulate the PTP (inducers are indicated in orange, inhibitors in blue): 1) CyPD phosphorylation by GSK3β facilitates PTP opening, and GSK3β activity is abrogated by induction of the Ser/Thr kinase ERK; 2) phosphorylated STAT3 binds to CyPD and inhibits PTP opening; and 3) CyPD acetylation sensitizes the PTP to opening and is prevented by AMPK activation of the SIRT3 deacetylase. In addition, the Ser/Thr kinase PINK1 inhibits PTP opening through poorly defined mechanisms. [Modified from Rasola and Bernardi (490) with permission.]
CyPD is not the only molecular chaperone involved in PTP regulation. In the last few years, the mitochondrial pools of Hsp90 and Hsp60 and the mitochondria-restricted TRAP1 chaperone were reported to prevent PTP opening in neoplastic cells following interaction with CyPD (213, 305). As the activity of all these molecules can be modulated by PTMs (491, 504), these observations raise the intriguing possibility that multichaperone platforms might maintain the appropriate folding and activity of pore components and regulators, dynamically adjusting PTP modulation to the requirements of the cell by conveying signaling through phosphorylation changes.
It is also possible that other kinase pathways can modulate the PTP through GSK3-independent regulation, even if mechanistic details are still missing. One example is provided by the Ser/Thr kinase PINK1. PINK1 can inhibit PTP-dependent cell death (617), whereas its ablation lowers the threshold for PTP opening (205). Importantly, kinase activity is always balanced by phosphatases, but information on mitochondrial phosphatases and on their role as PTP regulators is scarce. It was reported that a mitochondrial matrix-targeted protein phosphatase 2C family member, PP2Cm, blocks PTP opening and acts as a survival element (375), whereas PTP-mediated cell death is inducible by the protein phosphatases 2B (649) and SH2-containing tyrosine phosphatase 1 (286).
B. F-ATP Synthase as a Signaling Target
Our understanding of how F-ATP synthase enzymatic activity is finely tuned is still incomplete. The enzyme is regulated by CyPD, IF1, and a variety of regulatory peptides (62, 216, 294, 352), but nothing is known about PTMs that might regulate its transition and function as the PTP. Importantly, no tissue-specific isoforms except for the heart and liver type of γ subunit (396) have been reported for F-ATP synthase, but some evidence suggests that the enzyme cannot be regulated only by substrate availability. For instance, in the heart, ATP levels do not dramatically change with activity (mainly due to buffering by creatinine kinase), yet the F-ATP synthase fully responds to increased ATP expenditure, indicating that it can be regulated by biochemical events, such as PTMs, in vivo. Accordingly, a surprisingly high number of modifications have been identified in the various subunits of F-ATP synthase, including phosphorylation, acetylation, trimethylation, nitration, S-nitrosylation, and tryptophan oxidation. In the last few years, the utilization of powerful technical approaches, such as the combination of cell incubation with [32P]γ-ATP, followed by two-dimensional gel analysis, detection of phosphorylated residues with anti-phospho antibodies, and MALDI-MS/MS mapping of specific phosphorylation sites is further increasing the identification of PTMs (282). It was recently estimated that 67 different phosphorylation sites exist in 12 of the 16 subunits of mammalian F-ATP synthase (128). However, in most cases, it remains completely unknown what are the signaling pathways responsible for these PTMs, in which tissues or biological conditions these modifications occur, how they impact on the biochemical activity of the target protein and of the holoenzyme, and how much of the target protein can be modified (304). Indeed, most sites were detected by a targeted MS/MS approach, suggesting that the abundance of phosphopeptides was very low and raising doubts on their possible biological effects.
In spite of these limitations, some PTMs were reported to affect the F-ATP synthase enzymatic activity, while others might regulate its ability to form dimers and/or multimers, both changes that could impact on PTP modulation. Among PTMs that regulate F-ATP synthase activity, it was reported that 1) phosphorylation of the d subunit of the lateral stalk by PKCδ inhibits F-ATP synthase activity in neonatal cardiac myocytes (430); 2) cell stimulation with platelet-derived growth factor induced phosphorylation of the δ subunit (323); and 3) phosphorylation of Tyr213 in the β subunit might play a role in modulation of the F-ATP synthase, and it is deregulated in type 2 diabetes (272). Remarkably, most F-ATP synthase phosphorylation sites are found in the F1 sector, mainly in the hexamer formed by the α and β subunits as well as in the lateral stalk of the enzyme (OSCP, b, d, and F6 subunits) (128). However, little conformational changes during the catalytic cycle of the enzyme are observed in the upper region of the F1 hexamer. Yet, most phosphorylated residues were found in this region, with the exception of four phosphorylation residues located near the substrate binding pocket of subunit β (68), and four phosphorylation sites in subunit γ (128, 158). These PTMs could affect its rotation relative to the α-β hexamer (613).
The functional role of PTMs other than phosphorylations has also been investigated. Several F-ATP synthase subunits, including α, β, γ, b, d, g, OSCP, 8, and F6, are acetylated, and these acetylation events dynamically follow fasting/feeding cycles in mice, suggesting a functional effect that remains poorly understood (304). However, S-glutathionylation and S-nitrosation (also known as S-nitrosylation) of subunit α decrease the ATP hydrolytic activity of the enzyme under oxidative stress (104, 207, 562), with possible implications during ischemic conditions when these PTMs might contribute to preserve intracellular ATP levels (563, 621).
Dimerization and oligomerization of F-ATP synthase, together with the formation of respiratory supercomplexes including other OXPHOS components, might affect not only formation of the PTP, but also substrate channeling, stabilization of OXPHOS components, sequestration of reactive intermediates, and IMM morphology, crucially contributing to formation of cristae (634). It is therefore conceivable that such delicate biochemical activities might be finely regulated by PTMs. As already mentioned transmembrane helices of subunit a, subunits of the stator stalk and accessory subunits (e, g, b, h, and i) are predicted to form the structural basis for dimerization by stabilizing the monomer-monomer interface (19, 21, 58, 545, 632, 633). Accordingly, phosphorylation of a Ser residue of subunit g has been proposed to regulate F-ATP synthase dimerization (497), yet the signaling pathways that control subunit g phosphorylation remain unknown. In addition, it was shown that phosphorylation of a Tyr residue in the γ subunit of F1 might take part in the regulation of dimer formation, since it was only observed in the monomeric form of the F-ATP synthase complex of bovine heart by the use of phosphotyrosine antibodies and MS/MS (158). A further candidate potentially involved in F-ATP synthase dimerization is IF1, where two phosphorylation sites that may serve regulatory functions have been detected (128). In particular, phosphorylation of Ser39 (658) might interfere with the binding to the β subunit, as it is found inside F1 in the dimeric form (33). Future studies are needed to understand whether PTMs of IF1 could impact on PTP activity through modulation of F-ATP synthase dimeric conformation.
C. Redox Signaling
ROS are potent PTP inducers through several mechanisms: oxidants increase intracellular Ca2+ levels by modulating the activity of IP3 and ryanodine receptors on the ER/SR, and also of SERCA pumps and Ca2+ channels on the plasma membrane (92). Above a certain threshold level, mitochondrial Ca2+ or membrane hyperpolarization might also produce ROS through inhibition of respiratory complexes or displacement of cytochrome c from cardiolipin in the IMM (444). These events would induce ROS generation by decreasing the rate of respiration and by PTP induction, which completely stops electron flow along the respiratory chain and causes release of mitochondrial GSH (240). Transient PTP inductions might prompt short bursts of ROS and propagating waves of short PTP openings in the surrounding mitochondria. These events would produce additional ROS in the so-called ROS-induced ROS release (667), with possible signaling functions through the modulation of chaperones, kinases, and gene expression (490). Above a certain degree of mitochondrial ROS rise, this can augment the Ca2+ surge, exhausting the high intramitochondrial redox buffering capacity and resulting in an unrestrained elevation of mitochondrial ROS that eventually prompts prolonged PTP opening and cell death.
Changes in respiratory activity are an important factor in causing alterations of the redox equilibrium of a cell also through modulation of ROS production (328). Dozens of PTMs have been reported in OXPHOS complexes (128), which could contribute in tuning ROS production and eventually PTP opening. For instance, Tyr phosphorylation of both cytochrome oxidase and cytochrome c inhibits respiration, shielding mitochondria from hyperpolarization and the subsequent increase in ROS levels (282). Cytochrome c can trigger PTP induction also following reduction by the mitochondrial pool of phosphorylated p66Shc, which prompts electron transfer directly from cytochrome c to oxygen to generate hydrogen peroxide, which may lead to pore opening (215). Induction of IF1 expression, which is observed in some pathological conditions such as neoplastic transformation, promotes an increase in mitochondrial membrane potential, thus contributing to trigger ROS generation (512). Recent observations highlight the importance of respiratory complex II, succinate dehydrogenase (SDH), in ROS generation. SDH subunit A, the catalytic flavoprotein of the holoenzyme (312), can be phosphorylated by the Src-family tyrosine kinase Fgr in response to ROS stimulation (5), whereas SDH activity can be further enhanced independently of ROS by deacetylation on several lysine residues mediated by sirtuin 3 (188). The following increase in SDH enzymatic activity leads to a secondary wave of ROS generation, with important implications in metabolic adaptations of cells to conditions in which the substrate shift from glucose to fatty acids requires a greater electron flux from FAD, such as during hypoxia/reoxygenation or starvation (5).
In keeping with the importance of SDH in maintaining the redox equilibrium of the cell, we have recently shown that SDH activity is crucial in inducing ROS-dependent PTP opening following nutrient depletion in some tumor cell models. These cells escape death elicited through PTP induction by the mitochondrial chaperone TRAP1, which inhibits SDH activity (243). TRAP1 is a member of the Hsp90 protein family that displays an antioxidant activity, which prevents PTP opening following ischemic damage (641, 642). This is also observed in several tumor types, where TRAP1 expression is generally increased and correlates with malignant progression (491). TRAP1 was reported to inhibit respiration following interactions with both SDH (523) and cytochrome oxidase (651). Thus it is conceivable that TRAP1 might display multiple chaperone functions that converge on blocking PTP induction, following its interaction with several OXPHOS components and with CyPD (see above). The interplay between SDH activity and ROS generation has recently been demonstrated to be crucial in ischemic tissues, where an excess of succinate is produced. Indeed, during ischemia SDH can act in reverse, generating succinate from its usual downstream metabolite fumarate. When reperfusion occurs, succinate is rapidly oxidized causing over-reduction of the coenzyme Q pool and driving “reverse” electron flow towards respiratory complex I, which generates a high amount of ROS leading to ischemia/reperfusion injury (113). It is highly likely that cell death in these conditions is triggered by ROS-dependent PTP opening; accordingly, the complex I inhibitor rotenone acts as an important PTP inhibitor (361).
In vitro, F-ATP synthase is susceptible to ROS (120, 369, 657) and to oxidative/nitrative stress associated with disorders of the central nervous system (482, 561), caloric restriction (561), and aging (234, 264). Hydrogen peroxide selectively inactivates F1 from bovine heart through formation of iron-protein adducts that generate high ROS levels (369). A specific target of singlet oxygen and hydrogen peroxide has been recently identified in a methionine-cysteine cluster of the γ subunit of chloroplasts. This cluster is highly conserved and is essential for enzyme coupling, as shown by F-ATP synthase inactivation following oxidation (264). Since the cluster is conserved, it is tempting to speculate that it may also mediate ROS-dependent PTP opening. Other selective targets of ROS that could be involved in PTP formation are three Trp residues of d subunit in mammals (583).
V. PATHOPHYSIOLOGY
A. Potential Role in Ca2+ Homeostasis
Mitochondria are critical regulators of Ca2+ homeostasis (593). There is no question that the PTP can undergo fast cycles of opening/closure both in isolated mitochondria and intact cells (280, 281, 471, 473, 666), a likely result of the flickering detected by patch-clamp (9, 569, 572) and bilayer experiments (96, 218, 611). If a [Ca2+] concentration gradient exists between the matrix and the intermembrane space, PTP-dependent depolarization will be followed by Ca2+ release. Whether this event provides a pathway for physiological Ca2+ release from mitochondria, and therefore participates in physiological Ca2+ homeostasis, has been discussed in the field (46, 50, 239) but remains a difficult problem to address. Indeed, opening of individual pores is usually not synchronized, and polarized mitochondria may take up the Ca2+ released by depolarized mitochondria (280, 281). Furthermore, the fraction of F-ATP synthase units undergoing channel formation under “resting” conditions must be very small, and it is not easy to predict what level of inhibition is required to observe an effect. Since the large size of the pore allows charge compensation within the channel itself (41), flux is presumably limited by the Ca2+ concentration gradient, suggesting that most if not all PTP units may need to be inhibited to observe an effect on Ca2+ release. These considerations raise some doubts about the conclusions of a recent study where the problem has been tackled by monitoring cytosolic and mitochondrial Ca2+ transients after treatment of HeLa cells with siRNAs against the c subunit. No differences were observed, leading the authors to conclude that the PTP does not play a role in mitochondrial Ca2+ efflux under basal conditions (148). We note that the experimental evidence that the F-ATP synthase was actually downregulated is a single Western blot reporting a partial decrease of the precursor of subunit c against actin (148). This problem adds to our concerns about the experimental design and should induce some caution in considering the authors' far-reaching conclusions (148).
Since no pore blockers are available, most experiments addressing the role of the PTP in Ca2+ homeostasis have used CsA, and this demands additional cautions. The first problem is that the inhibitory effect of CsA obviously depends on the expression level of CyPD, which is rarely assessed. A study from the Fontaine laboratory has revealed that the variation can be extremely large with a matching difference in sensitivity to CsA, which for example was totally absent in NIH3T3 fibroblasts and HL60 cells (361). The second issue is the relative expression of CyPD and of F-ATP synthase. Cross-linking experiments in beef heart mitochondria indicate that there is much less CyPD than b, d, and OSCP subunits (216). If these results can be extrapolated to other tissues, many F-ATP synthase channels will be insensitive to CsA even if CyPD is expressed. Finally, not all the mitochondrial effects of CsA are mediated by PTP inhibition. Even when the most common artifacts are avoided (47, 48), inhibition of calcineurin can prevent mitochondrial fission by preventing Drp1 translocation (98). This event can affect mitochondrial shape, which in turn influences Ca2+ homeostasis (501). Thus, while with proper precautions an effect of CsA indicates that the PTP may be involved in the event under study, a negative result does not always allow safe conclusions to be made (48, 52).
In 1992, Altschuld et al. (13) reported that CsA increased net Ca2+ uptake and decreased Ca2+ efflux in isolated cardiomyocytes, through a mitochondrial effect that had no impact on cell morphology or viability. On the other hand, no effect on Ca2+ homeostasis was found in a study on hormonally stimulated rat livers perfused with CsA where the PTP was demonstrably inhibited after in vivo treatment (176). Ca2+ homeostasis has also been addressed in Ppif−/− cells and mice. The hearts of Ppif−/− mice displayed functional changes that are consistent with increased Ca2+ retention due to decreased PTP activity, such as decreased contractile reserve, increased shortening and relaxation times, and longer decay of cytosolic Ca2+ transients (173). Mitochondria displayed an increased Ca2+ content that was matched by larger Ca2+ transients in wild-type individuals treated with CsA. CsA also decreased the rise time required for Ca2+ accumulation and prolonged the recovery time, consistent with operation of the PTP as a Ca2+ release channel preventing Ca2+ overload (173). In keeping with this idea, in adult cortical neurons from wild-type and Ppif−/− mice, cytosolic [Ca2+] increases induced by either ATP or by depolarizing concentrations of KCl gave comparable, transient increases of mitochondrial [Ca2+], while application of the two stimuli together resulted in much higher levels of mitochondrial [Ca2+] in the Ppif−/− neurons. This result suggests that the threshold for PTP activation had been reached in the wild-type but not in the CyPD-null mitochondria in situ (30). Consequently, the regulatory role of PTP opening in Ca2+ homeostasis, which may be of relevance to synaptic plasticity (593), may become apparent only for relatively large mitochondrial Ca2+ loads (30). Mice expressing amyotrophic lateral sclerosis-linked mutants of superoxide dismutase 1 develop a motor neuron disease with many pathological hallmarks also seen in patients (241). Spinal chord mitochondria display decreased Ca2+ retention capacity long before onset of motor weakness and neuronal death (141), and this was corrected by ablation of the Ppif gene (454). In these mice, improved mitochondrial Ca2+ buffering was matched by improved ATP synthesis and reduced swelling, attenuation of glial activation, reduction of misfolded aggregates in the spinal cord, and significant suppression of motor neuron death throughout disease, although survival was not improved (454).
Our recent findings that the F-ATP synthase of Drosophila mitochondria forms 53 pS channels (611) responsible for mitochondrial Ca2+-induced Ca2+ release (610), and that the channels may be selective for Ca2+ and H+ (610), are of relevance to the present discussion. Indeed, while the mammalian PTP could be involved either in Ca2+ homeostasis or in apoptosis induction depending on its open time (473), due to its small size the Drosophila PTP could be involved only in Ca2+ homeostasis (50, 610). Consistent with this fact, the mitochondrial pathway is not essential in most cases of Drosophila apoptosis (164, 403, 439, 616, 643).
Transient PTP openings are also intimately linked to mitochondrial ROS production in the process of ROS-induced ROS release (666). This process overlaps with Ca2+-induced Ca2+ release, to which it appears to be mechanistically connected, as thoroughly discussed by Zorov et al. (667). “Flashes” related to transient PTP openings have also been observed in mitochondria expressing a circularly permuted yellow fluorescent protein (622). The flashes have been attributed to bursts of local production of the superoxide anion, but there is an ongoing debate on whether they are partially or totally due to changes of matrix pH (521, 522), although matrix alkalinization appears way too small to account for the observed fluorescence changes (629). Irrespective of the nature of the detected species, there is little doubt that the flashes document the occurrence of transient PTP openings, which have been also detected in living animals (183). In summary, the role of transient PTP openings in physiological Ca2+ and ROS homeostasis is supported by increasing experimental evidence and deserves further study.
B. Pore Opening in Cell Death
Persistent PTP opening may constitute the point of no return in cell commitment to death (489). As already mentioned, prolonged PTP opening causes mitochondrial depolarization by equilibration of the proton gradient, followed by loss of pyridine nucleotides and respiratory inhibition (157, 607). Equilibration of ions and of solutes with molecular masses below the pore size induces mitochondrial swelling, given the colloidal osmotic pressure exerted by matrix proteins, leading to breaches in the OMM and release of intermembrane, apoptogenic proteins like cytochrome c (472). In addition, Ca2+ equilibrates across the open pores, and mitochondria can no longer maintain a Ca2+ concentration gradient (50). The combined effect of these dramatic events initiates an apoptotic routine, if enough ATP is available to supply the required energy, or induces necrosis (15). PTP dysregulation is a crucial element in many pathological conditions, such as degenerative diseases, where excessive PTP opening induces tissue damage, and cancer, where insensitivity to PTP induction contributes to the cellular ability to escape apoptosis that characterizes neoplastic cells (243, 322). As a corollary, identification of drugs that modulate PTP induction is a promising area of investigation for the potential treatment of many diseases (182, 550, 587, 631). We will briefly cover diseases where an increased propensity to PTP opening is part of pathogenesis and discuss in some detail emerging mechanisms that may link PTP resistance to cancer onset and progression.
1. Diseases with increased propensity to pore opening
Treatment with CyPD inhibitors, or CyPD ablation, generally confers remarkable protection against ischemia-reperfusion injury of the heart (27, 231, 426, 480, 538), brain (327, 353, 363, 518, 594, 623), kidney (453), and liver (271, 422). In the heart, protection extends to preconditioning and postconditioning protocols (366) and organ failure (367); in brain to hypoglicemic damage (186, 197, 311), ammonia neurotoxicity (10, 435), and trauma (558, 559). It is easy to predict that these fields will enormously benefit from novel PTP inhibitors acting independently of CyPD (182) and from increased understanding of the posttranslational modifications of F-ATP synthase that may be the basis for preconditioning, where IF1 binding to F-ATP synthase persisted after the end of the short ischemia (159, 612), and postconditioning.
Muscular dystrophies are a group of genetic diseases characterized by progressive loss of muscle tissue, which leads to progressive skeletal muscle weakness, impairment of contractility, disability often associated to inability to walk, respiratory problems, and cardiomyopathy (488). A severe form of dystrophy, Ullrich congenital muscular dystrophy, and the milder Bethlem myopathy are caused by mutations that hamper maturation of collagen VI, a key component of the extracellular matrix of skeletal muscle. Collagen VI deficiency impairs anchoring of skeletal muscle fibers to the matrix, exposing muscles to extensive mechanical stress that eventually leads to death of muscle fibers (341). We have shown that PTP induction is the effector mechanism responsible for the loss of muscle fibers in collagen VI null mice (287) in a process that involves ROS production by mitochondrial monoamine oxidases (539). Mitochondrial dysfunction and cell death could be prevented both in mice and in myoblasts from Ullrich patients by treatment with CsA and other CyPD inhibitors devoid of immunosuppressive activity, paving the way to the use of PTP inhibitors for the treatment of patients (43, 407, 669). Several pieces of evidence point to a role of the PTP in the pathogenesis of other muscular dystrophies as well. Genetic ablation of CyPD reduces the dystrophyc phenotype in both skeletal muscle and heart of mice lacking δ-sarcoglycan, and the premature lethality associated with the absence of the α2 chain of laminin, which causes congenital muscular dystrophy (410). Moreover, treatment with CyPD inhibitors rescues mitochondrial dysfunction and muscle fiber necrosis in mdx mice, a model of Duchenne muscular dystrophy (498).
Deregulated Ca2+ influx into neurons through receptors for N-methyl-d-aspartate and glutamate causes excitotoxicity, leading to activation of degradative enzymes, loss of redox equilibrium, and eventually mitochondrial depolarization and cell death (436, 576), probably induced by PTP opening. Accordingly, several features of the PT can be observed during excitotoxic injury (481), which can be blocked by CsA (517). Moreover, after induction of excitotoxicity, a secondary rise of cytosolic Ca2+, named “delayed Ca2+ deregulation,” occurs (78), which probably leads to cell demise via stable PTP opening (481).
Many diseases of the central nervous system are characterized by derangements of Ca2+ and ROS homeostasis that induce PTP opening as an important pathological feature. Alzheimer's disease, the most common form of dementia in aged people, is characterized by deposition of amyloid plaques formed by the amyloid β peptide, a cleavage product of the amyloid precursor protein (244). It has been shown that amyloid β oligomers alter Ca2+ homeostasis (153). In addition, amyloid β can be imported in mitochondria (257), where it interacts with CyPD and enhances PTP induction, whereas neurons from CyPD knockout mice are protected from cell death induced by amyloid β-dependent PTP (166). Interestingly, a novel association with Alzheimer's disease risk, which reached genome-wide significance, has been recently identified in the F-ATP synthase ATP5H locus, which encodes subunit d of the lateral stalk (60).
Huntington disease is caused by mutations that lead to translation of an expanded polyglutamine tract in the gene encoding the protein huntingtin. Patients display progressive motor and cognitive deficits that are invariably lethal (342). Pathogenic huntingtin mutations cause enhanced mitochondrial sensitivity to Ca2+ that eventually can lead to PTP induction both in cells from patients (448) and in animal models of the disease (212, 580). Accordingly, neurons carrying the mutant huntingtin gene display mitochondrial Ca2+ elevation with increased oxidative stress and mtDNA damage (618) and increased PTP sensitivity to Ca2+ (365).
Amyotrophic lateral sclerosis is an appalling neurodegenerative disease characterized by loss of spinal motor neurons, which is rapidly progressive and lethal (456). In a small subset of patients, the disease is genetically inherited and caused by mutations in superoxide dismutase 1 (63). Transgenic mice expressing the mutant protein display mitochondrial alterations including swelling, respiratory inhibition, reduced Ca2+ buffering capacity and fragmentation, and an elevated generation of ROS (235, 309). Either CsA or genetic ablation of CyPD increases Ca2+ buffering capacity and ATP synthesis in mitochondria of mouse models of amyotrophic lateral sclerosis, indicating that PTP opening might be the effector of neuron cell death (63, 456). The PTP-protective effects initially reported for minocycline (660) led to a major trial on patients that unfortunately had to be terminated because of lack of efficacy and presence of serious side effects (224), which can probably be traced to the permeabilizing effects of the drug on mitochondria (338, 519).
In multiple sclerosis, the myelin sheath of neurons in the central nervous system is destroyed as a consequence of an idiopathic disease leading to a variety of neurological symptoms and to progressive disability (556). The disease is worsened by axonal degeneration, which is associated to mitochondrial Ca2+ overload and bioenergetic dysfunction (198). Experimental autoimmune encephalomyelitis is widely used as an animal model of multiple sclerosis, as mice develop an inflammatory demyelinating disease of the central nervous system after administration of myelin components. When CyPD-null mice are treated, they display a marked protection from axonal degeneration and a milder clinical picture despite a normal inflammatory response, thus providing strong evidence that PTP is involved in disease pathogenesis (193). A further indication of the central role played by the PTP in multiple sclerosis is provided by the observation that axonal damage of mice undergoing experimental autoimmune encephalomyelitis is reduced by genetic inactivation of p66Shc, which abrogates an important pathway of ROS-induced PTP opening (see above) (514).
The most frequent form of neurodegenerative disorder affecting movement, Parkinson's disease, is caused by death of dopaminergic neurons in the mesencephalic region called substantia nigra pars compacta. A fine tuning of intracellular Ca2+ fluctuations and of the Ca2+ storage capacity of mitochondria is particularly important in these neurons, as the release of the neurotransmitter dopamine is under the control of voltage-dependent L-type Ca2+ channels (630). Indeed, sensitization to PTP opening has been proposed as a major cause of neurodegeneration in several models of the disease characterized by altered homeostasis of intracellular Ca2+ (87, 376, 384), including the forms caused by complex I inhibition (229, 528) and by inactivation of the Ser/Thr kinase PINK1 (597), in which changes in Ca2+ storage capacity (8, 205), impairment of respiratory complex I (209), and altered mitophagy (307) are observed. Moreover, the PTP can be induced in dopaminergic neurons by inability to buffer increased intracellular ROS levels (79). Interestingly, the decline of motor scores is much slower in patients treated with dopamine agonists (such as pramipexole and ropinirole) rather than with l-dopa. This effect could be explained by a neuroprotective inhibition of the PTP by dopamine agonists, which has been reported in rat mitochondria (455, 515), and would be similar in mechanism to PTP-dependent protection of the penumbra zone in oxygen-glucose deprivation protocols by treatment with melatonin (14).
Mitochondrial dysfunction occurs in several lysosomal storage diseases (321), and some evidence is available to suggest that the PTP is involved. Loss of mitochondrial Ca2+ homeostasis was proposed as the unifying mechanism and, indeed, in a mouse model of human GM1-gangliosidosis, accumulation of GM1-ganglioside caused mitochondrial Ca2+ overload, PTP opening, and apoptosis (513). Mitochondrial dysfunction is also key to Gaucher's disease, the most common lysosomal storage disorder (443). This genetic disease causes loss of activity of glucocerebrosidase and, intriguingly, confers a high risk of developing Parkinson's disease (443).
The PTP is involved in liver failure models that mimic fulminant hepatitis (356). Protection by CsA has been reported after treatment with lipopolysaccharide of Escherichia coli (136–138) and heat-inactivated Propionibacterium acnes (625) or d-galactosamine (308, 542), with anti-Fas antibody (184), and in an animal model of α-1 antitrypsin deficiency with liver injury (584). Interestingly, encouraging results have also been obtained in one cohort of cases of fulminant viral hepatitis treated with CsA (650). Ca2+ deregulation and PTP opening are also involved in toxicity of diclofenac (391) and acetaminophen (258, 392), which accounts for about half of the cases of acute liver failure. Acetaminophen poisoning induces glutathione depletion and inhibition of mitochondrial respiratory complexes I and II, thereby increasing ROS generation and altering the mitochondrial ability to sequester Ca2+. Such events ultimately lead to PTP opening and to potentially fatal centrilobular hepatic necrosis (270). Notably, in cultured mouse hepatocytes, acetaminophen toxicity is abrogated by both CsA and by its nonimmunosuppressive analog NIM811 (326).
Mitochondrial dysfunction due to PTP dysregulation is considered to be an important effector mechanism in sepsis (137, 138, 344) and in the subsequent multiple organ dysfunction syndrome (142, 642), which derives from decreased oxygen utilization and may also be a consequence of damage to endothelial mitochondria (145). Finally, a causal link with the PTP is emerging in diabetes (336), which may be related both to endothelial damage and its vascular complications, and to the demise of insulin-secreting β cells (200, 201, 340, 379, 577).
2. Cancer and decreased propensity to pore opening
Cancer cells are extremely refractory to cell death following a variety of stress stimuli. PTP desensitization is emerging as an important strategy used by neoplastic cells to escape death. An example is provided by the induction of liver tumors in rats by administration of the arylamine 2-acetylaminofluorene, which closely mimics the diverse stress conditions (alcoholic liver disease, chronic hepatitis B and C, and cholestasis) that predispose to hepatocellular carcinoma in humans. We have found that 2-acetylaminofluorene-treated rats become resistant to liver apoptosis and necrosis by desensitizing the PTP before onset of transformation, which indicates that the pore is an early target of adaptive responses that promote tumorigenesis in vivo (322).
Changes in metabolic functions and in transduction pathways, together with their exposure to an environment where oxygen and nutrient resources continuously fluctuate (230), lead to important changes in the redox status of tumor cells. In particular, increased levels of ROS must be strictly controlled to avoid oxidative stress and eventually pore opening and cell death (91). A deeper understanding of how tumor cells protect themselves from ROS-induced PTP opening offers the possibility for novel selective antineoplastic strategies. Indeed, as cancer cells must walk a delicate tightrope between ROS generation and scavenging, they may become extremely sensitive to further oxidative insults that overtake their antioxidant defenses (225).
Liver cancer cells are protected from oxidative stress elicited by chemotherapeutic treatment by inhibiting respiratory complex I through overexpression and mitochondrial localization of SERPINB3, a serine protease inhibitor of the serpin family (223), thus blocking ROS generation and protecting cells from PTP opening (114). As reported above, mitochondrial binding of HK II delivers a survival signal that stabilizes the PTP in the closed conformation (109). We (449) and others (564, 639) have observed that mitochondrial HK II displays an antioxidant function that explains its survival function, independently of HK II enzymatic activity (discussed in Ref. 449). Notably, the kinase AKT has an extremely relevant role in installing the antiapoptotic phenotype of tumor cells (174) by phosphorylating HK II and thus favoring its OMM binding. In turn, this inhibits Ca2+-induced cytochrome c release and apoptosis (418). Thus the design of drugs that target mitochondrial HK II, such as membrane-permeable peptides that compete for HK II binding to the OMM, is highly warranted, as this approach could selectively kill neoplastic cells by ROS-dependent PTP induction.
Another important survival strategy employed by tumors that impacts on the PTP involves the mitochondrial chaperone TRAP1. Indeed, TRAP1 displays an antioxidant activity in several pathological conditions, such as cerebral (644) and heart ischemia (641). In addition, it inhibits PTP opening and death of hypoxic cardiomyocytes (641). In several tumor types, the expression of TRAP1 is increased during malignant progression (reviewed in Ref. 491). Augmented TRAP1 expression enhances tumor malignancy by protecting neoplastic cells from toxicity elicited by several antineoplastic agents (126, 393, 420), likely through inhibition of ROS-dependent PTP opening (243). Therefore, the design of compounds that selectively inhibit TRAP1 chaperone activity constitutes a promising antitumor approach. Interestingly, the use of molecular dynamics simulations has been applied to the closely related chaperone Hsp90, allowing the identification of potential allosteric inhibitors of its chaperone activity cycle (424). A similar strategy can be envisioned for TRAP1, exploiting structural differences between TRAP1 and Hsp90 (346) to design novel classes of allosteric inhibitors that selectively target the chaperone cycle TRAP1.
Inhibition of Kv1.3, a voltage-dependent K+ channel located in the inner mitochondrial membrane, induces ROS generation and PTP opening (570). It was recently shown that membrane-permeant, selective Kv1.3 inhibitors such as Psora-4, PAP-1, and clofazimine are effective on several cancer cell lines and on B lymphocytes derived from B-chronic lymphocytic leukemia (348, 349). Notably, clofazimine reduced by 90% the size of a melanoma in an orthotopic mouse model, without any side effects (348).
It is becoming increasingly clear that some widely used antineoplastic drugs are able to prompt a ROS surge that induces the PTP, which could explain at least in part the toxicity associated to their use. For instance, taxol (paclitaxel), which is known to induce apoptosis by stabilizing microtubules, also prompts ROS-dependent PTP induction in liver cancer cells (602). Cisplatin, on the other hand, causes the formation of adducts with DNA that induce apoptosis (204) and can also elicit CyPD-dependent necrosis in pancreatic cancer cells (102). Importantly, we have recently found that cisplatin, doxorubicin, and the BH3 mimetic EM20-25 (409) prompt a rapid ROS increase and PTP opening in hepatoma cells (114). These observations suggest that the efficacy of compounds designed on the structure of cisplatin will increase with the decrease of the toxic side effects (111). We have characterized one such compound, the Gold(III)-dithiocarbamato complex AUL12, that is endowed with high selectivity towards tumor cells and xenograft tumors but with extremely low nephrotoxicity and acute toxicity (386). We found that AUL12 strongly inhibits respiratory complex I, inducing a rapid burst of mitochondrial ROS levels that leads to activation of the mitochondrial pool of GSK3 and to the ensuing phosphorylation of CyPD (110). Therefore, these data provide a biochemical mechanism that connects chemotherapeutic activity with ROS generation, kinase activation, and PTP induction in mitochondria. Significantly, several plant-derived natural compounds that act by increasing intracellular ROS levels, thereby serving as PTP inducers, have been tested in tumor cells and in animal models, and some of them are currently undergoing clinical or preclinical trials (350, 557). A summary of the signaling pathways described in this paragraph can be found in Figure 4.
Figure 4.

Mechanisms of PTP regulation in tumor cells. A variety of factors control tumor cell viability by modulating PTP opening, mainly by modulating ROS levels. A ROS surge elicits PTP opening and cell death, whereas ROS inhibition keeps the pore locked and protects cells from noxious stimuli. PTP inducers are indicated in orange, PTP inhibitors in blue. I–IV, respiratory complexes; 2-AAF, 2-acetylaminofluorene; Q, coenzyme Q; cyt c, cytochrome c; HK II, hexokinase II; IMM, inner mitochondrial membrane; SB3, serpin B3; UCP-2, uncoupling protein 2. [From Rasola and Bernardi (494) with permission.]
VI. CONCLUSIONS
Major progress has been made in understanding the nature of the PTP. Conservation of the channel-forming property of F-ATP synthases from yeast to Drosophila and mammals, with distinctive conductance features, suggests a function that has been acquired in eukaryotes, and maintained and exploited in evolution. The PTP may have initially evolved as a Ca2+-release channel and then acquired a role in cell death that became part of the mitochondrial pathway to apoptosis. In this perspective, it is very striking that the F-ATP synthase, the point of integration of Mitchell's proton gradient with ATP synthesis or hydrolysis, may also be the point of integration of death signals. The finding that the PTP forms from F-ATP synthase is so recent that key mechanistic issues still need to be clarified. Yet, it is easy to predict that, 60 years after its discovery, the years to come will see a solution to the mystery of the PTP and the dissection of the molecular mechanisms that mediate the transition of the F-ATP synthase from an energy-conserving into an energy-dissipating device.
GRANTS
Research in our laboratories is supported by the following grants: Associazione Italiana Ricerca sul Cancro (AIRC) IG 13392, Italian Ministry for University and Research (MIUR) FIRB RBAP11S8C3 and PRIN 20107Z8XBW, Telethon Italy GGP14037 and GPP14187 (to P. Bernardi); AIRC IG 15863 and University of Padova CPDA123598 (to A. Rasola); and National Institutes of Health (USA) R01GM069883 and R03DA033978-01 (to M. Forte and P. Bernardi).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank Mario Zoratti and Ildikò Szabò (Univ. of Padova) for helpful comments and Federico Fogolari (Univ. of Udine) for help with computer modeling of F-ATP synthases.
Address for reprint requests and other correspondence: P. Bernardi, Dept. of Biomedical Sciences, Univ. of Padova, Via Ugo Bassi 58/B, I-35131 Padova, Italy (e-mail: bernardi@bio.unipd.it); G. Lippe, Dept. of Food Science, Univ. of Udine, I-33100 Udine, Italy (e-mail: giovanna.lippe@uniud.it).
REFERENCES
- 1.Abrahams JP, Buchanan SK, van Raaij MJ, Fearnley IM, Leslie AG, Walker JE. The structure of bovine F1-ATPase complexed with the peptide antibiotic efrapeptin. Proc Natl Acad Sci USA 93: 9420–9424, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370: 621–628, 1994. [DOI] [PubMed] [Google Scholar]
- 3.Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci USA 104: 18091–18096, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL, Schlame M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J 100: 2184–2192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Acin-Pérez R, Carrascoso I, Baixauli F, Roche-Molina M, Latorre-Pellicer A, Fernández-Silva P, Mittelbrunn M, Sanchez-Madrid F, Pérez-Martos A, Lowell CA, Manfredi G, Enríquez JA. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab 19: 1020–1033, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adachi K, Oiwa K, Nishizaka T, Furuike S, Noji H, Itoh H, Yoshida M, Kinosita K Jr. Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell 130: 309–321, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Adams V, Bosch W, Schlegel J, Wallimann T, Brdiczka D. Further characterization of contact sites from mitochondria of different tissues: topology of peripheral kinases. Biochim Biophys Acta 981: 213–225, 1989. [DOI] [PubMed] [Google Scholar]
- 8.Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, Sullivan PG, Bueler H. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6: e16038, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 111: 10580–10585, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albrecht J, Norenberg MD. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44: 788–794, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature. In press. [DOI] [PubMed] [Google Scholar]
- 12.Allen RD. Membrane tubulation and proton pumps. Protoplasma 189: 1–8, 1995. [Google Scholar]
- 13.Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol 262: H1699–H1704, 1992. [DOI] [PubMed] [Google Scholar]
- 14.Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J 18: 869–871, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15: 961–973, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Appleby RD, Porteous WK, Hughes G, James AM, Shannon D, Wei YH, Murphy MP. Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur J Biochem 262: 108–116, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Arakaki N, Ueyama Y, Hirose M, Himeda T, Shibata H, Futaki S, Kitagawa K, Higuti T. Stoichiometry of subunit e in rat liver mitochondrial H+-ATP synthase and membrane topology of its putative Ca2+-dependent regulatory region. Biochim Biophys Acta 1504: 220–228, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Arechaga I, Butler PJ, Walker JE. Self-assembly of ATP synthase subunit c rings. FEBS Lett 515: 189–193, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schägger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J 17: 7170–7178, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arselin G, Giraud MF, Dautant A, Vaillier J, Brèthes D, Coulary-Salin B, Schaeffer J, Velours J. The GxxxG motif of the transmembrane domain of subunit e is involved in the dimerization/oligomerization of the yeast ATP synthase complex in the mitochondrial membrane. Eur J Biochem 270: 1875–1884, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Arselin G, Vaillier J, Salin B, Schaeffer J, Giraud MF, Dautant A, Brèthes D, Velours J. The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J Biol Chem 279: 40392–40399, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Azarashvili TS, Tyynelä J, Odinokova IV, Grigorjev PA, Baumann M, Evtodienko YV, Saris NE. Phosphorylation of a peptide related to subunit c of the F0F1-ATPase/ATP synthase and relationship to permeability transition pore opening in mitochondria. J Bioenerg Biomembr 34: 279–284, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynelä J, Saris NEL. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 55: 69–77, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Azzi A, Azzone GF. Swelling and shrinkage phenomena in liver mitochondria. I. Large amplitude swelling induced by inorganic phosphate and by ATP. Biochim Biophys Acta 105: 253–264, 1965. [DOI] [PubMed] [Google Scholar]
- 25.Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett 584: 2504–2509, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azzone GF, Azzi A. Volume changes in liver mitochondria. Proc Natl Acad Sci USA 53: 1084–1089, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker LA, Watt IN, Runswick MJ, Walker JE, Rubinstein JL. Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc Natl Acad Sci USA 109: 11675–11680, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barsukova A, Komarov A, Hajnoczky G, Bernardi P, Bourdette D, Forte M. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci 33: 831–842, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bason JV, Montgomery MG, Leslie AG, Walker JE. Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F1-ATPase. Proc Natl Acad Sci USA 111: 11305–11310, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bason JV, Montgomery MG, Leslie AG, Walker JE. How release of phosphate from mammalian F1-ATPase generates a rotary substep. Proc Natl Acad Sci USA 112: 6009–6014, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bason JV, Runswick MJ, Fearnley IM, Walker JE. Binding of the inhibitor protein IF1 to bovine F1-ATPase. J Mol Biol 406: 443–453, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem 280: 18558–18561, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Basso E, Petronilli V, Forte MA, Bernardi P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem 283: 26307–26311, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belogrudov GI. Recent advances in structure-functional studies of mitochondrial factor B. J Bioenerg Biomembr 41: 137–143, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belogrudov GI, Tomich JM, Hatefi Y. Membrane topography and near-neighbor relationships of the mitochondrial ATP synthase subunits e, f, and g. J Biol Chem 271: 20340–20345, 1996. [DOI] [PubMed] [Google Scholar]
- 39.Bergeaud M, Mathieu L, Guillaume A, Moll UM, Mignotte B, Le Floch N, Vayssiere JL, Rincheval V. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F1F0-ATP synthase. Cell Cycle 12: 2781–2793, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem 267: 8834–8839, 1992. [PubMed] [Google Scholar]
- 41.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127–1155, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Bernardi P, Azzone GF. Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J Biol Chem 256: 7187–7192, 1981. [PubMed] [Google Scholar]
- 43.Bernardi P, Bonaldo P. Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harb Perspect Biol 5: a011387, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore: a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr 26: 509–517, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Bernardi P, Giorgio V. EBEC 2012: an energetic time in Freiburg. EMBO Rep 14: 7–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr 28: 131–138, 1996. [DOI] [PubMed] [Google Scholar]
- 47.Bernardi P, Petronilli V, Di Lisa F, Forte M. A mitochondrial perspective on cell death. Trends Biochem Sci 26: 112–117, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem 264: 687–701, 1999. [DOI] [PubMed] [Google Scholar]
- 49.Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabó I, Zoratti M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267: 2934–2939, 1992. [PubMed] [Google Scholar]
- 50.Bernardi P, von Stockum S. The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52: 22–27, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol 4: 95, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 273: 2077–2099, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Berson A, Descatoire V, Sutton A, Fau D, Maulny B, Vadrot N, Feldmann G, Berthon B, Tordjmann T, Pessayre D. Toxicity of alpidem, a peripheral benzodiazepine receptor ligand, but not zolpidem, in rat hepatocytes: role of mitochondrial permeability transition and metabolic activation. J Pharmacol Exp Ther 299: 793–800, 2001. [PubMed] [Google Scholar]
- 54.Bertina RM, Slater EC. The effects of phosphate and electron transport on the carbonyl cyanide m-chlorophenylhydrazone-induced ATPase of rat-liver mitochondria. Biochim Biophys Acta 376: 492–504, 1975. [DOI] [PubMed] [Google Scholar]
- 55.Beutner G, Rück A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett 396: 189–195, 1996. [DOI] [PubMed] [Google Scholar]
- 56.Bisetto E, Comelli M, Salzano AM, Picotti P, Scaloni A, Lippe G, Mavelli I. Proteomic analysis of F1F0-ATP synthase super-assembly in mitochondria of cardiomyoblasts undergoing differentiation to the cardiac lineage. Biochim Biophys Acta 1827: 807–816, 2013. [DOI] [PubMed] [Google Scholar]
- 57.Bisetto E, Di Pancrazio F, Simula MP, Mavelli I, Lippe G. Mammalian ATPsynthase monomer versus dimer profiled by blue native PAGE and activity stain. Electrophoresis 28: 3178–3185, 2007. [DOI] [PubMed] [Google Scholar]
- 58.Bisetto E, Picotti P, Giorgio V, Alverdi V, Mavelli I, Lippe G. Functional and stoichiometric analysis of subunit e in bovine heart mitochondrial F0F1ATP synthase. J Bioenerg Biomembr 40: 257–267, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Blatt NB, Bednarski JJ, Warner RE, Leonetti F, Johnson KM, Boitano A, Yung R, Richardson BC, Johnson KJ, Ellman JA, Opipari AW Jr, Glick GD. Benzodiazepine-induced superoxide signals B cell apoptosis: mechanistic insight and potential therapeutic utility. J Clin Invest 110: 1123–1132, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boada M, Antúnez C, Ramírez-Lorca R, DeStefano AL, González-Pérez A, Gayán J, López-Arrieta J, Ikram MA, Hernández I, Marin J, Galán JJ, Bis JC, Mauleón A, Rosende-Roca M, Moreno-Rey C, Gudnasson V, Morón FJ, Velasco J, Carrasco JM, Alegret M, Espinosa A, Vinyes G, Lafuente A, Vargas L, Fitzpatrick AL, Launer LJ, Sáez ME, Vázquez E, Becker JT, López OL, Serrano-Rios M, Tárraga L, van Duijn CM, Real LM, Seshadri S, Ruiz A. ATP5H/KCTD2 locus is associated with Alzheimer's disease risk. Mol Psychiatry 19: 682–687, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105: 771–785, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol 27: 4365–4373, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52: 39–59, 2006. [DOI] [PubMed] [Google Scholar]
- 64.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34: 1475–1486, 2015. [DOI] [PubMed] [Google Scholar]
- 66.Borel JF, Feurer C, Magnee C, Stahelin H. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology 32: 1017–1025, 1977. [PMC free article] [PubMed] [Google Scholar]
- 67.Bowler MW, Montgomery MG, Leslie AG, Walker JE. How azide inhibits ATP hydrolysis by the F-ATPases. Proc Natl Acad Sci USA 103: 8646–8649, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowler MW, Montgomery MG, Leslie AG, Walker JE. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 Å resolution. J Biol Chem 282: 14238–14242, 2007. [DOI] [PubMed] [Google Scholar]
- 69.Boyer PD. The binding change mechanism for ATP synthase–some probabilities and possibilities. Biochim Biophys Acta 1140: 215–250, 1993. [DOI] [PubMed] [Google Scholar]
- 70.Boyer PD. The ATP synthase–a splendid molecular machine. Annu Rev Biochem 66: 717–749, 1997. [DOI] [PubMed] [Google Scholar]
- 71.Boyle GM, Roucou X, Nagley P, Devenish RJ, Prescott M. Modulation at a distance of proton conductance through the Saccharomyces cerevisiae mitochondrial F1F0-ATP synthase by variants of the oligomycin sensitivity-conferring protein containing substitutions near the C-terminus. J Bioenerg Biomembr 32: 595–607, 2000. [DOI] [PubMed] [Google Scholar]
- 72.Braig K, Menz RI, Montgomery MG, Leslie AG, Walker JE. Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ ADP and aluminium fluoride. Structure 8: 567–573, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B, Andrews D, Duclohier H, Reed JC, Kroemer G. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene 19: 329–336, 2000. [DOI] [PubMed] [Google Scholar]
- 74.Brenner-Holzach O, Raaflaub J. Die korrelation zwischen der schwellung isolierter mitochondrien und dem abbau intramitochondrialen adenosinnucleotide (ATP, ADP, AMP, CoA). Helv Physiol Pharmacol Acta 12: 242–252, 1954. [PubMed] [Google Scholar]
- 75.Broekemeier KM, Carpenter Deyo L, Reed DJ, Pfeiffer DR. Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett 304: 192–194, 1992. [DOI] [PubMed] [Google Scholar]
- 76.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem 264: 7826–7830, 1989. [PubMed] [Google Scholar]
- 77.Broekemeier KM, Pfeiffer DR. Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry 34: 16440–16449, 1995. [DOI] [PubMed] [Google Scholar]
- 78.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004. [DOI] [PubMed] [Google Scholar]
- 79.Brundin P, Li JY, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson's disease pathology spread. Nat Rev Neurosci 9: 741–745, 2008. [DOI] [PubMed] [Google Scholar]
- 80.Bruni A, Luciani S, Contessa AR. Inhibition by atractyloside of the binding of adenine nucleotides to rat-liver mitochondria. Nature 201: 1219–1220, 1964. [DOI] [PubMed] [Google Scholar]
- 81.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 35: 8483–8488, 1996. [DOI] [PubMed] [Google Scholar]
- 82.Brustovetsky N, Tropschug M, Heimpel S, Heidkamper D, Klingenberg M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry 41: 11804–11811, 2002. [DOI] [PubMed] [Google Scholar]
- 83.Bucheler K, Adams V, Brdiczka D. Localization of the ATP/ADP translocator in the inner membrane and regulation of contact sites between mitochondrial envelope membranes by ADP. A study on freeze-fractured isolated liver mitochondria. Biochim Biophys Acta 1056: 233–242, 1991. [DOI] [PubMed] [Google Scholar]
- 84.Buchet K, Godinot C. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted ρ°Cells. J Biol Chem 273: 22983–22989, 1998. [DOI] [PubMed] [Google Scholar]
- 85.Bulygin VV, Vinogradov AD. Interaction of Mg2+ with F0F1 mitochondrial ATPase as related to its slow active/inactive transition. Biochem J 276: 149–156, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bustos DM, Velours J. The modification of the conserved GXXXG motif of the membrane-spanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. J Biol Chem 280: 29004–29010, 2005. [DOI] [PubMed] [Google Scholar]
- 87.Büttner S, Habernig L, Broeskamp F, Ruli D, Vögtle FN, Vlachos M, Macchi F, Küttner V, Carmona-Gutierrez D, Eisenberg T, Ring J, Markaki M, Taskin AA, Benke S, Ruckenstuhl C, Braun R, Van den Haute C, Bammens T, van der Perren A, Fröhlich KU, Winderickx J, Kroemer G, Baekelandt V, Tavernarakis N, Kovacs GG, Dengjel J, Meisinger C, Sigrist SJ, Madeo F. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson's disease. EMBO J 32: 3041–3054, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Buzhynskyy N, Sens P, Prima V, Sturgis JN, Scheuring S. Rows of ATP synthase dimers in native mitochondrial inner membranes. Biophys J 93: 2870–2876, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cabezon E, Arechaga I, Jonathan P, Butler G, Walker JE. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. J Biol Chem 275: 28353–28355, 2000. [DOI] [PubMed] [Google Scholar]
- 90.Cabezon E, Butler PJ, Runswick MJ, Carbajo RJ, Walker JE. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J Biol Chem 277: 41334–41341, 2002. [DOI] [PubMed] [Google Scholar]
- 91.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 11: 85–95, 2011. [DOI] [PubMed] [Google Scholar]
- 92.Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol 291: C1082–C1088, 2006. [DOI] [PubMed] [Google Scholar]
- 93.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab 8: 13–25, 2008. [DOI] [PubMed] [Google Scholar]
- 94.Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF1: setting the pace of the F1Fo-ATP synthase. Trends Biochem Sci 34: 343–350, 2009. [DOI] [PubMed] [Google Scholar]
- 95.Carafoli E, Lehninger AL. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem J 122: 681–690, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carraro M, Giorgio V, Šileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabó I, Bernardi P. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 289: 15980–15985, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carroll J, Fearnley IM, Wang Q, Walker JE. Measurement of the molecular masses of hydrophilic and hydrophobic subunits of ATP synthase and complex I in a single experiment. Anal Biochem 395: 249–255, 2009. [DOI] [PubMed] [Google Scholar]
- 98.Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105: 15803–15808, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chanoit G, Lee S, Xi J, Zhu M, McIntosh RA, Mueller RA, Norfleet EA, Xu Z. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3β. Am J Physiol Heart Circ Physiol 295: H1227–H1233, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chappell JB, Crofts AR. Calcium ion accumulation and volume changes of isolated liver mitochondria. Calcium ion-induced swelling. Biochem J 95: 378–386, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chelli B, Falleni A, Salvetti F, Gremigni V, Lucacchini A, Martini C. Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol 61: 695–705, 2001. [DOI] [PubMed] [Google Scholar]
- 102.Chen B, Xu M, Zhang H, Wang JX, Zheng P, Gong L, Wu GJ, Dai T. Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. Biochem Biophys Res Commun 437: 526–531, 2013. [DOI] [PubMed] [Google Scholar]
- 103.Chen C, Ko Y, Delannoy M, Ludtke SJ, Chiu W, Pedersen PL. Mitochondrial ATP synthasome: three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J Biol Chem 279: 31761–31768, 2004. [DOI] [PubMed] [Google Scholar]
- 104.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen R, Runswick MJ, Carroll J, Fearnley IM, Walker JE. Association of two proteolipids of unknown function with ATP synthase from bovine heart mitochondria. FEBS Lett 581: 3145–3148, 2007. [DOI] [PubMed] [Google Scholar]
- 106.Cheng EHY, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513, 2003. [DOI] [PubMed] [Google Scholar]
- 107.Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. Eur J Biochem 238: 623–630, 1996. [DOI] [PubMed] [Google Scholar]
- 108.Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci USA 109: 20491–20496, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One 3: e1852, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chiara F, Gambalunga A, Sciacovelli M, Nicolli A, Ronconi L, Fregona D, Bernardi P, Rasola A, Trevisan A. Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3α, β and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis 3: e444, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chiara F, Rasola A. GSK-3 and mitochondria in cancer cells. Front Oncol 3: 16, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chinopoulos C, Adam-Vizi V. Modulation of the mitochondrial permeability transition by cyclophilin D: Moving closer to F0--F1 ATP synthase? Mitochondrion 12: 41–45, 2012. [DOI] [PubMed] [Google Scholar]
- 113.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ciscato F, Sciacovelli M, Villano G, Turato C, Bernardi P, Rasola A, Pontisso P. SERPINB3 protects from oxidative damage by chemotherapeutics through inhibition of mitochondrial respiratory complex I. Oncotarget 5: 2418–2427, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241: 779–786, 1996. [DOI] [PubMed] [Google Scholar]
- 116.Cleary J, Johnson KM, Opipari AW Jr, Glick GD. Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg Med Chem Lett 17: 1667–1670, 2007. [DOI] [PubMed] [Google Scholar]
- 117.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357: 695–697, 1992. [DOI] [PubMed] [Google Scholar]
- 118.Collinson IR, Runswick MJ, Buchanan SK, Fearnley IM, Skehel JM, van Raaij MJ, Griffiths DE, Walker JE. Fo membrane domain of ATP synthase from bovine heart mitochondria: purification, subunit composition, and reconstitution with F1-ATPase. Biochemistry 33: 7971–7978, 1994. [DOI] [PubMed] [Google Scholar]
- 119.Collinson IR, van Raaij MJ, Runswick MJ, Fearnley IM, Skehel JM, Orriss GL, Miroux B, Walker JE. ATP synthase from bovine heart mitochondria. In vitro assembly of a stalk complex in the presence of F1-ATPase and in its absence. J Mol Biol 242: 408–421, 1994. [DOI] [PubMed] [Google Scholar]
- 120.Comelli M, Lippe G, Mavelli I. Differentiation potentiates oxidant injury to mitochondria by hydrogen peroxide in Friend's erythroleukemia cells. FEBS Lett 352: 71–75, 1994. [DOI] [PubMed] [Google Scholar]
- 121.Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J 284: 381–385, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J 302: 321–324, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Selective inhibition of the mitochondrial permeability transition pore at the oxidation-reduction sensitive dithiol by monobromobimane. FEBS Lett 362: 239–242, 1995. [DOI] [PubMed] [Google Scholar]
- 124.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem 271: 6746–6751, 1996. [DOI] [PubMed] [Google Scholar]
- 125.Costantini P, Colonna R, Bernardi P. Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochim Biophys Acta 1365: 385–392, 1998. [DOI] [PubMed] [Google Scholar]
- 126.Costantino E, Maddalena F, Calise S, Piscazzi A, Tirino V, Fersini A, Ambrosi A, Neri V, Esposito F, Landriscina M. TRAP1, a novel mitochondrial chaperone responsible for multi-drug resistance and protection from apoptotis in human colorectal carcinoma cells. Cancer Lett 279: 39–46, 2009. [DOI] [PubMed] [Google Scholar]
- 127.Couoh-Cardel SJ, Uribe-Carvajal S, Wilkens S, Garcia-Trejo JJ. Structure of dimeric F1F0-ATP synthase. J Biol Chem 285: 36447–36455, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Covian R, Balaban RS. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am J Physiol Heart Circ Physiol 303: H940–H966, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Crichton PG, Harding M, Ruprecht JJ, Lee Y, Kunji ER. Lipid, detergent, and Coomassie Blue G-250 affect the migration of small membrane proteins in blue native gels: mitochondrial carriers migrate as monomers not dimers. J Biol Chem 288: 22163–22173, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 341: 233–249, 1999. [PMC free article] [PubMed] [Google Scholar]
- 131.Crompton M. Mitochondria and aging: a role for the permeability transition? Aging Cell 3: 3–6, 2004. [DOI] [PubMed] [Google Scholar]
- 132.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 84: 143–152, 2002. [DOI] [PubMed] [Google Scholar]
- 133.Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem 178: 489–501, 1988. [DOI] [PubMed] [Google Scholar]
- 134.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255: 357–360, 1988. [PMC free article] [PubMed] [Google Scholar]
- 135.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: 729–735, 1998. [DOI] [PubMed] [Google Scholar]
- 136.Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med 30: 276–284, 2002. [DOI] [PubMed] [Google Scholar]
- 137.Crouser ED, Julian MW, Huff JE, Joshi MS, Bauer JA, Gadd ME, Wewers MD, Pfeiffer DR. Abnormal permeability of inner and outer mitochondrial membranes contributes independently to mitochondrial dysfunction in the liver during acute endotoxemia. Crit Care Med 32: 478–488, 2004. [DOI] [PubMed] [Google Scholar]
- 138.Crouser ED, Julian MW, Joshi MS, Bauer JA, Wewers MD, Hart JM, Pfeiffer DR. Cyclosporin A ameliorates mitochondrial ultrastructural injury in the ileum during acute endotoxemia. Crit Care Med 30: 2722–2728, 2002. [DOI] [PubMed] [Google Scholar]
- 139.D'Alessandro M, Melandri BA. ATP hydrolysis in ATP synthases can be differently coupled to proton transport and modulated by ADP and phosphate: a structure based model of the mechanism. Biochim Biophys Acta 1797: 755–762, 2010. [DOI] [PubMed] [Google Scholar]
- 140.Dabbeni-Sala F, Lippe G, Sorgato MC. Structural and functional modifications induced by diamide on the F0 sector of the mammalian ATP synthase. FEBS Lett 281: 47–50, 1991. [DOI] [PubMed] [Google Scholar]
- 141.Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem 96: 1349–1361, 2006. [DOI] [PubMed] [Google Scholar]
- 142.Dare AJ, Phillips AR, Hickey AJ, Mittal A, Loveday B, Thompson N, Windsor JA. A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic Biol Med 47: 1517–1525, 2009. [DOI] [PubMed] [Google Scholar]
- 143.Dautant A, Velours J, Giraud MF. Crystal structure of the Mg·ADP-inhibited state of the yeast F1c10-ATP synthase. J Biol Chem 285: 29502–29510, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Davidson AM, Halestrap AP. Partial inhibition by cyclosporin A of the swelling of liver mitochondria in vivo and in vitro induced by sub-micromolar [Ca2+], but not by butyrate. Evidence for two distinct swelling mechanisms. Biochem J 268: 147–152, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res 100: 1128–1141, 2007. [DOI] [PubMed] [Google Scholar]
- 146.Davies KM, Anselmi C, Wittig I, Faraldo-Gomez JD, Kühlbrandt W. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA 109: 13602–13607, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA 108: 14121–14126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.De Marchi E, Bonora M, Giorgi C, Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56: 1–13, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.De Marchi U, Basso E, Szabó I, Zoratti M. Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol Membr Biol 23: 521–530, 2006. [DOI] [PubMed] [Google Scholar]
- 150.De Marchi U, Campello S, Szabó I, Tombola F, Martinou JC, Zoratti M. Bax does not directly participate in the Ca2+-induced permeability transition of isolated mitochondria. J Biol Chem 279: 37415–37422, 2004. [DOI] [PubMed] [Google Scholar]
- 151.De Marchi U, Szabò I, Cereghetti GM, Hoxha P, Craigen WJ, Zoratti M. A maxi-chloride channel in the inner membrane of mammalian mitochondria. Biochim Biophys Acta 1777: 1438–1448, 2008. [DOI] [PubMed] [Google Scholar]
- 152.De Stefani D, Raffaello A, Teardo E, Szabó I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285: 12463–12468, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J 176: 899–906, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Devenish RJ, Prescott M, Boyle GM, Nagley P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J Bioenerg Biomembr 32: 507–515, 2000. [DOI] [PubMed] [Google Scholar]
- 156.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene 26: 3279–3290, 2007. [DOI] [PubMed] [Google Scholar]
- 157.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571–2575, 2001. [DOI] [PubMed] [Google Scholar]
- 158.Di Pancrazio F, Bisetto E, Alverdi V, Mavelli I, Esposito G, Lippe G. Differential steady-state tyrosine phosphorylation of two oligomeric forms of mitochondrial F0F1ATPsynthase: a structural proteomic analysis. Proteomics 6: 921–926, 2006. [DOI] [PubMed] [Google Scholar]
- 159.Di Pancrazio F, Mavelli I, Isola M, Losano G, Pagliaro P, Harris DA, Lippe G. In vitro and in vivo studies of F0F1ATP synthase regulation by inhibitor protein IF1 in goat heart. Biochim Biophys Acta 1659: 52–62, 2004. [DOI] [PubMed] [Google Scholar]
- 160.Dickson VK, Silvester JA, Fearnley IM, Leslie AG, Walker JE. On the structure of the stator of the mitochondrial ATP synthase. EMBO J 25: 2911–2918, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dienhart M, Pfeiffer K, Schägger H, Stuart RA. Formation of the yeast F1F0-ATP synthase dimeric complex does not require the ATPase inhibitor protein, Inh1. J Biol Chem 277: 39289–39295, 2002. [DOI] [PubMed] [Google Scholar]
- 162.Diez M, Zimmermann B, Borsch M, König M, Schweinberger E, Steigmiller S, Reuter R, Felekyan S, Kudryavtsev V, Seidel CA, Gräber P. Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat Struct Mol Biol 11: 135–141, 2004. [DOI] [PubMed] [Google Scholar]
- 163.Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett 579: 633–637, 2005. [DOI] [PubMed] [Google Scholar]
- 164.Dorstyn L, Mills K, Lazebnik Y, Kumar S. The two cytochrome c species, DC3 and DC4, are not required for caspase activation and apoptosis in Drosophila cells. J Cell Biol 167: 405–410, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42, 2000. [DOI] [PubMed] [Google Scholar]
- 166.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 14: 1097–1105, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Duchen MR. Roles of mitochondria in health and disease. Diabetes 53 Suppl 1: S96–102, 2004. [DOI] [PubMed] [Google Scholar]
- 168.Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res 27: 1790–1794, 1993. [DOI] [PubMed] [Google Scholar]
- 169.Dudkina NV, Sunderhaus S, Braun HP, Boekema EJ. Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett 580: 3427–3432, 2006. [DOI] [PubMed] [Google Scholar]
- 170.Dyer MR, Walker JE. Sequences of members of the human gene family for the c subunit of mitochondrial ATP synthase. Biochem J 293: 51–64, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Eble KS, Coleman WB, Hantgan RR, Cunningham CC. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem 265: 19434–19440, 1990. [PubMed] [Google Scholar]
- 172.Ekert PG, Silke J, Connolly LM, Reid GE, Moritz RL, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53, 2000. [DOI] [PubMed] [Google Scholar]
- 173.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120: 3680–3687, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9: 550–562, 2009. [DOI] [PubMed] [Google Scholar]
- 175.Eriksson O, Fontaine E, Bernardi P. Chemical modification of arginines by 2,3-butanedione and phenylglyoxal causes closure of the mitochondrial permeability transition pore. J Biol Chem 273: 12669–12674, 1998. [DOI] [PubMed] [Google Scholar]
- 176.Eriksson O, Pollesello P, Geimonen E. Regulation of total mitochondrial Ca2+ in perfused liver is independent of the permeability transition pore. Am J Physiol Cell Physiol 276: C1297–C1302, 1999. [DOI] [PubMed] [Google Scholar]
- 177.Ernst S, Düser MG, Zarrabi N, Dunn SD, Börsch M. Elastic deformations of the rotary double motor of single FoF1-ATP synthases detected in real time by Forster resonance energy transfer. Biochim Biophys Acta 1817: 1722–1731, 2012. [DOI] [PubMed] [Google Scholar]
- 178.Ernster L, Hundal T, Sandri G. Resolution and reconstitution of F0F1-ATPase in beef heart submitochondrial particles. Methods Enzymol 126: 428–433, 1986. [DOI] [PubMed] [Google Scholar]
- 179.Everard-Gigot V, Dunn CD, Dolan BM, Brunner S, Jensen RE, Stuart RA. Functional analysis of subunit e of the F1Fo-ATP synthase of the yeast Saccharomyces cerevisiae: importance of the N-terminal membrane anchor region. Eukaryot Cell 4: 346–355, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Faccenda D, Tan CH, Seraphim A, Duchen MR, Campanella M. IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ 20: 686–697, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fan J, Lindemann P, Feuilloley MG, Papadopoulos V. Structural and functional evolution of the translocator protein (18 kDa). Curr Mol Med 12: 369–386, 2012. [DOI] [PubMed] [Google Scholar]
- 182.Fancelli D, Abate A, Amici R, Bernardi P, Ballarini M, Cappa A, Carenzi G, Colombo A, Contursi C, Di Lisa F, Dondio G, Gagliardi S, Milanesi E, Minucci S, Pain G, Pelicci PG, Saccani A, Storto M, Thaler F, Varasi M, Villa M, Plyte S. Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. J Med Chem 57: 5333–5347, 2014. [DOI] [PubMed] [Google Scholar]
- 183.Fang H, Chen M, Ding Y, Shang W, Xu J, Zhang X, Zhang W, Li K, Xiao Y, Gao F, Shang S, Li JC, Tian XL, Wang SQ, Zhou J, Weisleder N, Ma J, Ouyang K, Chen J, Wang X, Zheng M, Wang W, Zhang X, Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res 21: 1295–1304, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Feldmann G, Haouzi D, Moreau A, Durand SA, Bringuier A, Berson A, Mansouri A, Fau D, Pessayre D. Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology 31: 674–683, 2000. [DOI] [PubMed] [Google Scholar]
- 185.Feniouk BA, Rebecchi A, Giovannini D, Anefors S, Mulkidjanian AY, Junge W, Turina P, Melandri BA. Met23Lys mutation in subunit gamma of FOF1-ATP synthase from Rhodobacter capsulatus impairs the activation of ATP hydrolysis by protonmotive force. Biochim Biophys Acta 1767: 1319–1330, 2007. [DOI] [PubMed] [Google Scholar]
- 186.Ferrand-Drake M, Friberg H, Wieloch T. Mitochondrial permeability transition induced DNA-fragmentation in the rat hippocampus following hypoglycemia. Neuroscience 90: 1325–1338, 1999. [DOI] [PubMed] [Google Scholar]
- 187.Fillingame RH, Steed PR. Half channels mediating H+ transport and the mechanism of gating in the Fo sector of Escherichia coli F1Fo ATP synthase. Biochim Biophys Acta 1837: 1063–1068, 2014. [DOI] [PubMed] [Google Scholar]
- 188.Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 6: e23295, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337: 476–478, 1989. [DOI] [PubMed] [Google Scholar]
- 190.Fischer S, Gräber P, Turina P. The activity of the ATP synthase from Escherichia coli is regulated by the transmembrane proton motive force. J Biol Chem 275: 30157–30162, 2000. [DOI] [PubMed] [Google Scholar]
- 191.Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex I. J Biol Chem 273: 12662–12668, 1998. [DOI] [PubMed] [Google Scholar]
- 192.Formentini L, Pereira MP, Sánchez-Cenizo L, Santacatterina F, Lucas JJ, Navarro C, Martínez-Serrano A, Cuezva JM. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J 33: 762–778, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci USA 104: 7558–7563, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr 19: 297–303, 1987. [DOI] [PubMed] [Google Scholar]
- 195.Frasch WD. The participation of metals in the mechanism of the F1-ATPase. Biochim Biophys Acta 1458: 310–325, 2000. [DOI] [PubMed] [Google Scholar]
- 196.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci 25: 319–324, 2000. [DOI] [PubMed] [Google Scholar]
- 197.Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci 18: 5151–5159, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med 354: 942–955, 2006. [DOI] [PubMed] [Google Scholar]
- 199.Fronzes R, Weimann T, Vaillier J, Velours J, Brèthes D. The peripheral stalk participates in the yeast ATP synthase dimerization independently of e and g subunits. Biochemistry 45: 6715–6723, 2006. [DOI] [PubMed] [Google Scholar]
- 200.Fujimoto K, Chen Y, Polonsky KS, Dorn GW. Targeting cyclophilin D and the mitochondrial permeability transition enhances β-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci USA 107: 10214–10219, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fujimoto K, Ford EL, Tran H, Wice BM, Crosby SD, Dorn GW, Polonsky KS. Loss of Nix in Pdx1-deficient mice prevents apoptotic and necrotic β cell death and diabetes. J Clin Invest 120: 4031–4039, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Futai M, Nakanishi-Matsui M, Okamoto H, Sekiya M, Nakamoto RK. Rotational catalysis in proton pumping ATPases: from E. coli F-ATPase to mammalian V-ATPase. Biochim Biophys Acta 1817: 1711–1721, 2012. [DOI] [PubMed] [Google Scholar]
- 203.Gallerne C, Touat Z, Chen ZX, Martel C, Mayola E, Sharaf El Dein O, Buron N, Le Bras M, Jacotot E, Borgne-Sanchez A, Lemoine A, Lemaire C, Pervaiz S, Brenner C. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int J Biochem Cell Biol 42: 623–629, 2010. [DOI] [PubMed] [Google Scholar]
- 204.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 31: 1869–1883, 2012. [DOI] [PubMed] [Google Scholar]
- 205.Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33: 627–638, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Garcia JJ, Morales-Rios E, Cortés-Hernandez P, Rodriguez-Zavala JS. The inhibitor protein IF1 promotes dimerization of the mitochondrial F1F0-ATP synthase. Biochemistry 45: 12695–12703, 2006. [DOI] [PubMed] [Google Scholar]
- 207.Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem 285: 39646–39654, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Garzón JI, Kovacs J, Abagyan R, Chacón P. ADP_EM: fast exhaustive multi-resolution docking for high-throughput coverage. Bioinformatics 23: 427–433, 2007. [DOI] [PubMed] [Google Scholar]
- 209.Gautier CA, Giaime E, Caballero E, Nunez L, Song Z, Chan D, Villalobos C, Shen J. Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener 7: 22, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gavin PD, Devenish RJ, Prescott M. FRET reveals changes in the F1-stator stalk interaction during activity of F1F0-ATP synthase. Biochim Biophys Acta 1607: 167–179, 2003. [DOI] [PubMed] [Google Scholar]
- 211.Gavin PD, Prescott M, Devenish RJ. F1F0-ATP synthase complex interactions in vivo can occur in the absence of the dimer specific subunit e. J Bioenerg Biomembr 37: 55–66, 2005. [DOI] [PubMed] [Google Scholar]
- 212.Gellerich FN, Gizatullina Z, Nguyen HP, Trumbeckaite S, Vielhaber S, Seppet E, Zierz S, Landwehrmeyer B, Riess O, von Horsten S, Striggow F. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem 283: 30715–30724, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ghosh JC, Siegelin MD, Dohi T, Altieri DC. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res 70: 8988–8993, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gibbons C, Montgomery MG, Leslie AG, Walker JE. The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat Struct Biol 7: 1055–1061, 2000. [DOI] [PubMed] [Google Scholar]
- 215.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233, 2005. [DOI] [PubMed] [Google Scholar]
- 216.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284: 33982–33988, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Giorgio V, Soriano ME, Basso E, Bisetto E, Lippe G, Forte MA, Bernardi P. Cyclophilin D in mitochondrial pathophysiology. Biochim Biophys Acta 1797: 1113–1118, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110: 5887–5892, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Giraud MF, Paumard P, Sanchez C, Brèthes D, Velours J, Dautant A. Rotor architecture in the yeast and bovine F1-c-ring complexes of F-ATP synthase. J Struct Biol 177: 490–497, 2012. [DOI] [PubMed] [Google Scholar]
- 220.Giraud MF, Paumard P, Soubannier V, Vaillier J, Arselin G, Salin B, Schaeffer J, Brèthes D, di Rago JP, Velours J. Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim Biophys Acta 1555: 174–180, 2002. [DOI] [PubMed] [Google Scholar]
- 221.Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci USA 104: 13632–13637, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gonzalvez F, D'Aurelio M, Boutant M, Moustapha A, Puech JP, Landes T, Arnaune-Pelloquin L, Vial G, Taleux N, Slomianny C, Wanders RJ, Houtkooper RH, Bellenguer P, Moller IM, Gottlieb E, Vaz FM, Manfredi G, Petit PX. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta 1832: 1194–1206, 2013. [DOI] [PubMed] [Google Scholar]
- 223.Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem 78: 147–176, 2009. [DOI] [PubMed] [Google Scholar]
- 224.Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, Neville HE, Boylan K, Mozaffar T, Belsh JM, Ravits J, Bedlack RS, Graves MC, McCluskey LF, Barohn RJ, Tandan R. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 6: 1045–1053, 2007. [DOI] [PubMed] [Google Scholar]
- 225.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947, 2013. [DOI] [PubMed] [Google Scholar]
- 226.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 305: 626–629, 2004. [DOI] [PubMed] [Google Scholar]
- 227.Green DW, Grover GJ. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochim Biophys Acta 1458: 343–355, 2000. [DOI] [PubMed] [Google Scholar]
- 228.Green DR, Galluzzi L, Kroemer G. Metabolic control of cell death. Science 345: 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson's disease. IUBMB Life 52: 135–141, 2001. [DOI] [PubMed] [Google Scholar]
- 230.Grek CL, Tew KD. Redox metabolism and malignancy. Curr Opin Pharmacol 10: 362–368, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol 25: 1461–1469, 1993. [DOI] [PubMed] [Google Scholar]
- 232.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307: 93–98, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Grimm S, Brdiczka D. The permeability transition pore in cell death. Apoptosis 12: 841–855, 2007. [DOI] [PubMed] [Google Scholar]
- 234.Groebe K, Krause F, Kunstmann B, Unterluggauer H, Reifschneider NH, Scheckhuber CQ, Sastri C, Stegmann W, Wozny W, Schwall GP, Poznanovic S, Dencher NA, Jansen-Durr P, Osiewacz HD, Schrattenholz A. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp Gerontol 42: 887–898, 2007. [DOI] [PubMed] [Google Scholar]
- 235.Grosskreutz J, Van Den Bosch L, Keller BU. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47: 165–174, 2010. [DOI] [PubMed] [Google Scholar]
- 236.Guérin B, Bunoust O, Rouqueys V, Rigoulet M. ATP-induced unspecific channel in yeast mitochondria. J Biol Chem 269: 25406–25410, 1994. [PubMed] [Google Scholar]
- 237.Guex N, Diemand A, Peitsch MC. Protein modelling for all. Trends Biochem Sci 24: 364–367, 1999. [DOI] [PubMed] [Google Scholar]
- 238.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol Cell Physiol 267: C313–C339, 1994. [DOI] [PubMed] [Google Scholar]
- 239.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol 258: C755–C786, 1990. [DOI] [PubMed] [Google Scholar]
- 240.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta 1787: 1291–1308, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264: 1772–1775, 1994. [DOI] [PubMed] [Google Scholar]
- 242.Gutierrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol 72: 316–325, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Guzzo G, Sciacovelli M, Bernardi P, Rasola A. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget 5: 11897–11908, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol 8: 101–112, 2007. [DOI] [PubMed] [Google Scholar]
- 245.Habersetzer J, Larrieu I, Priault M, Salin B, Rossignol R, Brèthes D, Paumard P. Human F1F0 ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: a (super-)complex matter? PLoS One 8: e75429, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Habersetzer J, Ziani W, Larrieu I, Stines-Chaumeil C, Giraud MF, Brèthes D, Dautant A, Paumard P. ATP synthase oligomerization: from the enzyme models to the mitochondrial morphology. Int J Biochem Cell Biol 45: 99–105, 2013. [DOI] [PubMed] [Google Scholar]
- 247.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40: 553–560, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415–424, 1995. [DOI] [PubMed] [Google Scholar]
- 249.Halestrap AP. Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature 430: 983, 2004. [DOI] [PubMed] [Google Scholar]
- 250.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. [DOI] [PubMed] [Google Scholar]
- 251.Halestrap AP. The C ring of the F1Fo ATP synthase forms the mitochondrial permeability transition pore: a critical appraisal. Front Oncol 4: 234, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res 61: 372–385, 2004. [DOI] [PubMed] [Google Scholar]
- 253.Halestrap AP, Davidson AM. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J 268: 153–160, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78: 129–141, 2015. [DOI] [PubMed] [Google Scholar]
- 255.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011. [DOI] [PubMed] [Google Scholar]
- 256.Hansford RG. Effect of micromolar concentrations of free Ca2+ ions on pyruvate dehydrogenase interconversion in intact rat heart mitochondria. Biochem J 194: 721–732, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA 105: 13145–13150, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Haouzi D, Cohen I, Vieira HL, Poncet D, Boya P, Castedo M, Vadrot N, Belzacq AS, Fau D, Brenner C, Feldmann G, Kroemer G. Mitochondrial permeability transition as a novel principle of hepatorenal toxicity in vivo. Apoptosis 7: 395–405, 2002. [DOI] [PubMed] [Google Scholar]
- 259.Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J 280: 561–573, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Harris DA, Gomez-Fernandez JC, Klungsoyr L, Radda GK. Specificity of nucleotide binding and coupled reactions utilising the mitochondrial ATPase. Biochim Biophys Acta 504: 364–383, 1978. [DOI] [PubMed] [Google Scholar]
- 261.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 12: 217–234, 2007. [DOI] [PubMed] [Google Scholar]
- 262.Havlickova V, Kaplanova V, Nuskova H, Drahota Z, Houštek J. Knockdown of F1 ε subunit decreases mitochondrial content of ATP synthase and leads to accumulation of subunit c. Biochim Biophys Acta 1797: 1124–1129, 2010. [DOI] [PubMed] [Google Scholar]
- 263.Haworth RA, Hunter DR. The Ca2+-induced membrane transition of rat liver mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195: 460–467, 1979. [DOI] [PubMed] [Google Scholar]
- 264.Haynes V, Traaseth NJ, Elfering S, Fujisawa Y, Giulivi C. Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. Am J Physiol Endocrinol Metab 298: E978–E987, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 512: 1–7, 2002. [DOI] [PubMed] [Google Scholar]
- 266.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem 277: 432–438, 2001. [DOI] [PubMed] [Google Scholar]
- 267.Hejzlarová Mrácek T, Vrbacky M, Kaplanová V, Karbanová V, Nusková H, Pecina P, Houštek J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol Res 63 Suppl 1: S57–S71, 2014. [DOI] [PubMed] [Google Scholar]
- 268.Henderson PJ, Lardy HA. Bongkrekic acid an inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem 245: 1319–1326, 1970. [PubMed] [Google Scholar]
- 269.Herick K, Krämer R, Lühring H. Patch clamp investigation into the phosphate carrier from Saccharomyces cerevisiae mitochondria. Biochim Biophys Acta 1321: 207–220, 1997. [DOI] [PubMed] [Google Scholar]
- 270.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol 369–405, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Hirakawa A, Takeyama N, Nakatani T, Tanaka T. Mitochondrial permeability transition and cytochrome c release in ischemia-reperfusion injury of the rat liver. J Surg Res 111: 240–247, 2003. [DOI] [PubMed] [Google Scholar]
- 272.Hojlund K, Wrzesinski K, Larsen PM, Fey SJ, Roepstorff P, Handberg A, Dela F, Vinten J, McCormack JG, Reynet C, Beck-Nielsen H. Proteome analysis reveals phosphorylation of ATP synthase β-subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J Biol Chem 278: 10436–10442, 2003. [DOI] [PubMed] [Google Scholar]
- 273.Houštek J, Andersson U, Tvrdik P, Nedergaard J, Cannon B. The expression of subunit c correlates with and thus may limit the biosynthesis of the mitochondrial F0F1-ATPase in brown adipose tissue. J Biol Chem 270: 7689–7694, 1995. [DOI] [PubMed] [Google Scholar]
- 274.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 134: 703–707, 2008. [DOI] [PubMed] [Google Scholar]
- 275.Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-β-subunit is a calcium-binding protein. FEBS Lett 391: 323–329, 1996. [DOI] [PubMed] [Google Scholar]
- 276.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195: 453–459, 1979. [DOI] [PubMed] [Google Scholar]
- 277.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys 195: 468–477, 1979. [DOI] [PubMed] [Google Scholar]
- 278.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem 251: 5069–5077, 1976. [PubMed] [Google Scholar]
- 279.Hunter FE Jr, Ford L. Inactivation of oxidative and phosphorylative systems in mitochondria by preincubation with phosphate and other ions. J Biol Chem 216: 357–369, 1955. [PubMed] [Google Scholar]
- 280.Hüser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J 343: 311–317, 1999. [PMC free article] [PubMed] [Google Scholar]
- 281.Hüser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophys J 74: 2129–2137, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Huttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta 1817: 598–609, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Huttemann M, Lee I, Samavati L, Yu H, Doan JW. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim Biophys Acta 1773: 1701–1720, 2007. [DOI] [PubMed] [Google Scholar]
- 284.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89: 1145–1153, 1997. [DOI] [PubMed] [Google Scholar]
- 285.Imberti R, Nieminen AL, Herman B, Lemasters JJ. Synergism of cyclosporin A and phospholipase inhibitors in protection against lethal injury to rat hepatocytes from oxidant chemicals. Res Commun Chem Pathol Pharmacol 78: 27–38, 1992. [PubMed] [Google Scholar]
- 286.Inoue T, Suzuki Y, Mizuno K, Nakata K, Yoshimaru T, Ra C. SHP-1 exhibits a pro-apoptotic function in antigen-stimulated mast cells: positive regulation of mitochondrial death pathways and negative regulation of survival signaling pathways. Mol Immunol 47: 222–232, 2009. [DOI] [PubMed] [Google Scholar]
- 287.Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35: 267–271, 2003. [DOI] [PubMed] [Google Scholar]
- 288.Jault JM, Dou C, Grodsky NB, Matsui T, Yoshida M, Allison WS. The α3β3γ subcomplex of the F1-ATPase from the thermophilic bacillus PS3 with the βT165S substitution does not entrap inhibitory MgADP in a catalytic site during turnover. J Biol Chem 271: 28818–28824, 1996. [DOI] [PubMed] [Google Scholar]
- 289.Johans M, Milanesi E, Franck M, Johans C, Liobikas J, Panagiotaki M, Greci L, Principato G, Kinnunen PKJ, Bernardi P, Costantini P, Eriksson O. Modification of permeability transition pore arginine(s) by phenylglyoxal derivatives in isolated mitochondria and mammalian cells: structure-function relationship of arginine ligands. J Biol Chem 280: 12130–12136, 2005. [DOI] [PubMed] [Google Scholar]
- 290.Johnson KM, Chen X, Boitano A, Swenson L, Opipari AW Jr, Glick GD. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem Biol 12: 485–496, 2005. [DOI] [PubMed] [Google Scholar]
- 291.Johnson KM, Cleary J, Fierke CA, Opipari AW Jr, Glick GD. Mechanistic basis for therapeutic targeting of the mitochondrial F1F0-ATPase. ACS Chem Biol 1: 304–308, 2006. [DOI] [PubMed] [Google Scholar]
- 292.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004. [DOI] [PubMed] [Google Scholar]
- 293.Joshi S, Hughes JB. Inhibition of coupling factor B activity by cadmium ion, arsenite-2,3-dimercaptopropanol, and phenylarsine oxide, and preferential reactivation by dithiols. J Biol Chem 256: 11112–11116, 1981. [PubMed] [Google Scholar]
- 294.Joshi S, Hughes JB, Shaikh F, Sanadi DR. On the role of coupling factor B in the mitochondrial Pi-ATP exchange reaction. J Biol Chem 254: 10145–10152, 1979. [PubMed] [Google Scholar]
- 295.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3β in cardioprotection. Circ Res 104: 1240–1252, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Jung DW, Bradshaw PC, Pfeiffer DR. Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J Biol Chem 272: 21104–21112, 1997. [DOI] [PubMed] [Google Scholar]
- 298.Junge W, Sielaff H, Engelbrecht S. Torque generation and elastic power transmission in the rotary FOF1-ATPase. Nature 459: 364–370, 2009. [DOI] [PubMed] [Google Scholar]
- 299.Kabaleeswaran V, Puri N, Walker JE, Leslie AG, Mueller DM. Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J 25: 5433–5442, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kabaleeswaran V, Shen H, Symersky J, Walker JE, Leslie AG, Mueller DM. Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides. J Biol Chem 284: 10546–10551, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kagawa R, Montgomery MG, Braig K, Leslie AG, Walker JE. The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J 23: 2734–2744, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Kagawa Y, Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. 8. Properties of a factor conferring oligomycin sensitivity on mitochondrial adenosine triphosphatase. J Biol Chem 241: 2461–2466, 1966. [PubMed] [Google Scholar]
- 303.Kanazawa H, Horiuchi Y, Takagi M, Ishino Y, Futai M. Coupling factor F1 ATPase with defective β subunit from a mutant of Escherichia coli. J Biochem 88: 695–703, 1980. [DOI] [PubMed] [Google Scholar]
- 304.Kane LA, Van Eyk JE. Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr 41: 145–150, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 131: 257–270, 2007. [DOI] [PubMed] [Google Scholar]
- 306.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2: e00772, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kawajiri S, Saiki S, Sato S, Hattori N. Genetic mutations and functions of PINK1. Trends Pharmacol Sci 32: 573–580, 2011. [DOI] [PubMed] [Google Scholar]
- 308.Kawakami T, Sato S, Suzuki K. Beneficial effect of Cyclosporin A on acute hepatic injury induced by galactosamine and lipopolysaccharide in rats. Hepatol Res 18: 284–297, 2000. [DOI] [PubMed] [Google Scholar]
- 309.Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev 131: 517–526, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Khananshvili D, Gromet-Elhanan Z. Evidence that the Mg-dependent low-affinity binding site for ATP and Pi demonstrated on the isolated beta subunit of the F0F1 ATP synthase is a catalytic site. Proc Natl Acad Sci USA 82: 1886–1890, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Khaspekov L, Friberg H, Halestrap A, Viktorov I, Wieloch T. Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons. Eur J Neurosci 11: 3194–3198, 1999. [DOI] [PubMed] [Google Scholar]
- 312.Kim HJ, Winge DR. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim Biophys Acta 1827: 627–636, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology 124: 494–503, 2003. [DOI] [PubMed] [Google Scholar]
- 314.Kim YH, Haidl G, Schaefer M, Egner U, Mandal A, Herr JC. Compartmentalization of a unique ADP/ATP carrier protein SFEC (Sperm Flagellar Energy Carrier, AAC4) with glycolytic enzymes in the fibrous sheath of the human sperm flagellar principal piece. Dev Biol 302: 463–476, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503, 1989. [DOI] [PubMed] [Google Scholar]
- 316.King TD, Clodfelder-Miller B, Barksdale KA, Bijur GN. Unregulated mitochondrial GSK3β activity results in NADH: ubiquinone oxidoreductase deficiency. Neurotox Res 14: 367–382, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kinnally KW, Campo ML, Tedeschi H. Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr 21: 497–506, 1989. [DOI] [PubMed] [Google Scholar]
- 318.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta 1813: 616–622, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW, Tedeschi H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci USA 90: 1374–1378, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kironde FA, Cross RL. Adenine nucleotide binding sites on beef heart F1-ATPase. Asymmetry and subunit location. J Biol Chem 262: 3488–3495, 1987. [PubMed] [Google Scholar]
- 321.Kiselyov K, Muallem S. Mitochondrial Ca2+ homeostasis in lysosomal storage diseases. Cell Calcium 44: 103–111, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Klöhn PC, Soriano ME, Irwin W, Penzo D, Scorrano L, Bitsch A, Neumann HG, Bernardi P. Early resistance to cell death and to onset of the mitochondrial permeability transition during hepatocarcinogenesis with 2-acetylaminofluorene. Proc Natl Acad Sci USA 100: 10014–10019, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Ko YH, Pan W, Inoue C, Pedersen PL. Signal transduction to mitochondrial ATP synthase: evidence that PDGF-dependent phosphorylation of the δ-subunit occurs in several cell lines, involves tyrosine, and is modulated by lysophosphatidic acid. Mitochondrion 1: 339–348, 2002. [DOI] [PubMed] [Google Scholar]
- 324.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol 300: H1327–H1335, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40: 1170–1179, 2004. [DOI] [PubMed] [Google Scholar]
- 327.Korde AS, Pettigrew LC, Craddock SD, Pocernich CB, Waldmeier PC, Maragos WF. Protective effects of NIM811 in transient focal cerebral ischemia suggest involvement of the mitochondrial permeability transition. J Neurotrauma 24: 895–908, 2007. [DOI] [PubMed] [Google Scholar]
- 328.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18, 1997. [DOI] [PubMed] [Google Scholar]
- 329.Kottke M, Adam V, Riesinger I, Bremm G, Bosch W, Brdiczka D, Sandri G, Panfili E. Mitochondrial boundary membrane contact sites in brain: points of hexokinase and creatine kinase location, and control of Ca2+ transport. Biochim Biophys Acta 935: 87–102, 1988. [DOI] [PubMed] [Google Scholar]
- 330.Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett 495: 12–15, 2001. [DOI] [PubMed] [Google Scholar]
- 331.Kramarova TV, Shabalina IG, Andersson U, Westerberg R, Carlberg I, Houštek J, Nedergaard J, Cannon B. Mitochondrial ATP synthase levels in brown adipose tissue are governed by the c-Fo subunit P1 isoform. FASEB J 22: 55–63, 2008. [DOI] [PubMed] [Google Scholar]
- 332.Krämer R. Mitochondrial carrier proteins can reversibly change their transport mode: the cases of the aspartate/glutamate and the phosphate carrier. Exp Physiol 83: 259–265, 1998. [DOI] [PubMed] [Google Scholar]
- 333.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1−/− mitochondria. Biochim Biophys Acta 1757: 590–595, 2006. [DOI] [PubMed] [Google Scholar]
- 334.Krestinina OV, Grachev DE, Odinokova IV, Reiser G, Evtodienko YV, Azarashvili TS. Effect of peripheral benzodiazepine receptor (PBR/TSPO) ligands on opening of Ca2+-induced pore and phosphorylation of 3.5-kDa polypeptide in rat brain mitochondria. Biochemistry 74: 421–429, 2009. [DOI] [PubMed] [Google Scholar]
- 335.Krishnamurthy P, Xie T, Schuetz JD. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther 114: 345–358, 2007. [DOI] [PubMed] [Google Scholar]
- 336.Kristal BS, Matsuda M, Yu BP. Abnormalities in the mitochondrial permeability transition in diabetic rats. Biochem Biophys Res Commun 222: 519–523, 1996. [DOI] [PubMed] [Google Scholar]
- 337.Kristian T, Bernardi P, Siesjö BK. Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J Neurotrauma 18: 1059–1074, 2001. [DOI] [PubMed] [Google Scholar]
- 338.Kupsch K, Hertel S, Kreutzmann P, Wolf G, Wallesch CW, Siemen D, Schönfeld P. Impairment of mitochondrial function by minocycline. FEBS J 276: 1729–1738, 2009. [DOI] [PubMed] [Google Scholar]
- 339.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 21: 1209–1217, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lablanche S, Cottet-Rousselle C, Lamarche F, Benhamou PY, Halimi S, Leverve X, Fontaine E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis 2: e134, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet 42: 673–685, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep 5: 958–963, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lapidus RG, Sokolove PM. Spermine inhibition of the permeability transition of isolated rat liver mitochondria: an investigation of mechanism. Arch Biochem Biophys 306: 246–253, 1993. [DOI] [PubMed] [Google Scholar]
- 344.Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, Chopin C, Neviere R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol 48: 377–385, 2006. [DOI] [PubMed] [Google Scholar]
- 345.Lau WC, Baker LA, Rubinstein JL. Cryo-EM structure of the yeast ATP synthase. J Mol Biol 382: 1256–1264, 2008. [DOI] [PubMed] [Google Scholar]
- 346.Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol Cell 53: 330–343, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Lê-Quôc K, Lê-Quôc D. Crucial role of sulfhydryl groups in the mitochondrial inner membrane structure. J Biol Chem 260: 7422–7428, 1985. [PubMed] [Google Scholar]
- 348.Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabó I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol Med 4: 577–593, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Leanza L, Trentin L, Becker KA, Frezzato F, Zoratti M, Semenzato G, Gulbins E, Szabó I. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 27: 1782–1785, 2013. [DOI] [PubMed] [Google Scholar]
- 350.Leanza L, Zoratti M, Gulbins E, Szabó I. Mitochondrial ion channels as oncological targets. Oncogene 33: 5569–5581, 2014. [DOI] [PubMed] [Google Scholar]
- 351.Lee J, Ding S, Walpole TB, Holding AN, Montgomery MG, Fearnley IM, Walker JE. Organisation of subunits in the membrane domain of the bovine F-ATPase revealed by covalent cross-linking. J Biol Chem 290: 13308–13320, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Lee JK, Belogrudov GI, Stroud RM. Crystal structure of bovine mitochondrial factor B at 0.96-A resolution. Proc Natl Acad Sci USA 105: 13379–13384, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Leger PL, De Paulis D, Branco S, Bonnin P, Couture-Lepetit E, Baud O, Renolleau S, Ovize M, Gharib A, Charriaut-Marlangue C. Evaluation of cyclosporine A in a stroke model in the immature rat brain. Exp Neurol 230: 58–66, 2011. [DOI] [PubMed] [Google Scholar]
- 354.Lehninger AL, Remmert LF. An endogenous uncoupling and swelling agent in liver mitochondria and its enzymic function. J Biol Chem 234: 2459–2464, 1959. [PubMed] [Google Scholar]
- 355.Lehninger AL. Reversal of various types of mitochondrial swelling by adenosine triphosphate. J Biol Chem 234: 2465–2471, 1959. [PubMed] [Google Scholar]
- 356.Leist M, Gantner F, Kunstle G, Wendel A. Cytokine-mediated hepatic apoptosis. Rev Physiol Biochem Pharmacol 133: 109–155, 1998. [DOI] [PubMed] [Google Scholar]
- 357.Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J Gastroenterol Hepatol 22 Suppl 1: S31–S37, 2007. [DOI] [PubMed] [Google Scholar]
- 358.Lenartowicz E, Bernardi P, Azzone GF. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J Bioenerg Biomembr 23: 679–688, 1991. [DOI] [PubMed] [Google Scholar]
- 359.Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta 1777: 946–952, 2008. [DOI] [PubMed] [Google Scholar]
- 360.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 283: 26312–26323, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Li B, Chauvin C, De Paulis D, De Oliveira F, Gharib A, Vial G, Lablanche S, Leverve X, Bernardi P, Ovize M, Fontaine E. Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim Biophys Acta 1817: 1628–1634, 2012. [DOI] [PubMed] [Google Scholar]
- 362.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412: 95–99, 2001. [DOI] [PubMed] [Google Scholar]
- 363.Li PA, Uchino H, Elmer E, Siesjö BK. Amelioration by cyclosporin A of brain damage following 5 or 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res 753: 133–140, 1997. [DOI] [PubMed] [Google Scholar]
- 364.Lim CH, Hamazaki T, Braun EL, Wade J, Terada N. Evolutionary genomics implies a specific function of Ant4 in mammalian and anole lizard male germ cells. PLoS One 6: e23122, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lim D, Fedrizzi L, Tartari M, Zuccato C, Cattaneo E, Brini M, Carafoli E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem 283: 5780–5789, 2008. [DOI] [PubMed] [Google Scholar]
- 366.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res 75: 530–535, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Lim SY, Hausenloy DJ, Arjun S, Price AN, Davidson SM, Lythgoe MF, Yellon DM. Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J Cell Mol Med 15: 2443–2451, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Linder MD, Morkunaite-Haimi S, Kinnunen PKJ, Bernardi P, Eriksson O. Ligand-selective modulation of the permeability transition pore by arginine modification. Opposing effects of p-hydroxyphenylglyoxal and phenylglyoxal. J Biol Chem 277: 937–942, 2002. [DOI] [PubMed] [Google Scholar]
- 369.Lippe G, Comelli M, Mazzilis D, Sala FD, Mavelli I. The inactivation of mitochondrial F1 ATPase by H2O2 is mediated by iron ions not tightly bound in the protein. Biochem Biophys Res Commun 181: 764–770, 1991. [DOI] [PubMed] [Google Scholar]
- 370.Lippe G, Dabbeni-Sala F, Sorgato MC. ATP synthase complex from beef heart mitochondria. Role of the thiol group of the 25-kDa subunit of Fo in the coupling mechanism between Fo and F1. J Biol Chem 263: 18627–18634, 1988. [PubMed] [Google Scholar]
- 371.Liu J, Farmer JDJ, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66: 807–815, 1991. [DOI] [PubMed] [Google Scholar]
- 372.Liu S, Charlesworth TJ, Bason JV, Montgomery MG, Harbour ME, Fearnley IM, Walker JE. The purification and characterization of ATP synthase complexes from the mitochondria of four fungal species. Biochem J 468: 167–175, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157, 1996. [DOI] [PubMed] [Google Scholar]
- 374.Loupatatzis C, Seitz G, Schönfeld P, Lang F, Siemen D. Single-channel currents of the permeability transition pore from the inner mitochondrial membrane of rat liver and of a human hepatoma cell line. Cell Physiol Biochem 12: 269–278, 2002. [DOI] [PubMed] [Google Scholar]
- 375.Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev 21: 784–796, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289: 21490–21507, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Mai MS, Allison WS. Inhibition of an oligomycin-sensitive ATPase by cationic dyes, some of which are atypical uncouplers of intact mitochondria. Arch Biochem Biophys 221: 467–476, 1983. [DOI] [PubMed] [Google Scholar]
- 378.Majima E, Ikawa K, Takeda M, Hashimoto M, Shinohara Y, Terada H. Translocation of loops regulates transport activity of mitochondrial ADP/ATP carrier deduced from formation of a specific intermolecular disulfide bridge catalyzed by copper-o-phenanthroline. J Biol Chem 270: 29548–29554, 1995. [DOI] [PubMed] [Google Scholar]
- 379.Malaguti C, La Guardia PG, Leite AC, Oliveira DN, de Lima Zollner RL, Catharino RR, Vercesi AE, Oliveira HC. Oxidative stress and susceptibility to mitochondrial permeability transition precedes the onset of diabetes in autoimmune non-obese diabetic mice. Free Radic Res 48: 1494–1504, 2014. [DOI] [PubMed] [Google Scholar]
- 380.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Manon S, Roucou X, Guerin M, Rigoulet M, Guerin B. Characterization of the yeast mitochondria unselective channel: a counterpart to the mammalian permeability transition pore? J Bioenerg Biomembr 30: 419–429, 1998. [DOI] [PubMed] [Google Scholar]
- 382.Martin J, Hudson J, Hornung T, Frasch WD. Fo-driven rotation in the ATP synthase direction against the force of F1 ATPase in the F0F1 ATP synthase. J Biol Chem 290: 10717–10728, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol 218: 333–346, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Martin LJ, Semenkow S, Hanaford A, Wong M. Mitochondrial permeability transition pore regulates Parkinson's disease development in mutant α-synuclein transgenic mice. Neurobiol Aging 35: 1132–1152, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Martinez LO, Najib S, Perret B, Cabou C, Lichtenstein L. Ecto-F-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis 238: 89–100, 2014. [DOI] [PubMed] [Google Scholar]
- 386.Marzano C, Ronconi L, Chiara F, Giron MC, Faustinelli I, Cristofori P, Trevisan A, Fregona D. Gold(III)-dithiocarbamato anticancer agents: activity, toxicology and histopathological studies in rodents. Int J Cancer 129: 487–496, 2011. [DOI] [PubMed] [Google Scholar]
- 387.Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med 187: 1261–1271, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Masaike T, Koyama-Horibe F, Oiwa K, Yoshida M, Nishizaka T. Cooperative three-step motions in catalytic subunits of F1-ATPase correlate with 80° and 40° substep rotations. Nat Struct Mol Biol 15: 1326–1333, 2008. [DOI] [PubMed] [Google Scholar]
- 389.Masgras I, Rasola A, Bernardi P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochim Biophys Acta 1817: 1860–1866, 2012. [DOI] [PubMed] [Google Scholar]
- 390.Massari S, Azzone GF. The equivalent pore radius of intact and damaged mitochondria and the mechanism of active shrinkage. Biochim Biophys Acta 283: 23–29, 1972. [DOI] [PubMed] [Google Scholar]
- 391.Masubuchi Y, Nakayama S, Horie T. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 35: 544–551, 2002. [DOI] [PubMed] [Google Scholar]
- 392.Masubuchi Y, Suda C, Horie T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J Hepatol 42: 110–116, 2005. [DOI] [PubMed] [Google Scholar]
- 393.Masuda Y, Shima G, Aiuchi T, Horie M, Hori K, Nakajo S, Kajimoto S, Shibayama-Imazu T, Nakaya K. Involvement of tumor necrosis factor receptor-associated protein 1 (TRAP1) in apoptosis induced by β-hydroxyisovalerylshikonin. J Biol Chem 279: 42503–42515, 2004. [DOI] [PubMed] [Google Scholar]
- 394.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25: 4777–4786, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta 1797: 1225–1230, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Matsuda C, Endo H, Ohta S, Kagawa Y. Gene structure of human mitochondrial ATP synthase γ-subunit. Tissue specificity produced by alternative RNA splicing. J Biol Chem 268: 24950–24958, 1993. [PubMed] [Google Scholar]
- 397.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J 180: 533–544, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773: 1263–1284, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA 89: 3170–3174, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.McGeoch JE, Guidotti GA. 0.1–700 Hz current through a voltage-clamped pore: candidate protein for initiator of neural oscillations. Brain Res 766: 188–194, 1997. [DOI] [PubMed] [Google Scholar]
- 401.McGeoch JE, Guidotti G. Batten disease and the control of the Fo subunit c pore by cGMP and calcium. Eur J Paediatr Neurol 5 Suppl A: 147–150, 2001. [DOI] [PubMed] [Google Scholar]
- 402.McGeoch JE, McGeoch MW, Mao R, Guidotti G. Opposing actions of cGMP and calcium on the conductance of the F0 subunit c pore. Biochem Biophys Res Commun 274: 835–840, 2000. [DOI] [PubMed] [Google Scholar]
- 403.Means JC, Muro I, Clem RJ. Lack of involvement of mitochondrial factors in caspase activation in a Drosophila cell-free system. Cell Death Differ 13: 1222–1234, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Meier T, Matthey U, Henzen F, Dimroth P, Müller DJ. The central plug in the reconstituted undecameric c cylinder of a bacterial ATP synthase consists of phospholipids. FEBS Lett 505: 353–356, 2001. [DOI] [PubMed] [Google Scholar]
- 405.Menz RI, Walker JE, Leslie AG. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell 106: 331–341, 2001. [DOI] [PubMed] [Google Scholar]
- 406.Menze MA, Hutchinson K, Laborde SM, Hand SC. Mitochondrial permeability transition in the crustacean Artemia franciscana: absence of a calcium-regulated pore in the face of profound calcium storage. Am J Physiol Regul Integr Comp Physiol 289: R68–R76, 2005. [DOI] [PubMed] [Google Scholar]
- 407.Merlini L, Angelin A, Tiepolo T, Braghetta P, Sabatelli P, Zamparelli A, Ferlini A, Maraldi NM, Bonaldo P, Bernardi P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci USA 105: 5225–5229, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Meyer B, Wittig I, Trifilieff E, Karas M, Schägger H. Identification of two proteins associated with mammalian ATP synthase. Mol Cell Proteomics 6: 1690–1699, 2007. [DOI] [PubMed] [Google Scholar]
- 409.Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, Semenzato G, Cesura AM, Pinard E, Bernardi P. The mitochondrial effects of small organic ligands of BCL-2: sensitization of BCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6-trione derivative. J Biol Chem 281: 10066–10072, 2006. [DOI] [PubMed] [Google Scholar]
- 410.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 14: 442–447, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Miller WL. Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol 379: 62–73, 2013. [DOI] [PubMed] [Google Scholar]
- 412.Minauro-Sanmiguel F, Wilkens S, Garcia JJ. Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc Natl Acad Sci USA 102: 12356–12358, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Mironova GD, Gateau-Roesch O, Levrat C, Gritsenko E, Pavlov E, Lazareva AV, Limarenko E, Rey C, Louisot P, Saris NE. Palmitic and stearic acids bind Ca2+ with high affinity and form nonspecific channels in black-lipid membranes. Possible relation to Ca2+-activated mitochondrial pores. J Bioenerg Biomembr 33: 319–331, 2001. [DOI] [PubMed] [Google Scholar]
- 414.Mitchell P. Keilin's respiratory chain concept and its chemiosmotic consequences. Science 206: 1148–1159, 1979. [DOI] [PubMed] [Google Scholar]
- 415.Miura T, Miki T. GSK-3β, a therapeutic target for cardiomyocyte protection. Circ J 73: 1184–1192, 2009. [DOI] [PubMed] [Google Scholar]
- 416.Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res 94: 181–189, 2012. [DOI] [PubMed] [Google Scholar]
- 417.Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res 88: 7–15, 2010. [DOI] [PubMed] [Google Scholar]
- 418.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ 15: 521–529, 2008. [DOI] [PubMed] [Google Scholar]
- 419.Mnatsakanyan N, Kemboi SK, Salas J, Weber J. The β subunit loop that couples catalysis and rotation in ATP synthase has a critical length. J Biol Chem 286: 29788–29796, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Montesano Gesualdi N, Chirico G, Pirozzi G, Costantino E, Landriscina M, Esposito F. Tumor necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress 10: 342–350, 2007. [DOI] [PubMed] [Google Scholar]
- 421.Moreno AJ, Moreira PI, Custodio JB, Santos MS. Mechanism of inhibition of mitochondrial ATP synthase by 17β-estradiol. J Bioenerg Biomembr 45: 261–270, 2013. [DOI] [PubMed] [Google Scholar]
- 422.Morin D, Pires F, Plin C, Tillement JP. Role of the permeability transition pore in cytochrome C release from mitochondria during ischemia-reperfusion in rat liver. Biochem Pharmacol 68: 2065–2073, 2004. [DOI] [PubMed] [Google Scholar]
- 423.Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 155: 89–97, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Morra G, Potestio R, Micheletti C, Colombo G. Corresponding functional dynamics across the Hsp90 Chaperone family: insights from a multiscale analysis of MD simulations. PLoS Comput Biol 8: e1002433, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Moyle J, Mitchell P. Active/inactive state transitions of mitochondrial ATPase molecules influenced by Mg2+, anions and aurovertin. FEBS Lett 56: 55–61, 1975. [DOI] [PubMed] [Google Scholar]
- 426.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. [DOI] [PubMed] [Google Scholar]
- 427.Nakamura J, Fujikawa M, Yoshida M. IF1, a natural inhibitor of mitochondrial ATP synthase, is not essential for the normal growth and breeding of mice. Biosci Rep 33: 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Nam K, Pu J, Karplus M. Trapping the ATP binding state leads to a detailed understanding of the F1-ATPase mechanism. Proc Natl Acad Sci USA 111: 17851–17856, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Nathanson L, Gromet-Elhanan Z. Mutations in the beta-subunit Thr159 and Glu184 of the Rhodospirillum rubrum F0F1 ATP synthase reveal differences in ligands for the coupled Mg2+- and decoupled Ca2+-dependent F0F1 activities. J Biol Chem 275: 901–905, 2000. [DOI] [PubMed] [Google Scholar]
- 430.Nguyen T, Ogbi M, Johnson JA. Delta protein kinase C interacts with the d subunit of the F1F0 ATPase in neonatal cardiac myocytes exposed to hypoxia or phorbol ester. Implications for F1F0 ATPase regulation. J Biol Chem 283: 29831–29840, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286: 40184–40192, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem 271: 2185–2192, 1996. [DOI] [PubMed] [Google Scholar]
- 433.Nicolli A, Petronilli V, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 32: 4461–4465, 1993. [DOI] [PubMed] [Google Scholar]
- 434.Noji H, Yasuda R, Yoshida M, Kinosita K Jr. Direct observation of the rotation of F1-ATPase. Nature 386: 299–302, 1997. [DOI] [PubMed] [Google Scholar]
- 435.Norenberg MD, Rama Rao KV, Jayakumar AR. Ammonia neurotoxicity and the mitochondrial permeability transition. J Bioenerg Biomembr 36: 303–307, 2004. [DOI] [PubMed] [Google Scholar]
- 436.Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int 50: 983–997, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Noumi T, Mosher ME, Natori S, Futai M, Kanazawa H. A phenylalanine for serine substitution in the β subunit of Escherichia coli F1-ATPase affects dependence of its activity on divalent cations. J Biol Chem 259: 10071–10075, 1984. [PubMed] [Google Scholar]
- 438.Nowikovsky K, Schweyen RJ, Bernardi P. Pathophysiology of mitochondrial volume homeostasis: potassium transport and permeability transition. Biochim Biophys Acta 1787: 345–350, 2009. [DOI] [PubMed] [Google Scholar]
- 439.Oberst A, Bender C, Green DR. Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ 15: 1139–1146, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Ohori K, Miura T, Tanno M, Miki T, Sato T, Ishikawa S, Horio Y, Shimamoto K. Ser9 phosphorylation of mitochondrial GSK-3β is a primary mechanism of cardiomyocyte protection by erythropoietin against oxidant-induced apoptosis. Am J Physiol Heart Circ Physiol 295: H2079–H2086, 2008. [DOI] [PubMed] [Google Scholar]
- 441.Ohsakaya S, Fujikawa M, Hisabori T, Yoshida M. Knockdown of DAPIT (diabetes-associated protein in insulin-sensitive tissue) results in loss of ATP synthase in mitochondria. J Biol Chem 286: 20292–20296, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Orriss GL, Leslie AG, Braig K, Walker JE. Bovine F1-ATPase covalently inhibited with 4-chloro-7-nitrobenzofurazan: the structure provides further support for a rotary catalytic mechanism. Structure 6: 831–837, 1998. [DOI] [PubMed] [Google Scholar]
- 443.Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, Waddington SN, Schapira AH, Duchen MR. Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson's disease. Cell Metab 17: 941–953, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99: 1259–1263, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Palma E, Tiepolo T, Angelin A, Sabatelli P, Maraldi NM, Basso E, Forte MA, Bernardi P, Bonaldo P. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum Mol Genet 18: 2024–2031, 2009. [DOI] [PubMed] [Google Scholar]
- 446.Palmer DN, Barry LA, Tyynelä J, Cooper JD. NCL disease mechanisms. Biochim Biophys Acta 1832: 1882–1893, 2013. [DOI] [PubMed] [Google Scholar]
- 447.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107: 436–441, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Panov AV, Lund S, Greenamyre JT. Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington's disease individuals. Mol Cell Biochem 269: 143–152, 2005. [DOI] [PubMed] [Google Scholar]
- 449.Pantic B, Trevisan E, Citta A, Rigobello MP, Marin O, Bernardi P, Salvatori S, Rasola A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis 4: e858, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Papadopoulos V, Amri H, Boujrad N, Cascio C, Culty M, Garnier M, Hardwick M, Li H, Vidic B, Brown AS, Reversa JL, Bernassau JM, Drieu K. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 62: 21–28, 1997. [DOI] [PubMed] [Google Scholar]
- 451.Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27: 402–409, 2006. [DOI] [PubMed] [Google Scholar]
- 452.Papageorgiou S, Melandri AB, Solaini G. Relevance of divalent cations to ATP-driven proton pumping in beef heart mitochondrial F0F1-ATPase. J Bioenerg Biomembr 30: 533–541, 1998. [DOI] [PubMed] [Google Scholar]
- 453.Park JS, Pasupulati R, Feldkamp T, Roeser NF, Weinberg JM. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol 301: F134–F150, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33: 4657–4671, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Parvez S, Winkler-Stuck K, Hertel S, Schönfeld P, Siemen D. The dopamine-D2-receptor agonist ropinirole dose-dependently blocks the Ca2+-triggered permeability transition of mitochondria. Biochim Biophys Acta 1797: 1245–1250, 2010. [DOI] [PubMed] [Google Scholar]
- 456.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7: 710–723, 2006. [DOI] [PubMed] [Google Scholar]
- 457.Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem 10: 1535–1551, 2003. [DOI] [PubMed] [Google Scholar]
- 458.Pastorino JG, Simbula G, Gilfor E, Hoek JB, Farber JL. Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J Biol Chem 269: 31041–31046, 1994. [PubMed] [Google Scholar]
- 459.Pastorino JG, Snyder JW, Serroni A, Hoek JB, Farber JL. Cyclosporin and carnitine prevent the anoxic death of cultured hepatocytes by inhibiting the mitochondrial permeability transition. J Biol Chem 268: 13791–13798, 1993. [PubMed] [Google Scholar]
- 460.Paumard P, Arselin G, Vaillier J, Chaignepain S, Bathany K, Schmitter JM, Brèthes D, Velours J. Two ATP synthases can be linked through subunits i in the inner mitochondrial membrane of Saccharomyces cerevisiae. Biochemistry 41: 10390–10396, 2002. [DOI] [PubMed] [Google Scholar]
- 461.Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brèthes D, di Rago JP, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J 21: 221–230, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Paumard P, Vaillier J, Napias C, Arselin G, Brèthes D, Graves PV, Velours J. Environmental study of subunit i, a Fo component of the yeast ATP synthase. Biochemistry 39: 4199–4205, 2000. [DOI] [PubMed] [Google Scholar]
- 463.Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ. A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys J 88: 2614–2625, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Pemberton TJ, Kay JE. Identification and comparative analysis of the peptidyl-prolyl cis/trans isomerase repertoires of H. sapiens, D. melanogaster, C. elegans, S. cerevisiae and S. pombe. Comp Funct Genomics 6: 277–300, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Penzo D, Petronilli V, Angelin A, Cusan C, Colonna R, Scorrano L, Pagano F, Prato M, Di Lisa F, Bernardi P. Arachidonic acid released by phospholipase A2 activation triggers Ca2+-dependent apoptosis through the mitochondrial pathway. J Biol Chem 279: 25219–25225, 2004. [DOI] [PubMed] [Google Scholar]
- 466.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467: 291–296, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Petit-Paitel A, Brau F, Cazareth J, Chabry J. Involvment of cytosolic and mitochondrial GSK-3β in mitochondrial dysfunction and neuronal cell death of MPTP/MPP-treated neurons. PLoS One 4: e5491, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Petronilli V, Cola C, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+. J Biol Chem 268: 1011–1016, 1993. [PubMed] [Google Scholar]
- 469.Petronilli V, Cola C, Massari S, Colonna R, Bernardi P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J Biol Chem 268: 21939–21945, 1993. [PubMed] [Google Scholar]
- 470.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem 269: 16638–16642, 1994. [PubMed] [Google Scholar]
- 471.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76: 725–734, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta 1187: 255–259, 1994. [DOI] [PubMed] [Google Scholar]
- 473.Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 276: 12030–12034, 2001. [DOI] [PubMed] [Google Scholar]
- 474.Petronilli V, Šileikyte J, Zulian A, Dabbeni-Sala F, Jori G, Gobbo S, Tognon G, Nikolov P, Bernardi P, Ricchelli F. Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochim Biophys Acta 1787: 897–904, 2009. [DOI] [PubMed] [Google Scholar]
- 475.Petronilli V, Szabó I, Zoratti M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 259: 137–143, 1989. [DOI] [PubMed] [Google Scholar]
- 476.Pfeiffer DR, Gunter TE, Eliseev R, Broekemeier KM, Gunter KK. Release of Ca2+ from mitochondria via the saturable mechanisms and the permeability transition. IUBMB Life 52: 205–212, 2001. [DOI] [PubMed] [Google Scholar]
- 477.Pfeiffer DR, Kuo TH, Tchen TT. Some effects of Ca2+, Mg2+, and Mn2+ on the ultrastructure, light-scattering properties, and malic enzyme activity of adrenal cortex mitochondria. Arch Biochem Biophys 176: 556–563, 1976. [DOI] [PubMed] [Google Scholar]
- 478.Pfeiffer DR, Schmid PC, Beatrice MC, Schmid HH. Intramitochondrial phospholipase activity and the effects of Ca2+ plus N-ethylmaleimide on mitochondrial function. J Biol Chem 254: 11485–11494, 1979. [PubMed] [Google Scholar]
- 479.Pinton P, Kroemer G. Cancer therapy: altering mitochondrial properties. Nat Chem Biol 10: 89–90, 2014. [DOI] [PubMed] [Google Scholar]
- 480.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481, 2008. [DOI] [PubMed] [Google Scholar]
- 481.Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J 277: 3622–3636, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Poon HF, Calabrese V, Calvani M, Butterfield DA. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by l-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal 8: 381–394, 2006. [DOI] [PubMed] [Google Scholar]
- 483.Prieto S, Bouillaud F, Rial E. The nature and regulation of the ATP-induced anion permeability in Saccharomyces cerevisiae mitochondria. Arch Biochem Biophys 334: 43–49, 1996. [DOI] [PubMed] [Google Scholar]
- 484.Prieto S, Bouillaud F, Ricquier D, Rial E. Activation by ATP of a proton-conducting pathway in yeast mitochondria. Eur J Biochem 208: 487–491, 1992. [DOI] [PubMed] [Google Scholar]
- 485.Raaflaub J. Die schwellung isolierter leberzell mitochondrien und ihre physikalisch beeinfluβarkeit. Helv Physiol Pharmacol Acta 11: 142–156, 1953. [PubMed] [Google Scholar]
- 486.Raaflaub J. Über den wirkungsmechanismus von adenosintriphosphat (ATP) als cofaktor isolierter mitochondrien. Helv Physiol Pharmacol Acta 11: 157–165, 1953. [PubMed] [Google Scholar]
- 487.Raghavan A, Sheiko T, Graham BH, Craigen WJ. Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim Biophys Acta 1818: 1477–1485, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 201: 499–510, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 12: 815–833, 2007. [DOI] [PubMed] [Google Scholar]
- 490.Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50: 222–233, 2011. [DOI] [PubMed] [Google Scholar]
- 491.Rasola A, Neckers L, Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol 24: 455–463, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci USA 107: 726–731, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Rasola A, Sciacovelli M, Pantic B, Bernardi P. Signal transduction to the permeability transition pore. FEBS Lett 584: 1989–1996, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 56: 437–445, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci USA 106: 21597–21601, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Rees DM, Montgomery MG, Leslie AG, Walker JE. Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc Natl Acad Sci USA 109: 11139–11143, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Reinders J, Wagner K, Zahedi RP, Stojanovski D, Eyrich B, van der Laan M, Rehling P, Sickmann A, Pfanner N, Meisinger C. Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol Cell Proteomics 6: 1896–1906, 2007. [DOI] [PubMed] [Google Scholar]
- 498.Reutenauer J, Dorchies OM, Patthey-Vuadens O, Vuagniaux G, Ruegg UT. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br J Pharmacol 155: 574–584, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Ricchelli F, Šileikyte J, Bernardi P. Shedding light on the mitochondrial permeability transition. Biochim Biophys Acta 1807: 482–490, 2011. [DOI] [PubMed] [Google Scholar]
- 500.Rigobello MP, Barzon E, Marin O, Bindoli A. Effect of polycation peptides on mitochondrial permeability transition. Biochem Biophys Res Commun 217: 144–149, 1995. [DOI] [PubMed] [Google Scholar]
- 501.Rizzuto R, Pozzan T. Microdomains of Intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408, 2006. [DOI] [PubMed] [Google Scholar]
- 502.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 25: 4683–4696, 2006. [DOI] [PubMed] [Google Scholar]
- 503.Robinson GC, Bason JV, Montgomery MG, Fearnley IM, Mueller DM, Leslie AG, Walker JE. The structure of F1-ATPase from Saccharomyces cerevisiae inhibited by its regulatory protein IF1. Open Biol 3: 120164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Rohl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci 38: 253–262, 2013. [DOI] [PubMed] [Google Scholar]
- 505.Rottenberg H, Marbach M. Regulation of Ca2+ transport in brain mitochondria. II. The mechanism of the adenine nucleotides enhancement of Ca2+ uptake and retention. Biochim Biophys Acta 1016: 87–98, 1990. [DOI] [PubMed] [Google Scholar]
- 506.Roucou X, Manon S, Guérin M. ATP opens an electrophoretic potassium transport pathway in respiring yeast mitochondria. FEBS Lett 364: 161–164, 1995. [DOI] [PubMed] [Google Scholar]
- 507.Roucou X, Manon S, Guérin M. Conditions allowing different states of ATP- and GDP-induced permeability in mitochondria from different strains of Saccharomyces cerevisiae. Biochim Biophys Acta 1324: 120–132, 1997. [DOI] [PubMed] [Google Scholar]
- 508.Roucou X, Manon S, Guérin M. Modulation of the electrophoretic ATP-induced K+-transport in yeast mitochondria by delta pH. Biochem Mol Biol Int 43: 53–61, 1997. [DOI] [PubMed] [Google Scholar]
- 509.Rubinstein JL, Walker JE, Henderson R. Structure of the mitochondrial ATP synthase by electron cryomicroscopy. EMBO J 22: 6182–6192, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 9: 971–988, 2010. [DOI] [PubMed] [Google Scholar]
- 511.Salet C, Moreno G, Ricchelli F, Bernardi P. Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J Biol Chem 272: 21938–21943, 1997. [DOI] [PubMed] [Google Scholar]
- 512.Sánchez-Cenizo L, Formentini L, Aldea M, Ortega ÁD, García-Huerta P, Sánchez-Aragó M, Cuezva JM. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 285: 25308–25313, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d'Azzo A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca2+-dependent mitochondrial apoptosis. Mol Cell 36: 500–511, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Savino C, Pelicci P, Giorgio M. The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxid Med Cell Longev 2013: 719407, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Sayeed I, Parvez S, Winkler-Stuck K, Seitz G, Trieu I, Wallesch CW, Schönfeld P, Siemen D. Patch clamp reveals powerful blockade of the mitochondrial permeability transition pore by the D2-receptor agonist pramipexole. FASEB J 20: 556–558, 2006. [DOI] [PubMed] [Google Scholar]
- 516.Scherer B, Klingenberg M. Demonstration of the relationship between the adenine nucleotide carrier and the structural changes of mitochondria as induced by adenosine 5’-diphosphate. Biochemistry 13: 161–170, 1974. [DOI] [PubMed] [Google Scholar]
- 517.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16: 6125–6133, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Schönfeld P, Siemen D, Kreutzmann P, Franz C, Wojtczak L. Interaction of the antibiotic minocycline with liver mitochondria: role of membrane permeabilization in the impairment of respiration. FEBS J 280: 6589–6599, 2013. [DOI] [PubMed] [Google Scholar]
- 520.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7: 295–308, 2007. [DOI] [PubMed] [Google Scholar]
- 521.Schwarzlander M, Logan DC, Fricker MD, Sweetlove LJ. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide “flashes.” Biochem J 437: 381–387, 2011. [DOI] [PubMed] [Google Scholar]
- 522.Schwarzlander M, Murphy MP, Duchen MR, Logan DC, Fricker MD, Halestrap AP, Muller FL, Rizzuto R, Dick TP, Meyer AJ, Sweetlove LJ. Mitochondrial “flashes”: a radical concept repHined. Trends Cell Biol 22: 503–508, 2012. [DOI] [PubMed] [Google Scholar]
- 523.Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, Calabrese F, Laudiero G, Esposito F, Landriscina M, Defilippi P, Bernardi P, Rasola A. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab 17: 988–999, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2: 55–67, 2002. [DOI] [PubMed] [Google Scholar]
- 525.Scorrano L, Nicolli A, Basso E, Petronilli V, Bernardi P. Two modes of activation of the permeability transition pore: the role of mitochondrial cyclophilin. Mol Cell Biochem 174: 181–184, 1997. [PubMed] [Google Scholar]
- 526.Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-α apoptotic signaling. J Biol Chem 276: 12035–12040, 2001. [DOI] [PubMed] [Google Scholar]
- 527.Scorrano L, Petronilli V, Bernardi P. On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J Biol Chem 272: 12295–12299, 1997. [DOI] [PubMed] [Google Scholar]
- 528.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res 809: 12–17, 1998. [DOI] [PubMed] [Google Scholar]
- 529.Seelert H, Dencher NA. ATP synthase superassemblies in animals and plants: two or more are better. Biochim Biophys Acta 1807: 1185–1197, 2011. [DOI] [PubMed] [Google Scholar]
- 530.Seidlmayer LK, Gomez-Garcia MR, Blatter LA, Pavlov E, Dedkova EN. Inorganic polyphosphate is a potent activator of the mitochondrial permeability transition pore in cardiac myocytes. J Gen Physiol 139: 321–331, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Selwyn MJ. Model reaction for mitochondrial adenosine triphosphatase. Nature 219: 490–493, 1968. [DOI] [PubMed] [Google Scholar]
- 532.Shirakihara Y, Leslie AG, Abrahams JP, Walker JE, Ueda T, Sekimoto Y, Kambara M, Saika K, Kagawa Y, Yoshida M. The crystal structure of the nucleotide-free αβ subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5: 825–836, 1997. [DOI] [PubMed] [Google Scholar]
- 533.Shulga N, Pastorino JG. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J Cell Sci 123: 4117–4127, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 534.Siemen D, Ziemer M. What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 65: 255–262, 2013. [DOI] [PubMed] [Google Scholar]
- 535.Sileikyte J, Blachly-Dyson E, Sewell R, Carpi A, Menabò R, Di Lisa F, Ricchelli F, Bernardi P, Forte M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (translocator protein of 18 kDa (TSPO)). J Biol Chem 289: 13769–13781, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Sileikyte J, Petronilli V, Zulian A, Dabbeni-Sala F, Tognon G, Nikolov P, Bernardi P, Ricchelli F. Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J Biol Chem 286: 1046–1053, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Sinha Roy S, Madesh M, Davies E, Antonsson B, Danial N, Hajnoczky G. Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol Cell 33: 377–388, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol 52: 1009–1018, 2012. [DOI] [PubMed] [Google Scholar]
- 539.Sorato E, Menazza S, Zulian A, Sabatelli P, Gualandi F, Merlini L, Bonaldo P, Canton M, Bernardi P, Di Lisa F. Monoamine oxidase inhibition prevents mitochondrial dysfunction and apoptosis in myoblasts from patients with collagen VI myopathies. Free Radic Biol Med 75C: 40–47, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Sorgato MC, Keller BU, Stühmer W. Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature 330: 498–500, 1987. [DOI] [PubMed] [Google Scholar]
- 541.Sorgato MC, Moran O, De Pinto V, Keller BU, Stühmer W. Further investigation on the high-conductance ion channel of the inner membrane of mitochondria. J Bioenerg Biomembr 21: 485–496, 1989. [DOI] [PubMed] [Google Scholar]
- 542.Soriano ME, Nicolosi L, Bernardi P. Desensitization of the permeability transition pore by cyclosporin A prevents activation of the mitochondrial apoptotic pathway and liver damage by Tumor Necrosis Factor-α. J Biol Chem 279: 36803–36808, 2004. [DOI] [PubMed] [Google Scholar]
- 543.Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A. An electron-transport system associated with the outer membrane of liver mitochondria. J Cell Biol 32: 415–438, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Soubannier V, Rusconi F, Vaillier J, Arselin G, Chaignepain S, Graves PV, Schmitter JM, Zhang JL, Mueller D, Velours J. The second stalk of the yeast ATP synthase complex: identification of subunits showing cross-links with known positions of subunit 4 (subunit b). Biochemistry 38: 15017–15024, 1999. [DOI] [PubMed] [Google Scholar]
- 545.Soubannier V, Vaillier J, Paumard P, Coulary B, Schaeffer J, Velours J. In the absence of the first membrane-spanning segment of subunit 4(b), the yeast ATP synthase is functional but does not dimerize or oligomerize. J Biol Chem 277: 10739–10745, 2002. [DOI] [PubMed] [Google Scholar]
- 546.Spannagel C, Vaillier J, Arselin G, Graves PV, Grandier-Vazeille X, Velours J. Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochim Biophys Acta 1414: 260–264, 1998. [DOI] [PubMed] [Google Scholar]
- 547.Spannagel C, Vaillier J, Chaignepain S, Velours J. Topography of the yeast ATP synthase F0 sector by using cysteine substitution mutants. Cross-linkings between subunits 4, 6, and f. Biochemistry 37: 615–621, 1998. [DOI] [PubMed] [Google Scholar]
- 548.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann NY Acad Sci 1147: 37–52, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Rad Biol Med 38: 687–697, 2005. [DOI] [PubMed] [Google Scholar]
- 550.Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med 200: 211–222, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Stelzer AC, Frazee RW, Van Huis C, Cleary J, Opipari AW Jr, Glick GD, Al-Hashimi HM. NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F1F0-ATPase. Biopolymers 93: 85–92, 2010. [DOI] [PubMed] [Google Scholar]
- 552.Stephens AN, Khan MA, Roucou X, Nagley P, Devenish RJ. The molecular neighborhood of subunit 8 of yeast mitochondrial F1F0-ATP synthase probed by cysteine scanning mutagenesis and chemical modification. J Biol Chem 278: 17867–17875, 2003. [DOI] [PubMed] [Google Scholar]
- 553.Stock D, Gibbons C, Arechaga I, Leslie AG, Walker JE. The rotary mechanism of ATP synthase. Curr Opin Struct Biol 10: 672–679, 2000. [DOI] [PubMed] [Google Scholar]
- 554.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science 286: 1700–1705, 1999. [DOI] [PubMed] [Google Scholar]
- 555.Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J 27: 1154–1160, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci 13: 507–514, 2012. [DOI] [PubMed] [Google Scholar]
- 557.Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front Oncol 3: 41, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 79: 231–239, 2005. [DOI] [PubMed] [Google Scholar]
- 559.Sullivan PG, Sebastian AH, Hall ED. Therapeutic window analysis of the neuroprotective effects of cyclosporine A after traumatic brain injury. J Neurotrauma 28: 311–318, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Sultan A, Sokolove PM. Free fatty acid effects on mitochondrial permeability: an overview. Arch Biochem Biophys 386: 52–61, 2001. [DOI] [PubMed] [Google Scholar]
- 561.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis 22: 76–87, 2006. [DOI] [PubMed] [Google Scholar]
- 562.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 101: 1155–1163, 2007. [DOI] [PubMed] [Google Scholar]
- 563.Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res 106: 285–296, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 28: 1007–1017, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 184: 1331–1341, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Suzuki T, Tanaka K, Wakabayashi C, Saita E, Yoshida M. Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat Chem Biol 10: 930–936, 2014. [DOI] [PubMed] [Google Scholar]
- 567.Symersky J, Osowski D, Walters DE, Mueller DM. Oligomycin frames a common drug-binding site in the ATP synthase. Proc Natl Acad Sci USA 109: 13961–13965, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Symersky J, Pagadala V, Osowski D, Krah A, Meier T, Faraldo-Gomez JD, Mueller DM. Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol 19: 485–491, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Szabó I, Bernardi P, Zoratti M. Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem 267: 2940–2946, 1992. [PubMed] [Google Scholar]
- 570.Szabó I, Bock J, Grassme H, Soddemann M, Wilker B, Lang F, Zoratti M, Gulbins E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc Natl Acad Sci USA 105: 14861–14866, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Szabó I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 330: 206–210, 1993. [DOI] [PubMed] [Google Scholar]
- 572.Szabó I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem 266: 3376–3379, 1991. [PubMed] [Google Scholar]
- 573.Szabó I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr 24: 111–117, 1992. [DOI] [PubMed] [Google Scholar]
- 574.Szabó I, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett 330: 201–205, 1993. [DOI] [PubMed] [Google Scholar]
- 575.Szabó I, Zoratti M. Mitochondrial channels: ion fluxes and more. Physiol Rev 94: 519–608, 2014. [DOI] [PubMed] [Google Scholar]
- 576.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium 47: 122–129, 2010. [DOI] [PubMed] [Google Scholar]
- 577.Taddeo EP, Laker RC, Breen DS, Akhtar YN, Kenwood BM, Liao JA, Zhang M, Fazakerley DJ, Tomsig JL, Harris TE, Keller SR, Chow JD, Lynch KR, Chokki M, Molkentin JD, Turner N, James DE, Yan Z, Hoehn KL. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol Metab 3: 124–134, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11: 621–632, 2010. [DOI] [PubMed] [Google Scholar]
- 579.Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337: 473–475, 1989. [DOI] [PubMed] [Google Scholar]
- 580.Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci USA 102: 2602–2607, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Tanigawara M, Tabata KV, Ito Y, Ito J, Watanabe R, Ueno H, Ikeguchi M, Noji H. Role of the DELSEED loop in torque transmission of F1-ATPase. Biophys J 103: 970–978, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Tapley DF. The effect of thyroxine and other substances on the swelling of isolated rat liver mitochondria. J Biol Chem 222: 325–339, 1956. [PubMed] [Google Scholar]
- 583.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J Biol Chem 278: 19587–19590, 2003. [DOI] [PubMed] [Google Scholar]
- 584.Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in α1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol 286: G851–G862, 2004. [DOI] [PubMed] [Google Scholar]
- 585.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH and light scattering. J Biol Chem 276: 2586–2599, 2001. [DOI] [PubMed] [Google Scholar]
- 586.Thomas D, Bron P, Weimann T, Dautant A, Giraud MF, Paumard P, Salin B, Cavalier A, Velours J, Brèthes D. Supramolecular organization of the yeast F1Fo-ATP synthase. Biol Cell 100: 591–601, 2008. [DOI] [PubMed] [Google Scholar]
- 587.Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, Baldari CT, Bonaldo P, Bernardi P. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1−/− myopathic mice. Br J Pharmacol 157: 1045–1052, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Tomasetig L, Di Pancrazio F, Harris DA, Mavelli I, Lippe G. Dimerization of F0F1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim Biophys Acta 1556: 133–141, 2002. [DOI] [PubMed] [Google Scholar]
- 589.Traba J, del Arco A, Duchen MR, Szabadkai G, Satrustegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca2+ buffering. Cell Death Differ 19: 650–660, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 21: 3872–3878, 2002. [DOI] [PubMed] [Google Scholar]
- 591.Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem 289: 27444–27454, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Tucker WC, Schwarz A, Levine T, Du Z, Gromet-Elhanan Z, Richter ML, Haran G. Observation of calcium-dependent unidirectional rotational motion in recombinant photosynthetic F1-ATPase molecules. J Biol Chem 279: 47415–47418, 2004. [DOI] [PubMed] [Google Scholar]
- 593.Uchi J, Pan S, Sheu SS. Perspectives on: SGP symposium on mitochondrial physiology and medicine: molecular identities of mitochondrial Ca2+ influx mechanism: updated passwords for accessing mitochondrial Ca2+-linked health and disease. J Gen Physiol 139: 435–443, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Uchino H, Elmer E, Uchino K, Li PA, He QP, Smith ML, Siesjö BK. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res 812: 216–226, 1998. [DOI] [PubMed] [Google Scholar]
- 595.Uribe-Carvajal S, Luévano-Martìnez LA, Guerrero-Castillo S, Cabrera-Orefice A, Corona-de-la-Peña NA, Gutiérrez-Aguilar M. Mitochondrial unselective cthroughout the eukaryotic domain. Mitochondrion 11: 382–390, 2011. [DOI] [PubMed] [Google Scholar]
- 596.Vaillier J, Arselin G, Graves PV, Camougrand N, Velours J. Isolation of supernumerary yeast ATP synthase subunits e and i. Characterization of subunit i and disruption of its structural gene ATP18. J Biol Chem 274: 543–548, 1999. [DOI] [PubMed] [Google Scholar]
- 597.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304: 1158–1160, 2004. [DOI] [PubMed] [Google Scholar]
- 598.Van Raaij MJ, Abrahams JP, Leslie AG, Walker JE. The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B. Proc Natl Acad Sci USA 93: 6913–6917, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Van Walraven HS, Scholts MJ, Zakharov SD, Kraayenhof R, Dilley RA. pH-dependent Ca2+ binding to the F0 c-subunit affects proton translocation of the ATP synthase from Synechocystis 6803. J Bioenerg Biomembr 34: 455–464, 2002. [DOI] [PubMed] [Google Scholar]
- 600.Vantourout P, Radojkovic C, Lichtenstein L, Pons V, Champagne E, Martinez LO. Ecto-F1-ATPase: a moonlighting protein complex and an unexpected apoA-I receptor. World J Gastroenterol 16: 5925–5935, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12: 120–125, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Varbiro G, Veres B, Gallyas F Jr, Sumegi B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med 31: 548–558, 2001. [DOI] [PubMed] [Google Scholar]
- 603.Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, Kim HS, Deng CX, Walker JE, Gius D. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal 21: 551–564, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Velours J, Stines-Chaumeil C, Habersetzer J, Chaignepain S, Dautant A, Brèthes D. Evidence of the proximity of ATP synthase subunits 6 (a) in the inner mitochondrial membrane and in the supramolecular forms of Saccharomyces cerevisiae ATP synthase. J Biol Chem 286: 35477–35484, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Venard R, Brèthes D, Giraud MF, Vaillier J, Velours J, Haraux F. Investigation of the role and mechanism of IF1 and STF1 proteins, twin inhibitory peptides which interact with the yeast mitochondrial ATP synthase. Biochemistry 42: 7626–7636, 2003. [DOI] [PubMed] [Google Scholar]
- 606.Vignais PV, Satre M. Recent developments on structural and functional aspects of the F1 sector of H+-linked ATPases. Mol Cell Biochem 60: 33–71, 1984. [DOI] [PubMed] [Google Scholar]
- 607.Vinogradov A, Scarpa A, Chance B. Calcium and pyridine nucleotide interaction in mitochondrial membranes. Arch Biochem Biophys 152: 646–654, 1972. [DOI] [PubMed] [Google Scholar]
- 608.Vinogradov AD, Gyurova ZS, Fitin AF. Effect of SH-reagents on the mitochondrial ATPase and induction of respiratory control in EDTA particles. FEBS Lett 54: 230–233, 1975. [DOI] [PubMed] [Google Scholar]
- 609.Vives-Bauza C, Magrané J, Andreu AL, Manfredi G. Novel role of ATPase subunit C targeting peptides beyond mitochondrial protein import. Mol Biol Cell 21: 131–139, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.von Stockum S, Basso E, Petronilli V, Sabatelli P, Forte MA, Bernardi P. Properties of Ca2+ transport in mitochondria of Drosophila melanogaster. J Biol Chem 286: 41163–41170, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.von Stockum S, Giorgio V, Trevisan E, Lippe G, Glick GD, Forte MA, Da-Rè C, Checchetto V, Mazzotta G, Costa R, Szabò I, Bernardi PF. ATPase of D. melanogaster forms 53 Picosiemen (53-pS) channels responsible for mitochondrial Ca2+-induced Ca2+ release. J Biol Chem 290: 4537–4544, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Vuorinen K, Ylitalo K, Peuhkurinen K, Raatikainen P, Ala-Rämi A, Hassinen IE. Mechanisms of ischemic preconditioning in rat myocardium. Roles of adenosine, cellular energy state, and mitochondrial F1F0-ATPase. Circulation 91: 2810–2818, 1995. [DOI] [PubMed] [Google Scholar]
- 613.Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans 41: 1–16, 2013. [DOI] [PubMed] [Google Scholar]
- 614.Walsh CT, Zydowsky LD, McKeon FD. Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J Biol Chem 267: 13115–13118, 1992. [PubMed] [Google Scholar]
- 615.Walter L, Miyoshi H, Leverve X, Bernardi P, Fontaine E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic Res 36: 405–412, 2002. [DOI] [PubMed] [Google Scholar]
- 616.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet 43: 95–118, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Wang HL, Chou AH, Yeh TH, Li AH, Chen YL, Kuo YL, Tsai SR, Yu ST. PINK1 mutants associated with recessive Parkinson's disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol Dis 28: 216–226, 2007. [DOI] [PubMed] [Google Scholar]
- 618.Wang JQ, Chen Q, Wang X, Wang QC, Wang Y, Cheng HP, Guo C, Sun Q, Chen Q, Tang TS. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J Biol Chem 288: 3070–3084, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Wang P, Heitman J. The cyclophilins. Genome Biol 6: 226.1–226.6, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Wang SB, Foster DB, Rucker J, O'Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res 109: 750–757, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Wang SB, Murray CI, Chung HS, Van Eyk JE. Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc Med 23: 14–18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 134: 279–290, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, Hagberg H. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci 29: 2588–2596, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Warburg O. On the origin of cancer cells. Science 123: 309–314, 1956. [DOI] [PubMed] [Google Scholar]
- 625.Wasaki S, Sakaida I, Uchida K, Kimura T, Kayano K, Okita K. Preventive effect of cyclosporin A on experimentally induced acute liver injury in rats. Liver 17: 107–114, 1997. [DOI] [PubMed] [Google Scholar]
- 626.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA 107: 16823–16827, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Weber J, Bowman C, Senior AE. Specific tryptophan substitution in catalytic sites of Escherichia coli F1-ATPase allows differentiation between bound substrate ATP and product ADP in steady-state catalysis. J Biol Chem 271: 18711–18718, 1996. [DOI] [PubMed] [Google Scholar]
- 628.Weber J, Senior AE. ATP synthase: what we know about ATP hydrolysis and what we do not know about ATP synthesis. Biochim Biophys Acta 1458: 300–309, 2000. [DOI] [PubMed] [Google Scholar]
- 629.Wei-Lapierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S, Wang X, Sheu SS, Cheng H, Dirksen RT, Wang W. Respective contribution of mitochondrial superoxide and pH to Mt-cpYFP flash activity. J Biol Chem 288: 10567–10577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 1802: 29–44, 2010. [DOI] [PubMed] [Google Scholar]
- 631.Wissing ER, Millay DP, Vuagniaux G, Molkentin JD. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul Disord 20: 753–760, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Wittig I, Meyer B, Heide H, Steger M, Bleier L, Wumaier Z, Karas M, Schägger H. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim Biophys Acta 1797: 1004–1011, 2010. [DOI] [PubMed] [Google Scholar]
- 633.Wittig I, Schägger H. Structural organization of mitochondrial ATP synthase. Biochim Biophys Acta 1777: 592–598, 2008. [DOI] [PubMed] [Google Scholar]
- 634.Wittig I, Schägger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta 1787: 672–680, 2009. [DOI] [PubMed] [Google Scholar]
- 635.Wittig I, Velours J, Stuart R, Schägger H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Mol Cell Proteomics 7: 995–1004, 2008. [DOI] [PubMed] [Google Scholar]
- 636.Wojtczak L, Lehninger AL. Formation and disappearance of an endogenous uncoupling factor during swelling and contraction of mitochondria. Biochim Biophys Acta 51: 442–456, 1961. [DOI] [PubMed] [Google Scholar]
- 637.Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 336: 287–290, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Woodfield KY, Price NT, Halestrap AP. cDNA cloning of rat mitochondrial cyclophilin. Biochim Biophys Acta 1351: 27–30, 1997. [DOI] [PubMed] [Google Scholar]
- 639.Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, Epting CL, Ardehali H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med 4: 633–646, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Wu YT, Lee HC, Liao CC, Wei YH. Regulation of mitochondrial FoF1ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977 bp deletion of mitochondrial DNA. Biochim Biophys Acta 1832: 216–227, 2013. [DOI] [PubMed] [Google Scholar]
- 641.Xiang F, Huang YS, Shi XH, Zhang Q. Mitochondrial chaperone tumour necrosis factor receptor-associated protein 1 protects cardiomyocytes from hypoxic injury by regulating mitochondrial permeability transition pore opening. FEBS J 277: 1929–1938, 2010. [DOI] [PubMed] [Google Scholar]
- 642.Xu C, Yi C, Wang H, Bruce IC, Xia Q. Mitochondrial nitric oxide synthase participates in septic shock myocardial depression by nitric oxide overproduction and mitochondrial permeability transition pore opening. Shock 37: 110–115, 2012. [DOI] [PubMed] [Google Scholar]
- 643.Xu D, Woodfield SE, Lee TV, Fan Y, Antonio C, Bergmann A. Genetic control of programmed cell death (apoptosis) in Drosophila. Fly 3: 78–90, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab 29: 365–374, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Yagi T, Hatefi Y. Thiols in oxidative phosphorylation: inhibition and energy-potentiated uncoupling by monothiol and dithiol modifiers. Biochemistry 23: 2449–2455, 1984. [DOI] [PubMed] [Google Scholar]
- 646.Yagi T, Hatefi Y. Thiols in oxidative phosphorylation: thiols in the F0 of ATP synthase essential for ATPase activity. Arch Biochem Biophys 254: 102–109, 1987. [DOI] [PubMed] [Google Scholar]
- 647.Yamada A, Yamamoto T, Yoshimura Y, Gouda S, Kawashima S, Yamazaki N, Yamashita K, Kataoka M, Nagata T, Terada H, Pfeiffer DR, Shinohara Y. Ca2+-induced permeability transition can be observed even in yeast mitochondria under optimized experimental conditions. Biochim Biophys Acta 1787: 1486–1491, 2009. [DOI] [PubMed] [Google Scholar]
- 648.Yan WL, Lerner TJ, Haines JL, Gusella JF. Sequence analysis and mapping of a novel human mitochondrial ATP synthase subunit 9 cDNA (ATP5G3). Genomics 24: 375–377, 1994. [DOI] [PubMed] [Google Scholar]
- 649.Yehuda-Shnaidman E, Kalderon B, Azazmeh N, Bar-Tana J. Gating of the mitochondrial permeability transition pore by thyroid hormone. FASEB J 24: 93–104, 2010. [DOI] [PubMed] [Google Scholar]
- 650.Yoshiba M, Sekiyama K, Inoue K, Fujita R. Interferon and cyclosporin A in the treatment of fulminant viral hepatitis. J Gastroenterol 30: 67–73, 1995. [DOI] [PubMed] [Google Scholar]
- 651.Yoshida S, Tsutsumi S, Muhlebach G, Sourbier C, Lee MJ, Lee S, Vartholomaiou E, Tatokoro M, Beebe K, Miyajima N, Mohney RP, Chen Y, Hasumi H, Xu W, Fukushima H, Nakamura K, Koga F, Kihara K, Trepel J, Picard D, Neckers L. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci USA 110: E1604–E1612, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Yu L, Fesik SW. pH titration of the histidine residues of cyclophilin and FK506 binding protein in the absence and presence of immunosuppressant ligands. Biochim Biophys Acta 1209: 24–32, 1994. [DOI] [PubMed] [Google Scholar]
- 653.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2: 67–71, 2001. [DOI] [PubMed] [Google Scholar]
- 654.Zanotti F, Guerrieri F, Capozza G, Fiermonte M, Berden J, Papa S. Role of F0 and F1 subunits in the gating and coupling function of mitochondrial H+-ATP synthase. The effect of dithiol reagents. Eur J Biochem 208: 9–16, 1992. [DOI] [PubMed] [Google Scholar]
- 655.Zanotti F, Raho G, Gaballo A, Papa S. Inhibitory and anchoring domains in the ATPase inhibitor protein IF1 of bovine heart mitochondrial ATP synthase. J Bioenerg Biomembr 36: 447–457, 2004. [DOI] [PubMed] [Google Scholar]
- 656.Zborowski J, Wojtczak L. Induction of swelling of liver mitochondria by fatty acids of various chain length. Biochim Biophys Acta 70: 596–598, 1963. [DOI] [PubMed] [Google Scholar]
- 657.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem 265: 16330–16336, 1990. [PubMed] [Google Scholar]
- 658.Zhao X, León IR, Bak S, Mogensen M, Wrzesinski K, Højlund K, Jensen ON. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol Cell Proteomics 10: M110, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Zharova TV, Vinogradov AD. Energy-linked binding of Pi is required for continuous steady-state proton-translocating ATP hydrolysis catalyzed by F0--F1 ATP synthase. Biochemistry 45: 14552–14558, 2006. [DOI] [PubMed] [Google Scholar]
- 660.Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu dC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417: 74–78, 2002. [DOI] [PubMed] [Google Scholar]
- 661.Zoccarato F, Nicholls DG. The role of phosphate in the regulation of the independent calcium-efflux pathway of liver mitochondria. Eur J Biochem 127: 333–338, 1982. [DOI] [PubMed] [Google Scholar]
- 662.Zoeteweij JP, van de Water B, de Bont HJ, Mulder GJ, Nagelkerke JF. Calcium-induced cytotoxicity in hepatocytes after exposure to extracellular ATP is dependent on inorganic phosphate. Effects on mitochondrial calcium. J Biol Chem 268: 3384–3388, 1993. [PubMed] [Google Scholar]
- 663.Zoratti M, De Marchi U, Gulbins E, Szabò I. Novel channels of the inner mitochondrial membrane. Biochim Biophys Acta 1787: 351–363, 2009. [DOI] [PubMed] [Google Scholar]
- 664.Zoratti M, Szabó I. Electrophysiology of the inner mitochondrial membrane. J Bioenerg Biomembr 26: 543–553, 1994. [DOI] [PubMed] [Google Scholar]
- 665.Zoratti M, Szabó I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta 1706: 40–52, 2005. [DOI] [PubMed] [Google Scholar]
- 666.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: 1001–1014, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res 83: 213–225, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Zulian A, Rizzo E, Schiavone M, Palma E, Tagliavini F, Blaauw B, Merlini L, Maraldi NM, Sabatelli P, Braghetta P, Bonaldo P, Argenton F, Bernardi P. NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum Mol Genet 23: 5353–5363, 2014. [DOI] [PubMed] [Google Scholar]