

Abstract
For a solid tumor to grow, it must be able to support the compressive stress that is generated as it presses against the surrounding tissue. Although the literature suggests a role for the cytoskeleton in counteracting these stresses, there has been no systematic evaluation of which filaments are responsible or to what degree. Here, using a three-dimensional spheroid model, we show that cytoskeletal filaments do not actively support compressive loads in breast, ovarian, and prostate cancer. However, modulation of tonicity can induce alterations in spheroid size. We find that under compression, tumor cells actively efflux sodium to decrease their intracellular tonicity, and that this is reversible by blockade of sodium channel NHE1. Moreover, although polymerized actin does not actively support the compressive load, it is required for sodium efflux. Compression-induced cell death is increased by both sodium blockade and actin depolymerization, whereas increased actin polymerization offers protective effects and increases sodium efflux. Taken together, these results demonstrate that cancer cells modulate their tonicity to survive under compressive solid stress.
Main Text
Solid tumors grow under stress from the local tissue. This growth results in further accumulation of compressive stress in the tumor. To remain viable and grow in vivo, the tumor cells must be able to survive under these compressive stresses, which correspond to mechanical loads of 10–100 mmHg (1,2). Previous work has shown that this solid stress prevents the growth of cancer cell spheroids (1). Moreover, as this stress accumulates in vivo, it can lead to further adverse effects such as blood vessel compression, hindering drug delivery (2). Despite the importance of solid stress in tumor growth, however, few studies have explored how cancer cells actually support this stress.
Most of the literature suggests that the ability of cells to resist deformation largely emanates from the cytoskeleton (3). A series of studies have demonstrated that actin, microtubules, and intermediate filaments all contribute to support external compressive stresses (4–6). In accord with these studies, in a genetically engineered model of the epithelial-mesenchymal transition, we recently found that mesenchymal cells could support less stress than their epithelial counterparts, which correlated with decreased polymerized actin and cytoplasmic stiffness (7). Here, we show that compressive stress is not actively supported by cytoskeletal filaments, but induces NHE1-dependent sodium efflux from the tumor cells. Although actin polymerization did not actively support solid stress, depolymerization of actin mitigated the ability of cells to efflux sodium. Taken together, these results show that regulation of intracellular tonicity is required for cells to maintain viability under compressive stress.
Supporters of solid stress
To analyze how cancer cells support compressive-solid stress, we cultured tumor spheroids from single MCF7 breast cancer cells in inert agarose gels, resulting in the accumulation of solid stress (1,8). Next, to elucidate the roles of cytoskeletal filaments in supporting this stress, we treated the spheroids with a variety of cytoskeleton-stabilizing and -destabilizing molecules, and tracked changes in spheroid cross-sectional areas using live-cell microscopy (for details, see Supporting Materials and Methods in the Supporting Material). Unexpectedly, depolymerization of actin filaments using cytochalasin D resulted in an increase in spheroid size, whereas increasing actin polymerization with Jasplakinolide had no effect, suggesting that actin was not primarily responsible for supporting solid stress (Fig. 1 A). Modulation of microtubule polymerization did not alter spheroid diameter (Fig. 1 B), and depolymerization of intermediate filaments with 4 mM of acrylamide (9) produced a slight increase in spheroid area (Fig. 1 C). Since none of these treatments produced significant decreases in spheroid area, indicating that they actively supported stress, we hypothesized that the stress may instead be supported by osmotic pressure. Decreasing media tonicity caused spheroid swelling, whereas increasing the media tonicity with a variety of solutes caused spheroids to shrink (Fig. 1 D). In addition to breast cancer MCF7 cells, this phenomenon was also observed in ovarian cancer OVCAR3 cells and prostate cancer DU145 cells (Fig. S1), suggesting it is conserved across other solid tumor models.
Figure 1.
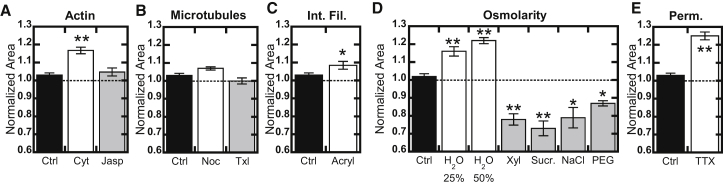
Change in spheroid area after 6 h of treatment, with all values normalized to the initial area before treatment. (A) Depolymerization of actin with cytochalasin D (Cyt) and enhanced polymerization with Jasplakinolide (Jasp). (B) Microtubule depolymerization with nocodazole (Noc) and stabilization with Taxol (Txl). (C) Depolymerization of intermediate filaments with acrylamide. (D) Altering media osmolarity with water (hypoosmotic) or 250 mM xylose, sucrose, NaCl, or PEG400 (hyperosmotic). (E) Spheroid permeabilization with 0.0025% Triton-X100 (TTX). Values given are mean ± SE; ∗p < 0.05, ∗∗p < 0.01.
To verify that these changes were due to solid stress and were not an artifact of the spheroid culture, we generated equal-sized spheroids by anchorage-independent growth in solution for 1 week before embedding them in agarose overnight for analysis in the absence of significant accumulated stress. These unstressed spheroids showed larger fold changes in response to tonicity, presumably because there was no initial stress, allowing for easier expansion against the gel. However, in contrast to the stressed spheroids, the unstressed spheroids collapsed after actin depolymerization (Fig. S2). This result shows that actin polymerization is required for cell structure under conditions of minimal solid stress, but primarily controls the ability of cells to efflux sodium under higher levels of solid stress.
Finally, to ascertain whether cells in steady-state stressed spheroids have higher or lower tonicity than their surrounding media, we permeabilized cells with 0.0025% Triton-X100. After permeabilization, the spheroids increased in area by >25% (Fig. 1 E). Based on osmotic models of cell size, this implies that cells within stressed spheroids have lower tonicity than their surrounding media, such that after permeabilization, extracellular ions enter the cells, inducing an inward water flux (and consequently spheroid swelling) to equilibrate (10).
Sodium efflux under compressive solid stress
To ascertain how the cells decreased their intracellular tonicity, we utilized a 2D model of solid stress. Here, instead of allowing spheroids to accumulate solid stress from growth, we used weights to directly apply a controlled 5 mmHg stress to a monolayer of cells (11), on the same order of magnitude as the stresses generated by spheroids (9.8 ± 0.3 mmHg). Quantification of intracellular sodium with CoroNa Green revealed that the cells effluxed sodium after mechanical compression (Fig. 2 A).
Figure 2.
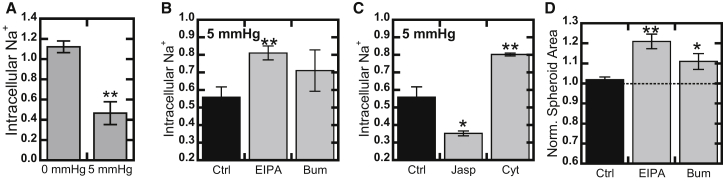
Cancer cells efflux sodium under solid stress. (A) A 2D monolayer of MCF7 cells was loaded with CoroNa Green sodium tracer and compressed with 5 mmHg solid stress for 2 h. CoroNa intensity was measured using a plate reader and values are reported relative to the zero time point. (B) Intracellular sodium under 5 mmHg stress in the presence of the NHE1 inhibitor EIPA and the NKCC1 inhibitor bumetanide (Bum). (C) Intracellular sodium under 5 mmHg stress in the presence of Jasplakinolide (Jasp) to enhance actin polymerization, and cytochalasin D (Cyt) to depolymerize actin. (D) Blockade of the sodium channels NHE1 (EIPA) and NKCC1 (Bum) in 3D tumor spheroids. Values given are mean ± SE, ∗p < 0.05, ∗∗p < 0.01.
Although blockade of sodium channel NKKC1 did not produce a statistically significant difference in sodium efflux under stress, inhibition of the sodium-hydrogen channel NHE1 with ethyl-isopropyl amiloride (EIPA) did significantly block sodium efflux (p < 0.05; Fig. 2 B). This is consistent with previous studies that showed that hydrostatic pressure induces sodium efflux (12). Repeating this experiment with modulation of actin filaments revealed that actin depolymerization blocked sodium efflux to a similar degree as NHE1 blockade. Consistent with this, actin stabilization with Jasplakinolide produced a slight but statistically insignificant (p = 0.063) increase in sodium efflux (Fig. 2 C). None of the inhibitors altered sodium levels in the absence of stress (Fig. S3). These results suggest that although polymerized actin may not actively support stress, it is required for cells to function properly under compression. This is consistent with recent studies that showed the importance of NHE1 for confined cell migration, where it colocalizes with polymerized actin (13).
To verify that these results would translate into our 3D spheroid model, we repeated the spheroid assay. Blockade of NKCC1 with bumetanide caused modest swelling, but consistent with the sodium efflux findings, NHE1 blockade with EIPA produced a larger increase in spheroid cross-sectional area (Figs. 2 D and S1), which was identical to that seen with actin depolymerization (Figs. 1 A and S1). This swelling was not observed in unstressed spheroids (Fig. S2).
Sodium efflux, tonicity, and actin polymerization contribute to cell viability under stress
To test whether the observed stress-induced sodium efflux was actually protective, we finally analyzed the viability of cells under compression. In our 2D system, we found that compression with 5 mmHg induced significant cell death (Fig. 3 A). Although cells in hypertonic media showed increased death without compression, 5 mmHg of compression did not significantly increase the amount of death in these cells, suggesting that it may have served to protect the surviving cells (Fig. 3 A). Increasing actin polymerization with Jasplakinolide was more protective under stress, significantly decreasing cell death relative to the compressed control. Conversely, decreased media tonicity, blockade of sodium efflux, and actin depolymerization all significantly increased the amount of cell death under compression (Fig. 3 A). Although Jasplakinolide pretreatment improved 2D sodium efflux and viability under stress, it was not sufficient to decrease the 3D spheroid size. This may be because spheroid growth selected for tumor cells that could optimally efflux sodium under stress and did not need additional polymerized actin to increase this efflux rate.
Figure 3.

Sodium efflux and actin polymerization are required for viability under solid stress. (A) Quantification of cell death based on the ratio of propidium iodide to calcein AM after 4 h of compression of a 2D monolayer. Hyper: 250 mM xylose; hypo: 25% H2O. (B) Cell death in 3D tumor spheroids versus radial stress. Stress-dependent toxicity is indicated by the slope value. Representative images of spheroids stained for live cells (calcein AM, green) and dead cells (propidium iodide, red) are given below. Scale bar is 50 μm. Values given are mean ± SE; # indicates significant difference compared with stressed control; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. To see this figure in color, go online.
To verify that these mechanisms were conserved in our 3D model, we repeated the analysis of viability after addition of EIPA and cytochalasin D. These results are presented as the resulting cell death as a function of radial stress (Fig. 3 B). If these molecules induce compressive stress-dependent toxicity, we should see increasing cell death as a function of radial stress in the spheroids. In the control condition, there is a weak positive correlation between radial stress and cell death, which is subsequently increased by NHE1 inhibition and even further increased by actin depolymerization, demonstrating that both are required for tumor cells to survive under stress.
Conclusions
Cells use ion pumps to maintain a constant volume by modulating their tonicity and subsequently their osmotic pressure (14). During mitosis, cells also use osmotic pressure to generate a rounding force, which is counteracted by actomyosin tension (15). Here, we document that under compressive stress, multicell spheroids as well as cell monolayers will decrease their intracellular tonicity by effluxing sodium, and that although cytoskeletal filaments do not actively support compressive stress, actin polymerization is required for osmotic regulation. This finding is consistent with the work of Hui et al. (12), who found that increased hydrostatic pressure induced active sodium efflux. Recent studies using C. elegans suggested that these organisms may have an absolute internal pressure set point (16). This could be consistent with the model of cell volume and pressure regulation presented by Jiang and Sun (10), which relates changes in cell volume to water flux controlled by differences in hydrostatic and osmotic pressures across the cell membrane. The lower steady-state intracellular tonicity under stress implies a negative pressure change across the cell membrane from osmotic pressure. This negative pressure change may help offset the compressive stress. Taken together, our results demonstrate that to survive under compressive stress, cells must be able to modulate their tonicity by effluxing sodium, and that polymerized actin is necessary for this process.
Author Contributions
D.J.M., K.M.M., C.P.B, N.R., and Q.M.N.K. performed experiments. D.J.M. designed research and analyzed data. D.J.M., K.M.M., and M.R.D. wrote the manuscript.
Acknowledgments
Funding for this work was provided by the National Science Foundation (grants 1032527 and DGE-0965945).
Editor: Sean Sun.
Footnotes
Supporting Materials and Methods and three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(15)00782-1.
Supporting Material
References
- 1.Helmlinger G., Netti P.A., Jain R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997;15:778–783. doi: 10.1038/nbt0897-778. [DOI] [PubMed] [Google Scholar]
- 2.Stylianopoulos T., Martin J.D., Jain R.K. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. 2013;73:3833–3841. doi: 10.1158/0008-5472.CAN-12-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fletcher D.A., Mullins R.D. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaudhuri O., Parekh S.H., Fletcher D.A. Reversible stress softening of actin networks. Nature. 2007;445:295–298. doi: 10.1038/nature05459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brangwynne C.P., MacKintosh F.C., Weitz D.A. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol. 2006;173:733–741. doi: 10.1083/jcb.200601060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendez M.G., Restle D., Janmey P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014;107:314–323. doi: 10.1016/j.bpj.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGrail D.J., Mezencev R., Dawson M.R. SNAIL-induced epithelial-to-mesenchymal transition produces concerted biophysical changes from altered cytoskeletal gene expression. FASEB J. 2015;29:1280–1289. doi: 10.1096/fj.14-257345. [DOI] [PubMed] [Google Scholar]
- 8.Roose T., Netti P.A., Jain R.K. Solid stress generated by spheroid growth estimated using a linear poroelasticity model. Microvasc. Res. 2003;66:204–212. doi: 10.1016/s0026-2862(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 9.Wang N., Butler J.P., Ingber D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 10.Jiang H., Sun S.X. Cellular pressure and volume regulation and implications for cell mechanics. Biophys. J. 2013;105:609–619. doi: 10.1016/j.bpj.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tse J.M., Cheng G., Munn L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA. 2012;109:911–916. doi: 10.1073/pnas.1118910109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hui T.H., Zhou Z.L., Gao H. Volumetric deformation of live cells induced by pressure-activated cross-membrane ion transport. Phys. Rev. Lett. 2014;113:118101. doi: 10.1103/PhysRevLett.113.118101. [DOI] [PubMed] [Google Scholar]
- 13.Stroka K.M., Jiang H., Konstantopoulos K. Water permeation drives tumor cell migration in confined microenvironments. Cell. 2014;157:611–623. doi: 10.1016/j.cell.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann E.K., Lambert I.H., Pedersen S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 15.Stewart M.P., Helenius J., Hyman A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature. 2011;469:226–230. doi: 10.1038/nature09642. [DOI] [PubMed] [Google Scholar]
- 16.Gilpin W., Uppaluri S., Brangwynne C.P. Worms under pressure: bulk mechanical properties of C. elegans are independent of the cuticle. Biophys. J. 2015;108:1887–1898. doi: 10.1016/j.bpj.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.