

Abstract
The current understanding on the role of microbiology on periodontitis causation is reviewed. An appraisal of the literature reveals several issues that have limited the attempts to investigate candidate periodontal pathogens as causes of periodontitis and confirms that only limited epidemiological evidence is available. Several aspects of the contemporary understanding on causal inference are discussed with examples for periodontitis.
Keywords: causality, cohort studies, epidemiology, germ theory of disease, infection, periodontitis
Background
Periodontal diseases are inflammatory conditions occurring in the tissues surrounding the teeth and, as it is the case for any disease, the list of causal components responsible for their onset and progression is large. Periodontal diseases require the presence of a tooth, a periodontal ligament, a living host with all its associated characteristics of an immune system, blood supply, tissue turnover, and of course a microflora. Key in causal investigations is to identify the component causes that are useful in terms of preventing and treating the disease. Extracting a tooth for instance will lead to an arrest of periodontal disease but is usually not a useful component cause from a clinical perspective.
The designation periodontal diseases includes gingivitis,1 necrotizing periodontal diseases (NPDs),2 and periodontitis.3 Gingivitis is an inflammatory reaction restricted to the gingival tissues, without signs of destruction of the supportive periodontal tissues, whereas NPDs2 and periodontitis3 are inflammatory conditions characterized by permanent loss of periodontal tissue. Even though evidence in the form of epidemiological cohort studies has been largely missing, both conditions are often defined as infectious diseases.1,4-9 Necrotizing periodontal diseases may be limited to destruction of the gingival tissues presenting with pain, gingival bleeding, ‘punched-out’ appearance of the interproximal papillae, fetid breath and pseudomembrane formation; or extend to compromise the supportive tissues of the teeth i.e., periodontal ligament and alveolar bone. NPDs are frequently observed among subjects with systemic conditions like malnutrition, periods of increased exposure to stress, and immunosuppression.2 The prevalence of NPDs has been reduced dramatically in developed countries during the last 2 decades, but remains an issue in less affluent populations, particularly in African countries.10
Periodontitis is the most frequent destructive periodontal condition, affecting in its severe stages approximately 10–15% of human populations across continents,11,12 and it is characterized by detachment and apical migration of the junctional epithelium with destruction of periodontal ligament and alveolar bone loss. The lesions are clinically characterized by loss of clinical attachment, accompanied by pocket formation and/or recession of the gingival tissue. These lesions are usually painless unless they present concurrently with its acute expression, the periodontal abscess.13
Focus of this review will be invested on a critical discussion of the current understanding of periodontitis causation and an appraisal of epidemiological evidence supporting putative periodontal pathogens as causes of periodontitis.
Two competing explanations have strongly influenced the way we define periodontitis and the strategies used for research into its etiology (for review see ref.14). One theory was that periodontitis represented an inflammatory condition initiated by a variety of systemic or remote determinants.15,16 This explanation saw periodontitis as the result of complex multifactorial etiology, which could involve a number of remote causes, such as metabolic syndrome, nutrition, use of tobacco, and other constitutional factors.14 The limited ability to treat remote causes like diabetes or an unwillingness on the part of the patient to modify behaviors such as tobacco smoking or sugar consumption created challenges in managing periodontitis. The second theory postulated that the causes of periodontitis were local to the tooth, involving factors such as occlusion, deposits and oral bacteria15-21 and hence recommended local treatments.19 This theory was developed simultaneously with Robert Koch's efforts to deliver experimental evidence for the germ theory of disease.22
With the development of microbiological methods, and the progressive identification of new microorganisms applying new techniques, the idea of infection, particularly a specific infection23 gained terrain and predominated as the main explanation for periodontitis for many decades14 becoming a defining feature of this disease.6,24,25 The influence of the germ theory,14 led to a narrow perspective of disease causation, namely, single agents relating one to one to specific periodontal disease categories.26,27
Undoubtedly, the extensive use of the infection discourse points to reluctance to acknowledge a causal character for determinants above and beyond putative periodontal pathogens.28 “Factors such as diabetes and smoking are commonly described as modifying factors …”and “…they are merely perceived as exogenous modulators of the hosts’ susceptibility to the causal infection. This view is maintained even though ‘less than 20% of the variability in periodontal disease expression can be explained by levels of specific microbes’.” (for review see ref.28).
In the 1960's, a series of small, uncontrolled studies were conducted on experimental induction of gingival inflammation in humans by avoidance of oral hygiene procedures and subsequent resolution of gingival inflammation when oral hygiene was reinstituted.29,30 The results of these experimental gingivitis studies – which were so small as to preclude statistical analyses – were extrapolated to the subsequent idea that if dental plaque development resulted in gingivitis, untreated gingivitis would invariably lead to periodontitis. This notion became a dominant paradigm in periodontology for many decades (for review see ref.31).
Notes on the definition of the periodontal outcomes
A significant issue hampering our understanding of the microbial – periodontitis relationship is the inconsistency in the characterization of periodontitis31,32 and a deeply rooted belief in the existence of various clinical periodontitis entities that may be caused by different microbial determinants.5,6,33 A current example of this is the proposed categorization of periodontitis as either chronic or aggressive.34-36
The issue of periodontal definitions and classifications is not new and has escorted the development of periodontal microbiology since its commencement as clearly documented back in 1877 during the 17th Annual Session of the American Dental Association when the periodontal outcome of interest was presented as “that formidable class of diseases of the gums which are difficult to classify."15 The debate has continued and is illustrated by the existence of at least 10 different systems for classification of periodontitis during the 1980's and 1990's.24,37-44
Heterogeneity in the definition of cases, and the lack of an agreed upon operational clinical definition of periodontitis that can be used for research purposes are not merely an academic conversation. They hinder comparison of research results, leading to overestimation or underestimation of disease occurrence,45,46 and probably more serious, they result in different and sometimes opposing results of analytical etiological research.46,47
Destructive periodontal disease occurring in otherwise apparently healthy subjects has been the subject of numerous reclassifications during the last 5 decades. The main categories alluded to in the literature comprise juvenile periodontitis,48 early onset periodontitis,49 and aggressive periodontitis.50 While for some the implications of reclassifications may appear trivial, reclassifications are not only changes of diseases names, but regrouping of subjects into partially overlapping disease categories; something that can have implications on the acquired evidence on diagnoses, etiology, effect of treatment and prognosis. With regards to its impact on our understanding of microbial causation of periodontitis it is unknown for example how the exclusion of subjects with evident supragingival biofilm from the category juvenile periodontitis51 may have influenced studies on the microbial etiology of this disease outcome and how evidence originating from these studies can be compared with similar studies comprising the alleged disease category aggressive periodontitis.36
Subsequently, 2 main local etiological theories for the occurrence of periodontitis emerged. The “non-specific plaque hypothesis," which claimed that the overall increase in numbers of subgingival microorganisms and their altered proportions were responsible for provoking inflammation and that, no single bacterial species was liable. Hence, different combinations of bacteria, rather than just a single species were considered to be accountable for the progression from gingivitis to destructive periodontitis.52 On the other hand, the “specific plaque hypothesis” supported the idea that certain forms of periodontitis appeared to be the result of overgrowth of specific indigenous plaque bacteria, warranting antimicrobial treatment targeting, based on the identification of these microorganisms upon diagnosis.53 In the early 1990's, the idea that the exposure of the dental microflora to microenvironmental changes can result in changes of its bacterial composition, which can then result in special susceptibility of the affected site to disease emerged. This notion is the cornerstone of the "ecological plaque hypothesis," which describes the relationship between the biofilms and the host response as a determining factor between maintenance of health and switch to disease.54,55
The prevailing paradigm periodontitis is an infectious disease inevitably resulted in the focus on microbiological control approaches as the main therapeutic strategy for controlling periodontitis56 (for review see refs.14,31) and regular mechanical disruption of biofilm development in the form of professional tooth cleanings as the standard of care.5,57 Even today, a simple search in PubMed using ‘periodontal AND infection’ restricted to articles published in English since 2013 yields 480 publications, suggesting that the notion of infection remains dominant in periodontal research.
Discussing epidemiological aspects of destructive periodontal diseases as infectious diseases today inevitably prompts the idea of framing this review with reference to ‘infectious disease epidemiology’ and focus on the expression ‘infectious diseases’, which are understood today as “caused by transmissible agents that replicate in the affected host."58 Paraphrasing Horsburgh and Mahon58 and struggling to make a case for periodontitis as an infectious disease, the human host should be exposed to the infectious agent/s; exposure must lead to invasion into the host tissues; and finally, this invasion must lead to the development of clinical signs and symptoms we recognize as periodontitis.
Numerous researchers have reported putative periodontal pathogens, such as Porphyromonas gingivalis, invading gingival tissues in vitro, and several case-series involving morphological, in situ hybridization, and immunohistochemistry techniques have identified microbial ‘invasion’ in periodontal lesion biopsies. Yet, it remains unclear whether these phenomena can be taken as evidence for infection or whether they hold a role in the etiology of periodontitis. A provoking counterintuitive argument is presented by evidence documenting the frequent occurrence of intracellular putative periodontal pathogens in periodontal tissues of healthy subjects, something that suggests that mucosal colonization with putative periodontal pathogens may be a widespread phenomenon in humans59 and by the notion that putative periodontal pathogens do not need to invade periodontal tissues in order to stimulate an inflammatory reaction.60 In fact, the periodontal pocket epithelium can be highly ulcerated at sites, allowing for the direct contact between the vessel-rich gingival connective tissue, and the biofilm or its secreted products.
Some periodontal microbiologists are moving away from the description of infection and now refer to periodontal disease as being caused by a dysbiosis.61-64 While the proponents of this hypothesis have moved away from the term ‘infection’ they still consider periodontal disease to be a microbiological problem and propose that a future approach to periodontal treatment could be the control of the growth or metabolic activity of the keystone pathogens.62
From an epidemiological perspective there are currently no cohort studies indicating that destructive periodontal diseases can qualify as infectious diseases. Three main issues related to infection are now discussed: (1) the occurrence and distribution of suspected periodontal pathogens in human populations, (2) the geographic variation in this distribution that may explain variation in the distribution of periodontitis, and (3) a review of the summarized epidemiological evidence supporting putative periodontal pathogens as causes of periodontitis.
Occurrence and Distribution of Putative Periodontal Pathogens
The methods used to identify and quantify the exposure
Many studies have reported information on the distribution of various subsets of putative periodontal pathogens from oral clinical samples in various human groups using traditional biochemical and phenotypical methods and different molecular DNA based techniques.4,65-67 A drawback of the earlier studies was the limited number of candidate organisms that were evaluated. Already 20 years ago Haffajee et al., suggested that information on a single species may not be informative in the context of the possibility of periodontal pockets representing mixed infections4 and it should be acknowledged today that most studies available in the periodontal literature have been conducted targeting a rather small repertoire of bacterial species, possibly representing less than 5% of the total number of organisms that can inhabit the periodontal niche.68 Several of the limitations encountered at the beginning of the enterprise have been amended with improved methods and remarkable technological development in microbiological research, particularly during the last 2 decades.4,6,65,66 Our understanding on the composition of the subgingival microbiota has expanded considerably during the last few years mainly as a consequence of technological advances in molecular methods including the availability of high-throughput analysis for large numbers of samples. This has sidestepped limitations of phenotypical and culture procedures and have allowed for a more efficient and comprehensive investigation of the distribution of subgingival microbial exposure making possible, for example, the simultaneous evaluation of numerous species from samples originating from several sites in the mouth from many subjects in clinical intervention studies and observational epidemiological studies.
While these earlier studies call for a cautious interpretation, they have provided useful information on the diversity and complexity of the subgingival microbiota while still focusing attention on some selected candidate organisms (for review see refs.66-69) That knowledge highlights that directing the scope for putative periodontal pathogens to a few bacterial species is not commensurate with available evidence on potential implicated species and their role.57,68 Taking into account that microbial species relate to each other suppressing, supplementing, and synergizing in complex systems it is reasonable to speculate about whether positive association findings between a few selected species and periodontitis is due to the influence of these selected identified bacterial species or the result of the effect of unmeasured alternative microbial covariates.
Despite the tremendous developments in oral microbiological research, major sources of variation remain in today's attempts to assess microbial exposure in etiological studies. Although molecular methods for the identification of putative oral pathogens have developed considerably, variation in the identification and recruitment of subjects and selection of sites for sampling as well as the various strategies for biological sampling advocated by different research groups hamper attempts to describe and compare the distribution of candidate periodontal pathogens in populations.57 Many studies have focused on obtaining microbial samples from periodontitis patients, and very limited information is available on subgingival microbiological profiles in human subjects representing the broad spectrum of periodontal health and disease in well-defined underlying populations.
Geographic Variation in the Distribution of Putative Periodontal Pathogens
The results of numerous studies demonstrate that a common subset of subgingival species is frequently found across study groups from different countries.70-74 Nevertheless, some reports have been interpreted as reflecting real geographical variation.73,75,76
A closer inspection of these latter studies reveals considerable sources of heterogeneity in the methods used that can well explain variability across geographical regions. These differences include variation in the methods used for identification of suspected pathogens, which for example can be restricted to the use of culture techniques70,74, biochemical and morphological methods,73,77 or involve the use of DNA based techniques,71,72,75,76,78,79 or differences in the strategies used for obtaining biological samples. Some authors have used curettes for scrapping biofilm from the subgingival root surfaces of the teeth72,76,78 whereas others have placed paper points subgingivally.73 Information on the sites selected for sampling and whether these samples have been pooled or not before laboratory analyses reveal additional sources of variation.70-74 In addition to this, the number of sites selected for sampling influences differences in prevalence estimates and distribution profiles because a larger number of sites included for sampling will necessarily increase the probability of finding the putative pathogens under investigation. Similarly, if the inclusion criteria for sampling is based on disease severity like for example with the selection of sites with deeper pocket depth,71,73,74,77 more advanced levels of attachment loss73 and/or positive bleeding on probing77 it is more likely that a selection of putative periodontal pathogens that ’like’ subgingival sites with these characteristics is overrepresented. While these sources of variation are important and may account for a significant disparity in the results reported, the most likely explanatory source of variation may be the identification and recruitment of study participants. Most studies have recruited convenience samples of patients with different periodontal diagnostic categories and are thus void of the strengths of well-defined epidemiological frameworks.70,73-76,79 This patient-selection does not mirror the distribution of the investigated species in underlying populations57 and it is such selection bias which may have caused apparent geographic variation. As a consequence, they cannot be seen as providing reliable evidence on microbiological profiles for comparisons across geographical locations. Only a few studies have comprised study groups sampled using epidemiological methods from well-defined underlying populations and the results of these studies do not reveal considerable variation in the distribution of selected putative periodontal species between different locations.72,78,80
This is not to say that bacterial clones may not vary across ethnic groups and geographic places (for review see ref.81 for instance, possibly a highly leukotoxin clone of Aggregatibacter actinomycetemcomitans (JP2 clone), can be associated with progression of periodontal destruction in selected populations.82,83 Such findings raise interesting questions about the unexpected high occurrence of periodontitis in some specific populations. It might be valuable to investigate the alluded variation systematically with standardized methods across well-defined study populations. New initiatives aiming to address this topic ought to consider careful standardization of methods used including the selection of study groups, strategies for subgingival sampling, selection of sites to be sampled, subsets of candidate agents to be investigated as well as methods for their identification.
Putative Periodontal Pathogens as Causes of Periodontitis
How to pinpoint pathogenic microorganisms
For many decades, Henle-Koch's postulates22,84 were considered key references to recognize a suspected pathogen as a cause of disease. According to these postulates, the agent must be isolated from every case of the disease by isolation in pure culture, it must not be recovered from cases of other forms of disease or among healthy animals, and after isolation and repeated growth in pure culture the pathogen must induce disease in experimental animals. Finally, the agent must be recovered from the experimental disease produced.85 These postulates were important as references to distinguish agents that could be identified with the microbiological techniques available at that time. Nevertheless they presented important challenges for periodontal researchers, some of whom proposed alternative criteria back in 1979,7 later amended by Haffajee and Socransky.4 The reasons for the inability of the application of Koch's postulates in the identification of specific periodontal pathogens include, but are not restricted to, the fact that more than half of the biofilm microbiota is as-yet uncultivable by conventional methods. According to the modified criteria, (a) the suspected microorganism should be associated with periodontitis, (b) its elimination should reduce the clinical signs of the disease, (c) it should display evidence of a host response to a pathogen (i.e., in in vitro models), (d) when applied to an animal model, it should reproduce the signs of the disease, and (e) it should actively produce virulence factors that can generate a pathogenic effect on the affected tissues.7 Based on these criteria, a number of bacterial species that can colonize a subgingival biofilm were characterized as putative causative agents of periodontal disease, including the “red complex” species (P. gingivalis, Tannerella forsythia, Treponema denticola)86 and A. actinomycetemcomitans.87
Socransky's criteria represented much lower challenge to establishing causality when compared to Henle and Koch's postulates. In addition, these postulates remained closely linked to a mono-causal etiological explanation exclusively focused on the tooth that is not compatible with our current understanding of the dominant role of systemic factors such as smoking in the etiology of periodontitis.31 Substantial disadvantages of the recommended criteria include parochial definition of causality, which applies to one human disease – periodontitis,88,89 the exclusion of concepts such as the Bradford Hill's criteria for causal inference90 and the seminal Rothman paper on causality published in 1976.91 In this latter study, Rothman presented a working definition for causation and discussed etiology in terms of sufficient causes (a.k.a. causal mechanisms) and their causal components (Fig. 1). The model embraces key principles of causation like (1) multi-causality, (2) the dependence of the strength of component causes on the distribution of complementary causes, and (3) the interaction between component causes; all aspects of causation that also apply to periodontitis. Briefly, Rothman and Greenland defined a cause of a disease as “… an event, condition, or characteristic that preceded the disease event and without which the disease event either would not have occurred at all or would not have occurred until some later time.”92 In this model, a sufficient cause is a complete causal mechanism, “a set of minimal conditions and events that inevitably produce disease.”92 The completion of a sufficient cause is equivalent to the onset of the earliest stage of the disease process. Each pie of component causes in Figure 1 is minimally sufficient to produce periodontitis. Identification of all causal components in a sufficient cause is not required for prevention, because elimination of a single causal component would stop that mechanism and prevent the occurrence of all events explained by that sufficient cause.91 If there is a causal component, which is a member of every causal mechanism, such a component is known a necessary cause.91 In the hypothetical models presented in Figure 1, P. gingivalis is pictured as a necessary cause because it appears as a member of each sufficient cause.
Figure 1.
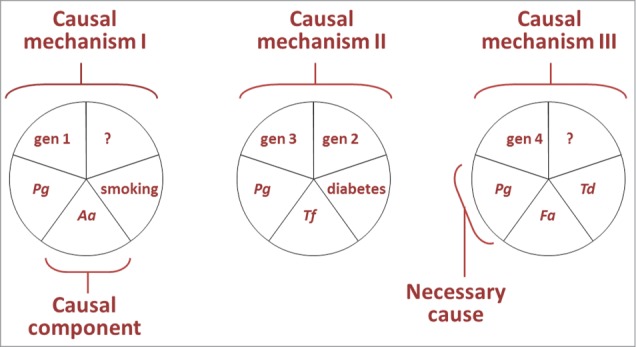
Three hypothetical sufficient causes of disease are pictured representing 3 different mechanisms in the etiology of periodontitis. In these models Porphyromonas gingivalis (Pg), Aggregatibacter actinomycetemcomitans (Aa), Tanerella forsythia (Tf), Filifactor alocis (Fa), Treponema denticola (Td), smoking, diabetes, and gene mutations 1, 2, 3, and 4 represent causal components for periodontitis; whereas Pg is pictured as a necessary cause for periodontitis. Modified from Kenneth Rothman's model of causation91 with approval from Oxford University Press and the author.
We tend to think that strong causes are strong because of their internal properties, but the strength of a causal component depends of the prevalence of its complementary component causes for periodontitis. The model of Figure 1 illustrates how the idea of causes being inherently ‘strong’ or ‘weak’ has no universal foundation. The first causal mechanism depicted in Figure 1 illustrates that the strength of A. actinomycetemcomitans (Aa) as a cause depends of the distribution of complementary causes working in the same sufficient cause. If the complementary causes for A. actinomycetemcomitans are not prevalent, for example if we assume that gene mutation 1 occurs in one out of 10.000 subjects, A. a actinomycetemcomitans will be a ‘weak’ cause that “modifies the probability of the outcome only slightly.”91 On the other hand, a causal component that needs, to complete a sufficient cause, other components that are ubiquitous is a ‘strong’ cause and will increase risk of periodontitis considerably.91 For example, suppose that gene mutation 4, in the third sufficient cause of Figure 1, represents a mutation in the cathepsin C gene (CTSC).93 CTSC encodes the lysosomal protease cathepsin C and has been reliably associated with Papillon-Lefèvre syndrome (PLS),93 a rare autosomal recessive disorder characterized clinically by palmoplantar hyperkeratosis and severe generalized early periodontitis. Nearly all subjects with PLS develop severe periodontitis, refractory to periodontal treatment. In this model ‘CTSC mutation’ can be considered a ‘strong’ cause because the frequent occurrence of regular commensal species would be enough to complete this sufficient cause. Targeting complementary causes of this mechanism for prevention or treatment of periodontitis may be irrelevant because it may be impossible to reduce their presence to levels that will prevent the completion of the causal mechanism in individuals with this systemic mutation. In this context, it is interesting to note that studies on the microbiologic profile of subjects with PLS suggest that periodontitis lesions in subjects with PLS appear to hold a considerably broader microbial diversity94 that includes opportunistic species when compared to periodontitis lesions in subjects without PLS.
The model in Figure 1 is a simple hypothetical model with only 3 sufficient causes, with 5 causal components each. The real picture involves many causal components; most of them unknown, interacting to complete each sufficient cause; and many sufficient causes (biological mechanisms), each explaining part of the occurrence of a common outcome, periodontitis.
The interaction between component causes – biological interactions
The ecological plaque hypothesis embraces that it is the interplay between host and microbial factors that can define the switch from health to disease. It has been proposed that periodontitis is caused by dysbiosis. According to this, it is not ‘selected’ periodontal pathogens that initiate the disease, but the disruption of the ecological balance leads to the synergistic interaction of variable members of the microbial community (or their specific gene combinations), that can be considered as disease-provoking.61 A combination of various virulence factors that derive from different members of the microbial community, which can yet complement each other, may be required to elicit an overall pathogenic host response. Certain bacterial species may display an “inflammophilic” profile and thrive under a degenerated inflammatory-propagating host response.95 This may in turn generate a vicious cycle of community dysbiosis and disease progression. Hence, the context of causality, an interaction between an advantaged microbial constitution and disadvantaged host response is required for disease to occur or to progress.
On a broader microbiological perspective, it is argued that the binary view on a microorganism being either a pathogen or not, is inconclusive. Attempts to classify microbes as pathogens or non, are perhaps out of scope since they misattribute a microbial property to a function that is actually a multi-variable interaction with the host.96 A recent review by Mèthot and Alizon highlights the paradigm shift toward a process-oriented model of host-parasite interactions.97 As such, there are no clear-cut unique pathogens, while the commensal, parasitic or mutualistic interactions of microbes with each other and with the host should be viewed as a continuum without clear borders. These notions are strengthened by findings on large-scale sequencing in health and disease that reveal a large genetic diversity of microbes within and between hosts, as well as by acknowledging microbial ecology and evolution as key components of the crosstalk between microbiota and their host. The results of a recently published systematic review support this and suggest a positive association of at least 17 novel species or phylotypes including the phyla Bacteroidetes, Candidatus Saccharibacteria, Firmicutes, Proteobacteria, Spirochaetes, and Synergistetes with periodontitis.98
Longitudinal epidemiological evidence for an infectious etiology of periodontitis
From Hill's 9 criteria90 particularly one, temporality, can strongly influence our understanding and weighting of the scientific evidence on putative periodontal pathogens. As long as the sequence of the events in an association between an exposure and an outcome has not been established there is no evidence for causation. While not everything that precedes an event can be considered a cause of it,99 a cause must always precede the effect. This necessarily calls for the use of prospective cohort evidence when disentangling the pathogenic nature of putative agents.
Even though many research groups have investigated associations between selected subgroups of putative periodontal pathogens and periodontitis during the last 5 decades using various methods and approaches, a recently published systematic review of this evidence highlights that only a few studies have employed methods that could be considered to provide prospective longitudinal evidence for a causal relationship.100 The review found 3 studies conducted in predominantly non-Caucasian disadvantaged pediatric populations supporting the infection hypothesis for one putative periodontal pathogen: A. actinomycetemcomitans.80,82,101 Several cohort studies evaluating A. actinomycetemcomitans did not support the infection hypothesis. None of the studies supported the infection hypothesis among adult groups, Caucasian subjects, or in population residing in socioeconomically wealthier populations.100
The weight of progression studies
A significant portion of the studies available in the literature document what could be described as studies on the progression of periodontitis.100 While it may be tempting to interpret the positive associations between bacteria and periodontitis reported in many of these studies, it should be kept in mind that a requirement for prospective cohort studies is that exposed subjects are disease-free at baseline102 and even mild severity levels of the outcome should be avoided in the cohort under investigation. Positive associations between progression of periodontal destruction and subsets of putative periodontal pathogens may well reflect that early periodontitis provided favorable niches for the development of certain suspected pathogenic candidates. In consequence, these progression studies provide weak evidence on putative periodontal pathogens as causes of periodontitis. If these putative pathogens are causally related to periodontitis, the recognized presence of the implicated pathogen/s must antecede the signs of periodontitis.
A recent update of the electronic search conducted by Hujoel et al.,100 was run in September 2014 and an updated assessment was conducted for inclusion in this review. The new search added 3 new candidate publications to the original yield.83,103,104 A closer inspection of the clinical criteria employed in the publications now included80,82,83,101,103,104 revealed that periodontitis could not be excluded at baseline and that strictly these studies could also be considered progression studies. In the study by Van der Velden et al.,80 subjects labeled healthy at the starting point could present with 2 mm of attachment loss in several teeth and/or 3 mm of attachment loss in one tooth or 2 adjacent teeth. Similarly, in the studies by Haubek et al.,82 and Aberg et al.,83,104 young subjects categorized as healthy at baseline may have present with 2 mm of attachment loss. According to the case definition by Fine et al.,101 from 2007 and Fine et al.,103 from 2013 adolescents subjects were examined for the occurrence of destruction of the supportive tissues of the teeth in the form of attachment loss only if they presented with pockets deeper than 5 mm. This means that subjects with 4 mm pocket depth and various levels of clinical attachment loss would be considered healthy at baseline. This decision was possibly based on the questionable assumption that periodontitis is characterized by deepening of the pocket and disregards that screening for cases of early periodontitis based on deepening of the pockets results in a considerable number of subjects with periodontitis, being overlooked. Lopez et al.,105 found, in a Chilean adolescent population, that at least 57 % of sites with attachment ≥ 3 mm in young subjects with periodontitis did not present pocket depth > 2 mm due to retraction of the gingival tissues.
Conclusions
The heterogeneity of the methods used in the studies available hinders reasonable comparisons of the distribution of putative periodontal pathogens across age and ethnic populations or geographic locations. The results of a handful of studies suggested an association between selected putative pathogens and progression of periodontitis. These studies identified different organisms, used different definitions of periodontal outcomes, and typically used sites as experimental unit of analysis without proper accounting of correlation. The literature on the evidence of microbial agents as a primary etiology of periodontitis is essentially barren for prospective cohort studies including validated assessment of exposure in periodontitis-free study populations at baseline.
From an epidemiological perspective understanding periodontitis as a complex inflammatory syndrome characterized by destruction of the supporting tissues of the teeth may provide a better frame for causal inference. The inflammatory model can be understood as the result of the possible interaction of many constellations of causal components where microbial components may be adopted, without this indicating that the researchers are devoted to a single microbial theory of destructive periodontal disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. American Academy of P Glossary of periodontal terms, 4th Edition Chicago, Illinois: The American Academy of Periodontology, 2001. [Google Scholar]
- 2. Lang N, Soskolne AW, Greenstein G, Cochran D, Corbet E, Meng WX, Newman M, Novak MJ, Tenenbaum H. 1999 International Workshop for a Classification of Periodontal Diseases and Conditions. Papers. Oak Brook, Illinois, October 30-November 2, 1999. Consensus report: Necrotizing periodontal diseases. Ann Periodontol 1999; 4:78; http://dx.doi.org/ 10.1902/annals.1999.4.1.78 [DOI] [PubMed] [Google Scholar]
- 3. Flemmig TF. Periodontitis. Ann Periodontol 1999; 4:32-7; PMID:10863373; http://dx.doi.org/ 10.1902/annals.1999.4.1.32 [DOI] [PubMed] [Google Scholar]
- 4. Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000 1994; 5:78-111; PMID:9673164; http://dx.doi.org/ 10.1111/j.1600-0757.1994.tb00020.x [DOI] [PubMed] [Google Scholar]
- 5. Loesche WJ, Grossman NS. Periodontal disease as a specific, albeit chronic, infection: diagnosis and treatment. Clin Microbiol Rev 2001; 14:727-52; PMID:11585783; http://dx.doi.org/ 10.1128/CMR.14.4.727-752.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Socransky SS, Haffajee AD. Evidence of bacterial etiology: a historical perspective. Periodontol 2000 1994; 5:7-25; PMID:9673160; http://dx.doi.org/ 10.1111/j.1600-0757.1994.tb00016.x [DOI] [PubMed] [Google Scholar]
- 7. Socransky SS. Criteria for the infectious agents in dental caries and periodontal disease. J Clin Periodontol 1979; 6:16-21; PMID:295292; http://dx.doi.org/ 10.1111/j.1600-051X.1979.tb02114.x [DOI] [PubMed] [Google Scholar]
- 8. Schroeder HE. Discussion: Pathogenesis of periodontitis. J Clin Periodontol 1986; 13:426-8; http://dx.doi.org/ 10.1111/j.1600-051X.1986.tb01486.x [DOI] [Google Scholar]
- 9. 'There is an overuse of implants in the world and an underuse of teeth as targets for treatment'. Br Dent J 2014; 217:396-7; http://dx.doi.org/ 10.1038/sj.bdj.2014.930 [DOI] [PubMed] [Google Scholar]
- 10. Baelum V, Scheutz F. Periodontal diseases in Africa. Periodontol 2000 2002; 29:79-103; PMID:12102704; http://dx.doi.org/ 10.1034/j.1600-0757.2002.290105.x [DOI] [PubMed] [Google Scholar]
- 11. Dye BA. Global periodontal disease epidemiology. Periodontol 2000 2012; 58:10-25; PMID:22133364; http://dx.doi.org/ 10.1111/j.1600-0757.2011.00413.x [DOI] [PubMed] [Google Scholar]
- 12. Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990-2010: A systematic review and meta-regression. J Dent Res 2014; 93:1045-53; PMID:25261053; http://dx.doi.org/ 10.1177/0022034514552491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meng HX. Periodontal abscess. Ann Periodontol 1999; 4:79-83; PMID:10863378; http://dx.doi.org/ 10.1902/annals.1999.4.1.79 [DOI] [PubMed] [Google Scholar]
- 14. Hujoel PP, Zina LG, Cunha-Cruz J, Lopez R. Historical perspectives on theories of periodontal disease etiology. Periodontol 2000 2012; 58:153-60; PMID:22133374; http://dx.doi.org/ 10.1111/j.1600-0757.2011.00423.x [DOI] [PubMed] [Google Scholar]
- 15. Rehwinkel FH. Proceedings of dental societies. American Dental Association – seventeenth annual session. Dental Cosmos 1877; 19:567-79. [Google Scholar]
- 16. Miller WD. The micro-organisms of the human mouth. The local and general diseases which are caused by them. Philadelphia, PA: The S.S. White Dental Mfg. Co., 1890. [Google Scholar]
- 17. Harris GB. The treatment of pyorrhea by bacterial vaccines, and the results of animal experimentation. Dental Cosmos 1913; 55:388-92. [Google Scholar]
- 18. Harlan AW. A review of recent literature on the loose tooth or pyorrhea problem. Dental Cosmos 1900; 42:401-8. [Google Scholar]
- 19. Harlan AW. Treatment of pyorrhea alveolaris. Dental Cosmos 1883; 25:517-21. [PMC free article] [PubMed] [Google Scholar]
- 20. Talbot ES. Pyorrhea alveolaris. Dental Cosmos 1886; 28:692. [Google Scholar]
- 21. Talbot ES. Pyorrhea alveolaris. Dental Cosmos 1896; 38:310-21. [Google Scholar]
- 22. Koch R. Ueber bakteriologische Forschung In. Verh X Int Med Congr Berlin 1890. 1892:35. [Google Scholar]
- 23. Barreto ML, Teixeira MG, Carmo EH. Infectious diseases epidemiology. J Epidemiol Community Health 2006; 60:192-5; PMID:16476746; http://dx.doi.org/ 10.1136/jech.2003.011593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Page RC, Schroeder HE. Periodontitis in Man and other Animals. A Comparative Review. Basel: Karger, 1982. [Google Scholar]
- 25. American contributions to the new age of dental research Bethesda, Md. : U.S. Dept. of Health and Human Services, National Institutes of Health, National Institute of Dental Research, National Library of Medicine, 1988. [Google Scholar]
- 26. Susser M, Susser E. Choosing a future for epidemiology: I. Eras and paradigms. Am J Public Health 1996; 86:668-73; PMID:8629717; http://dx.doi.org/ 10.2105/AJPH.86.5.668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evans AS. Causation and disease: the Henle-Koch postulates revisited. Yale J Biol Med 1976; 49:175-95; PMID:782050 [PMC free article] [PubMed] [Google Scholar]
- 28. Lopez R, Baelum V. Contesting conventional periodontal wisdom: implications for periodontal classifications. Community Dent Oral Epidemiol 2012; 40:385-95; PMID:22360496; http://dx.doi.org/ 10.1111/j.1600-0528.2012.00677.x [DOI] [PubMed] [Google Scholar]
- 29. Löe H, Theilade E, Jensen SB. Experimental gingivitis in man. J Periodontol 1965; 36:177-87; PMID:14296927; http://dx.doi.org/ 10.1902/jop.1965.36.3.177 [DOI] [PubMed] [Google Scholar]
- 30. Theilade E, Wright WH, Jensen SB, Löe H. Experimental gingivitis in man. II. A longitudinal clinical and bacteriological investigation. J Periodontal Res 1966; 1 1-13; PMID:4224181; http://dx.doi.org/ 10.1111/j.1600-0765.1966.tb01842.x [DOI] [PubMed] [Google Scholar]
- 31. Baelum V, Lopez R. Periodontal disease epidemiology – learned and unlearned? Periodontol 2000 2013; 62:37-58; PMID:23574463; http://dx.doi.org/ 10.1111/j.1600-0757.2012.00449.x [DOI] [PubMed] [Google Scholar]
- 32. Baelum V, Lopez R. Defining a periodontitis case: analysis of a never-treated adult population. J Clin Periodontol 2012; 39:10-9; PMID:22093052; http://dx.doi.org/ 10.1111/j.1600-051X.2011.01812.x [DOI] [PubMed] [Google Scholar]
- 33. Armitage GC. Classifying periodontal diseases – a long-standing dilemma. Periodontol 2000 2002; 30:9-23; PMID:12236892; http://dx.doi.org/ 10.1034/j.1600-0757.2002.03002.x [DOI] [PubMed] [Google Scholar]
- 34. Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol 1999; 4:1-6; PMID:10863370; http://dx.doi.org/ 10.1902/annals.1999.4.1.1 [DOI] [PubMed] [Google Scholar]
- 35. Lindhe J, Ranney R, Lamster I, Charles A, Chung CP, Flemmig T, Kinane D, Listgarten M, Löe H, Schoor R, et al. . 1999 International Workshop for a Classification of Periodontal Diseases and Conditions. Papers. Oak Brook, Illinois, October 30-November 2, 1999. Consensus report: Chronic periodontitis. Ann Periodontol 1999; 4:38; http://dx.doi.org/ 10.1902/annals.1999.4.1.38 [DOI] [Google Scholar]
- 36. Lang N, Bartold PM, Cullinan M, Jeffcoat M, Mombelli A, Murakami S, Page R, Papapanou P, Tonetti M, Van Dyke T. 1999 international workshop for a classification of periodontal diseases and conditions. Papers. Oak Brook, Illinois, October 30-November 2, 1999. Consensus report: Aggressive periodontitis. Ann Periodontol 1999; 4:53; http://dx.doi.org/ 10.1902/annals.1999.4.1.53 [DOI] [Google Scholar]
- 37. American Academy of P Proceedings of the World Workshop in Clinical Periodontics. Consensus report, Discussion section I. Periodontal diagnosis and diagnostic aids. Princeton, NJ, 1989
- 38. Johnson NW, Griffiths GS, Wilton JMA, Maiden MFJ, Curtis MA, Gillet IR, Wilson DT, Sterne JAC. Detection of high-risk groups and individuals for periodontal diseases. J Clin Periodontol 1988; 15:276-82; PMID:3292592; http://dx.doi.org/ 10.1111/j.1600-051X.1988.tb01584.x [DOI] [PubMed] [Google Scholar]
- 39. Suzuki JB. Diagnosis and classification of the periodontal diseases. Dent Clin North Am 1988; 32:195-216; PMID:3288510 [PubMed] [Google Scholar]
- 40. American Academy of P . Glossary of periodontic terms. J Periodontol 1986; 57:1-31; PMID:3484785; http://dx.doi.org/ 10.1902/jop.1986.57.1.1 [DOI] [PubMed] [Google Scholar]
- 41. Topic B. Classification of periodontal diseases. Int Dent J 1990; 40 171-5; PMID:2194974 [PubMed] [Google Scholar]
- 42. Ranney RR. Classification of periodontal diseases. Periodontol 2000 1993; 2:13-25; PMID:9673177; http://dx.doi.org/ 10.1111/j.1600-0757.1993.tb00216.x [DOI] [PubMed] [Google Scholar]
- 43. Attström R, Van der Velden U. Consensus report session I. In: Lang NP, Karring T, eds. Proceedings of the 1st European Workshop on Periodontology: Quintessence Publishing Co, 1994:120-6.
- 44. 1999 International Workshop for a Classification of Periodontal Diseases and Conditions. Papers. Oak Brook, Illinois, October 30-November 2, 1999. Ann Periodontol 1999; 4:1-112; PMID:10863370; http://dx.doi.org/ 10.1902/annals.1999.4.1.1 [DOI] [PubMed] [Google Scholar]
- 45. Costa FO, Guimaraes AN, Cota LO, Pataro AL, Segundo TK, Cortelli SC, Costa JE. Impact of different periodontitis case definitions on periodontal research. J Oral Sci 2009; 51:199-206; PMID:19550087; http://dx.doi.org/ 10.2334/josnusd.51.199 [DOI] [PubMed] [Google Scholar]
- 46. Lopez R, Baelum V. Classifying periodontitis among adolescents: implications for epidemiological research. Community Dent Oral Epidemiol 2003; 31:136-43; PMID:12641595; http://dx.doi.org/ 10.1034/j.1600-0528.2003.00022.x [DOI] [PubMed] [Google Scholar]
- 47. Manau C, Echeverria A, Agueda A, Guerrero A, Echeverria JJ. Periodontal disease definition may determine the association between periodontitis and pregnancy outcomes. J Clin Periodontol 2008; 35:385-97; PMID:18341599; http://dx.doi.org/ 10.1111/j.1600-051X.2008.01222.x [DOI] [PubMed] [Google Scholar]
- 48. Butler JH. A familial pattern of juvenile periodontitis (periodontosis). J Periodontol 1969; 40:115-8; PMID:5251414; http://dx.doi.org/ 10.1902/jop.1969.40.2.115 [DOI] [PubMed] [Google Scholar]
- 49. Vandesteen GE, Williams BL, Ebersole JL, Altman LC, Page RC. Clinical, microbiological and immunological studies of a family with a high prevalence of early-onset periodontitis. J Periodontol 1984; 55:159-69; PMID:6584592; http://dx.doi.org/ 10.1902/jop.1984.55.3.159 [DOI] [PubMed] [Google Scholar]
- 50. Parameter on Aggressive Periodontitis . American academy of periodontology. J Periodontol 2000; 71 (suppl):867-9; PMID:10875695 [DOI] [PubMed] [Google Scholar]
- 51. Baer PN. The case for periodontosis as a clinical entity. J Periodontol 1971; 42:516-20; PMID:5284178; http://dx.doi.org/ 10.1902/jop.1971.42.8.516 [DOI] [PubMed] [Google Scholar]
- 52. Theilade E. The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol 1986; 13 905-11; PMID:3540019; http://dx.doi.org/ 10.1111/j.1600-051X.1986.tb01425.x [DOI] [PubMed] [Google Scholar]
- 53. Loesche WJ. Clinical and microbiological aspects of chemotherapeutic agents used according to the specific plaque hypothesis. J Dent Res 1979; 58:2404-12; PMID:41862; http://dx.doi.org/ 10.1177/00220345790580120905 [DOI] [PubMed] [Google Scholar]
- 54. Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology 2003; 149:279-94; PMID:12624191; http://dx.doi.org/ 10.1099/mic.0.26082-0 [DOI] [PubMed] [Google Scholar]
- 55. Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res 1994; 8:263-71; PMID:7865085 [DOI] [PubMed] [Google Scholar]
- 56. Lindhe J, Nyman S. The effect of plaque control and surgical pocket elimination on the establishment and maintenance of periodontal health. A longitudinal study of periodontal therapy in cases of advanced disease. J Clin Periodontol 1975; 2:67-79; PMID:1055729; http://dx.doi.org/ 10.1111/j.1600-051X.1975.tb01727.x [DOI] [PubMed] [Google Scholar]
- 57. Palmer RJ, Jr. Composition and development of oral bacterial communities. Periodontol 2000 2014; 64:20-39; PMID:24320954; http://dx.doi.org/ 10.1111/j.1600-0757.2012.00453.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Horsburgh CR, Jr., Mahon BE. Infectious Disease Epidemiology. Modern Epidemiology. Philadelphia: Lippincott Williams & Wilkins, 2008:549-63 [Google Scholar]
- 59. Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infect Immun 2001; 69:2700-7; PMID:11254637; http://dx.doi.org/ 10.1128/IAI.69.4.2700-2707.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hajishengallis G, Moutsopoulos NM. Etiology of leukocyte adhesion deficiency-associated periodontitis revisited: not a raging infection but a raging inflammatory response. Expert Rev Clin Immunol 2014; 10:973-5; PMID:24931458; http://dx.doi.org/ 10.1586/1744666X.2014.929944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 2012; 27:409-19; PMID:23134607; http://dx.doi.org/ 10.1111/j.2041-1014.2012.00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol 2012; 10:717-25; PMID:22941505; http://dx.doi.org/ 10.1038/nrmicro2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zaura E, Nicu EA, Krom BP, Keijser BJ. Acquiring and maintaining a normal oral microbiome: current perspective. Front Cell Infect Microbiol 2014; 4:85; PMID:25019064; http://dx.doi.org/ 10.3389/fcimb.2014.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nibali L, Henderson B, Sadiq ST, Donos N. Genetic dysbiosis: the role of microbial insults in chronic inflammatory diseases. J Oral Microbiol 2014; 6; PMID:24578801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Paster BJ, Dewhirst FE. Molecular microbial diagnosis. Periodontol 2000 2009; 51:38-44; PMID:19878468; http://dx.doi.org/ 10.1111/j.1600-0757.2009.00316.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000 2006; 42:80-7; PMID:16930307; http://dx.doi.org/ 10.1111/j.1600-0757.2006.00174.x [DOI] [PubMed] [Google Scholar]
- 67. Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000 2005; 38:135-87; PMID:15853940; http://dx.doi.org/ 10.1111/j.1600-0757.2005.00107.x [DOI] [PubMed] [Google Scholar]
- 68. Curtis MA. Periodontal microbiology-the lid's off the box again. J Dent Res 2014; 93:840-2; PMID:25074493; http://dx.doi.org/ 10.1177/0022034514542469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kilian M, Frandsen EV, Haubek D, Poulsen K. The etiology of periodontal disease revisited by population genetic analysis. Periodontol 2000 2006; 42:158-79; PMID:16930310; http://dx.doi.org/ 10.1111/j.1600-0757.2006.00159.x [DOI] [PubMed] [Google Scholar]
- 70. Cao CF, Aeppli DM, Liljemark WF, Bloomquist CG, Bandt CL, Wolff LF. Comparison of plaque microflora between Chinese and Caucasian population groups. J Clin Periodontol 1990; 17:115-8; PMID:2303572; http://dx.doi.org/ 10.1111/j.1600-051X.1990.tb01072.x [DOI] [PubMed] [Google Scholar]
- 71. Dowsett SA, Kowolik MJ, Archila LA, Eckert GJ, LeBlanc DJ. Subgingival microbiota of indigenous Indians of Central America. J Clin Periodontol 2002; 29:159-67; PMID:11895544; http://dx.doi.org/ 10.1034/j.1600-051x.2002.290211.x [DOI] [PubMed] [Google Scholar]
- 72. Papapanou PN, Teanpaisan R, Obiechina NS, Pithpornchaiyakul W, Pongpaisal S, Pisuithanakan S, Baelum V, Fejerskov O, Dahlén G. Periodontal microbiota and clinical periodontal status in a rural sample in southern Thailand. Eur J Oral Sci 2002; 110:345-52; PMID:12664464; http://dx.doi.org/ 10.1034/j.1600-0722.2002.21361.x [DOI] [PubMed] [Google Scholar]
- 73. Sanz M, Van Winkelhoff AJ, Herrera D, Dellemijn-Kippuw N, Simón R, Winkel EG. Differences in the composition of the subgingival microbiota of two periodontitis populations of different geographical origin. A comparison between Spain and The Netherlands. Eur J Oral Sci 2000; 108:383-92; PMID:11037754; http://dx.doi.org/ 10.1034/j.1600-0722.2000.108005383.x [DOI] [PubMed] [Google Scholar]
- 74. Yano-Higuchi K, Takamatsu N, He T, Umeda M, Ishikawa I. Prevalence of Bacteroides forsythus, Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans in subgingival microflora of Japanese patients with adult and rapidly progressive periodontitis. J Clin Periodontol 2000; 27:597-602; PMID:10959786; http://dx.doi.org/ 10.1034/j.1600-051x.2000.027008597.x [DOI] [PubMed] [Google Scholar]
- 75. Haffajee AD, Bogren A, Hasturk H, Feres M, Lopez NJ, Socransky SS. Subgingival microbiota of chronic periodontitis subjects from different geographic locations. J Clin Periodontol 2004; 31:996-1002; PMID:15491316; http://dx.doi.org/ 10.1111/j.1600-051X.2004.00597.x [DOI] [PubMed] [Google Scholar]
- 76. Haffajee AD, Japlit M, Bogren A, Kent RL, Goodson JM, Socransky SS. Differences in the subgingival microbiota of Swedish and USA subjects who were periodontally healthy or exhibited minimal periodontal disease. J Clin Periodontol 2005; 32:33-9; PMID:15642056; http://dx.doi.org/ 10.1111/j.1600-051X.2004.00624.x [DOI] [PubMed] [Google Scholar]
- 77. Timmerman MF, Van der Weijden GA, Armand S, Abbas F, Winkel EG, Van Winkelhoff AJ, Van der Velden U. Untreated periodontal disease in Indonesian adolescents. Clinical and microbiological baseline data. J Clin Periodontol 1998; 25:215-24; PMID:9543192; http://dx.doi.org/ 10.1111/j.1600-051X.1998.tb02431.x [DOI] [PubMed] [Google Scholar]
- 78. Papapanou PN, Baelum V, Luan WM, Madianos PN, Chen X, Fejerskov O, Dahlén G. Subgingival microbiota in adult Chinese: prevalence and relation to periodontal disease progression. J Periodontol 1997; 68:651-66; PMID:9249637; http://dx.doi.org/ 10.1902/jop.1997.68.7.651 [DOI] [PubMed] [Google Scholar]
- 79. Colombo AP, Teles RP, Torres MC, Souto R, Rosalem WJ, Mendes MC, Uzeda M. Subgingival microbiota of Brazilian subjects with untreated chronic periodontitis. J Periodontol 2002; 73:360-9; PMID:11990436; http://dx.doi.org/ 10.1902/jop.2002.73.4.360 [DOI] [PubMed] [Google Scholar]
- 80. Van der Velden U, Abbas F, Armand S, Loos BG, Timmerman MF, Van der Weijden GA, Van Winkelhoff AJ, Winkel EG. Java project on periodontal diseases. The natural development of periodontitis: risk factors, risk predictors and risk determinants. J Clin Periodontol 2006; 33:540-8; PMID:16899096; http://dx.doi.org/ 10.1111/j.1600-051X.2006.00953.x [DOI] [PubMed] [Google Scholar]
- 81. Rylev M, Kilian M. Prevalence and distribution of principal periodontal pathogens worldwide. J Clin Periodontol 2008; 35:346-61; PMID:18724862; http://dx.doi.org/ 10.1111/j.1600-051X.2008.01280.x [DOI] [PubMed] [Google Scholar]
- 82. Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet 2008; 371:237-42; PMID:18207019; http://dx.doi.org/ 10.1016/S0140-6736(08)60135-X [DOI] [PubMed] [Google Scholar]
- 83. Hoglund Aberg C, Kwamin F, Claesson R, Dahlen G, Johansson A, Haubek D. Progression of attachment loss is strongly associated with presence of the JP2 genotype of Aggregatibacter actinomycetemcomitans: a prospective cohort study of a young adolescent population. J Clin Periodontol 2014; 41:232-41; PMID:24304011; http://dx.doi.org/ 10.1111/jcpe.12209 [DOI] [PubMed] [Google Scholar]
- 84. Henle J. On Miasmata and contagia. Baltimore: Johns Hopkings Press, 1938. [Google Scholar]
- 85. Porta M. (ed.). A dictionary of epidemiology. Oxford, UK: Oxford University Press, 2001. [Google Scholar]
- 86. Socransky SS, Haffajee AD, Cugini MA, Smith C. Microbial complexes in subgingival plaque. J Clin Periodontol 1998; 25:134-44; PMID:9495612; http://dx.doi.org/ 10.1111/j.1600-051X.1998.tb02419.x [DOI] [PubMed] [Google Scholar]
- 87. Genco RJ. Current view of risk factors for periodontal diseases. J Periodontol 1996; 67:1041-9; PMID:8910821; http://dx.doi.org/ 10.1902/jop.1996.67.10s.1041 [DOI] [PubMed] [Google Scholar]
- 88. Evans AS. Causation and disease: a chronological journey. The Thomas Parran Lecture. Am J Epidemiol 1978; 108:249-58; PMID:727194 [DOI] [PubMed] [Google Scholar]
- 89. Huebner RJ. Criteria for etiologic association of prevalent viruses with prevalent diseases; the virologist's dilemma. Ann N Y Acad Sci 1957; 67:430-8; PMID:13411978; http://dx.doi.org/ 10.1111/j.1749-6632.1957.tb46066.x [DOI] [PubMed] [Google Scholar]
- 90. Hill AB. The environment and disease: association or causation? Proc R Soc Med 1965; 58:295-300; PMID:14283879 [PMC free article] [PubMed] [Google Scholar]
- 91. Rothman KJ. Causes. Am J Epidemiol 1976; 104:587-92; PMID:998606 [DOI] [PubMed] [Google Scholar]
- 92. Rothman KJ, Greenland S. Causation and causal inference in Epidemiology. Am J Public Health 2005; 95 (suppl 1):S144-S50; PMID:16030331; http://dx.doi.org/ 10.2105/AJPH.2004.059204 [DOI] [PubMed] [Google Scholar]
- 93. Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, Hewitt C, Moynihan L, Roberts E, Woods CG, et al. . Loss-of-function mutation in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nature Genetics 1999; 23:421-4; PMID:10581027; http://dx.doi.org/ 10.1038/70525 [DOI] [PubMed] [Google Scholar]
- 94. Albandar JM, Khattab R, Monem F, Barbuto SM, Paster BJ. The subgingival microbiota of Papillon-Lefevre syndrome. J Periodontol 2012; 83:902-8; PMID:22141356; http://dx.doi.org/ 10.1902/jop.2011.110450 [DOI] [PubMed] [Google Scholar]
- 95. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol 2014; 29:248-57; PMID:24976068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pirofski LA, Casadevall A. Q&A: what is a pathogen? A question that begs the point. BMC Biol 2012; 10; PMID:22293325; http://dx.doi.org/ 10.1186/1741-7007-10-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Méthot P-O, Alizon S. What is a pathogen? Towards a process view of host-parasite interactions. Virulence 2014; 5:775-85; http://dx.doi.org/ 10.4161/21505594.2014.960726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Perez-Chaparro PJ, Goncalves C, Figueiredo LC, Faveri M, Lobao E, Tamashiro N, Duarte P, Feres M. Newly identified pathogens associated with periodontitis: a systematic review. J Dent Res 2014; 93:846-58; PMID:25074492; http://dx.doi.org/ 10.1177/0022034514542468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hughes W, Lavery J. Critical thinking. An introduction to the basic skills. Ontario: Broadview press Ltd, 2004. [Google Scholar]
- 100. Hujoel PP, Zina LG, Cunha-Cruz J, Lopez R. Specific infections and the etiology of destructive periodontal disease: A systematic review. Eur J Oral Sci 2013; 121:2-6; PMID:23331417; http://dx.doi.org/ 10.1111/eos.12011 [DOI] [PubMed] [Google Scholar]
- 101. Fine DH, Markowitz KV, Furgang D, Fairlie K, Ferrandiz J, Nasri C, McKiernan M, Gunsolley J. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol 2007; 45:3859-69; PMID:17942658; http://dx.doi.org/ 10.1128/JCM.00653-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rothman KJ, Greenland S. Cohort Studies. Modern Epidemiology. Philadelphia: Lippincott Williams & Wilkins, 2008:100-10. [Google Scholar]
- 103. Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrendiz J, Furgang D, Paster BJ, Dewhirst FE. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol 2013; 51:2850-61; PMID:23784124; http://dx.doi.org/ 10.1128/JCM.00729-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hoglund Aberg C, Antonoglou G, Haubek D, Kwamin F, Claesson R, Johansson A. Cytolethal distending toxin in isolates of Aggregatibacter actinomycetemcomitans from Ghanaian adolescents and association with serotype and disease progression. PLoS One 2013; 8:e65781; PMID:23922633; http://dx.doi.org/ 10.1371/journal.pone.0065781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lopez R, Frydenberg M, Baelum V. Clinical features of early periodontitis. J Periodontol 2009; 80:749-58; PMID:19405828; http://dx.doi.org/ 10.1902/jop.2009.080463 [DOI] [PubMed] [Google Scholar]