

ABSTRACT
The extracellular aggregation of proteins into proteotoxic oligomers and amyloid fibrils is implicated in the onset and pathology of numerous diseases referred to as amyloid diseases. All of the proteins that aggregate extracellularly in association with amyloid disease pathogenesis originate in the endoplasmic reticulum (ER) and are secreted through the secretory pathway. Disruptions in ER protein homeostasis or proteostasis (i.e., ER stress) can facilitate the aberrant secretion of misfolded protein conformations to the extracellular space and exacerbate pathologic protein aggregation into proteotoxic species. Activation of an ER stress-responsive signaling pathway, the Unfolded Protein Response (UPR), restores ER proteostasis through the transcriptional regulation of ER proteostasis pathways. In contrast, the functional role for the UPR in regulating extracellular proteostasis during ER stress is poorly defined. We recently identified ERdj3 as a UPR-regulated secreted chaperone that increases extracellular proteostasis capacity in response to ER stress, revealing a previously-unanticipated direct mechanism by which the UPR impacts extracellular proteostasis. Here, we discuss the functional implications of ERdj3 secretion on extracellular proteostasis maintenance and define the mechanisms by which ERdj3 secretion coordinates intra- and extracellular proteostasis environments during ER stress.
Keywords: unfolded protein response, extracellular chaperones, proteostasis, amyloid disease, protein aggregation
Abbreviations
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- AD
Alzheimers' disease
- CJD
Creutzfeldt-Jakob disease
- SA
systemic amyloidoses
- TTR
transthyretin
- ROS
reactive oxygen species
- TPrP
toxic prion protein
Introduction
The extracellular aggregation of proteins or peptides is causatively associated with the onset and pathology of over 30 diseases including Alzheimer disease (AD), Creutzfeldt-Jakob disease (CJD), and the systemic amyloidoses (SA).1–3 In each of these so-called amyloid diseases, a specific protein fails to form or maintain a well-folded structure, facilitating its aggregation into proteotoxic soluble oligomers and amyloid fibrils. These aggregates deposit on post-mitotic tissues such as neurons and the heart, disrupting organ function and ultimately resulting in death. The misfolding or misassembly of proteins that leads to proteotoxic aggregation in these diseases reflects an imbalance in extracellular protein homeostasis (or proteostasis), providing motivation to identify pathways that regulate and maintain extracellular proteostasis in the context of human disease.
The most direct mechanism by which extracellular proteostasis is maintained is through the activity of secreted chaperones.4 These include well-established secreted chaperones such as clusterin and abundant chaperone-like proteins such as albumin.5,6 Unlike the intracellular environment, where intricate proteostasis networks rely on the activity of ATP-dependent chaperones, 103- to 106-fold lower levels of ATP7,8 in the extracellular environment necessitate that secreted chaperones regulate extracellular proteostasis through a mechanism independent of ATP. As such, secreted chaperones bind to misfolded proteins in extracellular environments such as the blood or cerebral spinal fluid through an ATP-independent ‘holdase’ activity, preventing the proteotoxic aggregation of misfolded proteins. Furthermore, binding to secreted chaperones promotes the removal of misfolded proteins from the extracellular environment through increased targeting to endocytic pathways and subsequent lysosomal degradation in cells such as macrophages.4,9,10 Other chaperones including cytosolic Hsp70s, the ER Hsp70 chaperone BiP, the ER lectin calreticulin, and the mitochondrial Hsp60 chaperonin are presented on the cell surface, although the extracellular populations of these chaperones appear to primarily function for immunological signaling as opposed to extracellular proteostasis maintenance.11–15
Significant genetic and clinical evidence demonstrate the importance of secreted chaperones in the pathogenesis of amyloid diseases. For example, alterations in clusterin activity have been implicated in the pathologic aggregation of the amyloidogenic Aβ peptide and the amyloidogenic protein transthyretin (TTR) in mouse models of AD and SA, respectively.16,17 Similarly, altered clusterin activity is associated with the pathogenesis of AD and SA in patients.17–19 Clusterin also accumulates in protein aggregates observed in patients suffering from amyloid diseases including AD, CJD, and SA.18,20,21 These results indicate that secreted chaperones are critically important for regulating extracellular proteostasis in the pathology of amyloid diseases.
Apart from secreted chaperones, extracellular proteostasis is maintained by the activity of the endoplasmic reticulum (ER) – the first organelle of the secretory pathway.22–24 The ER is responsible for the folding and trafficking of nearly all proteins found in extracellular environments, including all secreted proteins that aggregate in association with amyloid diseases. In the ER, these proteins engage ER-localized chaperones and folding enzymes that facilitate their folding into native 3-dimensional conformations that are then trafficked to downstream environments of the secretory pathway including the extracellular space. Proteins unable to attain a folded conformation in the ER are targeted to degradation pathways that remove misfolded proteins from the ER lumen and direct them to proteasomal or lysosomal degradation. The partitioning of proteins between ER protein folding/trafficking and degradation pathways, also called ER quality control, is the mechanism by which the ER prevents the aberrant secretion of misfolded proteins to the extracellular environment where they could aggregate into proteotoxic oligomers.22–24
Although ER quality control efficiently attenuates the secretion of highly-destabilized proteins, disease-associated amyloidogenic proteins are typically only moderately destabilized, allowing them to evade ER quality control and be efficiently secreted to the extracellular space.24 Upon secretion into the serum, these destabilized proteins are then subject to misfolding and subsequent aggregation into soluble oligomers and amyloid fibrils, the pathogenic species responsible for distal proteotoxicity.1,24–26 The inability for ER quality control to identify destabilized, amyloidogenic proteins and reduce their secretion has been shown to influence the severity and pathogenesis of TTR amyloid disease in patients, demonstrating the critical importance of ER quality control in dictating amyloid disease pathogenesis in vivo.27
Proteostasis capacity has been demonstrated to influence the accumulation and trafficking of misfolded proteins.28–31 Declines in proteostasis capacity, in particular, are associated with an impaired ability for cells and organisms to prevent amyloidogenesis.32,33 Pathologic insults that induce the accumulation of misfolded proteins in the ER, i.e., ER stress, present a particular challenge to the extracellular proteostasis environment. When ER stress overwhelms the capacity for ER quality control pathways to maintain ER proteostasis, protein secretion to the extracellular space can become dysregulated. Furthermore, ER stress increases intracellular ROS that damage the cellular proteome, increasing aggregation propensity and further challenging ER proteostasis maintenance.34 Impaired ER quality control can then impact downstream environments of the secretory pathway, including the extracellular space, through increased trafficking of misfolded or damaged proteins. While increased trafficking during ER stress allows some misfolded proteins, such as the prion protein, to access a distinct quality control mechanism leading to lysosomal degradation,35 misfolded or damaged proteins that are secreted to the extracellular space could aggregate into proteotoxic conformations. Indeed, ER stress is associated with the pathology of numerous amyloid diseases.24,36
In order to protect the secretory pathway from ER stress, cells activate the Unfolded Protein Response (UPR). The UPR consists of 3 integrated signaling pathways activated downstream of the ER stress sensing proteins IRE1, ATF6 and PERK.24,37 The activation of these signaling pathways restores ER proteostasis by reducing the import of newly-synthesized proteins that enter the ER (i.e., reducing ER proteostasis load) and by activating downstream transcriptional programs that induce proteins involved in ER protein folding, trafficking and degradation pathways. UPR-dependent activation of these pathways enhances ER quality control and has been shown to attenuate the secretion and extracellular aggregation of amyloidogenic proteins associated with multiple amyloid diseases.24,38–40
While it is clear that the UPR can indirectly influence extracellular proteostasis through the regulation of ER quality control, the involvement of the UPR in regulating the extracellular concentration and activity of secreted chaperones is poorly understood. Interestingly, none of these well-established secreted chaperones is a transcriptional target of the UPR. In contrast, the secretion of the prominent secreted chaperone clusterin decreases in response to ER stress.41 Considering that ER stress has the potential to facilitate the aberrant secretion of misfolded protein conformations to the extracellular space, it was surprising that the UPR had no known mechanism to increase extracellular proteostasis capacity to protect the extracellular environment from damaged or misfolded proteins. In a recent manuscript,42 we addressed this issue by identifying a UPR regulated secreted chaperone that increases extracellular proteostasis capacity, revealing a previously unanticipated mechanism through which the UPR can prevent the potentially toxic aggregation of misfolded proteins that escape ER quality control and accumulate in the extracellular space during ER stress.
ERdj3 is a UPR-Regulated Secreted Chaperone
In order to identify potential UPR-regulated secreted chaperones, we identified ER proteins that: 1) are known transcriptional targets of UPR signaling pathways, 2) lack an ER retention motif, and 3) are known to bind misfolded protein conformations through an ATP-independent ‘holdase’ activity. Through this analysis, we identified the ER HSP40 co-chaperone ERdj3 as a potential UPR-regulated secreted chaperone. Intracellularly, ERdj3 functions as a canonical HSP40 co-chaperone for the ER HSP70 BiP.43,44 In this role, ERdj3 delivers misfolded proteins to BiP for ATP-dependent chaperoning and thus directly influences ER proteostasis for numerous secreted proteins including glucocerebrosidase and immunoglobulin light and heavy chains.43,45
Despite this intracellular role for ERdj3, we found that ERdj3 is efficiently secreted to the extracellular space in response to ER stress. The addition of ER stressors such as thapsigargin to cultured cells increases extracellular ERdj3 levels without significantly affecting intracellular ERdj3 levels. Similarly, inducing hepatic ER stress in mice increases ERdj3 serum levels from 25 nM to 50 nM, indicating that ER stress also promotes ERdj3 secretion in vivo. Extracellular ERdj3 levels are similarly increased by the stress-independent activation of the UPR-associated transcription factor ATF6, demonstrating that extracellular ERdj3 is regulated by the canonical UPR signaling pathway activated downstream of ATF6. Interestingly, metabolic labeling experiments show that a substantial fraction (>40 %) of newly-synthesized ERdj3 is trafficked to the extracellular space following ER stress or ATF6 activation. This high level of secretion is quite surprising for a protein predicted to primarily function in intracellular ER protein folding pathways and is more consistent with secreted proteins that have extracellular functions. By contrast, only minor fractions of other ER chaperones (e.g., BiP) are trafficked to the plasma membrane in the presence or absence of ER stress.12,13 This suggests the intriguing possibility that ERdj3 has an important extracellular function during conditions of ER stress. Consistent with this prediction, genetic perturbation of ERdj3 did not influence cellular viability or UPR activation in response to ER stress, indicating that ERdj3 is not a critical regulator of ER proteostasis under these conditions.
ERdj3 binds misfolded proteins in the ER.44–46 Thus, we evaluated the potential for secreted ERdj3 to promote extracellular proteostasis through the binding of misfolded, aggregation-prone proteins. We found that secreted ERdj3 maintains the capacity to bind misfolded proteins through an ATP-independent ‘holdase’ activity common to all secreted chaperones.4 Furthermore, ERdj3 inhibits the aggregation of amyloidogenic Aβ1-40 at substoichiometric concentrations. Other secreted chaperones such as clusterin also inhibit Aβ aggregation at substoichiometric concentrations, preventing further aggregation into proteotoxic conformations through a mechanism involving binding to Aβ oligomers.47–49 The substoichiometric inhibition of Aβ aggregation by ERdj3 is likely also mediated through binding to oligomeric conformations, although this mechanism remains to be explicitly demonstrated. Regardless, our results indicate that ERdj3 is a secreted chaperone that functions to promote extracellular proteostasis during ER stress through a mechanism similar to that observed for other secreted chaperones.
We next sought to evaluate the potential for secreted ERdj3 to attenuate the proteotoxicity of a misfolded extracellular protein. Recently, a particularly toxic conformation of the prion protein, denoted TPrP, was identified.50 TPrP is a primarily α-helical and protease-resistant monomer that can be chromatographically purified from recombinant prion protein subjected to denaturation and refolding. Incubation of TPrP on neurons in vitro and in vivo recapitulates the molecular signatures, including cellular vacuolization, observed in animal models and patients with prion disease.50 The structural characterization and highly potent cellular toxicity (20 nM) of TPrP make it an attractive model for evaluating proteotoxicity in cell culture. We evaluated whether media conditioned on cells overexpressing ERdj3 could influence this TPrP-induced vacuolization. We found a significant reduction in vacuolization when cells were treated with TPrP in conditioned media containing about 4 times the concentration of secreted ERdj3 to that observed in mouse serum. In contrast, TPrP added to neuroblastoma cells in media conditioned on cells RNAi-depleted of ERdj3 showed an exacerbated vacuolization phenotype, as compared to cells treated with TPrP added to media containing ERdj3 at concentrations ∼1/6 that observed in serum. These results indicate that even modest serum concentrations of ERdj3 could prevent proteotoxicity of amyloidogenic proteins in vivo.
Collectively, our results support a model wherein ERdj3 is a secreted chaperone that attenuates aggregation and proteotoxicity of extracellular proteins during conditions of ER stress.42 The capacity of the UPR to respond to ER stress by inducing ERdj3 secretion provides a previously unanticipated mechanism to enhance extracellular proteostasis capacity and prevent pathological aggregation of aberrantly secreted misfolded proteins under conditions where the integrity of the extracellular proteome is threatened, (i.e., when misfolded protein accumulates in the secretory pathway) (Fig. 1).
Figure 1.

ERdj3 secretion restores extracellular proteostasis during ER stress. In response to ER stress, the intracellular accumulation of misfolded proteins in the ER can facilitate the aberrant secretion of misfolded proteins to extracellular environments. The UPR-dependent increase in ERdj3 provides a mechanism to increase extracellular chaperoning capacity and attenuate the potentially proteotoxic accumulation and aggregation of these misfolded protein conformations in the extracellular space.
ERdj3 Secretion links Intra- and Extracellular Proteostasis During ER Stress
ERdj3 is an important component of the ER BiP chaperoning pathway, yet our results indicate that its UPR-induced secretion is a mechanism to regulate extracellular proteostasis. We wondered how these 2 important yet distinct functions might be coordinated during ER stress. Since ERdj3 can bind to misfolded protein conformations in the ER and also traffic through the secretory pathway, we hypothesized that ERdj3 could perhaps be co-secreted in complex with misfolded protein substrates that evade ER quality control during ER stress. This co-secretion mechanism would avoid the need for ERdj3 to form encounter complexes with substrates in the extracellular space, and thus preemptively protect the extracellular environment from potentially toxic misfolded protein conformations.
By using a transfection paradigm that allowed us to distinguish between ERdj3-substrate complexes formed intracellularly, as opposed to extracellularly, we showed that ERdj3 is co-secreted with destabilized amyloidogenic proteins including the disease-associated A25T variant of TTR (TTRA25T) and amyloid precursor protein (the precursor to amyloidogenic Aβ peptides). Interestingly, this co-secretion is dependent on the destabilization of the protein fold. ERdj3 does not co-secrete with wild-type TTR, which is much more stable than TTRA25T. Similarly, the addition of small molecules that bind and stabilize TTRA25T in mammalian cells also prevents ERdj3-TTRA25T co-secretion. These results show that ERdj3-substrate co-secretion requires substrate destabilization. One surprising characteristic of these co-secreted ERdj3-substrate complexes is that they are resistant to washing in high-detergent buffer, indicating that ERdj3-substrate co-secretion offers a mechanism to provide preemptive chaperoning to destabilized secreted proteins and also enhances the integrity of the complexes to avoid disassociation following secretion.
Critically, the capacity for ERdj3 to co-secrete with destabilized proteins does not promote the secretion of terminally misfolded proteins to the extracellular space. For example, ERdj3 overexpression does not increase extracellular levels of the highly-destabilized, non-secreted TTR variant TTRD18G. In contrast, clusterin overexpression substantially increases extracellular levels of TTRD18G, indicating that clusterin can promote the secretion of terminally misfolded protein conformations to the extracellular space. This latter result suggests an adaptive reason for why clusterin secretion is reduced during ER stress, as the secretion of clusterin in complex with highly-destabilized, misfolded proteins could overwhelm the extracellular proteostasis environment and promote proteotoxic protein aggregation.
The differential capacity for ERdj3 and clusterin to co-secrete with terminally misfolded proteins suggests distinct biological mechanisms are involved in regulating the trafficking of these complexes through the secretory pathway. Since ERdj3 functions in the intracellular BiP chaperoning pathway by delivering misfolded proteins to BiP, while clusterin is not known to associate with any intracellular ER proteostasis pathway, we predicted that ERdj3-substrate co-secretion could be regulated through the activity of BiP. Consistent with this prediction, increasing BiP activity through either activation of ATF6 (the transcription factor responsible for inducing BiP during UPR activation38) or BiP overexpression reduces co-secretion of ERdj3-substrate complexes to the extracellular space. Alternatively, increasing BiP activity has no effect on the co-secretion of substrates bound to the ERdj3 H53Q variant – an ERdj3 mutant that disrupts functional interactions between ERdj3 and BiP.43 These results indicate that ERdj3 co-secretes with destabilized substrates under conditions where BiP activity is overwhelmed, such as occurs during ER stress.
These co-secretion results reveal a unique mechanism to regulate extracellular proteostasis during conditions of stress through ERdj3 secretion (Fig. 2). In response to ER stress where BiP chaperoning pathways are overwhelmed, ERdj3 can be co-secreted in complex with misfolded proteins that could challenge the extracellular proteostasis environment, preemptively protecting the extracellular space from proteotoxic protein aggregates. As BiP activity is increased through the activity of the UPR, ERdj3 is still secreted to the extracellular environment as an unbound protein that can alleviate the pathologic protein misfolding of proteins aberrantly secreted during the initial phases of ER stress, providing another level of extracellular proteostasis regulation through the UPR.
Figure 2.
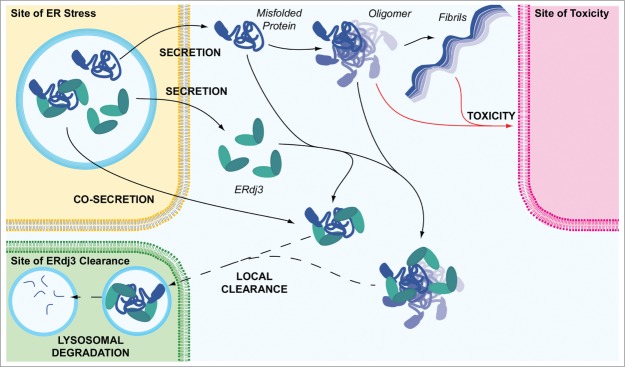
ERdj3 segregates proteotoxic protein conformations to protect the extracellular environment. ERdj3 can be secreted either as an unbound protein or bound to a destabilized protein. In the extracellular environment, unbound ERdj3 can complex with misfolded protein conformations or oligomers, preventing proteotoxic protein aggregation into soluble oligomers or amyloid fibrils that induce cellular toxicity. Based on comparisons with other secreted chaperones, ERdj3-substrate complexes are likely cleared from extracellular environments through a mechanism involving endocytosis and subsequent lysosomal degradation. ER stress and consequently increased secretion of misfolded, aggregation-prone proteins could have a particular impact on the cell types near the site of ER stress. Thus, increased ERdj3 secretion through the UPR could represent a mechanism to protect these local extracellular environments from proteotoxic protein conformations. In this model, ERdj3 and ERdj3-substrate complexes could potentially be locally cleared to protect the environment most threatened by the secretion of misfolded protein conformations to the extracellular space.
Outstanding Questions and Perspectives
Our initial report identified UPR-induced ERdj3 secretion as a mechanism by which extracellular proteostasis capacity is directly enhanced during conditions of ER stress. As we continue to explore the implications of extracellular proteostasis regulation by the UPR, important questions emerge. Below we highlight 4 of these questions and discuss their implications in defining the functional role for ERdj3 secretion in extracellular proteostasis maintenance.
What are the Endogenous, Extracellular Substrates of ERdj3? We have shown that ERdj3 binds to destabilized, misfolding prone proteins, including disease-associated TTR variants and APP, but the endogenous, extracellular proteins bound by ERdj3 in the presence or absence of stress have not been identified. Since ER stress and proteotoxic protein misfolding are intricately associated in amyloid disease pathogenesis, it is likely that ERdj3 binds secreted destabilized, misfolding-prone proteins associated with amyloid diseases and other protein aggregation disorders. This would suggest that ERdj3 activity has an important role in defining extracellular proteotoxic protein aggregation involved in amyloid disease pathology (see below). Similarly, ERdj3 secretion might also be one of the mechanisms by which organisms maintain extracellular proteostasis under physiological stress conditions such as those that occur during development, preventing the potentially toxic accumulation of misfolded proteins in extracellular environments. As endogenous, extracellular ERdj3 substrates are identified, the role for ERdj3 in regulating extracellular proteostasis during ER stress in these and other biologic pathways will become clear, providing significant insights into the physiologic roles for UPR-dependent ERdj3 secretion in organismal physiology.
What is the Fate of ERdj3-Substrate Complexes in the Extracellular Environment? The moderate levels of ERdj3 in serum are inconsistent with the efficient secretion of ERdj3 from mammalian hepatic and neuronal cells, suggesting that ERdj3 is efficiently cleared from extracellular environments. In particular, ERdj3-substrate complexes must be cleared from the extracellular space to prevent accumulation of the destabilized protein substrates. We predict that as with other secreted chaperones, ERdj3 and ERdj3-substrate complexes are removed from extracellular environments through increased targeting to endocytic pathways and subsequent lysosomal degradation (Fig. 2). Because ERdj3 secretion and co-secretion is local to the cells undergoing ER stress and/or secreting destabilized protein, it is conceivable that ERdj3 clearance is mediated by proximal cells or the same cells that secrete ERdj3, providing a mechanism to locally enhance extracellular proteostasis capacity around a specific tissue experiencing ER stress. This is opposed to a mechanism suggested for other secreted chaperones that globally regulate extracellular proteostasis environments such as the serum. Furthermore, in this local context, cell-surface expression of BiP and other ATP-dependent chaperones under ER stress conditions might allow functional interactions with ERdj3-substrate complexes, although this potential mechanism remains to be demonstrated. Identifying the underlying molecular mechanisms involved in ERdj3 clearance will be critical in defining the specific roles for ERdj3 in regulating local and global extracellular proteostasis in response to ER stress.
What is the Role for UPR-Dependent ERdj3 Secretion in the Pathophysiology of Amyloid Diseases?
Age is one of the most important risk factors for amyloid diseases. The relationship between aging and disease has led to the proteostasis hypothesis of amyloid disease – that with age, proteostasis networks lose their capacity to maintain the integrity of the proteome in response to proteotoxic stress.51,52 In particular, impaired UPR activation has been implicated in multiple amyloid diseases.36 Our results indicate that ERdj3 is a critical regulator of extracellular proteostasis during conditions of ER stress, suggesting that alterations in UPR-dependent ERdj3 secretion could exacerbate proteotoxic protein aggregation and amyloid disease onset. It will be interesting to monitor ERdj3 levels in cerebrospinal fluid and serum of amyloid disease patients to evaluate whether disease onset correlates with alterations in ERdj3 levels in these extracellular environments. Such a correlation would suggest that alterations in extracellular ERdj3 activity are a critical factor involved in disease pathogenesis, revealing a molecular mechanism that could contribute to the age-dependence of amyloid disease pathology.
Is There Therapeutic Potential for Attenuating Pathologic Protein Aggregation Involved in Amyloid Disease Pathology by Inducing UPR-Dependent Increases in Secreted ERdj3?
Small molecule approaches to intervene in amyloid diseases have suffered from the structural diversity of implicated proteins and the unclear etiology of individual pathologies. We have shown that stress-independent activation of ATF6 is a potential strategy to enhance ER quality control and reduce the secretion and subsequent aggregation of amyloidogenic proteins associated with multiple systemic amyloid diseases.38–40 Our new results show that ATF6 activation also directly promotes extracellular proteostasis through ERdj3 secretion. Hence, ATF6 activation offers a unique opportunity to combat proteotoxic extracellular protein aggregation through the complementary mechanisms of both increasing ER quality control stringency (thus reducing secretion of destabilized, aggregation-prone proteins) and increasing extracellular proteostasis capacity (thus attenuating extracellular proteotoxic aggregation of amyloid proteins). This potentially dual protective function of ATF6 suggests that activating this UPR signaling pathway could offer a broadly-applicable opportunity to ameliorate proteotoxic protein aggregation involved in amyloid disease pathology, further motivating ongoing efforts to identify small molecule activators of ATF6.
Concluding Remarks
Our initial manuscript identifies UPR-dependent increases in secreted ERdj3 as a direct mechanism to regulate extracellular proteostasis capacity in response to ER stress. As we continue to address the questions outlined above and others, we will learn more of the functional role for UPR activation and ERdj3 secretion in extracellular proteostasis maintenance, which we predict will reveal new insights into underlying mechanisms involved in amyloid disease pathology and new strategies to intervene in these devastating disorders.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Evan Powers at TSRI for critical reading of this perspective.
Funding>
RLW thanks NIH (NS079882 and DK102635), the Ellison Medical Foundation, and the Amyloidosis Foundation for funding. JCG thanks NIH (HL099245) and the American Heart Association for funding.
REFERENCES
- 1. Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem 2013; 82:745-74; PMID:23451869; http://dx.doi.org/ 10.1146/annurev-biochem-072611-130030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kraus A, Groveman BR, Caughey B. Prions and the potential transmissibility of protein misfolding diseases.Annu Rev Microbiol 2013; 67:543-64; PMID:23808331; http://dx.doi.org/ 10.1146/annurev-micro-092412-155735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 2014; 15(6):384-96; PMID:24854788; http://dx.doi.org/ 10.1038/nrm3810 [DOI] [PubMed] [Google Scholar]
- 4. Wyatt AR, Yerbury JJ, Ecroyd H, Wilson MR. Extracellular chaperones and proteostasis. Annu Rev Biochem 2013; 82:295-322; PMID:23350744; http://dx.doi.org/ 10.1146/annurev-biochem-072711-163904 [DOI] [PubMed] [Google Scholar]
- 5. Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A 1994; 91(18):8368-72; PMID:8078889; http://dx.doi.org/ 10.1073/pnas.91.18.8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finn TE, Nunez AC, Sunde M, Easterbrook-Smith SB. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J Biol Chem 2012; 287(25):21530-40; PMID:22549788; http://dx.doi.org/ 10.1074/jbc.M112.372961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 1994; 140(1):1-22; PMID:7877593; http://dx.doi.org/ 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- 8. Schwiebert EM, Zsembery A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim Biophys Acta 2003; 1615(1-2):7-32; PMID:12948585; http://dx.doi.org/ 10.1016/S0005-2736(03)00210-4 [DOI] [PubMed] [Google Scholar]
- 9. Nuutinen T, Huuskonen J, Suuronen T, Ojala J, Miettinen R, Salminen A. Amyloid-beta 1-42 induced endocytosis and clusterin/apoJ protein accumulation in cultured human astrocytes. Neurochem Int 2007; 50(3):540-7; PMID:17196306; http://dx.doi.org/ 10.1016/j.neuint.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 10. Wyatt AR, Yerbury JJ, Berghofer P, Greguric I, Katsifis A, Dobson CM, Wilson MR. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell Mol Life Sci 2011; 68(23):3919-31; PMID:21505792; http://dx.doi.org/ 10.1007/s00018-011-0684-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soltys BJ, Gupta RS. Immunoelectron microscopic localization of the 60-kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp Cell Res 1996; 222(1):16-27; PMID:8549659; http://dx.doi.org/ 10.1006/excr.1996.0003 [DOI] [PubMed] [Google Scholar]
- 12. Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S, Menger L, Michaud M, Zitvogel L, Kroemer G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann N Y Acad Sci 2010; 1209:77-82; PMID:20958319; http://dx.doi.org/ 10.1111/j.1749-6632.2010.05740.x [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem 2010; 285(20):15065-15075; PMID:20208072; http://dx.doi.org/ 10.1074/jbc.M109.087445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peters LR, Raghavan M. Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J Immunol 2011; 187(2):919-31; http://dx.doi.org/ 10.4049/jimmunol.1100690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 2014; 14(4):263-76; PMID:24658275; http://dx.doi.org/15181253 10.1038/nrc3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci 2004; 23(3):247-54; PMID:15181253; http://dx.doi.org/ 10.1385/JMN:23:3:247 [DOI] [PubMed] [Google Scholar]
- 17. Magalhaes J, Saraiva MJ. Clusterin overexpression and its possible protective role in transthyretin deposition in familial amyloidotic polyneuropathy. J Neuropathol Exp Neurol 2011; 70(12):1097-1106; PMID:22082661; http://dx.doi.org/ 10.1097/NEN.0b013e31823a44f4 [DOI] [PubMed] [Google Scholar]
- 18. Greene MJ, Sam F, Soo Hoo PT, Patel RS, Seldin DC, Connors LH. Evidence for a functional role of the molecular chaperone clusterin in amyloidotic cardiomyopathy. Am J Pathol 2011; 178(1):61-8; PMID:21224044; http://dx.doi.org/ 10.1016/j.ajpath.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Magalhaes J, Saraiva MJ. The heat shock response in FAP: the role of the extracellular chaperone clusterin. Amyloid 2012; 19 Suppl 1:3-4; PMID:22512538; http://dx.doi.org/ 10.3109/13506129.2012.675370 [DOI] [PubMed] [Google Scholar]
- 20. McGeer PL, Kawamata T, Walker DG. Distribution of clusterin in Alzheimer brain tissue. Brain Res 1992; 579(2):337-41; PMID:1378350; http://dx.doi.org/ 10.1016/0006-8993(92)90071-G [DOI] [PubMed] [Google Scholar]
- 21. Freixes M,B Puig A. Rodriguez B. Torrejon-Escribano R, Blanco I. Ferrer. Clusterin solubility and aggregation in Creutzfeldt-Jakob disease. Acta Neuropathol 2004; 108(4):295-301; PMID:15235804; http://dx.doi.org/ 10.1007/s00401-004-0891-6 [DOI] [PubMed] [Google Scholar]
- 22. Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 2011; 80:71-99; PMID:21495850; http://dx.doi.org/ 10.1146/annurev-biochem-062209-093836 [DOI] [PubMed] [Google Scholar]
- 23. Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 2012; 92(2):537-76; PMID:22535891; http://dx.doi.org/ 10.1152/physrev.00027.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryno LM, Wiseman RL, Kelly JW. Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr Opin Chem Biol 2013; 17(3):346-52; PMID:23647985; http://dx.doi.org/ 10.1016/j.cbpa.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol 2004; 16(1):67-75; PMID:14673392; http://dx.doi.org/ 10.1097/00002281-200401000-00013 [DOI] [PubMed] [Google Scholar]
- 26. Wiseman RL, Koulov A, Powers E, Kelly JW, Balch WE. Protein energetics in maturation of the early secretory pathway. Curr Opin Cell Biol 2007; 19(4):359-367; PMID:17686625; http://dx.doi.org/ 10.1016/j.ceb.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005; 121(1):73-85; PMID:15820680; http://dx.doi.org/ 10.1016/j.cell.2005.01.018 [DOI] [PubMed] [Google Scholar]
- 28. Gidalevitz T, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006; 311(5766):1471-4; PMID:16469881; http://dx.doi.org/ 10.1126/science.1124514 [DOI] [PubMed] [Google Scholar]
- 29. Hutt DM, Powers ET, Balch WE. The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett 2009; 583(16):2639-46; PMID:19708088; http://dx.doi.org/ 10.1016/j.febslet.2009.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009; 78:959-91; PMID:19298183; http://dx.doi.org/ 10.1146/annurev.biochem.052308.114844 [DOI] [PubMed] [Google Scholar]
- 31. Gidalevitz T, Kikis EA, Morimoto RI. A cellular perspective on conformational disease: the role of genetic background and proteostasis networks. Curr Opin Struct Biol 2010; 20(1):23-32; PMID:20053547; http://dx.doi.org/ 10.1016/j.sbi.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 2014; 9(3):1135-50; PMID:25437566; http://dx.doi.org/ 10.1016/j.celrep.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 2014; 24(9):506-14; PMID:24946960; http://dx.doi.org/ 10.1016/j.tcb.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 34. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013; 15(5):481-90; PMID:23624402; http://dx.doi.org/ 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Satpute-Krishnan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, Lippincott-Schwartz J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 2014; 158(3):522-33; PMID:25083867; http://dx.doi.org/ 10.1016/j.cell.2014.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 2014; 15(4):233-49; PMID:24619348; http://dx.doi.org/ 10.1038/nrn3689 [DOI] [PubMed] [Google Scholar]
- 37. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334(6059):1081-6; PMID:22116877; http://dx.doi.org/ 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 38. Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 2013; 3(4):1279-92; PMID:23583182; http://dx.doi.org/ 10.1016/j.celrep.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen JJ, Genereux JC, Qu S, Hulleman JD, Shoulders MD, Wiseman RL. ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem Biol 2014; 21:1564-74; PMID:25444553; http://dx.doi.org/ 10.1016/j.chembiol.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, Wiseman RL. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci U S A 2014; 111(36):13046-51; PMID:25157167; http://dx.doi.org/ 10.1073/pnas.1406050111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nizard P, Tetley S, Le Drean Y, Watrin T, Le Goff P, Wilson MR, Michel D. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 2007; 8(5):554-65; PMID:17451556; http://dx.doi.org/ 10.1111/j.1600-0854.2007.00549.x [DOI] [PubMed] [Google Scholar]
- 42. Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, Wiseman RL. Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J 2015; 34(1):4-19; PMID:25361606; http://dx.doi.org/ 10.15252/embj.201488896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell 2005; 16(1):40-50; PMID:15525676; http://dx.doi.org/ 10.1091/mbc.E04-05-0434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin Y, Awad W, Petrova K, Hendershot LM. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J 2008; 27(21):2873-82; PMID:18923428; http://dx.doi.org/ 10.1038/emboj.2008.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR, 3rd, Kelly JW. ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher's disease. Chem Biol 2014; 21(8):967-76; PMID:25126989; http://dx.doi.org/ 10.1016/j.chembiol.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Otero JH, Lizak B, Feige MJ, Hendershot LM. Dissection of structural and functional requirements that underlie the interaction of ERdj3 protein with substrates in the endoplasmic reticulum. J Biol Chem 2014; 289(40):27504-12; PMID:25143379; http://dx.doi.org/ 10.1074/jbc.M114.587147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yerbury JJ, Poon S, Meehan S, Thompson B, Kumita JR, Dobson CM, Wilson MR. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J 2007; 21(10):2312-22; PMID:17412999; http://dx.doi.org/ 10.1096/fj.06-7986com [DOI] [PubMed] [Google Scholar]
- 48. Yerbury JJ, Kumita JR, Meehan S, Dobson CM, Wilson MR. Alpha2-Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J Biol Chem 2009; 284(7):4246-54; PMID:19074141; http://dx.doi.org/ 10.1074/jbc.M807242200 [DOI] [PubMed] [Google Scholar]
- 49. Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, Meehan S, Wilson MR, Dobson CM, Klenerman D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat Struct Mol Biol 2012; 19(1):79-83; http://dx.doi.org/ 10.1038/nsmb.2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou M, Ottenberg G, Sferrazza GF, Lasmezas CI. Highly neurotoxic monomeric alpha-helical prion protein. Proc Natl Acad Sci U S A 2012; 109(8):3113-3118; PMID:22323583; http://dx.doi.org/ 10.1073/pnas.1118090109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 2011; 3(5):PMID:21441594; http://dx.doi.org/ 10.1101/cshperspect.a004440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Calamini B, Morimoto RI. Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr Top Med Chem 2012; 12(22):2623-40; PMID:23339312; http://dx.doi.org/ 10.2174/1568026611212220014 [DOI] [PMC free article] [PubMed] [Google Scholar]