

Abstract
ABSTRACT. Here we report a genetically confirmed case of Creutzfeldt-Jakob disease with a prion protein gene codon 180 mutation presenting atypical magnetic resonance imaging findings. The present case exhibited an acute onset and lateralized neurologic signs, and progressive cognitive impairment. No myoclonus or periodic synchronous discharges on electroencephalography were observed. Diffusion-weighted images revealed areas of high signal intensity in the right frontal and temporal cortices at onset that extended to the whole cortex and basal ganglia of the right cerebral hemisphere at 3 months. Although the cerebrospinal fluid (CSF) was initially negative for neuron specific enolase, tau protein, 14–3–3 protein, and abnormal prion protein, the CSF was positive for these brain-derived proteins at 3 months after onset.
Keywords: asymmetric cortical abnormalities, cerebrospinal fluid, creutzfeldt-Jakob disease, magnetic resonance imaging, V180I mutation
Abbreviations
- CSF
cerebrospinal fluid
- CJD
Creutzfeldt-Jakob disease
- PSD
periodic synchronous discharges
- EEG
electroencephalogram
- V180I CJD
CJD with the V180I mutation
- DWI
diffusion weighted imaging
- MRI
magnetic resonance imaging
- FLAIR
fluid-attenuated inversion recovery
- SPECT
single photon emission computed tomography
- WAIS-III
Wechsler Adult Intelligence Scale (third edition)
- IQ
intelligence quotient
- NSE
neuron specific enolase
- RT-QUIC
real-time quaking-induced conversion method
Introduction
Creutzfeldt-Jakob disease (CJD) is a transmissible and fatal neurodegenerative disease caused by prion protein. Clinical findings are characterized by rapidly progressive dementia, motor dysfunction, myoclonus, and periodic synchronous discharges (PSD) on electroencephalogram (EEG). The disease is classified into sporadic, genetic, and iatrogenic subtypes, and approximately 10% to 15% of all CJD is estimated to be the genetic form.1 The point mutation of valine to isoleucine at codon 180 in the prion protein gene is the most frequent mutation in Japan.2 The clinical and pathologic features of CJD with the V180I mutation (V180I CJD) differ from those of sporadic CJD.1,2 Therefore, patients with V180I CJD are often misdiagnosed with neurodegenerative disorders. Early diagnosis for patients with CJD is crucial for appropriate management of the disease and prevention of transmission to others. Diffusion-weighted imaging (DWI), which is a useful technique for early diagnosis of CJD, shows high signal intensity lesions in the cerebral cortex and deep gray matter.3,4 We describe a case with V180I CJD presenting with a strikingly asymmetric involvement of the cerebral hemisphere. The patient was initially diagnosed with acute infarctions, but genetic analysis confirmed V180I CJD. Moreover, while the cerebrospinal fluid (CSF) was initially negative for brain-derived proteins, including neuron specific enolase, tau protein, and 14–3–3 protein, it was positive for these proteins within 4 months. To our knowledge, repeated CSF analysis in cases with V180I CJD has not been reported. Here, we report an atypical case of V180I CJD diagnosed by genetic analysis and repeated CSF analysis.
Case Presentation
A 78-year-old, right-handed Japanese man presented with acute-onset mild weakness in his left extremities and gait disturbance without dementia and psychiatric symptoms. Ten days later, the patient was admitted to a local hospital with worsening of the gait disturbance. Brain magnetic resonance imaging (MRI) revealed high-signal intensity lesions in the right frontal and temporal cortices without involvement of the striatum and thalamus on DWI and fluid-attenuated inversion recovery (FLAIR) images (Fig. 1A, C). The apparent diffusion coefficient was decreased in areas corresponding to those with high-signal intensity (Fig. 1B). Although the patient was treated with anticoagulant therapy for suspected acute cerebral infarctions, his neurologic symptoms gradually worsened and his cognitive function began to decline. He was transferred to our hospital for further evaluation at 2 months after onset. The patient had no family history of prion disease or dementia. On admission, neurologic examination revealed mild weakness and clumsiness of the left hand, paratonia in the left extremities, and hyperreflexia in the left extremities without pathologic reflex, as well as gait disturbances. He did not present with cerebellar ataxia or myoclonus. Examination of cognitive function revealed disorientation and memory disturbance. His Mini-Mental State examination score was 24. The Wechsler Adult Intelligence Scale (third edition, WAIS-III) indicated a verbal intelligence quotient (IQ) of 93, a performance IQ of 63, and a full-scale IQ of 76. EEG showed generalized slow waves without PSD predominantly in the right cerebral hemisphere. Single photon emission computed tomography (SPECT) using99mTc-ethylcystinate dimer showed hypoperfusion in the right cerebral cortex (Fig. 2). CSF analysis on admission revealed normocytosis with an increased protein concentration of 76 mg/dl. The CSF was negative for 14–3–3 protein (500 > μg/ml), tau protein (150 > pg/ml), and neuron specific enolase (NSE: 12.9 ng/ml). The real-time quaking-induced conversion method (RT-QUIC) detected no abnormal prion protein. Follow-up MRI at 3 months after onset revealed high signal intensity in the entire right cerebral cortex, right head of the caudate nucleus and putamen, as well as in a part of the left posterior cingulate gyrus on DWI and FLAIR images (Fig. 1D, F). The apparent diffusion coefficient was decreased in areas corresponding to areas with high signal intensity (Fig. 1E). The abnormal lesions were mainly in the right cerebral hemisphere. The cerebellum and brainstem were not involved at this time. CSF analysis was repeated again at 3 months after onset, and it was positive for 14–3–3 protein (742 μg/ml), tau protein (726 pg/ml). The patient became unable to use his left upper extremity or to walk unassisted at 4 months after onset. Lateralization of the neurologic signs was preserved at this time. Follow-up testing with the WAIS-III at this time revealed a verbal IQ of 73, a performance IQ of 49, and a full-scale IQ of 59. Full-scale IQ, performance IQ, and verbal IQ had worsened. Follow-up CSF analysis at 4 months after onset showed a significant increase in 14–3–3 protein (3177.2 μg/ml), tau protein (2314 pg/ml), and NSE (20.1 ng/ml). The repeated RT-QUIC was negative for abnormal prion protein at this time. Informed consent was obtained from his closest relative and a prion protein gene analysis was performed. The results revealed a valine-to-isoleucine point mutation at codon 180. The polymorphic codon 129 was homozygous for methionine and codon 219 was homozygous for glutamate. The patient was diagnosed with CJD and a V180I mutation at this stage. He was bedridden at 5 months after onset. Myoclonus, startle reflex, and PSD on repeated EEGs were not observed over the follow-up period. The patient was transferred to a chronic care unit.
Figure 1.
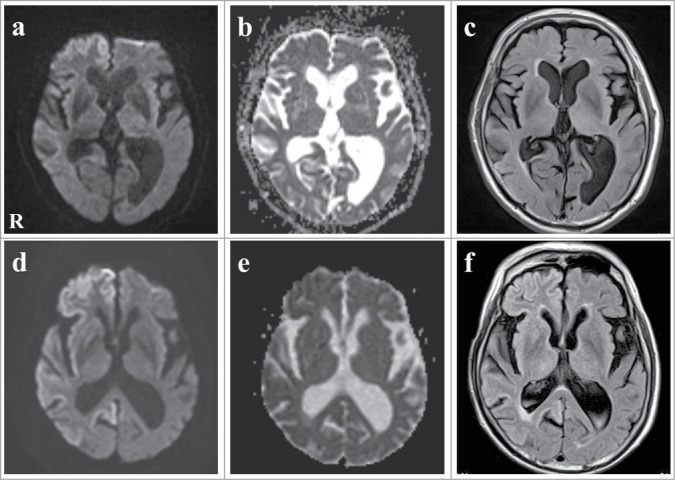
Serial images of axial diffusion-weighted imaging (DWI), apparent diffusion coefficient (ADC) map, and fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI). (A–C) Initial DWI and FLAIR images at onset showed high-signal intensity lesions in the right frontal and temporal cortex. ADC was decreased corresponding to the high-signal intensity areas. (D–F) Follow-up DWI and FLAIR images 3 months after onset showed that the high-signal intensity spread to the entire right cerebral cortex, the right head of the caudate nucleus and the putamen, as well as part of the left posterior cingulate gyrus. ADC was decreased corresponding to the high-signal intensity areas.
Figure 2.
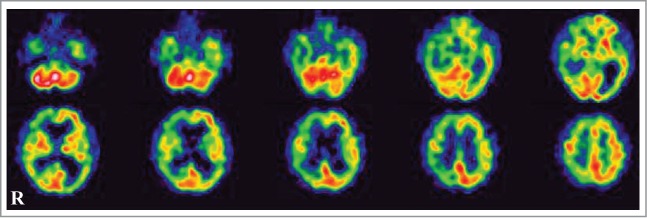
Single photon emission CT using99mTc-ethylcysteinate dimer at 2 months after onset showed decreased cerebral blood flow in the right cerebral hemisphere.
Discussion
We describe a genetically confirmed case of V180I CJD. The patient had an acute onset and lateralized neurologic signs, and progressive cognitive impairment, but no myoclonus and PSD on EEG. The initial CSF analysis was negative for NSE, tau protein, 14–3–3 protein, and prion protein Scrapie form (PrPSc). MRI showed high signal intensity, mainly in the right cerebral cortex on DWI and FLAIR images. The clinical features of V180I CJD are as follows: (1) older onset age; (2) slower disease progression compared to sporadic CJD; (3) a relatively low occurrence rate of symptoms, such as myoclonus, cerebellar symptoms, and visual disturbances; (4) lower positive rate of brain-specific proteins, such as NSE, total tau protein, and 14–3–3 protein in CSF; (5) lack of PSD on EEG throughout the disease course; and (6) no family history of prion disease or dementia.2 Detection of PrPSc in the CSF by real-time quaking-induced conversion method is useful for early diagnosis of CJD. The rate of PrPSc positivity in CSF, however, is much lower in patients with V180I CJD (39%), corresponding to the very weak accumulation of PrPSc in the brain.5 These clinical features make premortem diagnosis of V180I CJD difficult and genetic analysis is often required for a definite diagnosis. Although the clinical manifestations of our patient were consistent with the clinical features of V180I CJD, the present case was initially misdiagnosed with acute cerebral infarction based on the stroke-like presentation and the MRI findings.
The most interesting findings are the lateralized neurologic signs and strikingly asymmetric abnormalities on MRI. The present case showed acute monoparesis in the left extremity and high signal intensity lesions in the left frontal and temporal cortices on DWI at the onset. The cognitive impairment and motor dysfunction gradually progressed and high-signal intensity on DWI extended to the entire ipsilateral cerebral cortex at 3 months after onset. The lateralization of clinical and neuroradiologic findings, however, was preserved throughout the clinical course.
CJD is clinically characterized by rapidly progressive dementia, motor dysfunction, and myoclonus, and typical MRI findings of CJD are bilateral symmetric, diffuse abnormalities in the cerebral cortex and basal ganglia. Previous studies suggested that atypical CJD cases could show a stroke-like presentation and lateralized clinical signs, such as hemiparesis, aphasia or semicoma.3,4,6–8 Moreover, previous cases with V180I CJD showed the lateralized cortical lesions on DWI at the early stages of the disease.7,8 Although the reasons for the lateralization of the lesion remain unclear, we speculated that the lateralized cortical lesions on DWI may be characteristic findings in V180I CJD. The high signal intensity on DWI is related to the neuronal loss and spongiform changes.9 Microglial activation is also possible causes of DWI abnormalities. These findings lead us to hypothesize that pathologic changes started in only one hemisphere or that an unknown protective factor is associated with preservation of the other cerebral cortex.
Another interesting finding is the temporal profile of NSE, tau-protein, and 14–3–3 protein in CSF at the different disease stage over 4 months. These brain-derived proteins are well-known important diagnostic markers of CJD, which reflect the degree of neuronal damage.10 The neuropathological features of V180I CJD are diffuse spongiform change in the cerebral cortex and basal ganglia, and relatively mild neuronal loss. From this view point, the lower positive rate of brain-derived protein in the CSF may reflect the pathological process. Similarly, the initial analysis of brain-derived protein in CSF was negative in our patient. Follow-up analysis, however, revealed that the CSF was positive for NSE, total tau protein, and 14–3–3 protein. These results indicate that these surrogate markers increases corresponding to the progression of illness. Therefore, the present case suggests that repeated CSF analysis for brain-derived protein could provide additional supportive evidence for V180I CJD.
Conclusion
We describe a genetically confirmed case of V180I CJD with striking asymmetric abnormalities on MRI. V180I CJD can have a stroke-like presentation, lateralized clinical signs, and asymmetric cerebral lesions in the early stage. Therefore, genetic analysis is crucial for an accurate diagnosis of V180I CJD and appropriate care. Moreover, findings from the repeated CSF analysis in this case suggest that brain-derived protein in CSF may increase gradually with disease progression and thus repeated CSF analysis could provide additional supportive evidence for CJD.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Acknowledgments
The authors give thanks to Tetsuyuki Kitamoto, Department of Neurological Science, Tohoku University School of Medicine, for the prion protein gene analysis.
REFERENCES
- 1. Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F., et al. Prospective 10-year surveillance of human prion diseases in japan. Brain 2010; 133:3043-57; PMID:20855418; http://dx.doi.org/ 10.1093/brain/awq216 [DOI] [PubMed] [Google Scholar]
- 2. Qina T, Sanjo N, Hizume M, Higuma M, Tomita M, Atarashi R, Satoh K, Nozaki I, Hamaguchi T, Nakamura Y., et al. Clinical features of genetic creutzfeldt-Jakob disease with V180I mutation in the prion protein gene. BMJ Open 2014; 4(5):e004968; PMID:24838726; http://dx.doi.org/ 10.1136/bmjopen-2014-004968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cambier DM, Kantarci K, Worrell GA, Westmoreland BF, Aksamit AJ. Lateralized and focal clinical, EEG, and FLAIR MRI abnormalities in creutzfeldt-jakob disease. Clin Neurophysiol 2003;114:1724-8; PMID:12948802; http://dx.doi.org/ 10.1016/S1388-2457(03)00109-3 [DOI] [PubMed] [Google Scholar]
- 4. Yee AS, Simon JH, Anderson CA, Sze CI, Filley CM. Diffusion-weighted MRI of right-hemisphere dysfunction in creutzfeldt-jakob disease. Neurol 1999; 52:1514-5; PMID:10227651; http://dx.doi.org/ 10.1212/WNL.52.7.1514 [DOI] [PubMed] [Google Scholar]
- 5. Higuma M, Sanjo N, Satoh K, Shiga Y, Sakai K, Nozaki I, Hamaguchi T, Nakamura Y, Kitamoto T, Shirabe S., et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in japanese patients with genetic prion diseases. PLoS One 2013; 8:e60003; PMID:23555862; http://dx.doi.org/ 10.1371/journal.pone.0060003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamanouchi H, Budka H, Vass K, Sr. Unilateral creutzfeldt-jakob disease. Neurol 1986; 36:1517-20; PMID:3531919; http://dx.doi.org/ 10.1212/WNL.36.11.1517 [DOI] [PubMed] [Google Scholar]
- 7. Yeo MJ1, Lee SH, Lee SY, Jeon YC, Park SJ, Cho HJ, Choi KC, Kim YS, Kim SH. Familial creutzfeldt-jakob disease with a mutation at codon 180 presenting with an atypical phenotype. J Clin Neurosci 2013; 20:180-2; PMID:22999564; http://dx.doi.org/ 10.1016/j.jocn.2012.01.044 [DOI] [PubMed] [Google Scholar]
- 8. Kono S, Manabe Y, Fujii D, Sakai Y, Narai H, Omori N, Kitamoto T, Abe K. Serial diffusion-weighted MRI and SPECT findings in a creutzfeldt-jakob disease patient with V180I mutation. J Neurol Sci 2011; 301:100-3; PMID:21094959; http://dx.doi.org/ 10.1016/j.jns.2010.10.032 [DOI] [PubMed] [Google Scholar]
- 9. Mutsukura K, Satoh K, Shirabe S, Tomita I, Fukutome T, Morikawa M, Iseki M, Sasaki K, Shiaga Y, Kitamoto T., et al. Familial creutzfeldt-jakob disease with a V180I mutation: comparative analysis with pathological findings and diffusion-weighted images. Dement Geriatr Cogn Disord 2009; 28:550-7; PMID:20051687; http://dx.doi.org/ 10.1159/000254842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boesenberg-Grosse C, Schulz-Schaeffer WJ, Bodemer M, Ciesielczyk B, Meissner B, Krasnianski A, Bartl M, Heinemann U, Varges D, Eigenbrod S., et al. Brain-derived proteins in the CSF: do they correlate with brain pathology in CJD? BMC Neurol 2006; 21 6:35; PMID:16989662; http://dx.doi.org/ 10.1186/1471-2377-6-35 [DOI] [PMC free article] [PubMed] [Google Scholar]