

An engineered human carbonic anhydrase IX (CA IX) mimic was co-crystallized with the disaccharide sucrose. Its binding provides a novel ‘interaction’ site that can be used to design targeted inhibitors of CA IX, a key marker of hypoxic tumors.
Keywords: α-carbonic anhydrase, CA IX mimic, isoform-specific drug design, sugar approach, carbonic anhydrase IX, sucrose
Abstract
Human carbonic anhydrase (CA; EC 4.2.1.1) isoform IX (CA IX) is an extracellular zinc metalloenzyme that catalyzes the reversible hydration of CO2 to HCO3 −, thereby playing a role in pH regulation. The majority of normal functioning cells exhibit low-level expression of CA IX. However, in cancer cells CA IX is upregulated as a consequence of a metabolic transition known as the Warburg effect. The upregulation of CA IX for cancer progression has drawn interest in it being a potential therapeutic target. CA IX is a transmembrane protein, and its purification, yield and crystallization have proven challenging to structure-based drug design, whereas the closely related cytosolic soluble isoform CA II can be expressed and crystallized with ease. Therefore, we have utilized structural alignments and site-directed mutagenesis to engineer a CA II that mimics the active site of CA IX. In this paper, the X-ray crystal structure of this CA IX mimic in complex with sucrose is presented and has been refined to a resolution of 1.5 Å, an R cryst of 18.0% and an R free of 21.2%. The binding of sucrose at the entrance to the active site of the CA IX mimic, and not CA II, in a non-inhibitory mechanism provides a novel carbohydrate moiety binding site that could be further exploited to design isoform-specific inhibitors of CA IX.
1. Introduction
In many metastatic tumors, the rapidly proliferating cancer cells create an extracellular milieu that is characterized by a reduction in overall oxygen content (≤1.0%) and the acidification of the extracellular environment (Moulder & Rockwell, 1987 ▸; Peskin & Carter, 2008 ▸). This condition is termed tumor hypoxia and is owing to an imbalance between the demand of the proliferating cancer cells for oxygen and the capabilities of the vascular system surrounding them to supply it (Mahon & McKenna, 2013 ▸). As a result of hypoxic stress, tumor cells shift their energetic metabolism from mitochondrial oxidative phosphorylation to anaerobic glycolysis in the cytosol, a phenomenon known as the Warburg effect (Vander Heiden et al., 2009 ▸). This results in an increased amount of lactic acid being exported from the cells, leading to a decrease in the extracellular pH (pHe ∼6.5) and the upregulation of pH homeostasis factors that control the extracellular/intracellular pH (pHe/pHi) gradient (Racker, 1981 ▸).
Humans express 15 α-class carbonic anhydrase (CA) isoforms, each with numerous physiological roles and variable expression patterns, of which 12 are catalytically active and catalyze the reversible hydration of CO2 to HCO3 − (Frost, 2014 ▸). Two of these CAs (CA IX and XII), which are both extracellular, have been identified as being tumor-associated and play a role in the pH homeostasis of cancer cells (Liao et al., 2009 ▸; Benej et al., 2014 ▸). However, CA IX has been shown to be more prevalent in solid tumors compared with CA XII (Benej et al., 2014 ▸). In a non-disease state CA IX is almost exclusively expressed in the gut epithelium (Pastoreková et al., 1997 ▸; Liao et al., 2009 ▸; McDonald et al., 2012 ▸), but it is ectopically expressed in several tumors (including breast, liver, lung, colon/rectum and head/neck) in response to hypoxia (Wykoff et al., 2000 ▸; Mahon & McKenna, 2013 ▸). The major regulator of CA IX expression is hypoxia-induced factor 1, the upregulation of which is associated with hypoxia-responsive elements and the overall progression of cancer (Luo et al., 2014 ▸). As such, CA IX activity has been shown to be an important regulatory factor for tumor cells to maintain an acidic pHe yet to have a near-physiological pHi (Mahon et al., 2015 ▸). The limited expression of CA IX in healthy cells and its aberrant expression in numerous tumor types has led to its establishment as a general marker of tumor hypoxia as well as a potential drug-therapy target (Mahon & McKenna, 2013 ▸).
The inhibition mechanism of CA inhibitors (CAIs) is well established and sulfonamides are regarded as the most potent class of inhibitors. However, current CAIs are not specific owing to structural homology and high amino-acid conservation among CA isoforms (Aggarwal, Kondeti et al., 2013 ▸). Thus, they bind indiscriminately to CAs that are more abundant in the cytosol, such as CAs I, II and III (Tafreshi et al., 2012 ▸). In contrast, the catalytic domain of CA IX faces the extracellular milieu and presents an alternative method of targeting the enzyme in tumor cells (Barathova et al., 2008 ▸; Hilvo et al., 2008 ▸; Pinard et al., 2015 ▸). The incorporation of bulky membrane-impermeable chemical moieties appended to classic CAIs such as acetazolamide (AZM) or cationic sulfonamide derivatives is one such strategy (Mahon & McKenna, 2013 ▸).
A more recent class of CAIs, glycoconjugated sulfonamides, utilizes derivatives of the sulfonamide zinc-binding group (ZBG), for example secondary cyclic sulfonamides or benzene sulfonamides appended to a monosaccharide or disaccharide tail. The ZBG allows the CAI to bind directly to the zinc ion as seen in classical CAIs, while the bulky sugar moiety allows the CAI to maintain water solubility and maintain bioavailability (Meyer et al., 2011 ▸; Carroux et al., 2013 ▸). However, unlike previous designed bulky sulfonamide compounds, the high molecular weight of the glycoconjugated sulfonamides confers membrane impermeability onto the CAI. Moreover, studies have already shown that these CAIs show a >1000-fold selectivity for CA IX over CA II (Siebels et al., 2011 ▸; Moeker et al., 2014 ▸; Mahon et al., 2015 ▸).
At present, only one crystal structure of the catalytic domain of CA IX exists, with AZM. Despite the exploitation of structure-based drug design for other protein systems, difficulties in the expression and crystallization of CA IX have hindered the study of CA IX–inhibitor complexes. To overcome this, our group has engineered a CA IX mimic by mutating seven positionally equivalent amino acids in the active site (A65S, N67Q, E69T, I91L, F131V, K170E and L204A) using the cDNA of the much more easily crystallizable CA II as a structural template.
During efforts to study the structures of the CA IX mimic in complex with inhibitors using sucrose as a cryoprotectant, sucrose was serendipitously found to bind near the entrance to the active site of the CA IX mimic but not of CA II. The rationale for not using the more conventional glycerol as a cryoprotectant was its observed binding in the active site of CA II and therefore its possible effect on inhibitor binding (Aggarwal, Kondeti et al., 2013 ▸). Following this observation of a well ordered sucrose in the X-ray crystal structure of the CA IX mimic, binding studies using differential scanning fluorimetry (DSF) of sucrose in complex with CA II, the CA IX mimic and wild-type CA IX were performed. DSF studies of glucose and fructose, the monosaccharides constituting sucrose, in complex with CA II, the CA IX mimic and wild-type CA IX were also carried out for comparison.
Taken together, these data provide a detailed understanding of CA isoform-selective binding of sucrose near the active-site mouth and provide insight into the development of CAIs that incorporate a soluble sucrose moiety. Furthermore, the use of sucrose-based glycoconjugates over monosaccharides such as glucose or fructose would circumvent the issue of interactions with specific transporters since humans lack sucrose transporters.
2. Materials and methods
2.1. Expression and purification of the CA IX mimic and CA II
The CA IX mimic was prepared by site-directed mutagenesis of seven residues, A65S, N67Q, E69T, I91L, F131V, K170E and L204A, with the QuikChange mutagenesis kit from Stratagene using a CA II cDNA template. The DNA sequence was then confirmed for the entire coding region for the CA in the expression vector (Genis et al., 2009 ▸). The expression and purification of the CA IX mimic and CA II were performed as described previously (Pinard et al., 2013 ▸). A final protein concentration of ∼10 mg ml−1 was calculated for each protein by measuring the optical density at 280 nm and using a molar absorptivity of 5.4 × 104 M −1 cm−1.
2.2. Expression and purification of CA IX
A bacmid containing CA IX was graciously provided by our collaborators, Claudiu T. Supuran (University of Florence, Italy) and Seppo Parkkila (University of Tampere, Finland), and was optimized for Spodoptera frugiperda (Sf9) cells. The CA IX construct contained amino acids 1–377, which make up the catalytic domain (CA), the proteoglycan domain (PG) and a signal peptide linked to a poly-(8×)-histidine tag, similar to as described in Hilvo et al. (2008 ▸) and Alterio et al. (2009 ▸). CA IX was expressed using a baculovirus expression system (Invitrogen; Patterson et al., 1995 ▸). 1 l of Sf9 cells were grown in Sf-900 II SFM medium (Life Technologies, New York, USA) to a density of 2.0 × 106 cells ml−1 and were infected with baculovirus containing CA IX bacmid using a multiplicity of infection (MOI) of 5. The cells were then harvested by centrifugation after 72 h. Supernatant containing CA IX was diluted twofold in 20 mM sodium phosphate, 500 mM NaCl, 15 mM imidazole pH 7.8 and was purified by nickel column chromatography in a gravity column containing 15 ml Ni–NTA agarose (Life Technologies; Hilvo et al., 2008 ▸). Eluent containing CA IX was then concentrated by centrifugation to a total volume of 500 µl. The poly-(8×)-histidine tag was removed from the concentrated CA IX sample using a Thrombin CleanCleave Kit based on the manufacturer’s guidelines (Life Technologies). After removal of the poly-(8×)-histidine tag, the sample of CA IX was further purified and buffer-exchanged (50 mM Tris, 150 mM NaCl pH 7.4) by size exclusion using an ÄKTA pure protein-purification system (GE Healthcare) equipped with a Superdex 200 10/300 GL gel-filtration column (GE Healthcare). The eluted sample was collected in 1 ml fractions. The chromatogram (absorbance at 280 nm) of eluted CA IX revealed two individual peaks indicative of populations of monomeric and dimeric units (not shown). Peak fractions from both monomers and dimers of CA IX were pooled and concentrated by centrifugation. The final concentration of purified CA IX was estimated to be 3 mg ml−1 by UV–Vis spectroscopy (Gill & von Hippel, 1989 ▸) using an extinction coefficient of 5.2 × 104 M −1 cm−1.
2.3. Crystallization
Crystals of CA II and the CA IX mimic were grown at room temperature using the sitting-drop vapor-diffusion method. A crystallization screen was made using the Crystal Gryphon automated system (Art Robbins) with conditions ranging from 1.1 to 1.7 M sodium citrate and 50 mM Tris–HCl pH 7.0–9.0 as the precipitant solution. Crystals were obtained by mixing 0.3 µl protein solution (∼10 mg ml−1 in 50 mM Tris–HCl pH 7.8) with 0.3 µl precipitant solution. The drops were equilibrated against 60 µl precipitant solution. Crystals appeared within 7 d. Crystals formed using 1.6 M sodium citrate, 50 mM Tris–HCl pH 7.5 were used for data collection.
2.4. Diffraction data collection
X-ray diffraction data were collected on Cornell High Energy Synchrotron Source (CHESS) beamline F1 using an ADSC Quantum 210 detector at a wavelength of 0.918 Å. Crystals of CA II and the CA IX mimic were immersed in 20%(w/v) sucrose (∼0.6 M) cryoprotectant for approximately 5 s and immediately flash-cooled at 100 K for data collection. A total of 360 frames with an oscillation range of 0.5° each were collected with an exposure of 5 s. The data were indexed, integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▸). Diffraction data statistics for the CA IX mimic in complex with sucrose are highlighted in Table 1 ▸. In an attempt to complex sucrose to CA II, these soaks were repeated three more times, but this proved futile despite increasing the concentration of sucrose to 1.0 M.
Table 1. X-ray crystallographic data-collection and refinement statistics.
Values in parentheses are for the highest resolution bin.
| CA IX mimicsucrose | CA IX mimic | |
|---|---|---|
| PDB code | 4ywp | 4zao |
| Data-collection statistics | ||
| Temperature (K) | 100 | 100 |
| Wavelength () | 0.918 | 1.54 |
| Space group | P21 | P21 |
| Unit-cell parameters | ||
| a () | 41.8 | 41.8 |
| b () | 41.2 | 41.2 |
| c () | 72.3 | 72.0 |
| () | 103.8 | 103.8 |
| Reflections | ||
| Theoretical | 42836 | 22290 |
| Unique | 42193 | 21707 |
| Resolution () | 20.01.50 (1.501.45) | 20.01.80 (1.861.80) |
| R merge † (%) | 5.1 (10.3) | 4.5 (6.3) |
| I/(I) | 16.3 (11.7) | 17.3 (12.8) |
| Completeness (%) | 98.5 (99.7) | 97.5 (97.1) |
| Multiplicity | 3.7 (3.5) | 2.7 (2.7) |
| Model statistics | ||
| R cryst ‡ (%) | 18.0 | 17.5 |
| R free § (%) | 21.2 | 20.5 |
| Residue Nos. | 4261 | 4261 |
| No. of atoms¶ | ||
| Protein | 2132 | 2132 |
| Ligand | 27 | 0 |
| Water | 290 | 169 |
| R.m.s.d. | ||
| Bond lengths () | 0.009 | 0.006 |
| Bond angles () | 1.32 | 1.044 |
| Ramachandran statistics (%) | ||
| Favored | 89.0 | 94.8 |
| Allowed | 11.0 | 5.2 |
| Average B factors (2) | ||
| Main chain | 9.5 | 13.9 |
| Side chain | 13.4 | 18.6 |
| Ligand | 7.8 | |
| Solvent | 19.9 | 23.8 |
R
merge = 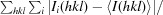
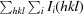 100.
100.
R
cryst = 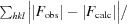
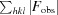 100.
100.
R free is calculated in the same manner as R cryst except that it uses 5% of the reflection data omitted from refinement.
Includes alternate conformations.
2.5. Model refinement
The initial phases were calculated using PDB entry 3ks3 (Avvaru et al., 2010 ▸) with waters removed and the residues unique to the CA IX active site (Ser65, Gln67, Thr69, Leu91, Val131, Leu135 and Glu170) mutated to alanines. The auto molecular-replacement (AutoMR) procedure in the PHENIX suite of programs (Adams et al., 2010 ▸) was used to carry out molecular replacement. The structure was refined using the PHENIX software suite (Adams et al., 2010 ▸) and manual refitting of the model based on the electron-density map was performed in the graphics program Coot (Emsley & Cowtan, 2004 ▸). A topology file for sucrose was generated using the PRODRG server (Schüttelkopf & van Aalten, 2004 ▸) and this file was used to model the sugar into the difference electron-density map (F obs − F calc). Iterative refinement of the structure was performed until the R work and R free were minimized and were within 5% of each other. PROCHECK (Laskowski et al., 1993 ▸) was then used to analyze the geometric restraints of the final model; the final model refinements are summarized in Table 1 ▸.
2.6. Enzyme kinetics
18O-Exchange mass spectrometry was used to measure the catalytic rate of the dehydration of HCO3 − to CO2 by the CA IX mimic in the presence of 2 M sucrose at 283 K using 18O-labeled HCO3 − as described previously (Silverman et al., 1980 ▸). In the first step of catalysis there is a random probability of labeling the active site of the enzyme with 18O (1). In the second step the protonation of the zinc-bound 18OH− via His64 results in the release of H2 18O into the bulk solvent. This method is dependent on the depletion of 18O from CO2 in solution. As the CO2 enters the semi-permeable membrane of an Extrel EXM-200 mass spectrometer, its 18O-labeled isotopic content is measured.
2.7. Differential scanning fluorimetry stability assay
Samples of CA II, the CA IX mimic and CA IX (at 0.25 mg ml−1) were incubated with varying sucrose concentrations from 0.1 to 1.0 M for 1 h in a 1:1(v:v) ratio. 2.5 µl of 1% SYPRO Orange dye (catalog No. S6651; Invitrogen Inc.) was added to each protein–ligand sample to bring the final volume of each reaction to 25 µl. The assays were conducted in a quantitative PCR (qPCR) instrument (RG-3000; Corbett Research) with temperature ramping from 30 to 99°C, increasing at the rate of 0.1°C every 6 s. Control samples of protein only at a concentration of 0.25 mg ml−1 were included in each run as a positive control. The melting temperature (T m) was defined as the maximum value of the first derivative (dF/dT; change in fluorescence/change in temperature) of the signal.
3. Results and discussion
3.1. Crystallographic studies
The CA IX mimic–sucrose complex crystal diffracted to 1.5 Å resolution and belonged to space group P21, with unit-cell parameters a = 41.8, b = 41.2, c = 72.1 Å, β = 103.8°. The structure was refined to an R cryst and R free of 18.0 and 21.2%, respectively. Other structural details are summarized in Table 1 ▸. An initial F o − F c map clearly showed a sucrose molecule bound near the entrance to the CA IX mimic active site (Fig. 1 ▸). The sucrose molecule interacts mostly with the residues forming the hydrophilic cleft, Asn62, His64, Gln67 and Gln92, and when superimposed onto an unbound CA IX mimic structure (PDB entry 4zao) showed that no ordered water molecules are displaced upon sucrose binding (Fig. 2 ▸). The sucrose makes significant hydrogen bonds to numerous solvent molecules within the active site, including those making up the conserved ordered water network observed in the unbound active site, further stabilizing the molecule in the active site (Mikulski et al., 2013 ▸). Weak van der Waals interactions were also seen between the sucrose and the side chains of Leu91 and Val131 from the hydrophobic cleft. Both of these residues are present in CA IX but not in CA II (Fig. 2 ▸).
Figure 1.

(a) Cartoon representation of the CA IX mimic in complex with sucrose. The gray sphere coordinated by three histidine residues represents the zinc ion. The electron density (grey mesh; 2F o − F c Fourier map) is contoured at 1.3σ. (b) Structure of sucrose. (c) Enlarged view of the CA IX mimic–sucrose complex.
Figure 2.

Overlay of interfacing residues in the CA IX mimic–sucrose crystal structure (green) and CA II (blue), showing conservation of the ordered water network in the CA IX mimic.
Sucrose could also serve as a potential ligand for CA II, but multiple attempts to crystallize CA II in the presence of sucrose proved futile, making sucrose an even more attractive isoform-specific ligand for CA IX since it appears to have low affinity for the off-target CA II.
3.2. Kinetics and differential scanning fluorimetry (DSF)
An inhibition constant for sucrose could not be measured since sucrose does not inhibit the enzymes. The ligand binds at the entrance to the active site, away from the substrate-binding site (PDB entry 3d92; Domsic et al., 2008 ▸), and does not disrupt the ordered water network (Mikulski et al., 2013 ▸).
Thermal stability assays were carried out using DSF for CA II, the CA IX mimic and CA IX in complex with sucrose, glucose and fructose. CA II exhibited the smallest change in T m among the three proteins, exhibiting a ΔT m of 3°C (Fig. 3 ▸ a). Similarly, a ΔT m of 3.5°C was observed for the CA IX mimic (Fig. 3 ▸ b), while CA IX showed the largest ΔT m of 5°C (Fig. 3 ▸ c). The T m values for each protein at the different sucrose concentrations given in Supplementary Table S1 indicate that increasing concentrations of sucrose stabilize each of these proteins. A set of distinct peaks were observed in the thermogram for CA IX (Fig. 3 ▸ c); unlike CA II and the CA IX mimic, CA IX is a dimer and consists of two domains. The two peaks indicate two different T m values owing to the unfolding of its different domains independently of each other. As mentioned previously, the catalytic domain of CA IX displays structural homology to those of the other α-CAs. Hence, at each sucrose concentration the initial (lower) T m is owing to the unfolding of the catalytic domain (similar in sequence to CA II), while the second (higher) T m is most likely owing to the unfolding of the proteoglycan domain of the enzyme. An alternative explanation is that these distinct peaks could be owing to the exposure of the hydrophobic dimer interface as the dimer separates and the enzyme is denatured.
Figure 3.

DSF measurements of fluorescence intensity during heating of (a) CA II, (b) the CA IX mimic and (c) CA IX in the presence of varying concentrations of sucrose. For each isoform, the data are normalized averages from three temperature-scan experiments. Each color represents a scan carried out at a different sucrose concentration (blue, 0.0 M; red, 0.2 M; green, 0.4 M, purple, 0.6 M; cyan, 0.8 M; orange, 1.0 M).
To estimate the dissociation constant (K d) of each sugar for either CA II, the CA IX mimic or CA IX, a simple saturation model was used. For some enzyme–ligand complexes saturation was not achieved and so an exact K d could not be determined. A K d of 0.08 ± 0.04 M was observed for CA IX in complex with sucrose as well as in complex with fructose. CA IX appears to have less affinity for glucose; the enzyme never fully saturated. For the CA IX mimic in complex with sucrose a K d of 1.2 ± 0.8 M was calculated.
One possible explanation for the similarity in the K d values for sucrose and fructose for both CA IX and the CA IX mimic could be inferred from the crystal structure of the CA IX mimic in complex with sucrose. When sucrose binds at the mouth of the active site, its fructose component binds closest to the active-site zinc (∼5 Å) and the fructose moiety makes the majority of its interactions with water molecules and amino acids in the active site (Fig. 2 ▸). On the other hand, the sucrose moiety extends out of the active site and interacts with a few sparse water molecules and makes weak interactions with Val131 and Leu91.
In the case of the CA II complexes, all of the sugars exhibited very little affinity for the enzyme. The enzyme was never saturated and no K d was calculated for CA II in complex with sucrose. Superimpositions of CA II onto the CA IX mimic–sucrose complex (r.m.s.d. of 0.4 Å) shows that the bulky side chain of Phe131 (compared with Val131 in the CA IX mimic) would interfere with sucrose binding, creating steric hindrance. This could be a possible explanation why sucrose is not able to bind in CA II. Another interesting interaction that sucrose makes is that with Gln67 in the CA IX mimic compared with Asn67 in CA II (Fig. 2 ▸). Glutamic acid contains an extra –CH2, which allows it to make two hydrogen bonds compared with the shorter Asn67 in CA II, which would probably only make one bond. This could be yet another reason for preferential binding.
4. Conclusion
The sugar approach to CA inhibition has become more popular with the search for more selective CA isoform inhibitors. The classical low-molecular-weight CAIs bind deep into the CA active-site cavity and make limited interactions with the amino acids that differ between isoforms in the hydrophilic and hydrophobic cleft surrounding the active site of the enzyme. These CAIs are mostly sulfonamides and, although potent inhibitors, they exhibit indiscriminatory inhibition profiles among the different isoforms. One way to overcome this difficulty and create isoform-selective CAIs is to elongate these inhibitors by using the ‘tail approach’ to append tails that will make extensive interactions with the surrounding amino acids to the ZBG moieties (Supuran, 2012 ▸). Additionally, linking a sugar moiety to a sulfonamide scaffold would alter the properties of this drug, making it more soluble yet impermeable to the cell membrane, thus eliminating any off-target effects.
The glycoconjugate class of inhibitors utilizes this approach, and in these compounds either a sulfonamide or sulfamate group is bound to the anomeric C atom of a carbohydrate. This class of inhibitors has been used primarily against the tumor-associated CA IX and XII. Initial studies carried out by the Poulsen group by screening anomeric sulfonamides using the CO2 hydration assay against the cytosolic CAs I and II, and the tumor-associated CAs IX and XII, showed that these inhibitors show no isoform selectivity (Lopez et al., 2009 ▸; Métayer et al., 2013 ▸). However, these studies did not include the use of sucrose as a carbohydrate. The anomeric sulfamides were shown to be much better CA IX inhibitors, with inhibition constants below 8 nM and with selectivity over CA I (480-fold to 1800-fold) and CA II (∼17-fold) (Winum et al., 2013 ▸). A third class of sugar inhibitors, the 6-sulfamoyl carbohydrates, are synthesized from monosaccharides or disaccharides through the selective sulfamoylation of the C6 hydroxyl group (Lopez et al., 2011 ▸). Studies by the Poulsen group have also shown that glucose derivatives were more selective inhibitors (in the nanomolar range) of CA IX (eightfold to 14-fold) compared with CA II (Lopez et al., 2011 ▸). A potential inhibitor could incorporate sucrose by sulfamoylation of its C6 hydroxyl group. Therefore, the addition of a ZBG derivative to the sucrose moiety could prove to be a formidable anticancer drug, and the use of structure-based drug design in an attempt to exploit the subtle differences between active sites of CAs would be critical in this effort.
The bound sucrose molecule (Fig. 2 ▸) gives insights into possible drug-design strategies using the ‘sugar approach’, which entails attaching a molecule moiety (acting as a ’tail’) to a known high-affinity zinc-binding motif. The DSF data suggest that the fructose component is what confers specificity and so the addition of a short linker between the fructose component and a potential ZBG would be one option. In addition, the crystal structure shows that the fructose component points towards the active site, while the glucose moiety extends out of the active site. The development of a drug that can selectively inhibit CA IX over off-target cytosolic CAs could be critical in slowing down tumor cell survival in the microenvironment created under hypoxic conditions. The potency and success of such a drug will be dependent on the interaction of its tail moiety with the residues near the active site. The mouth of a CA active site is the most variable region among the different isoforms and studies have shown that these ‘tails’ bind in these regions and could potentially lead to the development of isoform-selective CAIs (Aggarwal, Boone et al., 2013 ▸; Aggarwal, Kondeti et al., 2013 ▸).
Currently, the disaccharides being used in the synthesis of glycoconjugates utilize a galactose moiety and show stronger inhibition for CA II compared with CA IX. Although these compounds do not cross the membrane, they appear to be weak CA IX binders and so may not be valid drug candidates. Furthermore, although glycoconjugates constituting glucose moieties show stronger inhibition of CA IX versus CA II and present a lot of promise, these CAIs may unintentionally interact with glucose transporters in the body. Therefore, utilization of the disaccharide sucrose may prove to be a better option since humans lack a sucrose transporter, and our preliminary data already indicate that sucrose shows a preference for CA IX over CA II.
Supplementary Material
The Tm values for each protein at the different sucrose concentrations.. DOI: 10.1107/S2053230X1501239X/no5083sup1.pdf
PDB reference: carbonic anhydrase IX mimic, 4zao
PDB reference: complex with sucrose, 4ywp
Acknowledgments
This work was supported by a grant from the NIH (GM25154). RM would like to thank the Center of Structural Biology for support of the X-ray facility at UF. We would also like to thank the MacCHESS staff for their help during X-ray diffraction data collection at the Cornell High Energy Synchrotron (CHESS) Facility, Ithaca. MA is funded by the Department of Energy and Shull Fellowship at Oak Ridge National Laboratory.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aggarwal, M., Boone, C. D., Kondeti, B. & McKenna, R. (2013). J. Enzyme Inhib. Med. Chem. 28, 267–277. [DOI] [PubMed]
- Aggarwal, M., Kondeti, B. & McKenna, R. (2013). Bioorg. Med. Chem. 21, 1526–1533. [DOI] [PMC free article] [PubMed]
- Alterio, V., Hilvo, M., Di Fiore, A., Supuran, C. T., Pan, P., Parkkila, S., Scaloni, A., Pastorek, J., Pastorekova, S., Pedone, C., Scozzafava, A., Monti, S. M. & De Simone, G. (2009). Proc. Natl Acad. Sci. USA, 106, 16233–16238. [DOI] [PMC free article] [PubMed]
- Avvaru, B. S., Kim, C. U., Sippel, K. H., Gruner, S. M., Agbandje-McKenna, M., Silverman, D. N. & McKenna, R. (2010). Biochemistry, 49, 249–251. [DOI] [PMC free article] [PubMed]
- Barathova, M., Takacova, M., Holotnakova, T., Gibadulinova, A., Ohradanova, A., Zatovicova, M., Hulikova, A., Kopacek, J., Parkkila, S., Supuran, C. T., Pastorekova, S. & Pastorek, J. (2008). Br. J. Cancer, 98, 129–136. [DOI] [PMC free article] [PubMed]
- Benej, M., Pastorekova, S. & Pastorek, J. (2014). Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, edited by S. C. Frost & R. McKenna, pp. 199–219. Dordercht: Springer.
- Carroux, C. J., Rankin, G. M., Moeker, J., Bornaghi, L. F., Katneni, K., Morizzi, J., Charman, S. A., Vullo, D., Supuran, C. T. & Poulsen, S.-A. (2013). J. Med. Chem. 56, 9623–9634. [DOI] [PubMed]
- Domsic, J. F., Avvaru, B. S., Kim, C. U., Gruner, S. M., Agbandje-McKenna, M., Silverman, D. N. & McKenna, R. (2008). J. Biol. Chem. 283, 30766–30771. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Frost, S. C. (2014). Subcell. Biochem. 75, 9–30. [DOI] [PubMed]
- Genis, C., Sippel, K. H., Case, N., Cao, W., Avvaru, B. S., Tartaglia, L. J., Govindasamy, L., Tu, C., Agbandje-McKenna, M., Silverman, D. N., Rosser, C. J. & McKenna, R. (2009). Biochemistry, 48, 1322–1331. [DOI] [PMC free article] [PubMed]
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem. 182, 319–326. [DOI] [PubMed]
- Hilvo, M. et al. (2008). J. Biol. Chem. 283, 27799–27809. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Liao, S.-Y., Lerman, M. I. & Stanbridge, E. J. (2009). BMC Dev. Biol. 9, 22. [DOI] [PMC free article] [PubMed]
- Lopez, M., Paul, B., Hofmann, A., Morizzi, J., Wu, Q. K., Charman, S. A., Innocenti, A., Vullo, D., Supuran, C. T. & Poulsen, S.-A. (2009). J. Med. Chem. 52, 6421–6432. [DOI] [PubMed]
- Lopez, M., Trajkovic, J., Bornaghi, L. F., Innocenti, A., Vullo, D., Supuran, C. T. & Poulsen, S.-A. (2011). J. Med. Chem. 54, 1481–1489. [DOI] [PubMed]
- Luo, D., Wang, Z., Wu, J., Jiang, C. & Wu, J. (2014). Biomed Res. Int. 2014, 409272. [DOI] [PMC free article] [PubMed]
- Mahon, B. P. & McKenna, R. (2013). Curr. Top. Biochem. Res. 15(2), 1–21.
- Mahon, B. P., Pinard, M. A. & McKenna, R. (2015). Molecules, 20, 2323–2348. [DOI] [PMC free article] [PubMed]
- McDonald, P. C., Winum, J.-Y., Supuran, C. T. & Dedhar, S. (2012). Oncotarget, 3, 84–97. [DOI] [PMC free article] [PubMed]
- Métayer, B., Mingot, A., Vullo, D., Supuran, C. T. & Thibaudeau, S. (2013). Chem. Commun. 49, 6015–6017. [DOI] [PubMed]
- Meyer, H., Vitavska, O. & Wieczorek, H. (2011). J. Cell Sci. 124, 1984–1991. [DOI] [PubMed]
- Mikulski, R., West, D., Sippel, K. H., Avvaru, B. S., Aggarwal, M., Tu, C., McKenna, R. & Silverman, D. N. (2013). Biochemistry, 52, 125–131. [DOI] [PMC free article] [PubMed]
- Moeker, J., Mahon, B. P., Bornaghi, L. F., Vullo, D., Supuran, C. T., McKenna, R. & Poulsen, S.-A. (2014). J. Med. Chem. 57, 8635–8645. [DOI] [PMC free article] [PubMed]
- Moulder, J. E. & Rockwell, S. (1987). Cancer Metastasis Rev. 5, 313–341. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pastoreková, S., Parkkila, S., Parkkila, A. K., Opavský, R., Zelník, V., Saarnio, J. & Pastorek, J. (1997). Gastroenterology, 112, 398–408. [DOI] [PubMed]
- Patterson, R. M., Selkirk, J. K. & Merrick, B. A. (1995). Environ. Health Perspect. 103, 756–759. [DOI] [PMC free article] [PubMed]
- Peskin, B. & Carter, M. J. (2008). Med. Hypotheses, 70, 298–304. [DOI] [PubMed]
- Pinard, M. A., Boone, C. D., Rife, B. D., Supuran, C. T. & McKenna, R. (2013). Bioorg. Med. Chem. 21, 7210–7215. [DOI] [PubMed]
- Pinard, M. A., Mahon, B. & McKenna, R. (2015). Biomed Res. Int. 2015, 453543. [DOI] [PMC free article] [PubMed]
- Racker, E. (1981). Science, 213, 1313. [DOI] [PubMed]
- Schüttelkopf, A. W. & van Aalten, D. M. F. (2004). Acta Cryst. D60, 1355–1363. [DOI] [PubMed]
- Siebels, M., Rohrmann, K., Oberneder, R., Stahler, M., Haseke, N., Beck, J., Hofmann, R., Kindler, M., Kloepfer, P. & Stief, C. (2011). World J. Urol. 29, 121–126. [DOI] [PubMed]
- Silverman, D. N., Tu, C. K. & Wynns, G. C. (1980). Biophysics and Physiology of Carbon Dioxide, edited by C. Bauer, G. Gros & H. Bartels, pp. 254–261. Berlin, Heidelberg: Springer.
- Supuran, C. T. (2012). J. Enzyme Inhib. Med. Chem. 27, 759–772. [DOI] [PubMed]
- Tafreshi, N. K., Bui, M. M., Bishop, K., Lloyd, M. C., Enkemann, S. A., Lopez, A. S., Abrahams, D., Carter, B. W., Vagner, J., Grobmyer, S. R., Gillies, R. J. & Morse, D. L. (2012). Clin. Cancer Res. 18, 207–219. [DOI] [PMC free article] [PubMed]
- Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. (2009). Science, 324, 1029–1033. [DOI] [PMC free article] [PubMed]
- Winum, J.-Y., Colinas, P. A. & Supuran, C. T. (2013). Bioorg. Med. Chem. 21, 1419–1426. [DOI] [PubMed]
- Wykoff, C. C., Beasley, N. J., Watson, P. H., Turner, K. J., Pastorek, J., Sibtain, A., Wilson, G. D., Turley, H., Talks, K. L., Maxwell, P. H., Pugh, C. W., Ratcliffe, P. J. & Harris, A. L. (2000). Cancer Res. 60, 7075–7083. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Tm values for each protein at the different sucrose concentrations.. DOI: 10.1107/S2053230X1501239X/no5083sup1.pdf
PDB reference: carbonic anhydrase IX mimic, 4zao
PDB reference: complex with sucrose, 4ywp