

Abstract
Extracellular α-synuclein (α-syn) assemblies can be up-taken by neurons; however, their interaction with the plasma membrane and proteins has not been studied specifically. Here we demonstrate that α-syn assemblies form clusters within the plasma membrane of neurons. Using a proteomic-based approach, we identify the α3-subunit of Na+/K+-ATPase (NKA) as a cell surface partner of α-syn assemblies. The interaction strength depended on the state of α-syn, fibrils being the strongest, oligomers weak, and monomers none. Mutations within the neuron-specific α3-subunit are linked to rapid-onset dystonia Parkinsonism (RDP) and alternating hemiplegia of childhood (AHC). We show that freely diffusing α3-NKA are trapped within α-syn clusters resulting in α3-NKA redistribution and formation of larger nanoclusters. This creates regions within the plasma membrane with reduced local densities of α3-NKA, thereby decreasing the efficiency of Na+ extrusion following stimulus. Thus, interactions of α3-NKA with extracellular α-syn assemblies reduce its pumping activity as its mutations in RDP/AHC.
Keywords: misfolding disease, Parkinson’s disease, protein aggregation and clustering, single particle tracking, super-resolution imaging
See also: PJ Kahle et al (October 2015)
Introduction
Observations that embryonic dopamine neurons transplanted in PD patients developed α-syn-positive Lewy bodies 11–16 years after surgery (Kordower et al, 2008; Li et al, 2008) have led to the notion that α-syn assemblies released from neurons can propagate in a prion-like manner by seeding the assembly of endogenous α-syn (reviewed in Brettschneider et al, 2015). Several in vitro experiments (exposure to α-syn) and animal models (injection of α-syn) support the seeded aggregation and transmission of α-syn (Desplats et al, 2009; Hansen et al, 2011; Volpicelli-Daley et al, 2011; Luk et al, 2012; Mougenot et al, 2012; Rey et al, 2013; Holmqvist et al, 2014; Sacino et al, 2014; Peelaerts et al, 2015). The docking of infectious protein assemblies and binding to membrane proteins is critical for prion (Büeler et al, 1993) or amyloid beta (Aβ) oligomer (Renner et al, 2010)-mediated neurotoxicity. However, the interaction of exogenous α-syn with the plasma membrane and neuron-specific membrane proteins is not well documented.
In this study, we show that exogenously applied α-syn assemblies form clusters following lateral diffusion both in and outside synapses. In order to identify neuronal proteins that interact with exogenous α-syn, we exposed pure neuronal cultures to oligomeric and fibrillar α-syn for 10 min, pulled down the complex, and identified the associated proteins using a proteomic-based approach. Neuron-specific α3-subunit of plasma membrane-enriched enzyme Na+/K+-ATPase (NKA) or sodium pump (Azarias et al, 2013) was the only transmembrane protein identified to interact with both oligomeric and fibrillar α-syn. Interestingly, several mutations in the gene encoding α3-subunit of NKA (ATP1A3) are linked to rapid-onset dystonia Parkinsonism (RDP) (de Carvalho Aguiar et al, 2004; Rodacker et al, 2006; Clapcote et al, 2009; Kirshenbaum et al, 2011), alternating hemiplegia of childhood (AHC) (Heinzen et al, 2012; Rosewich et al, 2012) and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome (Demos et al, 2014), all of which are associated with motor dysfunction. Failure or reduction in pump activity leading to an increase in Na+ levels has been proposed to contribute to these diseases (Benarroch, 2011). In addition, reduced global expression (Ellis et al, 2003) and aggregation (Martin et al, 2007) of α3-NKA have been observed in mouse models of amyotrophic lateral sclerosis (ALS).
We postulate that alteration in the pumping activity of α3-NKA upon α-syn assemblies binding impairs the maintenance of Na+ gradient, thus disrupting neuronal function and initiating a detrimental signaling cascade. Using single particle tracking (SPT) in cultured neurons, we demonstrate that the otherwise freely diffusing α3-NKA were trapped within α-syn clusters and thus exhibit reduced mobility on the plasma membrane resulting from direct interaction involving the extracellular domain of α3-NKA. Quantitative super-resolution STORM (stochastic optical resolution microscopy) shows that α3-NKA exists as nanoclusters and that exposure to α-syn increases their size by recruiting additional α3-NKA molecules. We further demonstrate that rapid Na+ extrusion from neurons, an α3-NKA-specific function, is reduced upon interaction with α-syn.
Results
α-syn assemblies form clusters on the plasma membrane following lateral diffusion
All α-syn assemblies used in the study were characterized as in Pieri et al (2012), Bousset et al (2013), and Peelaerts et al (2015). We monitored in particular their assembly kinetics, their shape by using electron microscopy, and their composition by mass spectrometry (Appendix Fig S1). We assessed the distribution and localization of exogenously applied oligomeric and fibrillar α-syn assemblies on cultured striatal neurons. We observed that exogenous ATTO-550-labeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn form clusters on neuronal membranes (Fig1A). The fluorescence intensities of the clusters increased with the number of days neurons were maintained in culture (DIV: days in vitro) (Fig1A and B; Appendix Table S1). For oligomeric assemblies, clustering increased between DIV 14 and DIV 21, following neuronal maturation and an observed increase in number of synapses as reported previously (Weiss et al, 1986). α-syn clustering was observed on axons (tau immunoreactivity, Fig1C), dendrites (MAP2 immunoreactivity, Fig1C), and the cell body of neurons indicating that α-syn binds to the plasma membrane irrespective of the compartment.
Figure 1.

- A, B Neurons cultured for 7, 14, or 21 days in vitro (DIV) and exposed (60 min) to ATTO-550-labeled oligomeric (25 nM, blue bars) or fibrillar (0.03 nM, orange bars) α-syn. (A) Examples of cluster fluorescence (thresholded image). (B) Quantification of fluorescence intensity of clusters (mean ± SEM). Note a culture days-dependent increase in oligomeric α-syn clustering and weakly for fibrillar α-syn (t-test; **P < 0.01, ***P < 0.001, ns = non-significant; see also Appendix Table S1 for actual values). Scale bar: 5 μm.
- C DIV 21 neurons exposed (60 min) to ATTO-550-labeled fibrillar (0.03 nM) α-syn and immunolabeled to visualize dendrite (MAP2 antibody) or axons (TAU antibody). Note α-syn clusters both on dendrites and axons. Scale bar: 20 μm.
- D STORM imaging of α-syn-Alexa 647 (red) and PALM imaging of TMD-Dendra (green, transmembrane domain of syntaxin) visualizing the neuronal membrane and emphasizing that oligomeric and fibrillar clusters of α-syn are at the cell surface. Scale bar: 1 μm.
The location of oligomeric and fibrillar α-syn assemblies relative to the plasma membrane was assessed after transfection with TMD-Dendra plasmid (transmembrane domain of syntaxin protein) and two-color PALM (Dendra)/STORM (Alexa 647) super-resolution imaging (photoactivated localization microscopy/stochastic optical reconstruction microscopy). TMD-Dendra translocate to the plasma membrane, thus allowing a direct visualization of the neuronal silhouette (Ribrault et al, 2011). Neurons were then exposed to oligomeric (25 nM) or fibrillar (0.03 nM) Alexa 647-labeled α-syn. After exposure (1 h, Fig1D), both oligomeric and fibrillar α-syn clusters were detected along the plasma membrane. Overlay of the images showed that α-syn assemblies formed clusters adjacent to the plasma membrane. This was confirmed using immunolabeling with α-syn antibody on neurons pre-exposed to α-syn-Alexa 647 (Appendix Fig S2).
The amount of bound α-syn and the number of α-syn clusters exhibited concentration dependence (Appendix Fig S3). Notably, the binding and clustering of oligomeric α-syn was more pronounced than that of fibrillar α-syn (Appendix Fig S3, Appendix Table S2). More importantly, the fluorescence intensity of clusters (Fig2A) and the number of the clusters (Fig2B) depended also on the exposure time of oligomeric (25 nM) or fibrillar (0.03 nM) α-syn assemblies (also see Appendix Table S3). α-syn clusters could already be detected 5 min after exposure suggesting rapid kinetics of α-syn binding and clustering. The proportion of α-syn clusters associated with synapses (synapsin immunoreactivity) also increased with time (Fig2C and D). α-syn clustering was independent of synapse type and showed no preferential accumulation at excitatory (homer) or inhibitory (gephyrin) synapses (Appendix Table S4).
Figure 2.

- A, B Exposure of neurons cultured for 21 days to ATTO-550-labeled oligomeric (25 nM, blue) or fibrillar (0.03 nM, orange) α-syn for 5 or 60 min. Note the exposure time-dependent increase in α-syn clusters fluorescence intensity (A, mean ± SEM, t-test) and number/μm2 (B, mean ± SEM, t-test) (***P < 0.001; see Appendix Table S3 for actual values).
- C, D Neurons (21 DIV) exposed (5 or 60 min) to ATTO-550-labeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn (red), and synapsin-immunolabeled synapses (blue). Note the synaptic apposition of α-syn clusters (C). Time-dependent increase in α-syn clusters association with synapses (D, mean ± SEM, four independent experiments on four independent cultures, n = 75 fields of view, t-test, ***P < 0.001). Scale bar: 1 μm.
- E Neurons exposed (60 min) to biotin and ATTO-550 dual-labeled oligomeric α-syn (25 nM, red, left panel) followed by single-molecule streptavidin quantum dot (QD) labeling and subsequent single particle tracking (white, left panel). Single QDs diffused freely (outside, n = 491 QDs) or are trapped within (over, n = 33 QDs) α-syn clusters (red). Averaged mean square displacement (MSD) plot, respectively, displays Brownian and confined diffusion “outside” and “over” α-syn clusters (right panel). Scale bar: 1 μm.
- F, G Neurons exposed to biotin-labeled α-syn oligomers (25 nM) or fibrils (0.03 nM) for 5 or 60 min. Representative single-molecule trajectories are shown in red and synapses identified using FM4-64 dye in blue (F). Cumulative diffusion coefficient plots comparing exposure time (5 or 60 min) dependence of oligomeric (blue) and fibrillar (orange) α-syn at synaptic or extra-synaptic sites (G). Note a time-dependent diffusional slowdown of α-syn assemblies both at synaptic and extra-synaptic location (Kolmogorov–Smirnov test, number of QDs indicated in brackets, **P < 0.01; ***P < 0.001, three experiments). Scale bar: 5 μm.
The rapid binding on the plasma membrane and time-dependent increase in the clustering suggests a mechanism compatible with the recruitment of α-syn assemblies via lateral diffusion. To determine whether this is the case, 16–18 DIV old neurons were exposed (60 min) to biotin and ATTO550 dual-labeled oligomeric (25 nM) α-syn (Fig2E). Single molecules (white trajectories, Fig2E, left) were visualized together with α-syn clusters (red, ATTO-550 fluorescence, Fig2E, left). α-syn-QD displayed free Brownian diffusion outside clusters and confined diffusion over clusters characterized by respective linear or bent MSD (mean square displacement) versus time plot (Fig2E, right). This demonstrates that freely diffusing single molecules of α-syn are trapped within self-composed clusters on the neuronal plasma membrane.
Next, we assessed whether lateral diffusion accounts for time-dependent synaptic clustering as observed above (Fig2C and D). We performed SPT-QD studies on neurons exposed to biotin-labeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn for 5 min or 60 min (Fig2F and G). Cell surface-bound biotin-α-syn assemblies were then tracked following their labeling with streptavidin-QD-605 nm (Fig2F, trajectories: red), and the synapses were identified using FM4-64 dye (Fig2F, blue) labeling. Single-molecule trajectories overlapping with FM4-64 labeling were considered as synaptic. A global slowdown in the diffusion coefficient of α-syn assemblies was observed between 5 and 60 min after exposure (Fig2G). These data favor the notion that α-syn assemblies form clusters on plasma membrane following lateral diffusion and that they experience molecular interactions within clusters.
Identification of proteins that interact with extracellularly applied oligomeric and fibrillar α-syn assemblies
A proteomic screening was performed to identify membrane proteins interacting with extracellularly applied α-syn assemblies. Recombinant α-syn (oligomeric and fibrillar forms) with a C-terminal S-tag (Appendix Fig S1) that binds with high affinity to ribonuclease S-protein (Raines et al, 2000) were used to identify specific partners of extracellularly applied α-syn in neurons as illustrated (Fig3A). Pure cultures of cortical neurons were exposed to oligomeric or fibrillar S-tagged α-syn for 10 min. α-syn-S-tag and associated proteins were pulled down from whole-cell lysates using S-protein agarose beads, trypsin digested, and the resulting peptides were identified by nanoLC-MS/MS (LC: liquid chromatography; MS: mass spectrometry). Control samples were prepared from neurons unexposed to α-syn. Protein abundance was assessed by a label-free quantitative proteomic method using spectral counting. We identified 32 and 178 neuronal protein partners for oligomeric and fibrillar α-syn, respectively (Appendix Tables S5 and S6, column 1–4). Several intracellular proteins were identified in the screen due to the interaction following endocytosis of α-syn (Hansen et al, 2011; Volpicelli-Daley et al, 2011; Holmes et al, 2013) and/or interaction following cell disruption during protein extraction. Among the identified candidates, α3-subunit of NKA was picked for further study because of the confirmatory results of a hypothesis-driven approach: the pull-down data indicate that α3-NKA is the only transmembrane protein of our list with extracellularly exposed domains and was identified both with oligomeric and fibrillar α-syn. The mass spectrometry spectra of α3-NKA peptides are shown in Appendix Fig S4. In addition to α3-subunit of NKA, fibrillar α-syn also pulled down the β1-subunit of NKA (Appendix Table S6) that co-assembles with α-subunit to form a functional pump. Pull-down/MS studies were also performed on pure astrocytes culture following exposure (10 min) to α-syn assemblies (Appendix Table S7A and B). Astrocyte-specific α2-NKA was not identified as α-syn interacting partner.
Figure 3.

- A Overview of the strategy used to purify and identify membrane proteins that interact specifically with α-syn assemblies. Rat pure cortical neuron cultures were incubated for 10 min with oligomeric or fibrillar α-syn-S-tag (at a particle concentration of 1 μM and 4.8 nM, respectively, equivalent to 40 μM monomeric α-syn). Fresh protein extracts from those primary neurons were incubated with S-protein agarose beads to pull down α-syn-S-tag assemblies together with their specific protein partners. Unexposed primary neuron extracts incubated with S-protein agarose beads were used as a control. Proteins bound to the S-protein agarose beads were subjected to trypsin digestion and subsequently identified and quantified by nanoLC-MS/MS analysis, using a nanoLC-LTQ-Orbitrap.
- B Co-immunoprecipitation of α3-NKA and α-syn from cortical neurons exposed to monomeric, oligomeric or fibrillar α-syn (10 min). α3-NKA was immunoprecipitated using a specific antibody (Santa Cruz #sc-16052) and protein A-sepharose beads. The presence of α-syn within the immunoprecipitate was assessed by Western blot using the anti-α-syn antibody (BD Biosciences #610787). The molecular weight markers are indicated on the left. “α3-NKA” refers to immunoprecipitate with anti-α3-NKA antibody, “Pre” to immunoprecipitate with pre-immune antibody. A 2.4- and 4-fold enrichment of oligomeric and fibrillar α-syn, respectively, in α3-NKA immunoprecipitation, as compared to controls with the pre-immune antibody was observed. The blot was stripped and re-probed with anti-α-tubulin antibody (Abcam #ab7291) in order to ascertain that the each lane corresponding to the input material contains identical concentrations of a reference protein.
The interaction between α3-NKA and α-syn was confirmed using chemical cross-linking/MS (Appendix Tables S5 and S6, column 5). For this, membrane proteins were cross-linked using the cleavable cross-linker DTSSP (3,3′-dithiobis(sulfosuccinimidylpropionate)) in neurons exposed (10 min) to fibrillar α-syn-S-tag. Proteins interacting with α-syn-S-tag were pulled down, reduced, alkylated, cleaved with trypsin, and subjected to MS as described above. We performed a α3-NKA-specific peptide targeted identification strategy using SEQUEST search engine. This allowed us to identify nine peptides (FigEV1A) from α3-NKA as exemplified by the MS spectra for two of them (884–901 and 903–928, FigEV1B and C). One of these peptides spanning α3-NKA residues 903–928 was cross-linked through K928 or S915 to α-syn. Thus, cross-link/MS (FigEV1C) confirms the pull-down/MS results that α3-NKA binds α-syn assemblies. The interaction between α-syn and α3-NKA was further confirmed using co-immunoprecipitation experiments (Fig3B). When α3-NKA was immunoprecipitated using a specific antibody, α-syn was found associated. Fibrillar α-syn co-precipitated more than oligomeric isoforms, while monomeric α-syn was not co-precipitated suggesting conformation specificity. This experiment mirrors α3-NKA pull-down with S-tagged α-syn and clearly demonstrates that the two proteins interact.
Figure EV1.

- List of α3-NKA peptides identified from neurons exposed for 10 min to fibrillar α-syn, cross-linked with DTSSP, pulled-down, reduced and alkylated, digested by trypsin, and analyzed by MS/MS through an α3-NKA-targeted identification with SEQUEST.
- MS/MS spectrum of α3-NKA peptide TVNDLEDSYGQQWTYEQR [884-901].
- MS/MS spectrum of α3-NKA peptide VVEFTCHTAFFVSIVVVQWADLIICK [903-928] with one N-terminal acetylation (+42 Da), two carbamidomethyl cysteines (Cys908 and Cys927, +57 Da), and one DTSSP cross-linked residue (S915 or K928, +145 Da).
Trapping of α3-NKA within α-syn clusters
In order to probe the interaction between α-syn assemblies and α3-subunit containing Na+ pump, we performed live-cell single particle tracking experiments using quantum dots (SPT-QD) (Triller & Choquet, 2008). Indeed, SPT-QD allows probing of transient and local protein–protein interaction in non-invasive imaging conditions (Ribrault et al, 2011; Specht et al, 2011). First, the diffusion dynamics of α3-NKA over (inside) or outside (outside) α-syn clusters were analyzed using SPT-QD. Experiments were performed on DIV 16 neurons expressing pHluorin-α3-NKA and following 10 min exposure to ATTO-550-labeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn. pHluorin-α3-NKA molecules were labeled using a GFP antibody pre-coupled to QDs (see experimental procedures). Trapping of a freely diffusing α3-NKA single QD over fibrillar α-syn cluster can be seen (Fig4A; Movie EV1). The diffusion coefficients of pHluorin-α3-NKA over α-syn clusters (inside, dashed lines) were slower than outside clusters (outside, solid lines) (Fig4B; Appendix Table S8). These results suggest that free-diffusing α3-NKA were trapped within α-syn clusters and evident from the downward bent of MSD plots (Fig4C). Notably, pHluorin-α3-NKA molecules not diffusing over α-syn clusters (outside, Fig4B) exhibited slower diffusion coefficients for fibrillar (orange, solid line) than oligomeric α-syn (blue, solid line). This suggests that freely diffusing trajectories are already bound to α-syn and that they may be affected by the quaternary structure or the molecular weight of non-clustered α-syn molecules.
Figure 4.

- A–C SPT-QD trajectory (blue) shows trapping of a freely diffusing α3-NKA molecule over α-syn clusters (red) (A, see also Movie EV1; scale bar: 1 μm). Cumulative diffusion plot (B) and MSD plot (C) following exposure (10 min) to ATTO-550-labeled α-syn oligomers (blue, 25 nM) or fibrils (orange, 0.03 nM). Note that α3-NKA molecules diffuse faster (B) and are less confined (C) outside (outside, solid line) than those over (inside, dashed) α-syn clusters (see Appendix Table S8 for actual values).
- D, E SPT-QD of α3-NKA on pHluorin-α3-NKA-transfected neurons with (5 or 60 min, solid or dashed lines, respectively) or without (black line) exposure to unlabeled α-syn (oligomer: 25 nM, fibril: 0.03 nM). Synapses were labeled with FM4-64. Note the oligomeric (Da, Db, blue) α-syn exposure time-dependent slowdown of α3-NKA diffusion. Weaker but significant effect for fibrillar forms (Dc, Dd, Orange) (Kolmogorov–Smirnov test; refer to Appendix Table S9 for median values and percentage change). (E) Time-dependent decrease in surface area explored by α3-NKA trajectories following exposure of α-syn oligomers (blue) or fibrils (orange) (median ± interquartile range, Kolmogorov–Smirnov test, *P < 0.05; **P < 0.01; ***P < 0.001, ns = non-significant; refer to Appendix Table S9 for median values and percentage change).
The synaptic and extra-synaptic α3-NKA diffusion parameters were affected in a time-dependent manner (Fig4D and E). pHluorin-α3-NKA-transfected neurons were exposed to unlabeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn for 5 or 60 min and synapses identified using FM4-64 uptake. A global time-dependent slowdown in diffusion coefficient of α3-NKA was observed following exposure of neurons to oligomeric (blue) or fibrillar (orange) α-syn as compared to control neurons (Fig4D; Appendix Table S9). To determine the relative change in confinement, the area explored by α3-NKA trajectories was computed within a given time period (extracted from MSD, see Materials and Methods) (Fig4E; Appendix Table S9). A time-dependent reduction in average synaptic and extra-synaptic surface area explored by α3-NKA trajectories was observed, thus confirming an increased confinement. Thus, the concomitant slowdown of α-syn (Fig2G) and α3-NKA (Fig4) suggested a diffusion-trap mechanism resulting from their interactions. As a control of specificity, monomeric α-syn (50 nM) exposure (60 min) led to no or very subtle changes (see Fig EV2A), suggesting that the interaction and slowdown is specific to higher-order α-syn assemblies. In addition, the diffusion of α3-NKA was not modified following exposure (60 min) to amyloid-β (Aβ) oligomers (200 nM, Fig EV2B) that form clusters on the neuronal plasma membrane (Renner et al, 2010).
Figure EV2.
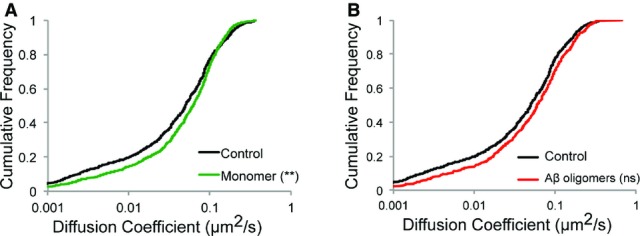
- A, B Monomeric α-syn-exposed (50 nM, 60 min, green) neurons showed no slowdown in diffusion coefficient of pHluorin-α3-NKA (A) (number of QDs: control, 674; monomer, 865; Kolmogorov–Smirnov test, **P < 0.01). (B) No slowdown in the diffusion coefficient of α3-NKA following exposure (200 nM, 60 min) to Aβ oligomers (number of QDs: control, 674; Aβ oligomers, 450; Kolmogorov–Smirnov test, ns = non-significant).
Selective interaction of α3-NKA and α-syn assemblies
In order to identify the α3-NKA extracellular binding site for α-syn, we took advantage of the fact that the α3-subunit but not the α1-subunit of NKA was identified in our MS experiments. The α-subunit of NKA has 10 transmembrane regions (TM1–TM10) and 5 extracellular loops of which four domains are short (Kaplan, 2002). Based on structural data, we focused on the 52 amino acid long extracellular loop between TM7 and TM8. It is ˜80% homologous between α3- and α1-NKA (Fig5A, blue and red). In fact, the central part of the TM7–TM8 loop harbors eight out of 11 primary sequence mismatches between α3- and α1-subunit. This region may thus contribute to α3-NKA but not α1-NKA interaction with α-syn assemblies. Three chimeric α3-/α1-NKA (boxed region, Fig5A), referred to as α3/α1-NKA-a, b, and c, were prepared by replacing α3-specific amino acids (blue) with the corresponding α1-specific amino acids (red) and SPT performed following exposure of cells expressing chimeric α3/α1-NKA to fibrillar α-syn. The three chimeric constructs had membrane expression similar to that of non-chimeric α3-NKA (Appendix Fig S5). As observed previously (Fig4D), the median diffusion coefficient of wild-type α3-NKA was reduced by 43% (Fig5B) upon addition of fibrillar α-syn. Chimeric constructs α3/α1-NKA-a (Fig5C) and α3/α1-NKA-c (Fig5E) showed a reduction of 35 and 28% in diffusion coefficient, respectively. In contrast, no reduction of the median diffusion coefficient of α3/α1-NKA-b (Fig5D) was observed in the presence of α-syn fibrils, suggesting that the interaction between chimeric α3/α1-NKA and fibrillar α-syn was abolished. Thus, amino acid Leu(L)878 and Asn(N)879 in the loop between TM7 and TM8 of α3-subunit of NKA (Fig5A) are likely to play a key role in α3-NKA interaction with α-syn assemblies.
Figure 5.
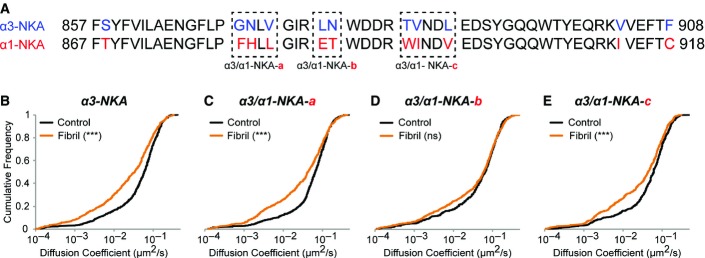
- A Sequence of the TM7-TM8 extracellular loop of α3-subunit (upper row) and α1-subunit (lower row) of NKA. Note the 11 amino acid difference between α3-NKA (blue) and α1-NKA (red) primary structures within this loop. Chimeric α3-/α1-NKA (boxed region; a/b/c) generated by replacing 2-3 amino acid residues in α3-subunit (blue) by the corresponding residues specific to α1-subunit (red) in the pHluorin-α3-NKA plasmid.
- B–E SPT-QD on α3-NKA (non-chimeric) and chimeric α3-NKA-a/b/c. Fibrillar α-syn exposure (60 min) reduced the diffusion of α3-NKA (B), α3-NKA-a (C), and α3-NKA-c (E) but not of α3-NKA-b (D) (three independent experiments, Kolmogorov–Smirnov test, ***P < 0.001, ns = non-significant).
Redistribution of α3-NKA and co-clustering with α-syn assemblies
The trapping of cell surface α3-NKA within α-syn clusters may alter their organization and localization. In order to examine this possibility, striatal neurons were exposed (5 or 60 min) to oligomeric (25 nM) or fibrillar (0.03 nM) ATTO-550-α-syn and immunolabeled with α3-NKA and synapsin antibodies (Fig6A). α3-NKA-associated immunoreactivity was evenly distributed over the surface of the neurons along with more fluorescent closely spaced clusters (Fig6A, green). Following exposure, several oligomeric and fibrillar α-syn clusters (red) were associated with α3-NKA clusters and their proportion increased with exposure time (38 and 47% association with oligomers after 5 and 60 min exposure respectively; 32 and 52% association with fibrils after 5 and 60 min, respectively; four experiments). Quantification of the α3-NKA fluorescence intensity revealed an increase in synaptic enrichment following exposure to α-syn (Fig6B; Appendix Table S10). For α3-NKA clusters not associated with synapses, fluorescence intensity was not modified (see Appendix Table S10). The level of association between α3-NKA and α-syn was assessed using the intensity correlation quotient (ICQ, Li et al, 2004). Variation of this parameter between −0.5 and 0 or between 0 and +0.5 indicates the respective absence or presence of association of the fluorophores over given pixels. ICQ gives an estimate of dependent staining, and is filtering or threshold independent. Using this non-biased quantification, α3-NKA and α-syn association increased with exposure time (Fig6C). Two-color STORM imaging was performed to visualize the co-localization between α3-NKA and α-syn at high resolution (Fig6D, see also Materials and Methods). High-resolution images (Fig6D, green) showed spatially distributed α3-NKA nanoclusters of variable sizes on the plasma membrane in agreement with a previous study (Blom et al, 2012). Large-sized nanoclusters of α3-NKA (Fig6D, green) were present where α-syn aggregated (Fig6D, red, arrows). The increased clustering and co-localization of α3-NKA was more prominent with fibrillar α-syn clusters than with oligomeric α-syn, thus being in line with results obtained with other methods (mass spectrometry and co-immunoprecipitation, Fig3).
Figure 6.

- A Cultured (21 DIV) striatal neurons unexposed or exposed (60 min) to ATTO-550-labeled oligomeric (25 nM) or fibrillar (0.03 nM) α-syn (red). α3-NKA (green) and synapsin (blue) identified by their immunoreactivities. Representative images show an increase in fluorescence of α3-NKA immunoreactivity after exposure to α-syn assemblies. Scale bar: 5 μm.
- B Averaged fluorescence intensity of synaptic α3-NKA clusters following exposure to oligomeric (blue) or fibrillar (orange) α-syn (normalized to control, black). Note α3-NKA enrichment at synapses following α-syn exposure (see also Appendix Table S10 for actual values for synaptic and extra-synaptic clusters) (mean ± SEM, one-way ANOVA with Dunnett’s test; *P < 0.05, **P < 0.01, ***P < 0.001).
- C Averaged intensity correlation quotient (ICQ) between ATTO-550-α-syn fluorescence and α3-NKA immunoreactivity. Note an exposure time-dependent increase in positive correlation for the two proteins (t-test; ***P < 0.001).
- D Two-color STORM super-resolution imaging of α-syn-Alexa 647 (red) and α3-NKA-ATTO488 (green). Note accumulation of α3-NKA where α-syn is clustered (arrows). Scale bar: 1 μm.
The α3-NKA clusters packaging and organization following exposure to α-syn were studied using super-resolution STORM (Fig7). Neurons were exposed to α-syn (unlabeled, oligomeric: 25 nM; fibrillar: 0.03 nM; 1 h) and were subsequently immunolabeled (Alexa 647) for α3-NKA. STORM images allowed the detection of single events corresponding to the detection of α3-NKA during the imaging period (Fig7A; see Appendix Supplementary Materials and Methods). Under control condition (unexposed), clustered and non-clustered α3-NKA could be detected (Fig7A). Following α-syn exposure, the distribution of detections was more clustered (Fig7A). The number of detection events per cluster (Fig7B) and inter-cluster distance (Fig7F) was estimated using intensity-based thresholding (see Materials and Methods). The number of detections of α3-NKA per cluster increased when neurons were exposed to α-syn (Fig7B). The inter-cluster distance (distance between centroid of two clusters) also increased (Fig7C) indicative of a spatial reorganization resulting from the recruitment of small nanoclusters or non-clustered α3-NKA to larger nanoclusters, thus leading to the formation of regions with reduced α3-NKA surface density on the plasma membrane.
Figure 7.

- A Representative super-resolution images showing individual detection of α3-NKA-Alexa 647. α3-NKA exists as nanometer-sized clusters as well as non-clustered on the plasma membrane. Scale bar: 1 μm.
- B Number of detections per α3-NKA nanocluster (after threshold) increases following exposure to α-syn (60 min). Top panel: Average of normalized values from three independent experiments (paired t-test: *P < 0.05). Bottom panel shows the distribution plot (median, quartile, and 10–90% distribution) for all α3-NKA clusters (number of nanoclusters, control = 7,265, oligomer = 7,549, fibril = 8,563; Kolmogorov–Smirnov test: ***P < 0.001).
- C α-syn exposure (60 min) increased the distance between α3-NKA nanoclusters (within a maximum distance of 0.3 μm). Top panel: Average of normalized values from three independent experiments (paired t-test: ns = non-significant). Bottom panel shows the distribution plot (median, quartile, and 10–90% distribution) for all α3-NKA clusters (n = control: 2,570, oligomer: 3,892, fibril: 4,022; Kolmogorov–Smirnov test: ***P < 0.001).
- D Unbiased density-based cluster analysis of all detection of α3-NKA (see Materials and Methods). α-syn exposure increased the clustering of α3-NKA. Plot shows mean ± SEM calculated from three independent experiments (Mann–Whitney U-test; ***P < 0.001).
Intensity-based threshold analysis is biased toward the center of highly dense clusters, and thus, no information is obtained from sparse regions. Therefore, we performed an unbiased cluster analysis on all detections to quantify the distribution of detections in and out of nanoclusters (Ester et al, 1996). This method organizes the detections based on a density-based clustering structure where the clustering parameter is defined by a minimal density, ρ0. Thus, objects with local density ρ0 or above are considered clustered. The plot shows the percentage of non-clustered detections (y-axis) versus density threshold (ρ0, x-axis). A lower value for a given density (ρ0) threshold implies a higher packing of detections (Fig7D). This unbiased approach confirms that α-syn assemblies mediate a tighter packing of α3-NKA by recruitment of additional non-clustered molecules. Altogether, these results demonstrate that exogenous α-syn assemblies impact the distribution of α3-NKA, with the formation of large α3-NKA nanoclusters.
Synaptic clustering and association with α3-NKA following in vivo injection of fluorescent α-syn
Prion-like propagation of α-syn has been studied following intra-striatal injection of α-syn (Luk et al, 2012; Mougenot et al, 2012; Sacino et al, 2014). It recapitulates PD pathology leading to Lewy body deposits (˜30 days), dopaminergic neuron loss (˜90–180 days), and impaired motor coordination (˜90–180 days). The observed pathological changes are likely to be a combination of several deleterious pathways during the course of 0–180 days. Thus, this model allowed us to investigate whether injected α-syn forms clusters on the plasma membrane and interacts with α3-NKA in vivo at an early time point post injection as observed in vitro.
Exogenous oligomeric or fibrillar α-syn assemblies (labeled with ATTO-550) were injected in one striatum of 10-week-old rats (as previously described in Luk et al, 2012; Rey et al, 2013). Eight hours after injection, oligomeric α-syn was widespread throughout the striatum (Fig8A) and clusters were seen at higher magnification (Fig8Ba-b). α-syn clusters were frequently associated with MAP2 (microtubule-associated protein 2)-positive processes thus identified as dendrites (Fig8C, green). Homer and gephyrin immunoreactivities identified excitatory and inhibitory synapses, respectively (Fig8Da-b). Several α-syn clusters were associated with these synaptic markers (Fig8Da-b, arrow): Nearly 50% of oligomeric α-syn clusters were associated with homer or gephyrin immunoreactivities (median: homer positive: 31%; gephyrin positive: 16%; n = 19 randomly selected regions around site of injection) (Fig8Dc).
Figure 8.

- A–E Injection of oligomeric α-syn-ATTO-550 (red) in rat striatum. Low magnification at a distance from injection site of α-syn (red) shows the spread (A) (delineated by dashed line). Confocal image at higher magnification shows oligomeric α-syn clusters (Ba and Bb). Note that the clusters are excluded from striato-pallidonigral axon bundles (*, Bb). Arrowhead (C) shows that most oligomeric α-syn clusters (red) were adjacent to dendrites (green, MAP2 immunoreactivity). (D) Double detection of α-syn oligomers (red) and homer (Da, green) or gephyrin (Db, blue) to identify excitatory and inhibitory synapses, respectively. Notably, some α-syn clusters (arrows) are adjacent to synapses. (Dc) Quantification of association between α-syn and homer/gephyrin (plot: median, quartile, min. to max. distribution). (E) Simultaneous detection of α-syn oligomers (red), α3-NKA (green), and synapsin (blue). (Ea) Diffused and clustered α3-NKA immunoreactivity. (Eb) Overlay of α-syn oligomers fluorescence (red) and α3-NKA immunoreactivity (green). (Ec) Overlay of α-syn oligomers fluorescence, α3-NKA, and synapsin immunoreactivities (boxed region in Ea and Eb). Note that α-syn oligomer clusters are often adjacent to α3-NKA ones (arrows, Eb, Ec) and sometimes at synapses (arrowhead, Ec). (Ed) Quantification of α3-NKA immunoreactivity: α3-NKA enrichment at synapse (synapsin) when associated with α-syn (normalized to total clusters, plot: median, quartile, and 10–90% distribution; Mann–Whitney U-test ***P < 0.001). Scale bar: 5 μm.
We also examined the fate of injected fibrillar α-syn in rat striatum after different time intervals (8 and 24 h). No spread could be detected after 8 h: Fibrillar α-syn remained at the site of injection (observation). After 24 h, very few small clusters of fibrillar α-syn were detected adjacent to the site of injection (see FigEV3A and B). Nearly 20% of these clusters were at synapses (FigEV3C) (median: homer positive: 12% (n = 21); gephyrin positive: 7% (n = 20)).
Figure EV3.
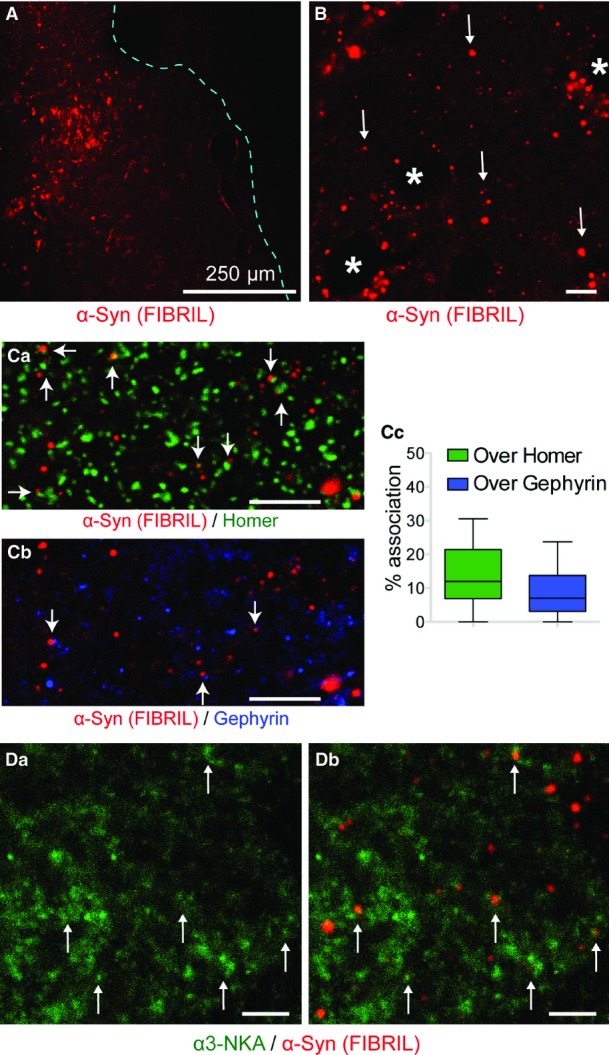
- A–D Distribution of fibrillar α-syn-ATTO-550 (red) 24 h after injection. (A) Limited spread (outlined by a dashed line) showing ATTO-550 signal. (B) Clusters of fibrillar α-syn (arrow). Note that the clusters are excluded from striato-pallidonigral axon bundles (*). Some of the fibrillar α-syn clusters co-localized with homer (Ca, Cc green) or gephyrin (Cb, Cc, blue) (plot: median, quartile, and min. to max. distribution). (Da-Db) Association of fibrillar α-syn clusters (red, Db) and α3-NKA (green, Da, Db) can be seen (arrow). Unless indicated, scale bars: 5 μm.
We then assessed α-syn assemblies and α3-NKA association. Following injection of oligomeric (Fig8E) or fibrillar (Fig EV3D) α-syn (red) in the rat striatum, α3-NKA (green) was immunolabeled. Synapses were identified by synapsin (blue) immunoreactivities. α3-NKA showed an uneven distribution with a diffuse labeling and several enriched spots/clusters (Fig8Ea, green). Few oligomeric (Fig8Eb-c) and fibrillar (Fig EV3Db) α-syn clusters were associated with α3-NKA clusters; some of them localized at synapses (Fig8Ec, arrowheads). The fluorescence intensities of α3-NKA clusters were measured (Fig8Ed, n = 103 randomly selected regions around the site of injection). The higher fluorescence intensity of α3-NKA spots over synapses (n = 102) or α-syn clusters (n = 103) suggests α3-NKA enrichment. Notably, an even stronger enrichment of α3-NKA was observed at synapses where α-syn clustered (n = 53). Thus, α-syn injected in the striatum forms clusters where α3-NKA molecules are enriched as observed in the in vitro experiments.
α-syn assemblies reduce α3-NKA-dependent Na+ efflux from neurons and increase glutamate-induced Ca2+ influx
α3-NKA subunit is responsible for the restoration of Na+ level in neurons after depolarization, while α1-NKA subunit maintains basal Na+ level (Azarias et al, 2013). Thus, the recovery rate of Na+ concentration following depolarization provides a specific quantification of α3-NKA subunit activity (Azarias et al, 2013). The basal Na+ level, which can be estimated using a calibration plot, allows the quantification of the α1-NKA subunit activity (Azarias et al, 2013). Neurons (DIV 16–20) were exposed or not (control) to α-syn assemblies (oligomer: 25 nM; fibril: 0.03 nM) and loaded with Na+ dye, ANG-2 (Asante NaTRIUM Green 2) (see Materials and Methods and Appendix Fig S6 for experimental design and calibrations). The basal level was monitored for 2–3 min; then, 0 mM K+ recording solution (˜10 ml) was added to mimic transient increase in Na+ reached during supra-threshold neuronal activity (Fig9A, red arrow). Subsequently, the 0 mM K+ recording solution was replaced with normal recording solution, thus allowing the visualization of Na+ efflux (Fig9A, green arrow). A representative control trace is shown in black. Calibration plot (Appendix Fig S6) was used to estimate the basal Na+ concentration in dendrites. α-syn assemblies did not modify the basal level of Na+ after 1 h exposure (˜20 mM Na+) suggesting no alteration in the overall α1-NKA activity. However, after 24 h of exposure, a higher basal Na+ level was observed (˜30 mM) suggesting large-scale perturbation in Na+ gradient. Following the 0 mM K+ recording solution depolarization, the Na+ concentration failed to recover to the basal level (Fig9A, black dashed lines) in neurons exposed 1 or 24 h to α-syn assemblies. A larger Na+ influx was observed after 24-h exposure to α-syn assemblies (Fig9B). Exposure to α-syn (fibrils or oligomers) reduced the recovery of Na+ to basal level (Fig9C) suggesting a decrease in α3-NKA activity. α3-NKA is responsible for the clearance of Na+ from neurons following depolarization (Azarias et al, 2013). In the presence of α-syn assemblies, we observed a decreased rate of the Na+ extrusion (Fig9D, see Materials and Methods), indicating a reduction of the α3-NKA-dependent Na+ efflux. Notably, monomeric α-syn had no effect on recovery of Na+ to basal level (Fig EV4A) or rate of Na+ extrusion (Fig EV4B).
Figure 9.
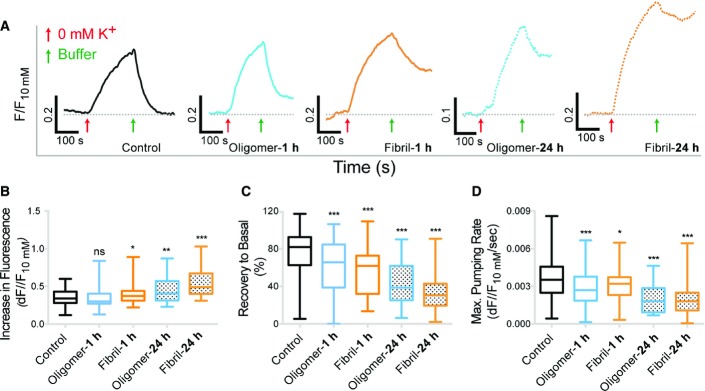
- A Representative traces showing Na+ dynamics in individual neurons exposed to unlabeled α-syn (oligomer: 25 nM (blue); fibril: 0.03 nM (orange)) for 1 or 24 h (refer to Appendix Fig S6 for the protocol used). Note that α-syn-exposed neurons failed to rapidly return to the basal level, whereas control neurons did (black dashed line).
- B–D Neurons exposed to α-syn assemblies for 24 h show increased Na+ influx (B) following 0 mM K+ recording solution application (red arrow, A). Neurons exposed to oligomeric or fibrillar α-syn failed to recover to basal level (C) and exhibit decreased Na+ pumping rate (D) after exchange of 0 mM K+ recording solution with normal recording solution (green arrow, A). Note α-syn exposure time-dependent decrease in Na+ recovery and pumping rate (plot: median, quartile, and 10–90% distribution; Mann–Whitney U-test: *P < 0.05, **P < 0.01, ***P < 0.001). Number of dendrites: control, n = 99; oligomer 1 h, n = 90; fibril 1 h, n = 85; oligomer 24 h, n = 38; fibril 24 h, n = 38.
Figure EV4.
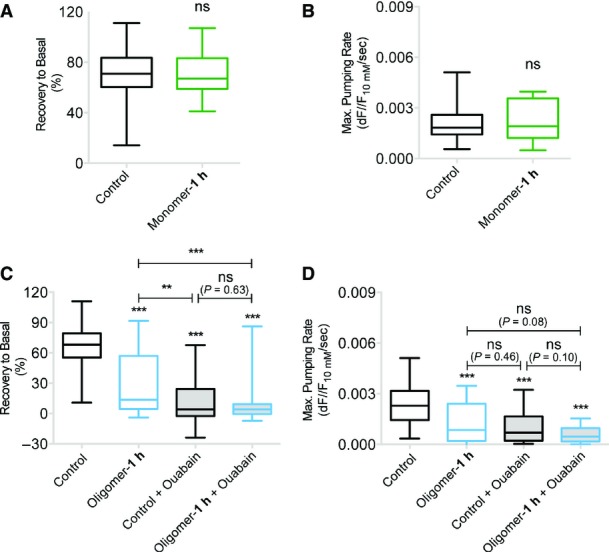
- A, B Neurons exposed to monomeric α-syn recover to basal level (A) and exhibit no change in Na+ pumping rate (B).
- C, D Na+ imaging in the presence of ouabain (1 μM, 3–5 min before 0 mM K+ application) on control or oligomeric α-syn (25 nM) exposed cells. Both oligomeric α-syn-exposed (column 2) and ouabain-treated (column 3) neurons show reduction in the recovery (C) and Na+ pumping rate (D). Note no additive effect of ouabain on oligomeric α-syn-exposed (column 4) compared to control (column 3) neurons.
We next performed experiments in the presence of ouabain to (i) quantify the extent of α3-NKA inhibition by α-syn and (ii) ascertain whether the reduced recovery of Na+ level from neurons in the presence of α-syn is due to α3-NKA and not other Na+ transporters. Nearly 3–5 min before depolarization, ouabain (1 μM) was added to completely inhibit α3-NKA (Azarias et al, 2013). As above, oligomeric α-syn (column 2) exposed (1 h) cells showed a reduction in recovery to basal level (53%, FigEV4C) and pumping rate (47%, FigEV4D) compared to control cells. Ouabain-treated cells (column 3) also showed a reduction in recovery to basal level (81%, FigEV4C) and pumping rate (57%, FigEV4D). These data show that α-syn partially suppresses α3-NKA-dependent Na+ efflux from neurons (recovery, 60%; pumping rate, 21%). No additive effect of ouabain was observed in neurons that were pre-exposed to oligomeric α-syn (column 4) compared to control neurons (column 3) (FigEV4C and D). This favors the notion that the reduction in the recovery of Na+ level in neurons following α-syn exposure is due to α3-NKA and not other Na+ transporters.
The activity of neuronal NKA may also have an effect on the Ca2+ response to glutamate release, because of its interaction with the NMDA receptor (Akkuratov et al, 2014) and/or the Na+/Ca2+ exchangers (Blaustein & Lederer, 1999). We observed a significantly larger Ca2+ influx in α-syn assembly-exposed neurons compared to unexposed neurons (FigEV5A and B). Thus, α-syn clustering and α3-NKA redistribution do not only compromise Na+ gradient in neurons, but may also make them more vulnerable to glutamate excitotoxicity.
Figure EV5.
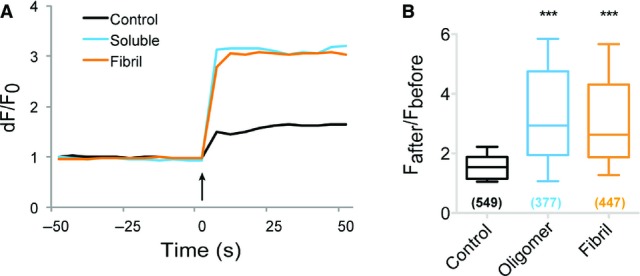
- A, B DIV 16–19 striatal neurons were exposed (1 h) to oligomeric (25 nM, blue) or fibrillar (0.03 nM, orange) unlabeled α-syn. Unexposed control is shown in black. Glutamate-evoked (arrow, 100 μM, A) Ca2+ rise was measured following Fluo-4-AM dye labeling. Note a larger Ca2+ influx in cells exposed to α-syn assemblies. Averaged normalized (dF/F0) fluorescence intensity for all cells is shown (B). Box plot in (B) shows the distribution of change in fluorescence intensity for all cells (box plot: median, quartile, and 10–90% distribution; Mann–Whitney U-test: ***P < 0.001; number of cells (n) is shown in parentheses in B; five independent experiments).
Discussion
We demonstrate that both oligomeric and fibrillar α-syn assemblies rapidly bind to the plasma membrane, laterally diffuse, and form clusters at synaptic as well as extra-synaptic locations. Using pull-down and MS on pure neuronal cultures, we identified α-syn interacting membrane proteins with extracellular domains. Among them, only the α3-subunit of NKA which is enriched in the plasma membrane directly interacts with both oligomeric and fibrillar α-syn assemblies and thus further studied here. The β1-subunit, which forms α3β1-NKA complex, was also identified in our fibrillar α-syn pull-down MS proteomic screen. In addition, several heparan sulfate proteoglycans, namely agrin, glypican, and testican, were present in the interactome of fibrillar α-syn assemblies. They may assist in the endocytosis of exogenous α-syn molecules as recently proposed (Holmes et al, 2013). A second transmembrane protein, neurexin (1α and 2α), involved in autism spectrum disorders (Etherton et al, 2009) was also pulled down as a fibrillar α-syn partner and needs further evaluation.
Clustering of α-syn assemblies within neuronal plasma membrane
The rapid binding of exogenous α-syn assemblies to neuronal membranes is consistent with what was described for Aβ oligomers (Renner et al, 2010) and prion proteins (Goold et al, 2011). Following binding and lateral diffusion on the neuronal plasma membranes, α-syn assemblies formed clusters in a time- and concentration-dependent manner. The fast diffusion (10−1 μm2/s) of oligomeric α-syn suggests that α-syn may be lipid-bound (Renner et al, 2009). The link between the size of α-syn clusters and the days the neurons were cultured is likely due to maturation-dependent changes in neuronal plasma membrane proteins and lipids composition. Unlike Aβ oligomers, which are primarily enriched at excitatory synapse (Renner et al, 2010), α-syn molecules clustered both at excitatory and inhibitory synapses and may also form clusters on the extra-synaptic membrane including axonal and dendritic membrane. Since α-syn diffusion was not significantly different in and out of synapse, it favors the notion that α-syn confinement and clustering is not dependent on the compartment where it occurs. The fast time dependence of cluster formation supports the notion that the plasma membrane serves as a biochemical reactor favoring α-syn inter-molecular interactions by reducing the diffusion space from 3D to 2D by increasing the confinement of the molecules.
Interaction between α3-NKA and α-syn assemblies
We show that α-syn clustering within the membrane leads to the redistribution of α3-NKA by diffusional trapping with a reduction of its ability to pump out Na+ from neurons, thus leading to an accumulation of Na+. The strength of the interaction between α3-NKA and α-syn likely depends on the type of assembly of α-syn rather than number of monomeric equivalents. This observation comes from the fact that fibrils exhibit much stronger association with α3-NKA (MS, co-immunoprecipitation, co-localization and SPT studies) as compared to their oligomeric counterparts. α-syn monomers did not interact with α3-NKA as indicated by co-immunoprecipitation, SPT, and Na+ imaging experiments.
Neurons express α1- and α3-subunit of NKA, α1-subunit is responsible for the maintenance of basal Na+ level and α3-subunit is involved in rapid Na+ extrusion after supra-threshold stimulus (Azarias et al, 2013). The basal Na+ level is unaltered after exposure of neurons to α-syn assemblies for 1 h, indicating that α1-NKA activity is unaffected within this time frame. The specificity of α-syn assemblies and α3-NKA interaction is further confirmed by the fact that only the α3, not the α1- or astrocyte-specific α2-subunit of NKA, was identified as partner of α-syn assemblies. This observation is important since several of the mutations within the α3-subunit of NKA, that lead to motor and cognitive disabilities, such as RDP and AHC, are associated with a reduction of Na+ affinity (Toustrup-Jensen et al, 2014). Alterations in Na+ affinity following α3-NKA interaction with α-syn should have profound effects on recovery after high neuronal activity, membrane potential, and hyperpolarization (Pulver & Griffith, 2010).
We found that α-syn led to α3-NKA redistribution and ultimately to α-syn-α3-NKA co-clustering in and outside synapses. Using super-resolution STED (simulated emission depletion) microscopy, it was recently shown that α3-NKA exist as nanoclusters on neuronal dendrites and spines (Blom et al, 2012). Reorganization and redistribution of α3-NKA bound to α-syn assemblies resulted in the formation of larger clusters by recruitment of non-clustered α3-NKA molecules. Trapping within α-syn clusters generates a non-uniform gradient with high and low density of α3-NKA molecules. This spatial redistribution of α3-NKA may contribute to the reduction in the capacity of neurons to rapidly pump out Na+ following supra-threshold stimulus. In addition, the binding of α-syn to the extracellular loop of α3-NKA may interfere with its turnover and transition between E1 and E2 forms, thus reducing pumping activity.
Restoration of intracellular Na+ concentration after high neuronal activity is an essential function for the α3-NKA, and recent studies have indicated a two-way interaction between NKA and the NMDA receptor (Akkuratov et al, 2014). Here we found that the Ca2+ response to glutamate was enhanced in α-syn assembly-exposed neurons, suggesting that the α-syn pathology may also result in an increased excitotoxicity. The mechanisms behind the increased calcium response remain to be elucidated, but might be explained by a change in the state of Src-mediated NMDA receptor phosphorylation. Src kinase is a hub for NMDA receptors signaling (Salter & Kalia, 2004), and NKA is a regulator of Src activity (Li & Xie, 2009). The reduced capacity of neurons to restore Na+ level in the α-syn-exposed neurons might also contribute to the enhanced Ca2+ response, since it would decrease and maybe revert the transport via the Na+/Ca2+ exchanger (Kiedrowski et al, 2004), the high capacity system for Ca2+ efflux.
Postsynaptic NKA also regulates NF-κB/CREB signaling (Desfrere et al, 2009; De Sá Lima et al, 2013) and AMPA receptor turnover (Zhang et al, 2009). In addition, α3-NKA is also enriched at presynaptic terminals where it controls the probability and precision of firing following post-tetanic hyperpolarization (Kim et al, 2007) as well as the availability of readily releasable pool of vesicles (Taruno et al, 2012). α3-NKA also putatively interacts with the glycine transporter (GlyT2) regulating the surface expression of the latter (De Juan-Sanz et al, 2013). Thus, an alteration in α3-NKA distribution and pumping activity will not only affect the ionic gradient in neurons, but also a plethora of other pre- and postsynaptic pathways in neurons. This is supported by our observation that α-syn binds and clusters on both axons and dendrites and also synaptic and extra-synaptic sites. Due to ubiquitous expression of α3-NKA on the plasma membrane, α-syn-α3-NKA co-clustering will not only alter local ion gradient but also NKA-dependent signaling pathways. Given that α3-NKA is more enriched at synapses (Figs6B and 8Ed), the effect will be important on synaptic ion concentration.
The most documented pathophysiological consequence of reduced/loss of NKA activity comes from studies on dystonia disease models resulting in hyperactivity, memory, and motor deficits (Clapcote et al, 2009; Kirshenbaum et al, 2011). Moreover, perfusion of ouabain, inhibiting α3-NKA, in the striatum leads to Parkinson-like dystonia (Calderon et al, 2011). Similarly, the interaction of α-syn assemblies with α3-NKA interfering with Na+ pumping out of neurons may contribute to α-synucleinopathies such as Parkinson’s disease.
Materials and Methods
An elaborated version of Materials and Methods is provided in the Appendix section.
Preparation and characterization of α-syn assemblies
Preparation and characterization of recombinant wild-type (WT) and C-terminally S-tagged (α-syn-S-tag) human α-syn is described in Appendix Supplementary Materials and Methods. For our preparation, the average number of constituting molecules measured for oligomeric and fibrillar α-syn was 40 and 8,333, respectively. The concentration of oligomeric α-syn was 25 nM and that of fibrillar α-syn was 0.03 nM, unless otherwise specified, corresponding to 1 or 0.25 μM monomeric α-syn, respectively.
Exposure to α-syn, single particle tracking, and analysis
For SPT of α-syn assemblies, biotin-labeled α-syn was used and identified by streptavidin-QD-605 nm. Following exposure to α-syn assemblies, cells were incubated with streptavidin-QD-605 nm (1:5,000, 2 min). For SPT of α3-NKA, cells transfected with pHluorin-α3-NKA plasmid were labeled using GFP antibody pre-coupled with QD-Fab-605 nm (Invitrogen). The center of the QD fluorescence spot was determined by Gaussian fitting, and maximum-likelihood approach was used to obtain trajectories. The mean square displacement (MSD) was calculated using MSD(ndt) = 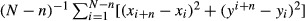 , where xi and yi are the coordinates of an object on frame I, N is the total number of steps in the trajectory, dt is the time between two successive frames, and ndt is the time interval over which displacement is averaged (Saxton & Jacobson, 1997). The diffusion coefficient D was calculated by fitting the first two to five points of the MSD plot versus time with the equation
, where xi and yi are the coordinates of an object on frame I, N is the total number of steps in the trajectory, dt is the time between two successive frames, and ndt is the time interval over which displacement is averaged (Saxton & Jacobson, 1997). The diffusion coefficient D was calculated by fitting the first two to five points of the MSD plot versus time with the equation 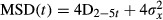 , with σx is the spot localization accuracy in one direction.
, with σx is the spot localization accuracy in one direction.
Pull-down of α-syn-S-tag-bound protein complexes and mass spectrometry (MS)
A detailed protocol is described in Appendix Supplementary Materials and Methods. Rat pure cortical neuron cultures were exposed (10 min) to α-syn-S-tag assemblies (40 μM monomer concentration). Unexposed neurons were used as control. Purified protein extracts were subjected to MS by nanoLC-MS/MS. Only proteins identified with at least two unique peptides in at least two replicates were quantified. Only proteins with a spectral count ratio, between the cells exposed to α-syn (either oligomeric or fibrillar) and the control cells, above 1.6 and a P-value < 0.05 were considered as significantly increased in the pull-down and thus considered as α-syn interacting proteins.
Proteomics data deposition
The mass spectrometry proteomics data (neurons and astrocytes) have been deposited to the ProteomeXchange Consortium (Vizcaíno et al, 2014) via the PRIDE partner repository with the dataset identifiers PXD002256 through PXD002263 and DOI 10.6019/PXD002256 through 10.6019/PXD002263.
Sodium dye loading, imaging and analysis
Sodium imaging was performed following loading of neurons with Na+-sensitive cytosolic ANG2 (Asante NaTRIUM Green 2) dye. The fluorescence intensity (F) of ANG2 on dendrites was normalized to the fluorescence levels of the calibration solutions containing 10 mM Na+ (F10 mM). The increase in fluorescence from basal level, the recovery to basal level after depolarization, and Na+ extrusion rate were measured as depicted in Appendix Fig S6.
Acknowledgments
We thank Karine Madiona for expert assistance with the analytical ultracentrifugation measurements. This work was supported by the Agence Nationale de la Recherche (ANR-09-MNPS-013-01 and ANR-11-BSV8-021-01), the ERC advanced research grant “PlasltInhib,” program “Investissements d’Avenir” (ANR-10-LABX-54 MEMO LIFE and ANR-11-IDEX-0001-02 PSL* Research University), Institut National de la Santé et de la Recherche Médicale (INSERM), France Alzheimer (Project R12035JJ), “Coup d’Elan a la Recherche Francaise” award from Fondation Bettencourt Schueller and Swedish Research Council. We thank Prof. Stuart J. Edelstein for carefully reading the manuscript. R.M. dedicates this work to late Prof. Paul Cohen.
Author contributions
ANS, AT, RM, and AA designed the research. ANS, VR, NF, and LP performed experiments. LB generated α-syn assemblies and characterized them. TL generated pHluorin-α3-NKA plasmid and optimized sodium imaging approach. MS and CL performed in vivo injections. LGA and MR developed new tools for image analysis. ANS, AT and RM wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Expanded View Figures PDF
Movie EV1
Review Process File
References
- Akkuratov EE, Lopacheva OM, Kruusmägi M, Lopachev AV, Shah ZA, Boldyrev AA, Liu L. Functional interaction between Na/K-ATPase and NMDA receptor in cerebellar neurons. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8975-3. doi: 10.1007/s12035-014-8975-3. [DOI] [PubMed] [Google Scholar]
- Azarias G, Kruusmägi M, Connor S, Akkuratov EE, Liu XL, Lyons D, Brismar H, Broberger C, Aperia A. A specific and essential role for Na, K-ATPase α3 in neurons co-expressing α1 and α3. J Biol Chem. 2013;288:2734–2743. doi: 10.1074/jbc.M112.425785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Na+, K+-ATPase: functions in the nervous system and involvement in neurologic disease. Neurology. 2011;76:287–293. doi: 10.1212/WNL.0b013e3182074c2f. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Blom H, Rönnlund D, Scott L, Spicarova Z, Rantanen V, Widengren J, Aperia A, Brismar H. Nearest neighbor analysis of dopamine D1 receptors and Na+-K+-ATPases in dendritic spines dissected by STED microscopy. Microsc Res Tech. 2012;75:220–228. doi: 10.1002/jemt.21046. [DOI] [PubMed] [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–365. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, Linazasoro G, Borg M, Tijssen MA, Bressman SB, Dobyns WB, Brashear A, Ozelius LJ. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–175. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Clapcote SJ, Duffy S, Xie G, Kirshenbaum G, Bechard AR, Rodacker Schack V, Petersen J, Sinai L, Saab BJ, Lerch JP, Minassian BA, Ackerley CA, Sled JG, Cortez MA, Henderson JT, Vilsen B, Roder JC. Mutation I810N in the alpha3 isoform of Na+, K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc Natl Acad Sci USA. 2009;106:14085–14090. doi: 10.1073/pnas.0904817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Juan-Sanz J, Núñez E, Villarejo-López L, Pérez-Hernández D, Rodriguez-Fraticelli AE, López-Corcuera B, Vázquez J, Aragón C. Na+/K+-ATPase is a new interacting partner for the neuronal glycine transporter GlyT2 that downregulates its expression in vitro and in vivo. J Neurosci. 2013;33:14269–14281. doi: 10.1523/JNEUROSCI.1532-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sá Lima L, Kawamoto EM, Munhoz CD, Kinoshita PF, Orellana AM, Curi R, Rossoni LV, Avellar MC, Scavone C. Ouabain activates NFκB through an NMDA signaling pathway in cultured cerebellar cells. Neuropharmacology. 2013;73:327–336. doi: 10.1016/j.neuropharm.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Demos MK, van Karnebeek CD, Ross CJ, Adam S, Shen Y, Zhan SH, Shyr C, Horvath G, Suri M, Fryer A, Jones SJ, Friedman JM FORGE Canada Consortium. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J Rare Dis. 2014;9:15. doi: 10.1186/1750-1172-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desfrere L, Karlsson M, Hiyoshi H, Malmersjö S, Nanou E, Estrada M, Miyakawa A, Lagercrantz H, El Manira A, Lal M, Uhlén P. Na, K-ATPase signal transduction triggers CREB activation and dendritic growth. Proc Natl Acad Sci USA. 2009;106:2212–2217. doi: 10.1073/pnas.0809253106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis DZ, Rabe J, Sweadner KJ. Global loss of Na, K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:43–51. doi: 10.1523/JNEUROSCI.23-01-00043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M, Kriegel H, Sander J, Xu X. 1996. A density-based algorithm for discovering clusters in large spatial databases with noise. Proc 2 Int Conf on Knowledge Discovery and Data Mining (KDD ‘96), Portland, Oregon, AAAI Press.
- Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci USA. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, Tabrizi SJ. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun. 2011;2:281. doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, Tiziano FD, Fontaine B, Walley NM, Heavin S, Panagiotakaki E. Fiori S, Abiusi E, Di Pietro L, Sweney MT, Newcomb TM, Viollet L, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. 2012;44:1030–1034. doi: 10.1038/ng.2358. , European Alternating Hemiplegia of Childhood (AHC) Genetics Consortium, Biobanca e Registro Clinico per l’Emiplegia Alternante (I.B.AHC) Consortium, European Network for Research on Alternating Hemiplegia (ENRAH) for Small and Medium-sized Enterpriese (SMEs) Consortium, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. 2013;110:E3138–E3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- Kaplan JH. Biochemistry of Na, K-ATPase. Annu Rev Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Czyz A, Baranauskas G, Li XF, Lytton J. Differential contribution of plasmalemmal Na/Ca exchange isoforms to sodium-dependent calcium influx and NMDA excitotoxicity in depolarized neurons. J Neurochem. 2004;90:117–128. doi: 10.1111/j.1471-4159.2004.02462.x. [DOI] [PubMed] [Google Scholar]
- Kim JH, Sizov I, Dobretsov M, von Gersdorff H. Presynaptic Ca2+ buffers control the strength of a fast post-tetanic hyperpolarization mediated by the alpha3 Na+/K+-ATPase. Nat Neurosci. 2007;10:196–205. doi: 10.1038/nn1839. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum GS, Clapcote SJ, Duffy S, Burgess CR, Petersen J, Jarowek KJ, Yücel YH, Cortez MA, Snead OC, 3rd, Vilsen B, Peever JH, Ralph MR, Roder JC. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+, K+-ATPase α3 sodium pump. Proc Natl Acad Sci USA. 2011;108:18144–18149. doi: 10.1073/pnas.1108416108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Li Q, Lau A, Morris TJ, Guo L, Fordyce C, Stanley EF. A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Li Z, Xie Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009;457:635–644. doi: 10.1007/s00424-008-0470-0. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano G, Van den Haute C, Melki R, Baekelandt V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulver SR, Griffith LC. Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nat Neurosci. 2010;13:53–59. doi: 10.1038/nn.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines RT, McCormick M, Van Oosbree TR, Mierendorf RC. The S-Tag fusion system for protein purification. Methods Enzymol. 2000;326:362–376. doi: 10.1016/s0076-6879(00)26065-5. [DOI] [PubMed] [Google Scholar]
- Renner M, Choquet D, Triller A. Control of the postsynaptic membrane viscosity. J Neurosci. 2009;29:2926–2937. doi: 10.1523/JNEUROSCI.4445-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human α–synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013;126:555–573. doi: 10.1007/s00401-013-1160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribrault C, Reingruber J, Petković M, Galli T, Ziv NE, Holcman D, Triller A. Syntaxin1A lateral diffusion reveals transient and local SNARE interactions. J Neurosci. 2011;31:17590–17602. doi: 10.1523/JNEUROSCI.4065-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodacker V, Toustrup-Jensen M, Vilsen B. Mutations Phe785Leu and Thr618Met in Na+, K+-ATPase, associated with familial rapid-onset dystonia parkinsonism, interfere with Na+ interaction by distinct mechanisms. J Biol Chem. 2006;281:18539–18548. doi: 10.1074/jbc.M601780200. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Thiele H, Ohlenbusch A, Maschke U, Altmüller J, Frommolt P, Zirn B, Ebinger F, Siemes H, Nürnberg P, Brockmann K, Gärtner J. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene- identification study. Lancet Neurol. 2012;11:764–773. doi: 10.1016/S1474-4422(12)70182-5. [DOI] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA. 2014;111:10732–10737. doi: 10.1073/pnas.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Saxton MJ, Jacobson K. Single-particle tracking: application to membrane dynamics. Annu Rev Biophys Biomol Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- Specht CG, Grünewald N, Pascual O, Rostgaard N, Schwarz G, Triller A. Regulation of glycine receptor diffusion properties and gephyrin interactions by protein kinase C. EMBO J. 2011;30:3842–3853. doi: 10.1038/emboj.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taruno A, Ohmori H, Kuba H. Inhibition of presynaptic Na+/K+-ATPase reduces readily releasable pool size at the avian end-bulb of Held synapse. Neurosci Res. 2012;72:117–128. doi: 10.1016/j.neures.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Toustrup-Jensen MS, Einholm AP, Schack VR, Nielsen HN, Holm R, Sobrido MJ, Andersen JP, Clausen T, Vilsen B. Relationship between intracellular Na+ concentration and reduced Na+ affinity in Na+, K+-ATPase mutants causing neurological disease. J Biol Chem. 2014;289:3186–3197. doi: 10.1074/jbc.M113.543272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–374. doi: 10.1016/j.neuron.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolomé S, et al. ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;30:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous a-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Pin JP, Sebben M, Kemp DE, Sladeczek F, Gabrion J, Bockaert J. Synaptogenesis of cultured striatal neurons in serum-free medium: a morphological and biochemical study. Proc Natl Acad Sci USA. 1986;83:2238–2242. doi: 10.1073/pnas.83.7.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hou Q, Wang M, Lin A, Jarzylo L, Navis A, Raissi A, Liu F, Man HY. Na, K-ATPase activity regulates AMPA receptor turnover through proteasome-mediated proteolysis. J Neurosci. 2009;29:4498–4511. doi: 10.1523/JNEUROSCI.6094-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Review Process File