

Abstract
African Americans (AA) with systemic sclerosis (SSc) have a worse prognosis compared to Americans of European descent (EA). We conducted the current study to test the hypothesis that AA patients with SSc have more severe disease and poorer outcomes compared to EA patients when afflicted with pulmonary arterial hypertension (PAH).
We studied 160 consecutive SSc patients with PAH diagnosed by right heart catheterization, comparing demographics, hemodynamics, and outcomes between AA and EA patients.
The cohort included 29 AA and 131 EA patients with similar baseline characteristics except for increased prevalence of diffuse SSc in AA. AA patients had worse functional class (FC) (80% FC III-IV vs 53%; p = 0.02), higher brain natriuretic peptide (NT-pro-BNP) (5729 ± 9730 pg/mL vs 1892 ± 2417 pg/mL; p = 0.02), more depressed right ventricular function, a trend toward lower 6-minute walk distance (263 ± 111 m vs 333 ± 110 m; p = 0.07), and worse hemodynamics (cardiac index 1.95 ± 0.58 L/min/m2 vs 2.62 ± 0.80 L/min/m2; pulmonary vascular resistance 10.3 ± 6.2 WU vs 7.6 ± 5.0 WU; p < 0.05) compared with EA patients. Kaplan-Meier survival estimates for AA and EA patients, respectively, were 62% vs 73% at 2 years and 26% vs 44% at 5 years (p > 0.05).
In conclusion, AA patients with SSc-PAH are more likely to have diffuse SSc and to present with significantly more severe PAH compared with EA patients. AA patients also appear to have poorer survival, though larger studies are needed to investigate this association definitively.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a chronic disorder of the pulmonary vasculature, characterized by a progressive increase in pulmonary vascular resistance (PVR) leading to right heart failure and death. PAH can be idiopathic (IPAH) or associated with underlying conditions, such as connective tissue diseases.35 Among these, PAH is most commonly associated with systemic sclerosis (SSc). While several measures of disease severity, including World Health Organization functional class (WHO-FC), exercise capacity, N-terminal pro-brain natriuretic peptide (NT-pro-BNP) and hemodynamics, have been associated with outcomes in SSc-associated PAH (SSc-PAH), other factors such as race have not been fully explored.
Limited data exist regarding racial differences in PAH, yet recent studies suggest race may be a risk factor for mortality in these patients. In a retrospective cohort study of IPAH patients, Kawut et al21 demonstrated a more than 4-fold increased risk of death for African American (AA) patients compared to European American (EA) patients. However, data from the REVEAL registry of over 3000 patients with PAH suggest that race is not associated with mortality.3
Several epidemiologic studies have illustrated significant geographic and racial disparities in the incidence, prevalence, and clinical manifestations of SSc.15,30 Within the American population of patients with SSc, racial disparities have long been noted, not only in clinical presentation, severity, and disease progression,15,29,30 but also in age at presentation, severity of organ involvement, and prognosis. Some studies have shown that AA patients have a younger age at disease onset, a higher frequency of diffuse skin involvement, and an overall worse prognosis compared to other racial groups.29
Based on racial differences in the epidemiology of SSc, we hypothesized that AA with SSc-PAH had more severe PAH disease compared to other racial groups, and therefore investigated the association of race with clinical severity, hemodynamics, and survival in a large cohort of SSc-PAH patients followed at a single referral center.
METHODS
Patient Population
The Johns Hopkins University Institutional Review Board approved this study. Consecutive patients with SSc diagnosed with PAH by right heart catheterization (RHC) and evaluated at the Johns Hopkins Pulmonary Hypertension Program from January 2000 through August 2012 were included in a prospective PAH registry after informed consent. The diagnosis of SSc was based on specific criteria,12 and all diagnoses were confirmed by rheumatologists with expertise in SSc (LKH and FMW). Date of onset of systemic sclerosis was defined as the date of first non-Raynaud symptom attributable to SSc. Date of onset of PAH was defined as the date of the diagnostic RHC. Racial classification was based on self-report.
A total of 251 patients with SSc referred to the Pulmonary Hypertension Clinic for suspected pulmonary hypertension underwent RHC. A diagnosis of PAH (defined as a resting mean pulmonary artery pressure [mPAP] ≥ 25 mm Hg and pulmonary capillary wedge pressure [PCWP] ≤ 15 mm Hg)1 was confirmed in 177 patients (Figure 1). Patients with other potential causes of pulmonary hypertension, including significant chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD), or severe sleep apnea syndrome, were excluded. Significant COPD was defined as a forced expiratory volume in 1 second (FEV1) to forced vital capacity (FVC) ratio <70% and a FEV1 <60% of predicted. ILD was defined by a total lung capacity (TLC) <60% or TLC of 60%–70% with the presence of significant radiologic abnormalities.12,24 Patients were also excluded if they had been previously treated with active drugs for PAH. The final study population included 160 patients with SSc-PAH (see Figure 1). The cohort included 29 AA and 131 EA patients (98% white and 2% Hispanic, considered together within the group of EA for simplicity).
FIGURE 1.
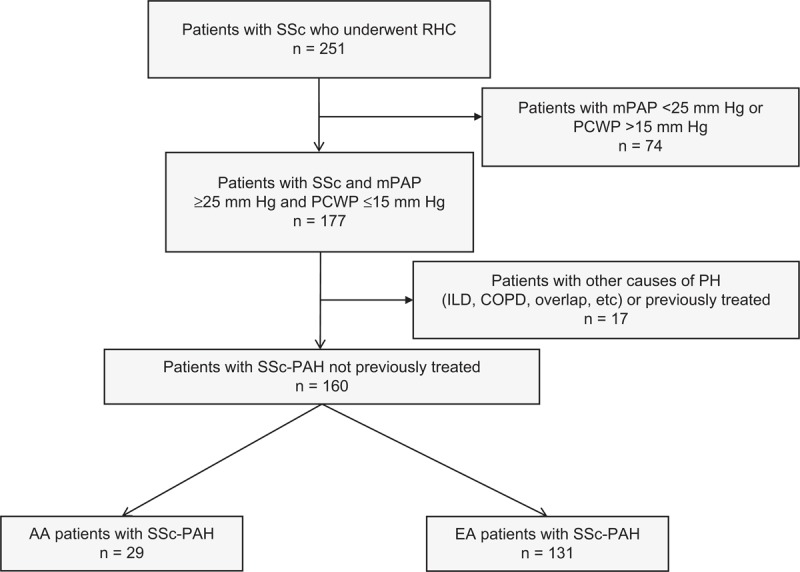
Flow chart of the study population. PH = pulmonary hypertension; WHO = World Health Organization.
RHC was performed as previously described,4 and standard measurements were obtained, including right atrial pressure (RAP), mPAP, cardiac index, PCWP, and PVR. Other hemodynamic measurements included stroke volume index (SVI), pulmonary arterial capacitance (SV/PP) calculated from stroke volume (SV) divided by the pulmonary arterial pulse pressure (PP), and right ventricle stroke work index (RVSWI) as calculated by SVI × (mPAP − RAP) × 0.0136.16
Echocardiograms performed as previously reported11 within 3 months of index RHC, serum autoantibodies, pulmonary function test results, computerized tomography, and WHO-FC were all obtained from the registry records. Renal function was assessed by the estimated glomerular filtration rate (eGFR) using the Modified Diet in Renal Disease equation.23 Renal dysfunction was defined as eGFR <60 mL/min/1.73 m2. Systemic hypertension was defined as average systolic blood pressure ≥ 140 mm Hg and/or diastolic BP ≥ 90 mm Hg at 2 or more visits, documented history of systemic hypertension by the patient’s primary care provider, or a history of anti-hypertensive medication use that was not described in any provider documentation as treatment for Raynaud.
Survival time was measured starting from the date of diagnosis of PAH. Death was determined from clinic and hospital records as well as from the Social Security Death Index. All causes of death were considered for survival analysis.
Statistical Analysis
For continuous variables, mean and standard deviation values were computed and group comparisons were made using the Student t test or Wilcoxon rank test as well as linear regression models where appropriate. For categorical variables, group comparisons were made using the chi square test or Fisher exact test as well as logistic regression models where appropriate. Survival analysis was performed using the Kaplan-Meier method. Comparisons between groups were made using the log-rank test. Univariable Cox and multivariable proportional hazards modeling was performed to investigate the relationship between race and outcome. All computations were performed using SPSS v. 15 (Chicago, IL) statistical software. A p value of < 0.05 was considered statistically significant.
RESULTS
Study Population
As described in Methods, a total of 160 patients with SSc-PAH were considered in the final analysis. Baseline characteristics of the study population are shown in Table 1.
TABLE 1.
Characteristics of the Study Population
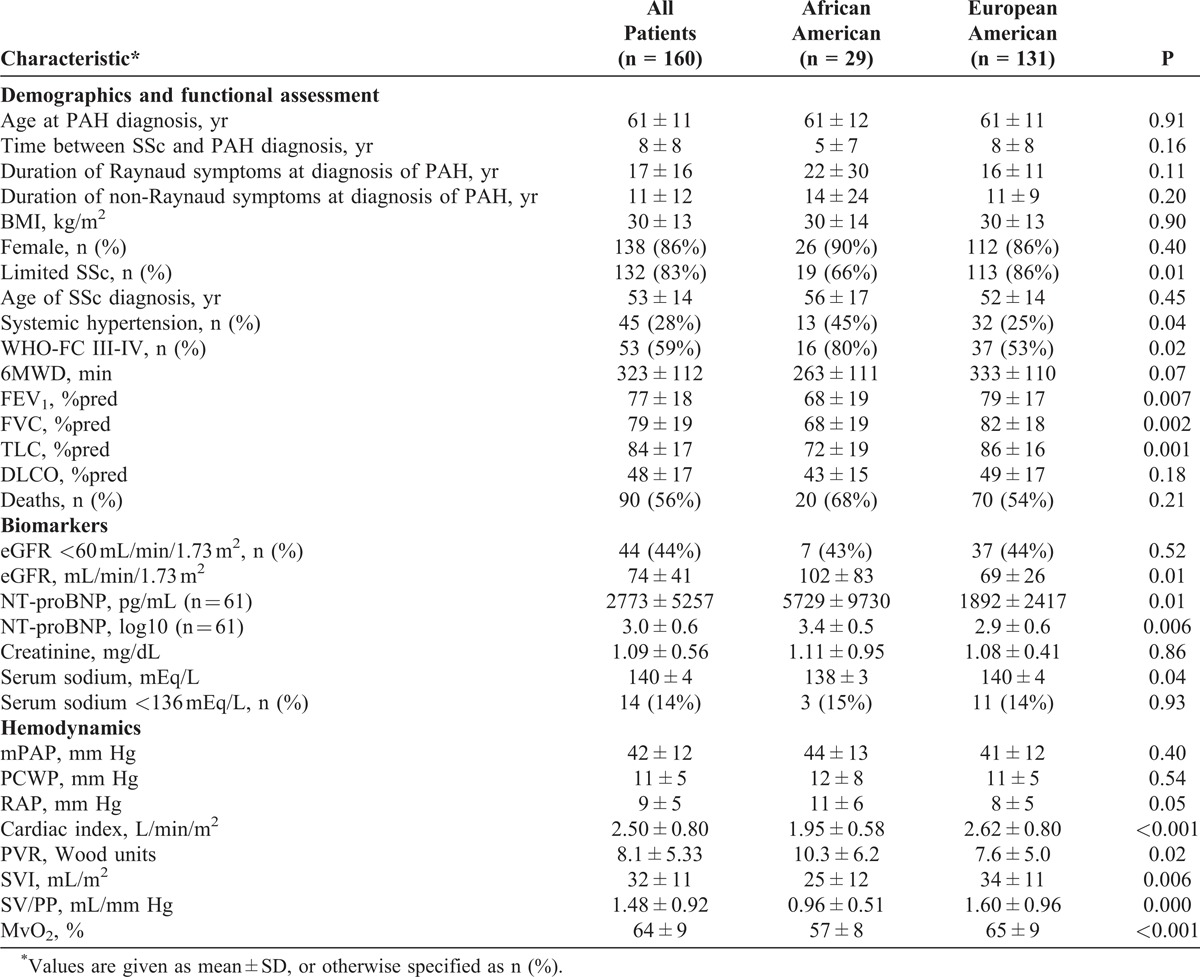
Most patients were white (82%), female (86%), and had limited SSc (83%). Mean ages in both groups were similar at SSc diagnosis and at PAH diagnosis. The mean duration of SSc before PAH diagnosis trended toward being shorter in the AA group (5 ± 7 yr for AA and 8 ± 8 yr for EA; p = 0.16). Onset of Raynaud phenomenon preceded PAH diagnosis by a median time of 18 years (range, 0–49 yr; no difference between groups). In 15 patients, the diagnoses of SSc and PAH were established in the same year. There were no significant differences in antibody profiles between the 2 groups (not shown). Although the limited form of SSc was the most prevalent presentation among both groups (83% overall), this trend was more pronounced in the EA group with diffuse SSc being significantly more common in the AA group compared to the EA group (44% vs 14%, respectively; p = 0.01).
On pulmonary function testing, patients showed moderate-to-severe reduction of diffusing capacity for carbon monoxide (DLCO) with no difference between the 2 groups. However, forced expiratory volume in the first second (FEV1), forced vital capacity (FVC) and total lung capacity (TLC) were significantly lower in the AA group (see Table 1).
At PAH diagnosis, AA patients presented with worse functional status; 80% of the AA group were in WHO-FC III-IV as compared to 53 EA patients (see Table 1). No notable differences in PAH symptomatology and signs were observed between the 2 groups (not shown). Systemic hypertension data were missing in 108 subjects. Out of the remaining 52, only 7 were AA patients, and thus an adequate comparison of prevalence by race was not possible. Angiotensin-converting enzyme inhibitors were used in both groups of patients with the same frequency: 3 of 29 AA (10%) and 17 of 131 EA (13%) (not significant). There were no other differences in comorbidities such as hyper- or hypothyroidism, obstructive sleep apnea syndrome, renal disease, diabetes mellitus, deep vein thrombosis, arrhythmia, asthma, COPD, or coronary artery disease.
Exercise Tolerance
Patients participating in the study showed moderate-to-severe impairment in exercise capacity as assessed by the 6-minute walk distance (6MWD) (323 ± 112 m). AA patients tended to walk relatively shorter distances (263 ± 111 m vs 333 ± 110 m; p = 0.07).
Serum Biomarkers and Renal Function
NT-pro-BNP levels were available in only 61 patients since this measurement was not routinely obtained at the time of initiation of the registry. Mean values were significantly higher in AA patients (5729 ± 9730 pg/mL [n = 14] in AA vs 1892 ± 2417 pg/mL in EA [n = 47]; p = 0.01), and persisted after log-transforming these values (3.4 ± 0.5 in AA vs 2.9 ± 0.6 in EA, p = 0.006).
Serum creatinine, sodium, and BUN levels obtained within 3 months of RHC were available for all patients. Serum creatinine (1.11 ± 0.95 mg/dL vs 1.08 ± 0.41 mg/dL; p = 0.86) and BUN (21 ± 10 mg/dL vs 22 ± 10 mg/dL; p = 0.86) were similar for AA compared with EA patients, respectively. Mean serum sodium levels were relatively lower (138 ± 3 mEq/L vs 140 ± 4 mEq/L; p = 0.04), but no differences were found in the proportions of patients with Na <136 mEq/L (15% vs 14%). Renal dysfunction, defined as eGFR <60 mL/min/1.73 m2, was found in 44 patients (no difference between groups).
Echocardiographic Studies
Results from baseline 2D-Doppler echocardiography are shown in Table 2. The majority of patients had evidence of right ventricle (RV) dysfunction; 76 patients (75%) had RV dilation, and 45 (45%) had a pericardial effusion. The maximal tricuspid regurgitation velocity was higher in AA patients (4.2 ± 0.7 m/s vs 3.7 ± 0.9 m/s; p = 0.05). RV function assessed by tricuspid annular plane systolic excursion (TAPSE) measurement revealed significantly lower values in AA patients (12.6 ± 3.5 mm vs 16.9 ± 5.4 mm; p < 0.01). However, TAPSE did not differ between groups when controlling for PVR, SV/PP, or cardiac index in multivariable linear regression models.
TABLE 2.
Echocardiographic Findings of the Study Population
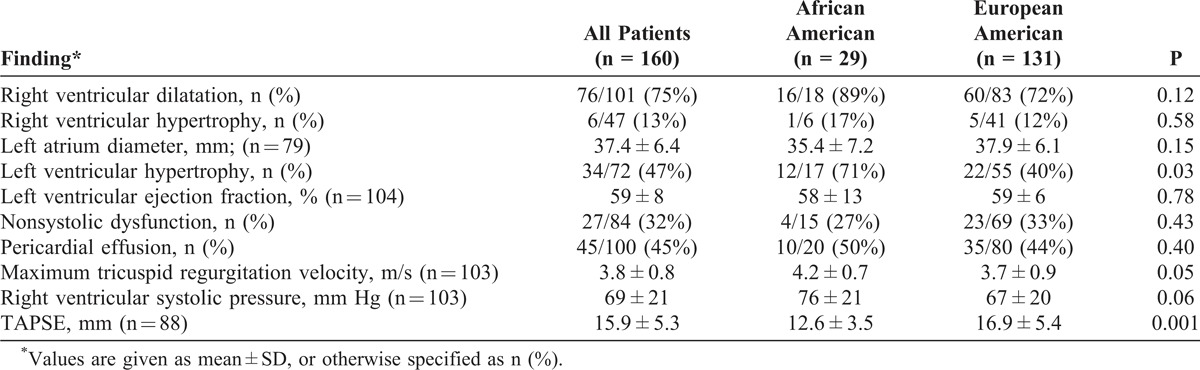
Left ventricular abnormalities were noted commonly. Twenty-seven percent of AA and 33% of EA patients had evidence of nonsystolic dysfunction of the left ventricle, similar to prior estimates in SSc populations.12,17 The mean estimated left ventricular ejection fraction was 58% ± 13% in AA and 59% ± 6% in EA patients (difference was not significant). Left ventricular hypertrophy was present in 71% of AA vs 40% of EA patients (p = 0.03), suggestive of a higher prevalence of systemic hypertension in the former group.
Hemodynamics
Hemodynamic measurements indicated moderate-to-severe PAH in both groups (mean mPAP, 44 ± 13 mm Hg and 41 ± 12 mm Hg among AA and EA patients, respectively [not significant]). Mean RAP was higher (11 ± 6 mm Hg vs 8 ± 5 mm Hg; p = 0.05), cardiac index was lower (1.95 ± 0.58 L/min/m2 vs 2.62 ± 0.80 L/min/m2; p < 0.01), PVR was higher (10.3 ± 6.2 Wood units vs 7.6 ± 5.0 Wood units; p = 0.02), and SVI, SV/PP, and RVSWI were lower (25 ± 12 mL/m2 vs 34 ± 11 mL/m2; 0.96 ± 0.51 mL/mm Hg vs 1.60 ± 0.96 mL/mm Hg; and 10.4 ± 4.4 g-m/m2 vs 14.1 ± 5.3 g-m/m2, respectively; p < 0.01) among AA patients compared with EA patients, indicating a significantly worse hemodynamic profile in the AA group.
Survival
All patients received similar PAH-specific treatments based on clinical guidelines and our Pulmonary Hypertension program’s recommended treatment algorithm.25 No differences were found in choices of initial therapy or anticoagulation treatment. Survival at 1, 2, and 5 years from PAH diagnosis was 83%, 62%, and 26% for AA patients vs 88%, 73%, and 44% for EA patients, respectively. Despite a consistent trend toward lower survival in AA patients, this was not statistically significant (p = 0.21) (Table 3, Figure 2). Overall, the median survival of the cohort was 4.0 years (95% confidence interval [CI], 3.7–4.4). AA patients had a median survival of 3.2 years (95% CI, 1.6–4.8) vs 4.2 years (95% CI, 3.2–5.3) for EA patients.
TABLE 3.
Survival Rates

FIGURE 2.
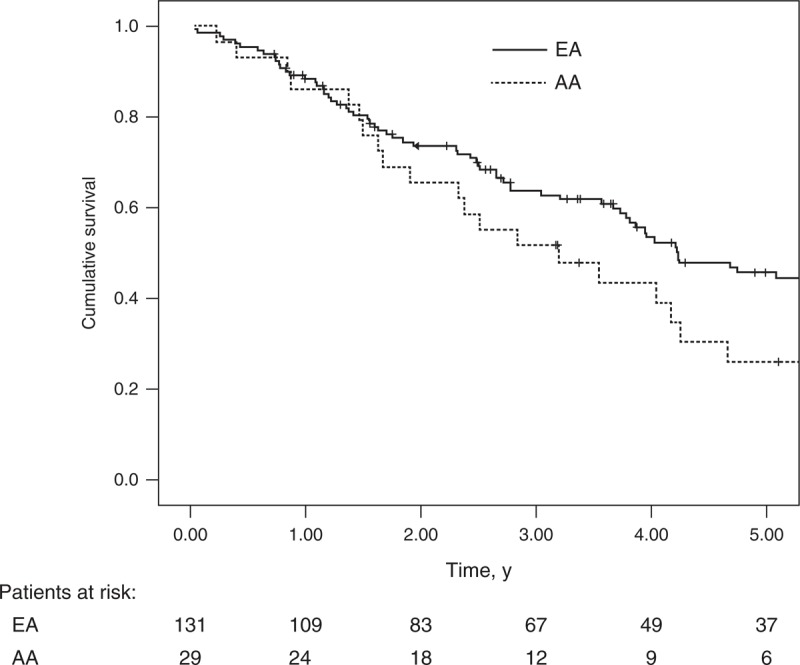
Survival curves according to racial group, African American and European American.
Univariable Cox proportional hazards model did not reveal significant association between race and outcomes. Multivariable models, adjusting for level of education (used as a marker of socioeconomic status), WHO-FC, hemodynamics, 6MWD, NT-pro-BNP levels, echocardiographic measures of RV function, systemic hypertension, and renal function yielded the same result. When examining the AA cohort alone, cardiac index predicted survival in univariable models (hazard ratio [HR], 0.43; 95% CI, 0.19–1.0; p = 0.05). However, other parameters previously demonstrated to be associated with outcomes in SSc-PAH such as RAP, SVI, and PVR were not found to be predictive.4
DISCUSSION
In the current study, we investigated the relationship between race and outcome in a large cohort of SSc-PAH patients followed at a single center. The results indicate that for both groups of patients mean ages at SSc and PAH diagnoses were similar. On the other hand, clinical characteristics such as SSc type and markers of disease severity including WHO-FC, biomarkers, RV function by echocardiography, and hemodynamics were significantly worse among AA patients. A trend toward worse survival, concordant with the less favorable hemodynamic status at diagnosis, was also observed.
Race may influence disease manifestation, severity, and response to therapy, particularly in cardiovascular diseases. Prior studies have shown racial differences in the risk of development of systemic hypertension,32 risk of fatal coronary heart disease,31 and in response to therapies for left heart failure.6 As demonstrated recently in the MESA-Right Ventricle Study, racial disparities in RV function may exist in persons without clinical evidence of cardiac disease; AA have lower RV mass and lower RV ejection fraction compared to age- and sex-matched EA.20 Differences in clinical and hemodynamic characteristics by race have also been reported in idiopathic PAH.8,21 On the other hand, race was not found to be associated with risk of death in the REVEAL Registry,3 which is the largest North American registry of PAH. However, this registry included Group 1 PAH of various etiologies, which makes it difficult to extrapolate the results to SSc-PAH. Beall et al2 have previously reported racial disparity in baseline clinical characteristics as well as progression of disease among SSc-PAH patients. To our knowledge, the current study is the first study performed at a pulmonary hypertension referral center to evaluate the association of race with hemodynamics and outcomes in a large cohort of rigorously phenotyped SSc-PAH patients who were all diagnosed by RHC.
Demographics and Functional Capacity
Epidemiologic studies have demonstrated that AA patients with SSc are younger at disease onset, are diagnosed at an earlier age, and more frequently have diffuse systemic sclerosis compared with patients of other races.27,30 However, the literature on whether the relationship between age at diagnosis of SSc and risk of development of PAH is modified by race is limited. One small study by Beall and colleagues2 reported significant differences in the age at PAH diagnosis, with AA patients developing PAH on average nearly 20 years earlier than their EA counterparts. However, duration of SSc prior to PAH diagnosis did not differ between groups. In a larger study, Schachna and colleagues33 showed age at SSc diagnosis to be a significant risk factor for development of PAH with a greater than 20% increased risk for every decade of age at SSc onset. In this study, despite the fact that the frequency of PAH was higher in the AA population, there was no modification of the direct relationship between age and risk of development of PAH based on race--that is, AA patients did not develop PAH at a younger age than other patients. Duration of SSc did not modify this association, either.
Although our study’s findings persisted when using various definitions of disease onset in SSc (age at first Raynaud symptom, age at first non-Raynaud symptom, and date of initial physician diagnosis of SSc), one cannot entirely exclude the possibility that the non-statistically significant trends toward longer duration of SSc symptoms among AA patients reflected truly delayed diagnosis in this group, which the study was too underpowered to uncover. In short, while the proportion of patients with diffuse disease was significantly higher in AA than EA patients (44% vs 14%, p = 0.01), there were no significant differences in age at disease onset, age at diagnosis of PAH, or duration of SSc prior to diagnosis of PAH. Additionally, patients in our cohort underwent the same screening algorithm for detection of pulmonary hypertension and were referred for definitive diagnosis of PAH (that is, RHC) based on this screening. Thus, it is unlikely that either lead-time bias or ascertainment bias for PAH contributed to the observed differences in disease severity between groups in this study.
Notably, AA patients tended to present with more significant functional impairment; 80% were in WHO-FC III/IV compared to 53% of EA patients. Similarly, AA patients showed a tendency to walk shorter distances on the 6MWT, again suggesting worse functional status. Although the 6MWT distance correlates with WHO-FC and hemodynamics and has prognostic value in IPAH, its relevance in SSc-PAH has been questioned.34 However, it is noteworthy that race may independently impact 6MWD as suggested by a recent study demonstrating decreased walk distances in African Brazilian patients with SSc-PAH compared to white patients with SSc-PAH.36
Biomarkers for PAH
Baseline serum levels of NT-pro-BNP have been shown to correlate with hemodynamics and exercise capacity22 and are independent predictors of mortality in PAH.28 We have previously shown that NT-pro-BNP levels are significantly higher in SSc-PAH compared to IPAH,24 despite comparatively less severe hemodynamic derangement, and are strong predictors of survival in this population. In our cohort, AA patients presented with higher serum NT-pro-BNP levels than EA patients, which was consistent with the worse hemodynamics in this group. Previous studies have shown hyponatremia to be a major determinant of survival in PAH13; however, we found no significant differences in the prevalence of hyponatremia between AA and EA patients.13
Echocardiography
While echocardiographic evidence of RV dysfunction was common, there were no differences in RV chamber dimensions between the 2 groups. Further, while pericardial effusion was noted frequently in the cohort (45%) in agreement with prior studies,5,12 its prevalence did not differ between groups. However, other measures of RV function were associated with race. Maximum tricuspid regurgitation velocity was significantly higher among AA patients. Similarly, TAPSE, a measure of global RV function, differed between groups, with AA patients having significantly lower values, consistent with the worse hemodynamics (higher RAP, lower cardiac index, and higher PVR) in AA patients at diagnosis.
AA patients showed a higher prevalence of left ventricular hypertrophy by echocardiography, probably as a response to long-standing systemic hypertension. Unfortunately, our limited data on the prevalence of systemic hypertension in this study cohort prevented us from studying the relationship between systemic hypertension and race. Indeed, systemic hypertension has been previously shown to have a higher prevalence among the general AA population.10 However, there was no difference in the prevalence of nonsystolic dysfunction between the 2 groups. Both LVH and nonsystolic dysfunction have been frequently found in SSc patients in general.9
Hemodynamics
Overall, our cohort had moderate-to-severe PAH. However, AA patients had a significantly worse hemodynamic profile at diagnosis as indicated by a lower cardiac index; higher RAP and PVR; and decreased SVI, SV/PP, and RVSWI compared to EA patients. While clinical parameters of disease severity, such as WHO-FC, 6MWD, and NT-pro-BNP, were similarly associated with AA race, the reasons for these differences remain unclear. Lead-time bias is unlikely but not definitively excluded, given that there were no significant differences between AA and EA patients in the duration of SSc-specific symptoms prior to diagnosis of PAH. Intrinsic differences in RV function between AA and EA patients have been recently reported.20 In a large multiracial cohort of more than 4000 patients without cardiovascular disease, Kawut et al20 reported significant differences in RV mass and RV ejection fraction as assessed by cardiac MRI, showing lower mass and ejection fraction in AA patients. These baseline differences in RV function may influence response to pulmonary vascular disease and could potentially explain differences in hemodynamic parameters noted in this study, although our findings indicate that, among AA patients, RV function was essentially altered in the setting of an excess RV afterload (compared to EA patients) at the time of presentation. Whether the combination of other cardiac factors, such as left ventricular hypertrophy for instance, affects outcome, remains unclear.
Survival
Kaplan-Meier survival estimates for the overall cohort were 87%, 71%, and 40% at 1, 2, and 5 years, respectively, with a median survival of 4 years. These findings are consistent with previously reported survival in SSc-PAH.4,7,18 The median survival was 3.2 years for AA and 4.2 for EA patients (p = 0.21). Although there was no significant difference in this outcome between the 2 groups, there was a consistent trend toward lower survival in AA patients throughout the 5 years, suggesting that a better-powered study may possibly uncover a statistically significant difference. Adjustment for various confounders including socioeconomic status (using level of education as a marker) did not significantly affect this relationship, which is unsurprising given the similar demographic profile of both groups such as similar ages at presentation and similar sex ratios. We note that the sole use of "education" as a marker of socioeconomic status is inadequate to exclude the latter as a confounder definitively. It is interesting that our findings parallel survival differences noted in epidemiologic studies of SSc patients in general; AA patients demonstrate a trend toward poorer survival (at least early in disease) and additionally, patients with diffuse SSc (which formed a bigger proportion of AA patients) may have poorer survival than patients with limited SSc. It is noteworthy that while our study does not address the specific prevalence of PAH in AA patients with SSc, it demonstrates that when present in AA patients, PAH is significantly worse and possibly associated with poorer survival.
The reasons for this trend toward poorer survival remain unclear. Recent studies in left heart failure have identified potential racial differences in heart failure outcomes and response to therapy. Impairment in nitric oxide homeostasis and nitric oxide-mediated cardiovascular effects in AA compared to EA patients may form the pathophysiologic basis for these differences.6 Similarly, within PAH, response to therapy may also differ by race. A recent analysis of patient-level data from 6 randomized placebo-controlled trials of endothelin receptor antagonist (ERA) therapies for the treatment of PAH suggests racial and sex differences in drug efficacy.14 In their analysis, Gabler and colleagues found a strong trend toward poorer response among AA as assessed by change in 6MWD over 12 weeks. While the mechanisms of this race-specific response to therapy remain to be elucidated, this could explain the trend in lower survival among AA patients in our cohort, at least in part. Other factors, including but not limited to socioeconomic differences and the remote possibility of differentially delayed diagnosis (considering the weak trend toward longer duration of SSc-specific symptoms prior to PAH diagnosis among AA patients), cannot be completely ruled out.
There are several limitations to the current study, including the fact that this is an observational study. Although subjects were mostly followed prospectively subsequent to enrollment in our registry, some data were collected retrospectively, resulting in occasional missing values. Despite the exclusion of all patients with clinically apparent ILD at the time of PAH diagnosis (because the pathogenesis, hemodynamics, and outcomes of pulmonary hypertension for these patients are quite different from those for patients with SSc-PAH26), we cannot exclude subclinical ILD and its potential effect on outcomes. Sociodemographic characteristics other than level of education were not recorded, which would have helped us carry out a more robust assessment of whether socioeconomic status was a confounding variable. There are disparities in progression and outcome that need to be further investigated to uncover the underlying social and/or biological factors. Another limitation is that we do not have data on echocardiographic findings, autoantibodies, and other parameters for the entire population; these data are available for only 50%–70% of the population; therefore, a certain bias related to these findings in our study cannot be excluded. Finally, our survival data report all-cause mortality as opposed to disease-specific mortality. When comparing this measure between 2 racial groups, the differences in their all-cause mortality rates for the age range in question at the population level cannot be ignored. Indeed, according to 2010 data from the National Center for Health Statistics (Atlanta, GA), black adults had a significantly higher hazard ratio for all-cause mortality than white adults (depending on the statistical model used, the hazard ratio varied from 1.11 to 1.36). 19
In conclusion, we have shown that AA patients with SSc-PAH present with more severe disease compared to EA patients, as assessed by functional status, biomarkers, echocardiography, and hemodynamics. Additionally, although not reaching statistical significance in this study, their survival appears to be worse. These findings suggest that race may modify the response to pulmonary vascular disease in patients with SSc-associated PAH, with possible pathophysiologic differences between groups. Further studies are needed to confirm these observations, identify mechanisms involved in these vascular responses, and definitively investigate the possibility of differentially delayed diagnosis (such as due to access of care issues) among AA patients.
Footnotes
Abbreviations: 6MWD = 6-minute walk distance, AA = African Americans, ACA = anticentromere autoantibody, ANA = antinucleolar autoantibody, CI = confidence interval, COPD = chronic obstructive pulmonary disease, EA = European Americans, eGFR = estimated glomerular filtration rate, FEV1 = FEV1 forced expiratory volume in 1 second, FVC = forced vital capacity, HR = hazard ratio, ILD = interstitial lung disease, IPAH = idiopathic pulmonary arterial hypertension, mPAP = mean pulmonary artery pressure, PAH = pulmonary arterial hypertension, PCWP = pulmonary capillary wedge pressure, Pro-BNP = NT-pro-brain natriuretic peptide, PVR = pulmonary vascular resistance, RAP = right atrial pressure, RHC = right heart catheterization, RV = right ventricle, SSc = systemic sclerosis, SSc-PAH = systemic sclerosis associated with pulmonary arterial hypertension, SV = stroke volume, SV/PP = pulmonary arterial capacitance, SVI = stroke volume index, TAPSE = tricuspid annular systolic plane excursion, TLC = total lung capacity, WHO-FC = World Health Organization functional class.
Financial support and conflicts of interest: Supported by Grants NHLBI P50 HL084946 (PMH); K23 HL092287 (SCM); and the Societat Catalana de Pneumologia (SOCAP), Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), University of Barcelona, and Fundación Alfonso Martín Escudero (IB). The authors have no conflicts of interest to disclose.
References
- 1.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009; 54:S55–S66. [DOI] [PubMed] [Google Scholar]
- 2.Beall AD, Nietert PJ, Taylor MH, Mitchell HC, Shaftman SR, Silver RM, Smith EA, Bolster MB. Ethnic disparities among patients with pulmonary hypertension associated with systemic sclerosis. J Rheumatol. 2007; 34:1277–1282. [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010; 122:164–172. [DOI] [PubMed] [Google Scholar]
- 4.Campo A, Mathai SC, Le PJ, Zaiman AL, Hummers LK, Boyce D, Housten T, Champion HC, Lechtzin N, Wigley FM, Girgis RE, Hassoun PM. Hemodynamic predictors of survival in scleroderma-related pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010; 182:252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, Miller DP, Nicolls MR, Zamanian RT. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest. 2010; 138:1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole RT, Kalogeropoulos AP, Georgiopoulou VV, Gheorghiade M, Quyyumi A, Yancy C, Butler J. Hydralazine and isosorbide dinitrate in heart failure: historical perspective, mechanisms, and future directions. Circulation. 2011; 123:2414–2422. [DOI] [PubMed] [Google Scholar]
- 7.Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, Das C, Elliot CA, Johnson M, DeSoyza J, Torpy C, Goldsmith K, Hodgkins D, Hughes RJ, Pepke-Zaba J, Coghlan JG. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009; 179:151–157. [DOI] [PubMed] [Google Scholar]
- 8.Davis KK, Lilienfeld DE, Doyle RL. Increased mortality in African Americans with idiopathic pulmonary arterial hypertension. J Natl Med Assoc. 2008; 100:69–72. [DOI] [PubMed] [Google Scholar]
- 9.de Groote P, Gressin V, Hachulla E, Carpentier P, Guillevin L, Kahan A, Cabane J, Frances C, Lamblin N, Diot E, Patat F, Sibilia J, Petit H, Cracowski JL, Clerson P, Humbert M. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008; 67:31–36. [DOI] [PubMed] [Google Scholar]
- 10.Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension. 2004; 44:398–404. [DOI] [PubMed] [Google Scholar]
- 11.Fisher MR, Forfia PR, Chamera E, Housten-Harris T, Champion HC, Girgis RE, Corretti MC, Hassoun PM. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am J Respir Crit Care Med. 2009; 179:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten-Harris T, Hummers L, Krishnan JA, Wigley F, Hassoun PM. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006; 54:3043–3050. [DOI] [PubMed] [Google Scholar]
- 13.Forfia PR, Mathai SC, Fisher MR, Housten-Harris T, Hemnes AR, Champion HC, Girgis RE, Hassoun PM. Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008; 177:1364–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabler NB, French B, Strom BL, Liu Z, Palevsky HI, Taichman DB, Kawut SM, Halpern SD. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest. 2012; 141:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greidinger EL, Flaherty KT, White B, Rosen A, Wigley FM, Wise RA. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest. 1998; 114:801–807. [DOI] [PubMed] [Google Scholar]
- 16.Her C. Right ventricular stroke-work. An index of distribution of pulmonary perfusion in acute respiratory failure. Chest. 1983; 84:719–724. [DOI] [PubMed] [Google Scholar]
- 17.Hinchcliff M, Desai CS, Varga J, Shah SJ. Prevalence Prognosis, and factors associated with left ventricular diastolic dysfunction in systemic sclerosis. Clin Exp Rheumatol. 2012; 30:S30–S37. [PMC free article] [PubMed] [Google Scholar]
- 18.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010; 36:549–555. [DOI] [PubMed] [Google Scholar]
- 19.Hummer RA, Chinn JJ. Race/ethnicity and U. S. adult mortality: progress, prospects, and new analyses. Du Bois Rev. 2011; 8:5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawut SM, Barr RG, Lima JA, Praestgaard A, Johnson WC, Chahal H, Ogunyankin KO, Bristow MR, Kizer JR, Tandri H. Bluemke DA. Right ventricular structure is associated with the risk of heart failure and cardiovascular death: the Multi-Ethnic Study of Atherosclerosis (MESA)-Right Ventricle Study. Circulation. 2012; 126:1681–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawut SM, Horn EM, Berekashvili KK, Garofano RP, Goldsmith RL, Widlitz AC, Rosenzweig EB, Kerstein D, Barst RJ. New predictors of outcome in idiopathic pulmonary arterial hypertension. Am J Cardiol. 2005; 95:199–203. [DOI] [PubMed] [Google Scholar]
- 22.Leuchte HH, Holzapfel M, Baumgartner RA, Ding I, Neurohr C, Vogeser M, Kolbe T, Schwaiblmair M, Behr J. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol. 2004; 43:764–770. [DOI] [PubMed] [Google Scholar]
- 23.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999; 130:461–470. [DOI] [PubMed] [Google Scholar]
- 24.Mathai SC, Bueso M, Hummers LK, Boyce D, Lechtzin N, Le Pavec J, Campo A, Champion HC, Housten T, Forfia PR, Zaiman AL, Wigley FM, Girgis RE, Hassoun PM. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur Respir J. 2010; 35:95–104. [DOI] [PubMed] [Google Scholar]
- 25.Mathai SC, Hassoun PM. Pulmonary arterial hypertension associated with systemic sclerosis. Expert Rev Respir Med. 2011; 5:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathai SC, Hummers LK, Champion HC, Wigley FM, Zaiman A, Hassoun PM, Girgis RE. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis Rheum. 2009; 60:569–577. [DOI] [PubMed] [Google Scholar]
- 27.Mayes MD, Lacey JV, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ. Schottenfeld D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003; 48:2246–2255. [DOI] [PubMed] [Google Scholar]
- 28.Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Kakishita M, Fukushima K, Okano Y, Nakanishi N, Miyatake K, Kangawa K. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000; 102:865–870. [DOI] [PubMed] [Google Scholar]
- 29.Reveille JD. Ethnicity and race and systemic sclerosis: how it affects susceptibility, severity, antibody genetics, and clinical manifestations. Curr Rheumatol Rep. 2003; 5:160–167. [DOI] [PubMed] [Google Scholar]
- 30.Reveille JD, Fischbach M, McNearney T, Friedman AW, Aguilar MB, Lisse J, Fritzler MJ, Ahn C, Arnett FC. Systemic sclerosis in 3 US ethnic groups: a comparison of clinical, sociodemographic, serologic, and immunogenetic determinants. Semin. Arthritis Rheum. 2001; 30:332–346. [DOI] [PubMed] [Google Scholar]
- 31.Safford MM, Brown TM, Muntner PM, Durant RW, Glasser S, Halanych JH, Shikany JM, Prineas RJ, Samdarshi T, Bittner VA, Lewis CE, Gamboa C, Cushman M, Howard V, Howard G. Association of race and sex with risk of incident acute coronary heart disease events. JAMA. 2012; 308:1768–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders E. Managing hypertension in African-American patients. J Clin Hypertens (Greenwich. 2004; 6:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schachna L, Wigley FM, Chang B, White B, Wise RA, Gelber AC. Age and risk of pulmonary arterial hypertension in scleroderma. Chest. 2003; 124:2098–2104. [DOI] [PubMed] [Google Scholar]
- 34.Schoindre Y, Meune C, nh-Xuan AT, Avouac J, Kahan A, Allanore Y. Lack of specificity of the 6-minute walk test as an outcome measure for patients with systemic sclerosis. J Rheumatol. 2009; 36:1481–1485. [DOI] [PubMed] [Google Scholar]
- 35.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing ZC, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009; 54:S43–S54. [DOI] [PubMed] [Google Scholar]
- 36.Villalba WO, Sampaio-Barros PD, Pereira MC, Cerqueira EM, Leme CA, Marques-Neto JF, Paschoal IA. Six-minute walk test for the evaluation of pulmonary disease severity in scleroderma patients. Chest. 2007; 131:217–222. [DOI] [PubMed] [Google Scholar]