

Abstract
Background
Two-component systems (TCSs) are abundant prokaryotic signaling pathways, whose evolution is of particular importance because of their role in bacterial pathogenicity. Comparative genomics can provide important insights into the evolution of these genes, but inferences are dependent on the relatedness of the compared genomes. This study investigated the relationship between evolutionary distance and TCS evolution in myxobacterial genomes, of which there are several sequenced examples, of varying relatedness, and which encode large numbers of TCSs.
Methods
Myxobacterial TCS gene sets were compared, orthologues defined, and changes in TCS properties such as gene organisation, domain architecture and size identified.
Results
Genome relatedness/evolutionary distance was found to have a large effect on the apparent frequency of evolutionary events affecting TCS genes, but not on the relative dominance of different types of mutations. Large (≥1 gene) indels were the most common changes, often giving rise to gene organisation changes. Smaller indels were also common, sometimes changing domain architecture, and/or leading to pseudogene formation. Individuality of myxobacterial TCS gene sets seems primarily due to lineage specific gene loss. However, there is also evidence of extensive acquisition of genes by lateral transfer, with gene duplication also creating new TCS genes.
Conclusions
This study provides catalogues of myxobacterial TCS gene sets and their orthology relationships, benchmarked against genome relatedness. It also provides insights into the relationship between evolutionary distance and the inference of TCS estudies of TCS evolution beyond the myxobacteriavolution, which may be important for studies of TCS evolutiThe online version of this articleon beyond the myxobacteria.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2018-y) contains supplementary material, which is available to authorized users.
Keywords: Myxobacteria, Myxococcus xanthus, Histidine kinase, Response regulator, Mutation, Comparative genomics
Background
Two-component systems (TCSs) are the dominant phosphorylation based signalling pathways in prokaryotes. The average eubacterial genome encodes ~50 TCS proteins [1, 2], and they regulate diverse and important behaviours, including pathComparative genomics and experimentalogenesis, cell cycle progression, sporulation and drug resistance [3]. Typical TCSs consist of two proteins – a histidine kinase (HK), which can sense an environmental parameter through an ‘input’ domain, and a response regulator (RR), which poComparative genomics and experimentalssesses an effector function (usually DNA-binding) via an ‘output’ domain. In response to an environmental signal the HK autophosphorylates within its ‘transmitter’ domain and the phosphoryl group is then transferred onto the ‘receiver’ domain of the partner RR, modulating its output domain effector activity. Common modifications to the basic scheme include hybrid kinases – proteins which contain both transmitter and receiver domains, and phosphotransfer proteins (PPs) - which can shuttle phosphoryl groups between receiver domains forming a phosphorelay. For comprehensive reviews of TCS biology, refer to the recent book by Gross and Beier [4] and the special issue of Current Opinion in Microbiology edited by Bourret and Silversmith [5].
Comparative genomics and experimental approaches have shed light on the general principles of TCS evolution [2, 6, 7]. For instance TCSs have evolved primarily by duplication and divergence of ancestral systems, with the contemporary diversity of TCS architecture generated by domain shuffling. This is important, as an understanding of natural variation due to evolution can inform the rational engineering of TCSs for biotechnological and research applications [8]. Nevertheless there are important questions left unanswered regarding TCS evolution and most studies have been forced to make several assumptions in their analyses. For instance, inferences regarding TCS evolution have been based on the comparison of various extant genomes. Common approaches are to use all available genomes, or to use multiple organisms from a single taxonomic unit. In both cases this typically results in comparisons between genomes which exhibit different relatedness to one another. Thus relatively distant and close genomes are considered equally, potentially distorting the conclusions drawn. This is particularly problematic for studies of TCS evolution, as it is thought that different evolutionary processes work at different timescales. For example, shuffling of HK input domains seems to occur rapidly post gene duplication, and gene gain/loss apparently dominates over gene fusion/fission at long timescales [9, 10]. In addition, studies are also often limited in power as there is sequencing bias within/between taxa, most organisms contain just dozens of TCS proteins, and at most taxonomic levels there is a relative shortfall in the number of available genomes, all of which can distort conclusions [1, 11–13].
Myxobacteria (order Myxococcales) are well known for their large numbers of TCSs, many of which have been characterised experimentally [14–16]. They exhibit complex developmental and predatory behaviours and are of ongoing importance to the biotechnology industry [17–20]. A major focus of myxobacterial research has been elucidation of the molecular mechanisms underpinning multicellular development, which are surprisingly plastic [21], and involve large numbers of TCS proteins [22].
At the time of analysis, genome sequences were available for 12 myxobacteria (Table 1). For ease of reference, genomes are abbreviated in the remaining text as shown in Table 1. Two of these genome sequences (Pp and Ca) are incomplete, but were included in the study anyway, as they were nearly complete, being composed of relatively small numbers of contigs (Table 1). Bdellovibrio bacteriovorus HD100 (Bb) was also included in the study as the bdellovibrionales are the closest known relatives of the myxobacteria.
Table 1.
Order Myxococcales – genome properties, taxonomy and ecology
| Organism | Size (Mbp) | Contigs | Proteins | %GC | Sub-order | Family | Fruits | Environment | Sequencing centre | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Myxococcus xanthus DK1622 (Mx) | 9.1 | 1 | 7316 | 68.9 | Cystobacterineae | Myxococcaceae | Yes | Aerobic, terrestrial mesophile | The Institute for Genomic Research | [52] |
| Myxococcus fulvus HW-1 (Mf) | 9.0 | 2a | 7284 | 70.6 | Cystobacterineae | Myxococcaceae | Yes | Aerobic, aquatic mesophile | Shandong University, China | [26] |
| Corallococcus coralloides DSM 2259 (Cc) | 10.1 | 1 | 8033 | 69.9 | Cystobacterineae | Myxococcaceae | Yes | Aerobic, terrestrial mesophile | Max Planck Institute, Marburg | [53] |
| Stigmatella aurantiaca DW4/3-1 (Sa) | 10.3 | 1 | 8352 | 67.5 | Cystobacterineae | Cystobacteraceae | Yes | Aerobic, terrestrial mesophile | Max Planck Institute, Marburg | [21] |
| Anaeromyxobacter sp. Fw109-5 (AF) | 5.3 | 1 | 4466 | 73.5 | Cystobacterineae | Myxococcaceae | No | Anaerobic, terrestrial mesophile | Department of Energy Joint Genome Institute | ncbi.nlm.nih.gov |
| Anaeromyxobacter sp. K (AK) | 5.1 | 1 | 4457 | 74.8 | Cystobacterineae | Myxococcaceae | No | Anaerobic, terrestrial mesophile | Department of Energy Joint Genome Institute | ncbi.nlm.nih.gov |
| Anaeromyxobacter dehalogenans 2CP-C (AdC) | 5.0 | 1 | 4346 | 74.9 | Cystobacterineae | Myxococcaceae | No | Facultative, terrestrial mesophile | Department of Energy Joint Genome Institute | [54] |
| Anaeromyxobacter dehalogenans 2CP-1 (Ad1) | 5.0 | 1 | 4473 | 74.7 | Cystobacterineae | Myxococcaceae | No | Facultative, terrestrial mesophile | Department of Energy Joint Genome Institute | [55] |
| Sorangium cellulosum So ce 56 (Sc) | 13.0 | 1 | 9374 | 71.4 | Sorangiineae | Polyangiaceae | Yes | Aerobic, terrestrial mesophile | Bielefeld University. | [56] |
| Chondromyces apiculatus DSM 436 (Ca) | 9.4 | 128 | 7633 | 69.2 | Sorangineae | Polyangiaceae | Yes | Aerobic, terrestrial mesophile | Institute of Microbial Technology | ncbi.nlm.nih.gov |
| Haliangium ochraceum DSM 14365 (Ho) | 9.4 | 1 | 6719 | 69.5 | Nannocystineae | Kofleriaceae | Yes | Aerobic, marine mesophile | Department of Energy Joint Genome Institute | [57] |
| Plesiocystis pacifica SIR-1 (Pp) | 10.6 | 238 | 8450 | 70.7 | Nannocystineae | Nannocystaceae | Yes | Aerobic, marine mesophile | J. Craig Venter Institute | [58] |
| Bdellovibrio bacteriovorus HD100 (Bb) | 3.8 | 1 | 3586 | 50.6 | Bdellovibrionaceae | Bdellovibrio | No | Aerobic, multiple habitat mesophile | Max Planck Institute, Tübingen | [59] |
Myxobacterial genomes are generally large, with a characteristically high %GC. Bdellovibrio bacteriovorus HD100 is included as a representative of the bdellovibrios – the closest known relatives of the myxobacteria
aThe M. fulvus genome includes a plasmid, which encodes no TCS proteins
This study sought to investigate how the apparent evolutionary mechanisms affecting myxobacterial TCSs change with timescale and/or phylogenetic distance. The analysis was restricted to myxobacterial genomes, as they possess large numbers of TCSs, and are available across a broad taxonomic range. Thus comparisons could be performed between organisms within the same species, genus, family, sub-order or order, with representative genomes available for all three sub-orders, and every family of the myxobacteria. Secondary aims of the study were to define myxobacterial TCS gene orthology relationships, and to identify core and accessory TCSs.
Results
Relatedness of sequenced genomes
To obtain a measure of the divergence of the sequenced myxobacterial genomes, the 16S rRNA genes of each genome were aligned and distances (substitution frequencies per nucleotide) between each pair of sequences calculated, allowing construction of a phylogenetic tree (see Methods). There was strong bootstrap support for the tree (Additional file 1: Figure S1), which also agreed well with that of Garcia et al., [23]. Distances clustered into four distinct groups, which largely shared particular taxonomic ranks (Fig. 1a). The ‘genus’ group is composed of distances between strains within the same genus (for example Mx vs. Mf, and Ad1 vs. AdC). Comparisons between genomes in the same formally-defined family (see Table 1 for family and sub-order membership) were mainly distributed into two groups of genetic distances, here denoted ‘family’ and ‘sub-order’ in Fig. 1. The final ‘order’ group was dominated by distances between genomes from the same order, but different sub-orders (see the caption for Fig. 1a for details of group membership). A redrawn phylogenetic tree with distances discretised according to the groupings defined above is shown in Fig. 1b.
Fig. 1.
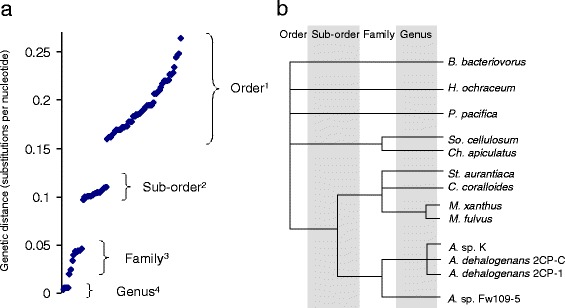
a Correlation between 16S rRNA gene sequence conservation and taxonomic classification. Pairwise genetic distances between Myxococcales 16S rRNA gene sequences (substitutions per nucleotide position), plotted in rank order. Distances cluster into four distinct groups: 1. Ho/Pp vs. Sc/Ca vs. Mx/Mf/Cc/Sa/AK/AF/Ad1/AdC and Ho vs. Pp (comparisons mainly between sub-orders), 2. AK/AF/Ad1/AdC vs. Mx/Mf/Cc/Sa (comparisons within a sub-order – across the extended Myxococcaceae family), 3. Sc vs. Ca, and AK/Ad1/AdC vs. AF, and Mx/Mf vs. Cc vs. Sa (comparisons within families), and 4. AK vs. Ad1 vs. AdC, and Mx vs. Mf (comparisons within a genus). See text for abbreviations. b Phylogenetic tree with distances discretised into four vertical bands as grouped in part A
In several instances, distance-based groupings of genome pairs were not consistent with the formal taxonomy of the myxobacteria. For instance: 1, Bb is only as distant from the Myxococcales as Pp is from Myxococcales of different sub-orders, 2, Ho and Pp should be assigned to separate sub-orders, 3, AF should be in a separate genus to the other Anaeromyxobacter, 4, Sa belongs to the Myxococcaceae family (as opposed to the Cystobacteraceae family as stated by the taxonomy database of the NCBI), and 5, The Anaeromyxobacter should be a separate family from the other Myxococcaceae.
For the rest of this study we have classified inter-genome relationships according to Fig. 1 rather than using the current taxonomic classification. Thus for our purposes AF is considered to be in a different genus (but the same family) to AK, Ad1 and AdC, while Ho and Pp are in separate sub-orders. Ak, AF, Ad1 and AdC are considered to be in the same sub-order, but in a different family from Mf, Mx, Cc and Sa, a suggestion previously made by Garcia et al. [23].
TCS protein sets
For each genome, a list of TCS proteins was inferred as described in the Methods section. Table 2 presents summary statistics for each TCS gene set, alongside myxobacterial mean values. Williams and Whitworth [24] provided a characterisation of the TCS genes from 1405 replicons as a pangenomic yardstick against which to compare TCS gene sets and these are included in Table 2 as bacterial mean values.
Table 2.
Two-component gene sets of the myxobacteria
| Genomea | TCS genes | Non-hybrid HKs | Hybrid HKs | PPs | RRs | % hybrid HKs | % TM HKs | % orphan | % paired | % complex | Intricate foci |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mx | 282 | 103 | 37 | 5 | 137 | 26.4 | 47 | 35 | 35 | 30 | 8 |
| Mf | 288 | 98 | 49 | 3 | 138 | 33.3 | 41 | 34 | 33 | 33 | 9 |
| Cc | 306 | 106 | 56 | 2 | 142 | 34.6 | 38 | 34 | 35 | 31 | 8 |
| Sa | 338 | 124 | 70 | 4 | 142 | 36.1 | 40 | 34 | 34 | 32 | 9 |
| AdC | 188 | 68 | 17 | 1 | 102 | 20.2 | 48 | 43 | 35 | 22 | 2 |
| Ad1 | 187 | 69 | 19 | 1 | 98 | 21.6 | 50 | 40 | 34 | 26 | 2 |
| AF | 207 | 72 | 35 | 0 | 100 | 33.3 | 50 | 28 | 33 | 39 | 5 |
| AK | 191 | 72 | 18 | 0 | 101 | 20.0 | 49 | 41 | 35 | 24 | 2 |
| Sc | 273 | 101 | 50 | 1 | 121 | 33.1 | 41 | 37 | 27 | 37 | 9 |
| Ca | 301 | 109 | 50 | 2 | 140 | 31.4 | 45 | N/A | N/A | N/A | N/A |
| Ho | 191 | 53 | 51 | 3 | 84 | 49.0 | 35 | 35 | 38 | 27 | 5 |
| Pp | 160 | 60 | 26 | 5 | 69 | 30.2 | 42 | N/A | N/A | N/A | N/A |
| Myxobacterial mean: | 242.7 | 86 | 39.8 | 2.3 | 114.5 | 30.8 | 44 | 36 | 34 | 30 | 5.9 |
| Bn | 92 | 32 | 16 | 2 | 42 | 33.3 | 73 | 37 | 48 | 18 | 4 |
| Bacterial mean:b | 31 | 14.8 | 3.3 | 0.6 | 15.6 | 18.2 | 63.5 | 46 | 40 | 14 | 0.3 |
For each genome, the numbers of TCS genes are presented, broken down by type. HKs are characterised according to the percentages that are hybrid proteins and that contain transmembrane helices. Genomic TCS gene organisation is also presented as the percentage of TCS genes which are orphaned, paired or in complex gene clusters. Values are presented for Bdellovibrio bacteriovorus (Bb), and the myxobacterial and bacterial means are also shown for comparison
a Ho = Haliangium ochraceum DSM 14365, Pp = Plesiocystis pacifica SIR-1, Sc = Sorangium cellulosum So ce 56, Ca = Chondromyces apiculatus DSM 436, Mx = Myxococcus xanthus DK1622, Mf = Myxococcus fulvus HW-1, Cc = Corallococcus coralloides DSM 2259, Sa = Stigmatella aurantiaca DW3/4-1, AK = Anaeromyxobacter sp. K, AF = Anaeromyxobacter sp. Fw109-5, Ad1 = Anaeromyxobacter dehalogenans 2CP-1, AdC = Anaeromyxobacter dehalogenans 2CP-C, and Bb = Bdellovibrio bacteriovorus HD100
bCalculated from Williams and Whitworth [24]
Differences between the myxobacterial and bacterial means are apparent for some metrics. Myxobacteria are characterised by large numbers of TCS genes, greater than expected even for organisms with such large genomes. Figure 2a shows the number of TCS as a function of genome size for 316 bacteria and the 12 myxobacteria. Myxobacteria also tend to have a high proportion of hybrid HKs and a high proportion of TCS genes in complex foci (with a corresponding lack of TCS genes in orphan or paired foci). They also exhibit exceptionally low proportions of TM HKs (Table 2 and Fig. 2b), implying they sense their internal state to an unusual degree [25]. Bb exhibits values intermediate between the myxobacterial and bacterial means, except that (in contrast to the myxobacteria) it has a substantially greater proportion of paired TCS genes and TM HKs than the average bacterium (Table 2).
Fig. 2.
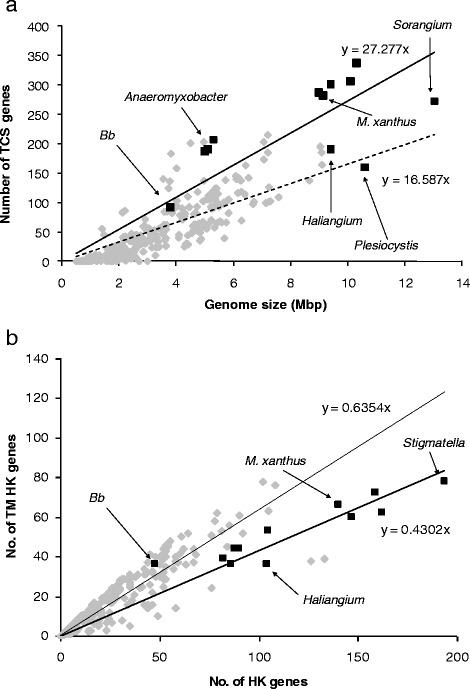
a Numbers of TCS genes as a function of genome size. Myxobacterial genomes (black spots) tend to have larger than expected numbers of TCS genes for genomes of their size. Bdellovibrio bacteriovorus HD100 (Bb) is also highlighted. A trendline is shown for all bacteria. b The proportion of transmembrane histidine kinases (TM HKs), as a function of total HK genes in myxobacterial (black) and bacterial (grey) genomes. Bdellovibrio bacteriovorus HD100 (Bb) is also highlighted. Trendlines and their equations are shown for bacteria (dashed) and myxobacteria (solid). Updated from [14]
On inspection of Table 2, some myxobacteria stand out as unusual. The Anaeromyxobacter strains generally exhibit percentages of hybrid kinases more similar to bacteria than myxobacteria, and also possess relatively high percentages of TM HKs. Sc, Ho and Pp have relatively small numbers of TCS genes for myxobacterial genomes of their size. Ho and Pp are marine organisms, and marine organisms generally exhibit reduced environmental responsiveness compared to their terrestrial counterparts [25]. Sc could be thought of as having an exceptionally large genome for a myxobacterium, rather than having relatively low numbers of TCSs for a myxobacterial genome of its size.
Orthology relationships between TCS proteins
TCS gene sets were clustered into groups of orthologous and paralagous proteins as described in the Methods section. Even at the lowest similarity cut-off used (30 % similarity), proteins clustered into pairs from Mx and Mf were also reciprocal highest-scoring BLAST hits, supporting the validity of this approach to define orthologues.
The percentage of proteins successfully grouped into clusters of orthologues decreased exponentially with increasing genetic distance (Fig. 3). A notable departure from the trend was the Ca and Sc comparison, which gave a remarkably low proportion of TCS genes being grouped as orthologues (6 %). This was the only pair-wise comparison which departed substantially from the trend, and the identified orthologues exhibited surprisingly low similarity. The cause of this extra variability is unclear.
Fig. 3.
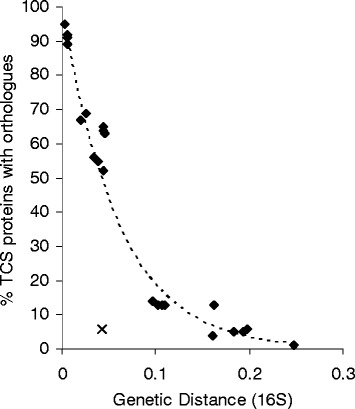
The percentage of orthologous TCS proteins varies exponentially with genetic distance. As genetic distance between two strains increases (substitution rate for the 16S rRNA gene), the percentage of proteins from either genome that are successfully clustered decreases exponentially. Each black diamond represents one pairwise comparison. Comparisons shown include all possible intra-genus and intra-family comparisons (ie. between AF, Ad1, AdC and AK, between Mx, Mf, Sa and Cc, and between Sc and Ca), and a representative sample of within sub-order and order comparisons (Mf vs. Ho/AF/AK, Sc vs. Sa/Ho/Bb, Ho vs. Pp, Mx vs. Ad1, Sa vs. AdC and Ca vs. AF). A notable departure from the trend is the comparison between Ca and Sc (black cross)
Within-genus orthologues
We began a detailed analysis of orthologues by comparing the gene set of Mf to that of Mx, the best characterised myxobacterium. Mx and Mf had 27 and 32 TCS genes respectively which were unique to that organism (singletons). The remaining TCS genes (89 %) all clustered into Mx/Mf pairs except for a single cluster of three proteins (LILAB_09435, LILAB_09385 and MXAN_7396), which seems to have arisen by a local duplication in the Mf lineage. A dot-plot of the relative location of TCS orthologues in the two genomes (Fig. 4a) recapitulated the global synteny observed between Mx and Mf, which is dominated by a single inversion [26]. There were no observable changes in relative genomic location of orthologues, suggesting that the relocation of individual TCS gene foci around a genome is a rare event, compared to re-location via large-scale chromosomal recombination. Five singleton TCS genes were found close to the putative inversion breakpoints (LILAB_00195, MXAN_1679/1680 and MXAN_5852/5853), indicating that gain/loss of those genes was potentially associated with the chromosomal inversion.
Fig. 4.
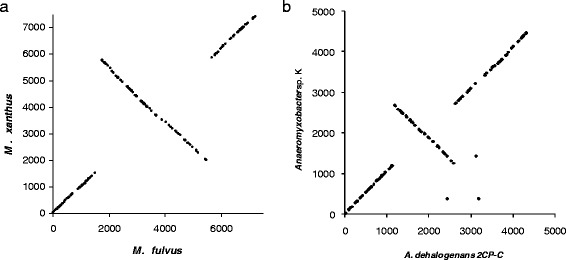
Dot-plots of TCS gene orthologues. Orthologues are plotted according to their genomic location (as inferred from the consecutive numbering of their locus tags) in both genomes. Each genome starts at the origin, with the dnaA gene being the first gene. A central line of orthologues extending from top left to bottom right indicates an inversion of a large section of the chromosome, centered around the origin/terminus. a Dot-plot of Mx/Mf TCS gene orthologues. b Dot-plot of AdC/AK orthologues
Within Mx/Mf paired clusters, TCS character (gene organisation, domain architecture, gene size and family membership) was conserved in the vast majority (247 of 254) of cases. In four pairs gene organisation was altered (MXAN_0259/LILAB_07355, MXAN_3738/LILAB_26865, MXAN_4580(Nla8)/LILAB_30920 and MXAN_5147(CheA3)/LILAB_33705), in two cases domain architecture/family was altered (MXAN_0727/LILAB_05150 and MXAN_6952/LILAB_11650A), and in one case both gene organisation and family membership changed between the two organisms (MXAN_4049/LILAB_28365A). Of the five observed changes in organisation, two were caused by gain/loss of intervening genes at a single focus, while the remaining three were caused by gain/loss of TCS genes. All three examples of family membership changes involved gain/loss of RR effector domains, and were associated with frameshift mutations. Intriguingly all three pairs included genes which were either mispredicted or annotated as pseudogenes, so it isn’t clear whether the observed differences represent true changes in family membership, or gene loss in progress.
There were seven cases of altered gene length (>100 aa difference) between Mx and Mf pairs, four of which involved gain/loss of effector/input domains. Two were changes of family membership mentioned previously (MXAN_4049/LILAB_28365A, MXAN_6952/LILAB_11650A), one was gain/loss of a duplicated PAS domain (MXAN_6586/LILAB_13600), and there was one instance of expansion/contraction of a poly-HAMP domain tract (MXAN_6735/LILAB_12790). The remaining three changes in gene length involved gain/loss of a C-terminal region (MXAN_4461(RomR)/LILAB_30440), extension/contraction of an N-terminal region (MXAN_0230/LILAB_07550), and gain/loss of an internal region (MXAN_4758/LILAB_31740), all of which may represent the gain/loss of currently uncharacterised domains.
Across the whole Mx/Mf genome, 23 % of protein-encoding genes were unique to either genome, whereas only 10–11 % of TCS genes were singletons, implying that TCS genes are a relatively conserved gene set. Mx TCS genes with names have published phenotypes, and historically Mx has been subjected to exhaustive genetic screens for developmental and motility mutations. The set of named TCS genes is therefore enriched for genes with significant phenotypes (particularly motility and/or development-related). Mx/Mf paired TCS genes were enriched for named genes –24 % of all M. xanthus TCS genes are named, however only two of the 27 singletons were named (7 %), and those two genes (nla1 and nla13) have minor phenotypes [27], suggesting that singletons are not subject to a strong retentive selective pressure. The Mx/Mf singletons were very different in character - Mx encoded eight hybrid kinases, 11 TCS genes with complex gene organisations and 11 paired TCS genes, whereas the Mf singletons included no hybrid kinases, only three complex TCS genes, but 17 paired genes.
To see whether Mx/Mf differences were typical of species-level comparisons, Anaeromyxobacter strains AK and AdC, were also compared. The AdC/AK genetic distance (16S) is identical to that between Mx and Mf, however the Anaeromyxobacter species are anaerobic, and non-fruiting, so any commonality between the AK/AdC and Mx/Mf comparisons is likely to be due to genetic similarity and independent of lifestyle. No duplications were observed, while 9 % of genes were singletons (16 and 19 in the two genomes). Six changes in gene organisation were observed (AnaeK_0372/Adeh_3183, AnaeK_0373/Adeh_2444, AnaeK_1885/Adeh_1993, AnaeK_1886/Adeh_1992, AnaeK_1889/Adeh_1989, AnaeK_4383/Adeh_4250), one change in size and domain architecture (AnaeK_2395/Adeh_1473) and one other gene size change (AnaeK_0940/Adeh_0892). A dot-plot of AK/AdC orthologues (Fig. 4b) shows global synteny, including across a large chromosomal inversion, albeit with three pairs of orthologues demonstrating re-location of TCS genes within the chromosome (AnaeK_1417/Adeh_3134 and AnaeK_0372/Adeh_3183), including one causing a change in gene organisation (AnaeK_0373/Adeh_2444).
Insertions/deletions (indels) of ≥1 gene at three foci (Adeh_2443, AnaeK_4382 and AnaeK_1888), resulted in four singletons, and the remaining five changes in gene organisation. One small (<1 gene) indel (AnaeK_2395, AnaeK_2396/Adeh_1473) resulted in one singleton, and a domain architecture change and gene size change, as the consequences of changing between a TR,T system and a TT system (domain organisation of a locus is represented using R, T and H to represent receiver, transmitter and HisKA/Hpt domains respectively, with commas separating genes, as explained in the Methods section). Finally, an indel resulted in an apparent gain/loss of two input domains from one HK (AnaeK_0940/Adeh_0892). Unlike with the Mx/Mf singletons, there were no obvious differences in character between the AdC and AK singletons.
In summary (Table 3), these two within-genus comparisons (in agreement with the other two comparisons Ad1 vs. AK/AdC – data not shown), suggest that inter-species differences are dominated by TCS gene gain/loss, occasionally causing changes in gene organisation. This also leads to ‘individuality’ within each genome, with organism-specific ‘accessory’ genes. Relatively infrequently, changes in domain architecture occur as a consequence of small indels or frameshift mutations. As expected, such alterations most frequently affect the output domains of RRs (changing family membership) and the sensory domains of HKs.
Table 3.
Mutational events and their consequences for TCS gene sets
| Apparent mutational event | Within-genus frequency (%) | Within-family frequency (%) | Typical consequences |
|---|---|---|---|
| Chromosomal Rearrangements | ~1-2 % | ~3 % | Gene gain/loss. |
| Large (≥1 gene) indels | ~10 % | ~20 | Gene gain/loss, with occasional changes in gene organisation (in ~10 % of cases). |
| - Duplications | <<1 % | ~3 % | Gene gain. |
| - Horizontal transfer | ~2 % | ~4 % | Gene gain. |
| Small (<1 gene) indels | ~1-2 % | ~3 % | Domain gain/loss, changes in family. |
| - Frameshifts | <1 % | ~1 % | Change in family (often through changes in RR output domains), pseudogene formation. |
Percentages of orthologous clusters which show evidence of evolutionary changes when comparing genomes from the same genus (within-genus) and the same family (within-family). Percentages obtained from pairwise comparisons between Cystobacterineae members
Within-family orthologues
To assess TCS gene changes between genera, three ‘within-family’ groups of comparisons were made; Cc/Sa/Mx/Mf, Ad1/AdC/AK/AF, and Sc/Ca.
In Cc/Sa/Mx/Mf comparisons, some clusters of homologues were observed with >4 members. One cluster of six proteins was observed due to an apparent double-duplication in Sa (STAUR_0029, STAUR_4565, STAUR_7210), while single duplications in Cc and Mf gave rise to two clusters of five proteins (COCOR_07667/COCOR_07814 and LILAB_09385/LILAB_09435 respectively). A final cluster of five proteins was composed entirely of Sa proteins (STAUR_1021, STAUR_1353, STAUR_5099, STAUR_7439 and STAUR_7639). 572 proteins were found in clusters of four (143), all but two of which included a single representative from each genome (one cluster contained a Mf duplication LILAB_11650A/LILAB_19280 while the second contained duplicated proteins from Cc and Sa COCOR_01454/COCOR_02086/STAUR_6958/STAUR_8144). Another 204 proteins were found in groups of three, with members from different genomes, however in two cases there was an apparent duplication in one of the genomes (COCOR_1905/COCOR_04361 and COCOR_07670/COCOR_07671). 206 proteins were paired, including five pairs of duplicated proteins from Sa (STAUR_0493/STAUR_2886, STAUR_1273/STAUR_4462, STAUR_1775/STAUR_5256, STAUR_1837/STAUR_4550 and STAUR_4571/STAUR_4572). This left 211 singletons, of which the majority belonged to Sa (124, 59 %), with large numbers also in Cc (64, 30 %), and relatively few in Mx (13) or Mf (10). The pattern of protein orthology can be seen in Additional file 2: Figure S2 as a Venn diagram, and numbers are consistent with genetic distance, with closer genomes sharing more orthologues and possessing fewer singletons. The same trends were observed for Ad1/AdC/AK/AF orthologues (Additional file 2: Figure S2).
Across the 318 Cc/Sa/Mx/Mf clusters with two or more members, in addition to the Mx/Mf changes described earlier, there were 43 cases of altered gene organisation (highlighted in Additional file 3), and three changes of domain architecture, all three of which involved hybrid kinases. A small indel caused a RRT/RT change (MXAN_6966 is the Mx orthologue), a TRT/TRTTRT change appears associated with a tandem duplication and fusion (STAUR_6958), while a TR/T change is associated with a large indel, also involving gain/loss of an adjacent gene (STAUR_7785). In addition to these changes in domain architecture, 21 cases of genes changing size by >100 aa were observed, of which the majority (16) affected HKs, mainly in their N-terminal regions (highlighted in Additional file 3).
For Ad1/AdC/AK/AF orthologues (196 clusters of two or more proteins), 17 instances of changes in gene organization were observed (highlighted in Additional file 3). These were mainly (15 cases) associated with large (>1 gene) indels, with five cases also involving gene rearrangements (adjacent to Ad1 genes A2cp1_1514, A2cp1_2989, A2cp1_2499 and A2cp1_3138 and A2cp1_3377), and two of the 17 were due to frameshift mutations generating pseudogenes (the AdC orthologue of A2cp1_1973, and the Ad1 homologue of Adeh_1992). Four changes in domain architecture were observed, each of which was associated with a change in gene size. In one case an N-terminal PilZ domain was gained/lost from a RR (Anae109_3862), while the other changes were due to gene fusion/fission events (T,TR/TT and T,R/TR changes involving Adeh_1473 and A2cp1_0976 respectively). Seven gene duplications at five loci were apparent (Anae109_0467/Anae109_3535, Anae109_0469/Anae109_3533, Anae109_0470/Anae109_3532, Anae109_1027/Anae109_2976, Anae109_1440/Anae109_3416, Anae109_1441/Anae109_3417, A2cp1_3106/A2cp1_3138), with an additional duplication also associated with a change in gene size (Anae109_1040/Anae109_2356). Finally, two additional changes in gene size were seen, both of which were due to gain/loss of C-terminal regions (one HK and one RR; Adeh_3180 and Anae109_0769).
The final within-family comparison possible is between Ca and Sc, however that comparison is characterised by an unusually low proportion of orthologous proteins (Fig. 3) and low similarity between orthologues. Of the 18 clusters of orthologues which included members from both organisms, there were no apparent changes in domain architecture, gene organisation or gene size. Further comparisons between more distant genomes were also uninformative due to the small number of orthologous protein clusters identified by CD-HIT or by bidirectional best-scoring BLAST hits. Thus, in summary, comparisons between same-family members show similar trends to those from same-genus comparisons albeit at higher frequencies (Table 3).
Complexity
Another notable feature of myxobacterial TCS proteins is their complexity – both in terms of their domain architectures, and their gene organisation [14, 15].
We define here the most complex systems as ‘intricate’, where at least two receiver and at least two transmitter domains are found in the same protein and/or genetic focus. TRTR proteins are encoded as singletons in four genomes (Cc, Ho, Sa and Sc; COCOR_05524, Hoch_3141, STAUR_7959 and sce3507), while Sa also encodes a TRTTRT protein (STAUR_6958). According to our analysis, the TRTR proteins are not orthologous to each other or other myxobacterial TCS proteins, while the TRTTRT protein appears to be a tandem duplication and fusion of two adjacent TRT proteins. In addition, myxobacterial genomes possessed between two and nine intricate multi-gene TCS foci (Table 2), considerably more than the bacterial mean.
The intricate foci of Ho, Sc and AF were largely unique to those genomes, otherwise orthologous relationships between the intricate foci were relatively clear at the genus and family level (Table S1). The foci highlighted exhibit several interesting properties. There seems to be a great deal of plasticity in these foci with many exhibiting orthology with non-intricate TCS foci as a consequence of gene gain/loss and/or recombination (Additional file 4: Table S1). In addition, a large proportion of the TCS domains encoded at intricate foci are found in hybrid kinases (111 of 145 domains, 77 %).
The proportion of foci containing large numbers of TCS domains are similar between bacteria and myxobacteria (data not shown). Nevertheless, when considering all TCS proteins (not just intricate foci/proteins), those of myxobacteria are generally more complex than other bacteria. Table 4 shows the relative frequencies of TCS proteins with different domain compositions for myxobacteria and bacteria. Myxobacteria possess far greater proportions of TCS proteins with two or more transmitter domains, and hybrid kinases with three or more receiver domains than other bacteria.
Table 4.
Transmitter and receiver domain composition of TCS proteins
| % myxobacteria / % bacteria | 0 R | 1 R | 2 R | 3 R | 4+ R |
|---|---|---|---|---|---|
| 0 T | 0.41 (28) | 0.92 (1340) | 1.58 (32) | 2.13 (2) | a(0) |
| 1 T | 0.97 (1026) | 1.42 (373) | 1.37 (54) | 3.73 (22) | 14.91 (4) |
| 2+ T | 7.45 (1) | 11.75 (26) | 5.73 (5) | 0† (0) | 0b (0) |
Values are the % of myxobacterial TCS proteins exhibiting the number of receiver (R) and transmitter (T) domains in question divided by the % of all such bacterial TCS proteins [24]
aThere are no such proteins in bacteria
bNo such proteins in myxobacteria (only 0.009 % of all TCS proteins in bacteria have 2+ transmitter and 3+ receiver domains). Numbers of such myxobacterial proteins are shown in parentheses
TCS gene expansion
Nearly two thirds (59 %) of all Mx/Mf singletons were found to have orthologues in Cc and/or Sa, implying that the formation of the majority of singletons was through progressive gene loss. However, genes can also be gained by duplication and divergence. Yet duplication is responsible for a relatively small amount of differences between TCS gene sets. For instance, clustering reveals only 12 lineage-specific duplications in Sa, four cases in Cc and two in Mf. Eight duplications were apparent within the Anaeromyxobacter strains (highlighted in Additional file 3).
Nevertheless, it appears that certain TCS families have been expanded hugely in the myxobacteria and this occurred relatively recently. For instance, NtrC family members are unusually numerous [14]. Indeed, eight of the 28 Mx NtrC family members exhibited more similarity to other Mx NtrC paralogues than to homologues in other organisms, suggesting origin by duplication after the divergence of contemporary genera of myxobacteria.
Another mechanism that can increase gene number is horizontal gene transfer (HGT). To test the frequency of horizontal acquisition, the Mx/Mf singletons lacking homologues in Cc/Sa/Mx/Mf were queried against the nr protein database using BLAST. Eight of the ten Mf singletons and six of the 13 Mx singletons gave BLAST hits across the Myxococcales, implying linear descent. However nine of the 23 Mx/Mf genes gave best non-self BLAST hits to non-Myxococcales genomes, implying horizontal acquisition. Such evidence of HGT is also abundant in the singletons of other myxobacteria (eg. for 34 of the 78 singletons of Ad1/AdC/AK/AF (Additional file 5: Table S2)).
Methods
Phylogenetic analysis
16S rRNA gene sequences were aligned using ClustalW [28], using default parameter settings, and neighbour-joining trees were created using PHYLIP [29] with 1000 bootstraps, excluding columns containing gaps.
Inference of TCS proteins sets
Eight myxobacterial genomes (Mx, Mf, Sc, Ho, AF, AK, Ad1 and AdC) and that of Bb are included within the latest version of the P2CS (Prokaryotic 2-Component Systems) database, available at www.p2cs.org [30, 31], which provides an automated and consistent pipeline for the identification and annotation of TCS genes. The same categorisation pipeline is available for unpublished sequences at www.p2rp.org [32] and this service was used to characterise the TCS gene sets for Sa, Cc, Pp and Ca nucleotide sequence files from www.ncbi.nlm.nih.gov. TCS gene sets were compared with those previously published for Mx, Sa, AdC and Sc [14], and discrepancies manually curated. For the TCS gene sets available through P2CS, potentially ‘mispredicted’ TCSs were identified, and these were validated manually and added to the TCS datasets (named after the closest gene, and given an ‘A’ suffix). These two processes resulted in the addition of five, six and 14 TCS proteins to the gene sets for Mx, Sc and AdC respectively, as originally described by Whitworth and Cock [14]. Three, one, one and three ‘mispredicted’ TCS genes were also validated for Mf, Ho, Ad1 and AF respectively. TCS datasets for each genome are available as additional data (Additional file 6).
Characterisation of TCS proteins
Domain architectures and the presence of transmembrane (TM) helices within TCS proteins were determined as described by Barakat et al. [33]. TCS gene organisation was defined using a classification scheme based on TCS domains (receiver, transmitter, Hpt/HisKA) encoded unidirectionally at a ‘focus’, as proposed by Williams and Whitworth [24]. If a transmitter and a receiver domain were found adjacent in the genome (whether within a single ‘hybrid kinase’ gene, or as separate HK and RR genes with no more than one intervening gene), and they had at least two non-TCS genes on each side, then the relevant genes were described as ‘paired’ (i.e. that focus appears to encode an entire TCS). If a gene contained a single transmitter, receiver or phosphotransfer domain and was found more than two genes from another TCS gene, it was described as ‘orphan’. TCS gene foci with any other domain/gene organisation were described as ‘complex’. The genomes of Pp and Ca are incomplete, so gene organisations were not defined for those organisms. Focus TCS domain organisation is described in the text using R, T and H to represent receiver, transmitter and HisKA/Hpt domains respectively, with commas separating genes. Thus a ‘RT,R’ organisation represents a hybrid kinase gene encoding an N-terminal receiver domain and a C-terminal transmitter domain, adjacent to a response regulator gene encoding a single receiver domain.
Sequence-based clustering
To define orthologues and paralogues, sets of TCS genes were clustered using CD-HIT [34]. A high similarity cut-off was used (up to 90 % similarity) to give high confidence clusters, and then a lower similarity (30 %) cut-off was used to identify more divergent clusters. Clusters obtained were then subjected to curation, whereby any large clusters containing multiple paralogues obtained using the 30 % cut-off were manually split into the high-confidence clusters obtained with a 90 % cut-off. Clusters which included proteins of significantly different length (>100 amino acid residues difference), or of varying domain architecture, family membership and/or gene organisation, or with varying numbers of members per genome were earmarked for further analysis.
Discussion
Genetic distance and inferred evolutionary events
Comparing the TCSs from organisms with differing degrees of relatedness allowed inference of the contribution of different types of mutation to the evolution of contemporary sets of TCSs. It appears that the relative impact of different mutational events are not affected by the relatedness of the organisms being compared, however, the apparent frequency of mutations is dependent on the relatedness of compared genomes (Table 3), echoing the findings of He et al. [35] in their study of clostridia. This means that when comparing the gene sets of multiple genomes, it is important to characterize the similarity of those genomes and compensate for differing degree of relatedness. It is even worth including organisms of diverse relatedness, to increase the power of analysis, as suggested by Eisen et al., [36]. In this study we used 16S rRNA gene sequence similarity as a straightforward indicator of genomic relatedness/distance and found that the ability to identify TCS orthologues between genomes was dependent on relatedness (Fig. 3). At low levels of relatedness, difficulties in the identification of orthologues reduced the power of comparative analysis making it difficult to infer evolutionary events. This difficulty was likely exacerbated by the large numbers of TCS in myxobacterial genomes, and should be less of a problem for the majority of prokaryotes.
There have been several studies comparing the TCS gene sets of related organisms, and although not noted, the phenomena we describe can often be seen in the TCS gene sets of diverse taxa. For instance, TCS gene complements can vary by 10–30 % when comparing organisms within the same family/genus, and the frequency of within-species variation is about half that observed when comparing organisms within the same genus [37–40].
Evolution of myxobacterial TCS gene sets
Our data suggest that most changes in myxobacterial TCS gene sets are due to large indels, some of which are associated with chromosomal recombination. There is evidence of frequent gene loss (which has also dominated TCS evolution in the Lactobacillaceae and Leuconostocaceae [39]), but also gene gain via HGT and occasional gene duplications. Some (~10 %) of these indels result in changes in gene organization, when the indel only affects part of a TCS locus. Small (within-gene) indels are less frequent and often result in domain gain/loss with associated changes in TCS family membership. Other studies have reported broadly consistent results. For example, in a study of recombination rates in Salmonella, Sun et al., [41] found that deletions are significantly more frequent than duplications. Both Mx and Bb exhibit genome-wide evidence of HGT [42, 43], and we find that HK and RR genes tend to be acquired/lost as pairs, which has been observed in several other taxa, including mutans streptococci and Xanthomonas spp. [40, 44].
Domain shuffling appears to have happened occasionally during TCS evolution, giving rise to HKs with a multiplicity of input domains, and RRs with diverse output domains [45]. Our methods of defining orthologues used whole gene sequences and may not have been capable of identifying shuffled domains, nevertheless we did observe several instances of input/output domain gain/loss. With domain shuffling being a rare evolutionary event, it seems probable that apparent domain shuffling is a consequence of domain loss followed by domain gain rather than direct shuffling between proteins.
We also did not observe significant relocation of TCS within genomes. The few cases observed (for example see Fig. 4b) were associated with breakpoints of chromosomal recombination. It is possible that those chromosomal relocations occurred due to recombination between homologous TCS genes at those points, however there is too little conservation of synteny in the break point regions to provide any evidence of this happening in myxobacterial genomes.
Myxobacteria were chosen as the subject of this study due, in part, to the large number of TCS genes they possess. Potentially, this could limit extrapolation of our conclusions to other organisms, as increased numbers of TCS genes may have promoted some mutagenic events (for example homologous recombination or gene conversion), and the large numbers of paralogous genes makes robust orthologue identification difficult.
In 2008 we described the TCS of four sequenced myxobacteria and used domain-based orthology searches to identify likely evolutionary events and peculiarities of myxobacteial TCS gene sets [14]. For this study we added a further eight myxobacterial genomes, dramatically increasing the number of inter-genome comparisons possible, and defined orthologues using whole gene sequence-based methods. The results obtained here agree well with that previous work, and provide a less anecdotal analysis of evolutionary events. Benchmarking our genome comparisons by relatedness also allows straightforward comparison with TCS gene sets from other genomes in the future.
The TCS genes of myxobacteria seem to be relatively conserved compared to non-TCS genes, and we also found that the Mx TCS genes with Mf orthologues were enriched for named genes, which tend to have observable phenotypes. This suggests that there is a ‘core’ set of TCS genes in myxobacteria responsible for regulating important functions, with other ‘accessory’ TCS genes being more variable between genomes, and with minor (if any) observable phenotypes.
The Ad1/AdC/AF/AK TCS gene sets exhibited much less variability than those of Mx/Mf/Cc/Sa (see text and Additional file 2: Figure S2), however this effect was proportional with the reduced numbers of TCS genes in the anaeromyxobacteria. It is possible that the fruiting myxobacteria need enhanced ‘individuality’ to allow easy discrimination of competing genotypes, but whether this is a selected trait, or purely a consequence of their increased numbers of TCS is uncertain. Among strain-specific genes, there is particularly good evidence of acquisition by HGT, which is known to have had a major effect on the shaping of contemporary myxobacterial genomes, alongside lineage-specific expansion [43].
Myxobacterial genomics
The field of myxobacterial genomics is currently hampered by the large number of genes encoded in each genome (and the consequent number of differences between genomes), the lack of easily assayable behavioural outputs (for instance fruiting body formation is typically just characterized by time, and % of wild-type spores produced), and pleiotropy of many genes. This means that phenotypic properties cannot easily be correlated with particular genomic differences.
However, further myxobacterial genomes are becoming available at an accelerating rate, and it is possible that in the near future, physiological characterisation of the new genome host organisms will enable correlations with genomic features. To that end, it is important to develop benchmarked catalogs of genomic differences. Taking into consideration the relative distance of compared genomes is an important feature of benchmarking, and this study provides a framework to develop such benchmarks using 16S ribosomal RNA gene sequence differences as indicators of genetic distance, which can be related to formal taxonomy and other systematic methods [46, 47].
During the course of this study draft genomes became available for strains of Cystobacter fuscus, Cystobacter violaceus, Enhygromyxa salina, Hyalangium minutum, Sandaracinus amylolyticus, and for two further strains of Myxococcus xanthus, including DZ2 and DZF1 [48, 49], however these genomes are not complete or scheduled for finishing. Thankfully, two complete myxobacterial genomes have also became available, Myxococcus stipitatus DSM 14675 and Sorangium cellulosum So0157-2 [50, 51]. Both of these genomes are now characterized in P2CS and seem typical members of their genera, albeit with slightly more TCS genes (309 in Sc So0157-2, 296 in M. stipitatus [Ms]), except that the LytTR family of RRs is expanded substantially in Ms (12 compared to two in each of Mx and Mf). As more genomes are completed it will become easier to identify more distant orthologues via ‘gap-bridging’ genomes. This will in turn improve deduction of the directionality of evolutionary changes such as indels, and allow us to create a more finely resolved timeline of myxobacterial TCS gene evolution.
Conclusion
Focusing on the TCS genes of myxobacteria, this study investigated the relationship between apparent evolutionary events, and genomic distance or ‘relatedness’. As genomic distance increases it becomes more difficult to identify TCS orthologues, and the relative frequency of evolutionary events increases. However, the relative frequency of evolutionary changes remain unaffected by scale (or lineage), and it seems that in the myxobacteria, as for other organisms, TCS gene evolution is dominated by large indels (often causing changes in gene organization), with smaller indels being less frequent (occasionally causing changes in domain architecture). TCS genes are ‘born’ by duplication and acquisition through HGT, but the individuality of TCS gene sets appears to be dominated by lineage-specific gene loss.
More generally, our results suggest that researchers should be mindful that the numbers of genomes compared can have a substantial effect on the apparent frequency of observed events. This is particularly true when comparing organisms that share different levels of ‘genetic relatedness’, which should not be assumed to always be consistent with the taxonomic assignments of compared organisms.
Abbreviations
- TCS
Two-component system
- HK
Histidine kinase
- RR
Response regulator
- TM
Transmembrane
Additional files
Genetic distance (substitutions per nucleotide position) and neighbour-joining tree inferred from myxobacterial 16S rRNA gene sequences. Bootstrap support for each clade was > 95 % (from 1000 bootstrap resamplings) in all cases except for the clade indicated by a filled circle (89 % bootstrap support). The tree is rooted with B. bacteriovorus. The bar represents 0.1 substitutions per nucleotide position. (PPT 119 kb)
Orthology relationships within-family. Venn diagrams showing the distribution of orthologous genes between organisms. Some orthologous clusters exhibited multiple members from the same organism, suggestive of lineage-specific gene duplications. For the purpose of these diagrams, duplicated proteins were treated as singletons. For instance, if a cluster contained members from Sa, Cc, Mx and Mf, but the Sa protein was duplicated, then 1 was entered in the Sa/Cc/Mx/Mf cell, and 1 into the Sa only cell. For a pair of Sa proteins, 2 was entered into the Sa cell. a: Sa/Cc/Mx/Mf, b: AF/AK/Ad1/AdC. (PDF 185 kb)
The sequence clusters of orthologues of every myxobacterial TCS protein. (DOC 279 kb)
Intricate multi-gene TCS foci. Each row shows all myxobacterial orthologues of intricate multi-gene foci, including non-intricate orthologues, arranged by organism. Foci in particular genomes are denoted by the number from the locus tag of the first protein within the focus. ‘//’ refers to an unspecified number (2 or more) of non-TCS genes. (PDF 39 kb)
Evidence of horizontal gene transfer (HGT) amongst cystobacterial TCS proteins. Singletons (TCS lacking orthologues) from comparisons between Ad1/AdC/AK/AF, and between Mx/Mf/Cc/Sa, are presented alongside the number of singletons acquired by HGT (as evidenced by highest-scoring BLAST hits to non-myxobacteria outside the organism’s own genus). Also tabulated are the number of TCS genes found in foci where every TCS gene was acquired by putative HGT and the number of those which constitute an ‘intact’ TCS (ie containing ≥1 transmitter domain and ≥1 receiver domain. (PDF 456 kb)
Datasets of the annotated TCS proteins from each genome. (XLS 586 kb)
Footnotes
Competing interests
The author declares that he has no competing interests.
References
- 1.Whitworth DE. Genomes and knowledge – a questionable relationship? Trends Microbiol. 2008;16:512–9. doi: 10.1016/j.tim.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Wuichet K, Cantwell BJ, Zhulin IB. Evolution and phyletic distribution of two-component signal transduction systems. Curr Opin Microbiol. 2010;13:219–25. doi: 10.1016/j.mib.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitworth DE. Two-component regulatory systems in prokaryotes. In: Filloux AAM, editor. Bacterial regulatory networks. Norfolk, UK: Caister Academic Press; 2012. pp. 191–222. [Google Scholar]
- 4.Gross R, Beier D, editors. Two-component systems in bacteria. Norfolk, UK: Caister Academic Press; 2012. [Google Scholar]
- 5.Bourret RB, Silversmith RE. Two-component signal transduction. Curr Opin Microbiol. 2010;13:113–5. doi: 10.1016/j.mib.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capra EJ, Laub MT. Evolution of two-component signal transduction systems. Ann Rev Microbiol. 2012;66:325–47. doi: 10.1146/annurev-micro-092611-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitworth DE, Cock PJA. Evolution of prokaryotic two-component systems: insights from comparative genomics. Amino Acids. 2009;47:459–66. doi: 10.1007/s00726-009-0259-2. [DOI] [PubMed] [Google Scholar]
- 8.Krueger B, Friedrich T, Förster F, Bernhardt J, Gross R, Dandekar T. Different evolutionary modifications as a guide to rewire two-component systems. Bioinform Biol Insights. 2012;6:97–128. doi: 10.4137/BBI.S9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alm E, Huang K, Arkin A. The evolution of two-component systems in bacteria reveals different strategies for niche adaptation. PLoS Comput Biol. 2006;2:e143. doi: 10.1371/journal.pcbi.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cock PJA, Whitworth DE. Evolution of prokaryotic two-component system signalling pathways: gene fusions and fissions. Mol Biol Evol. 2007;24:2355–7. doi: 10.1093/molbev/msm170. [DOI] [PubMed] [Google Scholar]
- 11.Pace NR. Mapping the tree of life: progress and prospects. Microbiol Mol Biol Rev. 2009;73:565–76. doi: 10.1128/MMBR.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dini-Andreote F, Andreote FD, Araújo WL, Trevors JT, van Elsas JD. Bacterial genomes: habitat specificity and uncharted organisms. Microb Ecol. 2012;64:1–7. doi: 10.1007/s00248-012-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:e1001920. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitworth DE, Cock PJA. Two-component systems of the myxobacteria: structure, diversity and evolutionary relationships. Microbiol. 2008;154:360–72. doi: 10.1099/mic.0.2007/013672-0. [DOI] [PubMed] [Google Scholar]
- 15.Whitworth DE, Cock PJA. Myxobacterial two-component systems. In: Whitworth DE, editor. Myxobacteria: multicellularity and differentiation. Washington, DC: ASM Press; 2008. pp. 169–89. [Google Scholar]
- 16.Keilberg D, Huntley S, Søgaard-Andersen L. Two-component systems involved in regulation of motility and development in Myxococcus xanthus. In: Gross R, Beier D, editors. Two-component systems in bacteria. Norfolk, UK: Caister Academic Press; 2012. pp. 293–314. [Google Scholar]
- 17.Whitworth DE. Myxobacteria: multicellularity and differentiation. Washington, D.C: ASM Press; 2008. [Google Scholar]
- 18.Wenzel SC, Müller R. Myxobacteria--‘microbial factories’ for the production of bioactive secondary metabolites. Mol Biosyst. 2009;5:567–74. doi: 10.1039/b901287g. [DOI] [PubMed] [Google Scholar]
- 19.Weissman KJ, Müller R. Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat Prod Rep. 2010;27:1276–95. doi: 10.1039/c001260m. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Higgs PI. Myxobacteria: genomics, cellular and molecular biology. Norfolk, UK: Caister Academic Press; 2014. [Google Scholar]
- 21.Huntley S, Hamann N, Wegener-Feldbrügge S, Treuner-Lange A, Kube M, Reinhardt R, et al. Comparative genomic analysis of fruiting body formation in Myxococcales. Mol Biol Evol. 2011;28:1083–97. doi: 10.1093/molbev/msq292. [DOI] [PubMed] [Google Scholar]
- 22.Shi X, Wegener-Feldbrugge S, Huntley S, Hamann N, Hedderich R, Sogaard-Andersen L. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 2008;190:613–24. doi: 10.1128/JB.01502-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia R, Gerth K, Stadler M, Dogma IJ, Jr, Müller R. Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol Phylogenet Evol. 2010;57:878–87. doi: 10.1016/j.ympev.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Williams RHN, Whitworth DE. The genetic organisation of prokaryotic two-component system signalling pathways. BMC Genomics. 2010;11:720. doi: 10.1186/1471-2164-11-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galperin MY. A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 2005;5:35. doi: 10.1186/1471-2180-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li ZF, Li X, Liu H, Liu X, Han K, Wu ZH, et al. Genome sequence of the halotolerant marine bacterium Myxococcus fulvus HW-1. J Bacteriol. 2011;193:5015–6. doi: 10.1128/JB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caberoy NB, Welch RD, Jakobsen JS, Slater SC, Garza AG. Global mutational analysis of NtrC-like activators in Myxococcus xanthus: identifying activator mutants defective for motility and fruiting body development. J Bacteriol. 2003;185:6083–94. doi: 10.1128/JB.185.20.6083-6094.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucl Acid Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felsenstein J. PHYLIP – Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- 30.Barakat M, Ortet P, Whitworth DE. P2CS: a database of prokaryotic two-component systems. Nucleic Acids Res. 2011;39(Database issue):D771–6. doi: 10.1093/nar/gkq1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ortet P, Whitworth DE, Santaella C, Achouak W, Barakat M. P2CS: updates of the prokaryotic two-component system database. Nucleic Acids Res. 2015;43(Database issue):D536–41. doi: 10.1093/nar/gku968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barakat M, Ortet P, Whitworth DE. P2RP: a web-based framework for the identification and analysis of regulatory proteins in prokaryotic genomes. BMC Genomics. 2013;14:269. doi: 10.1186/1471-2164-14-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barakat M, Ortet P, Jourlin-Castelli C, Mejean V, Whitworth DE. P2CS: a two-component system resource for prokaryotic signal transduction research. BMC Genomics. 2009;10:315. doi: 10.1186/1471-2164-10-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–2. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107:7527–32. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisen JA, Heidelberg JF, White O, Salzberg SL. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000;1:RESEARCH0011. doi: 10.1186/gb-2000-1-6-research0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doß S, Gröger C, Knauber T, Whitworth D, Treuner-Lange A. Comparative genomic analysis of signal transduction proteins in clostridia. In: Dürre P, editor. Handbook on clostridia. Boca Raton, FL: CRC Press-Taylor & Francis Group; 2005. pp. 561–82. [Google Scholar]
- 38.Kiil K, Ferchaud JB, David C, Binnewies TT, Wu H, Sicheritz-Pontén T, et al. Genome update: distribution of two-component transduction systems in 250 bacterial genomes. Microbiol. 2005;151:3447–52. doi: 10.1099/mic.0.28423-0. [DOI] [PubMed] [Google Scholar]
- 39.Zúñiga M, Gómez-Escoín CL, González-Candelas F. Evolutionary history of the OmpR/IIIA family of signal transduction two component systems in Lactobacillaceae and Leuconostocaceae. BMC Evol Biol. 2011;11:34. doi: 10.1186/1471-2148-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song L, Sudhakar P, Wang W, Conrads G, Brock A, Sun J, et al. A genome-wide study of two-component signal transduction systems in eight newly sequenced mutans streptococci strains. BMC Genomics. 2012;13:128. doi: 10.1186/1471-2164-13-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun S, Ke R, Hughes D, Nilsson M, Andersson DI. Genome-wide detection of spontaneous chromosomal rearrangements in bacteria. PLoS One. 2012;7:e42639. doi: 10.1371/journal.pone.0042639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gophna U, Charlebois RL, Doolittle WF. Ancient lateral gene transfer in the evolution of Bdellovibrio bacteriovorus. Trends Microbiol. 2006;14:64–9. doi: 10.1016/j.tim.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Goldman B, Bhat S, Shimkets LJ. Genome evolution and the emergence of fruiting body development in Myxococcus xanthus. PLoS One. 2007;2:e1329. doi: 10.1371/journal.pone.0001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian W, Han Z-J, He C. Two-component signal transduction systems of Xanthomonas spp.: a lesson from genomics. Mol. Plant Microbe Interact. 2008;21:151–61. doi: 10.1094/MPMI-21-2-0151. [DOI] [PubMed] [Google Scholar]
- 45.Reizer J, Saier MH., Jr Modular multidomain phosphoryl transfer proteins of bacteria. Curr Opin Struct Biol. 1997;7:407–15. doi: 10.1016/S0959-440X(97)80059-0. [DOI] [PubMed] [Google Scholar]
- 46.Ludwig W, Schleifer KH. Bacterial phylogeny based on 16S and 23S rRNA sequence analysis. FEMS Microbiol Rev. 1994;15:155–73. doi: 10.1111/j.1574-6976.1994.tb00132.x. [DOI] [PubMed] [Google Scholar]
- 47.Oren A. Prokaryote diversity and taxonomy: current status and future challenges. Philos Trans R Soc Lond B Biol Sci. 2004;359:623–38. doi: 10.1098/rstb.2003.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Müller S, Willett JW, Bahr SM, Scott JC, Wilson JM, Darnell CL, et al. Draft genome of a type 4 pilus defective Myxococcus xanthus strain, DZF1. Genome Announc. 2013;1:e00392–13. doi: 10.1128/genomeA.00392-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Müller S, Willett JW, Bahr SM, Darnell CL, Hummels KR, Dong CK, et al. Draft genome sequence of Myxococcus xanthus wild-type strain DZ2, a model organism for predation and development. Genome Announc. 2013;1:e00217–13. doi: 10.1128/genomeA.00217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han K, Li ZF, Peng R, Zhu LP, Zhou T, Wang LG, et al. Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci Rep. 2013;3:2101. doi: 10.1038/srep02101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huntley S, Kneip S, Treuner-Lange A, Søgaard-Andersen L. Complete genome sequence of Myxococcus stipitatus strain DSM 14675, a fruiting myxobacterium. Genome Announc. 2013;1:e0010013. doi: 10.1128/genomeA.00100-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA, et al. Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci U S A. 2006;103:15200–5. doi: 10.1073/pnas.0607335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huntley S, Zhang Y, Treuner-Lange A, Kneip S, Sensen CW, Søgaard-Andersen L. Complete genome sequence of the fruiting myxobacterium Corallococcus coralloides DSM 2259. J Bacteriol. 2012;194:3012–3. doi: 10.1128/JB.00397-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas SH, Wagner RD, Arakaki AK, Skolnick J, Kirby JR, Shimkets LJ, et al. The mosaic genome of Anaeromyxobacter dehalogenans strain 2CP-C suggests an aerobic common ancestor to the delta-proteobacteria. PLoS One. 2008;3:e2103. doi: 10.1371/journal.pone.0002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanford RA, Cole JR, Tiedje JM. Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl Environ Microbiol. 2002;68:893–900. doi: 10.1128/AEM.68.2.893-900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol. 2007;25:1281–9. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 57.Ivanova N, Daum C, Lang E, Abt B, Kopitz M, Saunders E, et al. Complete genome sequence of Haliangium ochraceum type strain (SMP-2) Stand Genomic Sci. 2010;2:96–106. doi: 10.4056/sigs.69.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iizuka T, Jojima Y, Fudou R, Hiraishi A, Ahn JW, Yamanaka S. Plesiocystis pacifica gen. nov., sp. nov., a marine myxobacterium that contains dihydrogenated menaquinone, isolated from the Pacific coasts of Japan. Int J Syst Evol Microbiol. 2003;53:189–95. doi: 10.1099/ijs.0.02418-0. [DOI] [PubMed] [Google Scholar]
- 59.Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, et al. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science. 2004;303:689–92. doi: 10.1126/science.1093027. [DOI] [PubMed] [Google Scholar]