

Abstract
Metastatic pheochromocytoma continues to be an incurable disease, and treatment with conventional cytotoxic chemotherapy offers limited efficacy. In the present study, we evaluated a novel topoisomerase I inhibitor, LMP-400, as a potential treatment for this devastating disease. We found a high expression of topoisomerase I in human metastatic pheochromocytoma, providing a basis for the evaluation of a topoisomerase 1 inhibitor as a therapeutic strategy. LMP-400 inhibited the cell growth of established mouse pheochromocytoma cell lines and primary human tumor tissue cultures. In a study performed in athymic female mice, LMP-400 demonstrated a significant inhibitory effect on tumor growth with two drug administration regimens. Furthermore, low doses of LMP-400 decreased the protein levels of hypoxia-inducible factor 1 (HIF-1α), one of a family of factors studied as potential metastatic drivers in these tumors. The HIF-1α decrease resulted in changes in the mRNA levels of HIF-1 transcriptional targets. In vitro, LMP-400 showed an increase in the growth-inhibitory effects in combination with other chemotherapeutic drugs that are currently used for the treatment of pheochromocytoma. We conclude that LMP-400 has promising antitumor activity in preclinical models of metastatic pheochromocytoma and its use should be considered in future clinical trials.
According to the World Health Organization tumor classification, pheochromocytomas (PHEOs) are tumors of neuroendocrine origin found in the adrenal glands. Closely related extraadrenal tumors found along the sympathetic or parasympathetic chain are referred to as paragangliomas (PGLs) (1). At least 35% of these tumors are of familial origin, caused by pathogenic mutations in several genes. Recently discovered mutations in the gene for hypoxia inducible factor 2 alpha (HIF2A) associated with multiple PHEOs/PGLs (2–5) opened another line of thinking about their future treatment options and how hereditary tumors could be linked to the HIF-signaling pathway (6, 7).
The only available curative treatment for PHEO/PGL is surgery. When the tumor is unresectable or metastases are present, systemic chemotherapy or radiopharmaceutical therapies are used (8–11). These treatment methods are aimed at stopping metastatic spread and decreasing tumor- and hormone-related events (eg, spinal instability, cardiovascular complications, etc) to improve quality of life and survival (12–14). The use of 131I-metaiodobenzylguanidine might help stabilize disease and lower tumor burden; however, 131I-metaiodobenzylguanidine can be used only when the tumor shows uptake (15). Many traditional chemotherapeutic agents and regimens are used for the treatment of metastatic PHEO, with the combination of cyclophosphamide, vincristine, and dacarbazine (CVD) being the most commonly used of these regimens. Patients receiving CVD often show initial benefit, but the disease usually recurs/progresses, leading to an overall poor prognosis (12, 16–19). A recent clinical review summarized the current and future therapeutic approaches for PHEO and PGL, dividing them into antiproliferative therapeutic strategies and proapoptotic strategies (12, 18, 19). As suggested, targeting topoisomerase I (Top1) may represent an interesting proapoptotic option (20).
Topoisomerases are ubiquitous enzymes essential for replication and transcription. They control DNA supercoiling and entanglement, which makes them attractive targets for anticancer and antibacterial treatment (21). Top1 inhibitors act as interfacial inhibitors by blocking Top1 functions, leading to DNA damage through the formation of double-strand breaks, which, if not repaired, lead to cell death (22, 23). The presence of Top1 is necessary for camptothecin and noncamptothecin Top1 inhibitors (eg, indenoisoquinolines) to exert their cytotoxic effects because Top1 is their primary target (24). However, there have been other effects reported in connection to Top1 inhibition, namely an effect on the hypoxia inducible factor (HIF)-1α protein and HIF-1 transcription targets (25–29). The HIF proteins (HIFs) function as transcription factors, physiologically responding to changes in oxygen levels. In cancer biology, HIFs play crucial roles in several processes such as cancer cell migration, invasiveness, metastasis, and resistance to radio- and chemotherapy (30, 31). The potential modulation of HIF-1α and HIF-2α expression might be of particular interest when treating PHEOs/PGLs because the hypoxic/pseudohypoxic pathway has been widely studied in these tumors (30, 32–35).
LMP-400/indotecan, an HCl salt of NSC 724998 developed by the National Cancer Institute (NCI), is currently undergoing clinical evaluation and represents one of the third-generation Top1 inhibitors (36). Indenoisoquinolines were developed to overcome certain limitations of camptothecin derivatives, which are the only group of Top1 inhibitors approved by the US Food and Drug Administration for the treatment of solid tumors (topotecan, irinotecan). The limitations of camptothecin derivatives include chemical instability, rapid diffusion from Top1-DNA cleavage complexes and active export from cells by efflux pumps. LMP-400 overcomes these limitations (23, 24, 37). Use of camptothecin derivatives has been recently shown as a possible treatment option in an in vitro study by our collaborative group (20).
Two main pharmacodynamic targets can be evaluated in connection with indenoisoquinoline treatment: Top1 and H2A histone family, member X (γ-H2AX). The measurement of pretreatment levels of Top1 in tumor tissue could be a predictive marker for response to indenoisoquinoline treatment and serve as a marker for patient selection. Correlation between Top1 levels and tumor response has been reported in previous studies (36, 38–40). Furthermore, an observed decrease in Top1 levels upon treatment with Top1 inhibitors might serve as a biomarker of target engagement, as proposed by Pfister et al (36, 41). Another pharmacodynamic target that has been extensively validated in connection with indenoisoquinoline treatment is histone γ-H2AX (24, 42). Phosphorylation of H2AX occurs shortly after the formation of DNA double-strand breaks, and the signal strength correlates with the number of breaks formed. Its detection assay was developed and validated by the NCI for use in clinical trials using LMP-400 and other DNA-damaging agents (43).
Our knowledge of signaling pathways involved in PHEO/PGL has been broadened over the past few years, leading to the identification of several promising molecular targets (12, 18, 44). The results of single, targeted molecular therapies seem to be inconclusive, as reported in recent reviews (12, 18). The lack of efficacy of certain agents may be due to compensatory signaling pathways (44). Combination approaches might overcome this issue as well as decreasing the likelihood of development of drug resistance. In the present study, we report our initial experience with LMP-400 both in vitro and in vivo on established animal PHEO cell lines and primary cell cultures from human tumor tissue. Testing included, among others, studies of tumor cell growth inhibition, animal models, drug synergism, and modulation of two pharmacodynamic targets. Additionally, we analyzed the expression of HIF-1α in treated cells because the HIF-1 is transcription factor important in PHEO/PGL tumor development. We conclude that LMP-400 is a promising treatment option for patients with metastatic PHEO and represents a potential candidate for future clinical trials involving patients with these tumors.
Materials and Methods
Cell lines and reagents
Mouse PHEO cell lines (MPC, MTT, and MTT-Luc) were maintained in DMEM supplemented with 10% fetal bovine serum, 5% horse serum, and antibiotics (Gibco-Life Technologies). A rat PHEO cell line (PC12) was maintained in DMEM supplemented with 10% fetal bovine serum and antibiotics (Gibco-Life Technologies). Cells were grown until 80% confluence and then detached using 0.05% trypsin/EDTA, resuspended, and counted to obtain the desired number.
Cells were grown in an incubator in a humidified atmosphere containing 5% CO2 at 37°C. For experiments in which cultivation under hypoxic conditions was necessary, the cells were cultured in a CO2/O2 incubator (MCO-5M; Panasonic), in which the volumes of O2 and CO2 were 1% and 5%, respectively.
LMP-400 (Indotecan, NSC 743400) was provided by the Division of Cancer Treatment and Diagnosis, NCI (Rockville, Maryland). Cisplatin, and vincristine (vincristine sulfate) were purchased from Tocris Bioscience (R&D Systems, Inc). All of the compounds were dissolved in dimethylsulfoxide; the stock solutions were stored at −20°C and were thawed prior to use. Control samples were treated with culture medium.
Cell proliferation assay
Cell proliferation was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, referred to as the MTT assay. Fifteen thousand cells per well were plated in 96-well plates and incubated for 24 hours before drug treatment. After 48 hours of drug treatment, MTT solution (1 mg/mL; Sigma Chemical Co) was added, and plates were incubated at 37°C for 3 hours before measuring the absorbance at 562 nm (Bio-TEK Instruments).
Human samples
Human PHEO/PGL tissue samples were obtained from patients who underwent surgery at our institution under the institutional review board-approved protocol 00-CH-0093 of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (NIH), and all patients gave written informed consent. Normal human adrenal medullas were obtained from anonymous organ donors without evidence of adrenal tumors from the Department of Urology, School of Medicine, Comenius University, Bratislava, Slovakia.
Primary tumor cultures and tyrosine hydroxylase immunocytochemistry
The procedure was performed as previously described (45). Briefly, dissociated cells were plated at low density in RPMI 1640 medium with 15% fetal bovine serum and antibiotics. Cultures in control media or dosed with various concentrations of LMP-400 were maintained for 10 days, with the media replaced every other day. The cells were then fixed and stained for tyrosine hydroxylase (TH). To measure drug-induced cytotoxicity, surviving TH-positive cells were counted.
Real-time PCR
MTT cells were grown to log phase (∼80% confluence) before treatment with indicated concentrations of LMP-400 or control in both hypoxic and normoxic conditions for 8 and 24 hours. Control samples were treated with media. Real-time PCR was performed on a ViiA7 real-time PCR system (Applied Biosystems) according to the manufacturer's recommendation using the TaqMan detection system. TaqMan gene expression assays for Hif1a, Epas1 (Hif2a), Hk2, Vegfa, and Slc2a1 were purchased from Applied Biosystems. 18S rRNA by Applied Biosystems was used as an endogenous control. ^^Cycle threshold values were plotted for the power.
Western blotting
MTT cells were grown to log phase before treatment with indicated concentrations of LMP-400 for 8 hours in hypoxia or normoxia. Control samples were treated with media. Cells were washed twice with ice-cold PBS and lysed in a cell lysis buffer (Cell Signaling Technology) supplemented with complete protease inhibitor cocktail (Roche) and a phosphatase inhibitor cocktail (Cell Signaling Technology). Protein concentrations were measured using the micro-BCA protein assay kit (Thermo Fisher Scientific Inc) according to the manufacturer's recommendation. Proteins were separated by 4%–20% gradient SDS-PAGE (Bio-Rad Laboratories) and transferred to a polyvinylidene fluoride membrane (Millipore). Antibodies against HIF-1α (H-206; Santa Cruz Biotechnology, Inc), phosphohistone γ-H2AX (Millipore), β-actin (Cell Signaling Technology), tyrosine hydroxylase (Immunostar), and topoisomerase I (BD Biosciences) were used. Proteins were visualized using the SuperSignal West Femto maximum sensitivity substrate and SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific Inc). Blots were analyzed using ImageJ 1.37v (Wayne Rasband, NIH).
Synergism analysis
Drug synergism was determined from median effect analysis equations developed by Chou (47). Cell proliferation data were analyzed using CalcuSyn software (Biosoft). The combination index (CI) indicates additivity when the CI is 0.8–1.2; synergism when CI is less than 0.8; and antagonism when CI is greater than 1.2. The dose reduction index (DRI) shows potential dose reduction of each single drug in synergistic combination at a given effect level achieved by combining these drugs (46).
Animal experiments and bioluminescence imaging
All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Animals and approved by the NIH Animal Care and Use Committee (Animal Study Proposal number 12-028 and Public Health Service assurance number A4149-01).
We used a spontaneously metastatic model of PHEO after an sc injection of MTT cells constitutively expressing luciferase (MTT-Luc) in the right flank of female athymic nude mice, as described previously (47). All bioluminescent data were collected and analyzed with a Xenogen IVIS system. The experiments were performed in the NIH Mouse Imaging Facility in accordance with Animal Care and Use Committee regulations. In the initial study, we administered LMP-400 or placebo (vehicle) once a day for 5 consecutive days with a dose of 12 mg/kg, starting 7 days after cell injection. In the following study, we administered 20 mg/kg once a week, starting 7 days after cell injection. The LMP-400 dilution, administration, and manipulation have been previously described (43).
Shortly after being received from vendor, mice in both (treated and control) groups developed an infection from Corynebacterium bovis and were equally treated with trimethoprim and sulfamethoxazole, followed by ampicillin in water.
Statistics
All in vitro experiments were repeated at least two times. After the ANOVA analyses, posttest pairwise comparisons were computed using either the Student-Newman-Keuls posttest (for all pairwise comparisons) or the Dunnett's test (comparing multiple treatments vs a single control), using Prism 6 software (GraphPad Software Inc). Time-course analyses of the animal experiments with bioluminescence imaging data were analyzed using mixed models on the log values to handle the repeating measurements over time, using Stata (release 12 software; StataCorp). These plots show 95% confidence intervals rather than SEMs. To analyze the caliper measurements of tumor size in these same experiments, at specific weeks, the treated and control group values were compared using the two-sample Student's t test on the arcsine-transformed values (necessary due to the substantial skewness of the values and the presence of 0 values). The data were plotted with SEM and considered significant when P < .05 (marked as asterisk).
Results
LMP-400 inhibits the proliferation of mouse and rat PHEO cell lines
Initially we evaluated the effect of LMP-400 on available PHEO cell lines and found that it inhibited the growth of established animal cell lines from mice (MPC and MTT cells) and rats (PC12 cells) in a dose-dependent manner (Figure 1). The MTT assay was additionally repeated for MPC and MTT cell lines in hypoxic conditions with minor changes in the IC50; in MPC cells, the change was from 0.025 μM in normoxia to 0.033 μM in hypoxia, whereas in MTT cells, IC50 changed from 0.04 μM to 0.094 μM (Supplemental Figure 1). We also tested the growth inhibition effect of LMP-400 given for various time intervals. After an 8-hour treatment of MTT cells with LMP-400, we did not observe an effect of the drug on more than 30% of cells at any concentration for the evaluated range (1, 0.1, and 0.01 μM). After 24 hours of LMP-400 treatment, the 1 μM concentration inhibited the growth of approximately 41% cells; for 0.1 and 0.01 μM concentrations, the cell inhibition remained below 30%. The IC50 for MTT cells after 24 hours of treatment was 0.58 μM (Supplemental Figure 2).
Figure 1.

Tumor cell growth inhibition by LMP-400. The tumor cell viability, measured by an MTT assay, after a 48-hour treatment of established animal PHEO cell lines with LMP-400 is shown.
Top1 levels in PHEO/PGL
First, we assessed the tumor content of the Top1 protein because it is known that Top1 protein is a criterion used successfully for the clinical application of LMP-400 (36). Thus, we evaluated the levels of Top1 in sporadic and genetically linked PHEOs/PGLs and compared them with levels of Top1 in normal human adrenal medulla. This analysis showed higher levels of Top1 in all the evaluated tumors compared to the adrenal medulla (see Figure 3A). Similar levels of Top1 expression were observed in sporadic and von Hippel-Lindau (VHL)-mutated tumors; the highest levels of Top1 expression were found in samples from succinate dehydrogenase subunit B (SDHB)-mutated tumors. These tumors are known to have a high metastatic potential and worse outcome than other known familial PHEOs/PGLs (2, 48).
Figure 3.
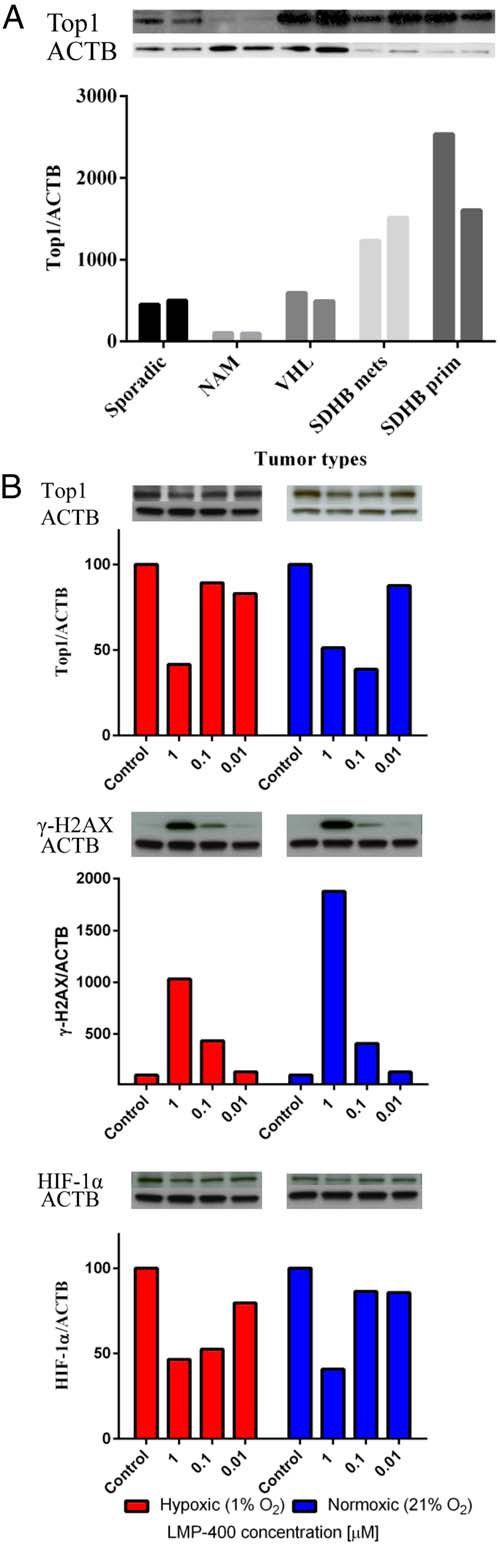
Protein analyses. A, Variable amounts of the Top1 protein in tumors with different genotypes when compared with normal adrenal medulla (NAM). B, The levels of Top1, γ-H2AX, and HIF-1α evaluated after 8 hours of treatment with LMP-400 in hypoxic (1% O2, red curve) and normoxic conditions (21% O2, blue curve). Each oxygen condition has its own control, as presented in the figure.
LMP-400 inhibits proliferation of primary PHEO cells
Based on the findings that human PHEOs/PGLs exhibit high levels of Top1, we initiated treatment with LMP-400 for 10 consecutive days using two independent primary human cancer cell cultures derived from two sporadic PHEOs. Immunostaining for TH, an enzyme necessary for catecholamine production, was used to distinguish chromaffin cells from other cells in the primary cell culture. Figure 2 shows a concentration-dependent decrease in cell proliferation for cells treated with LMP-400.
Figure 2.
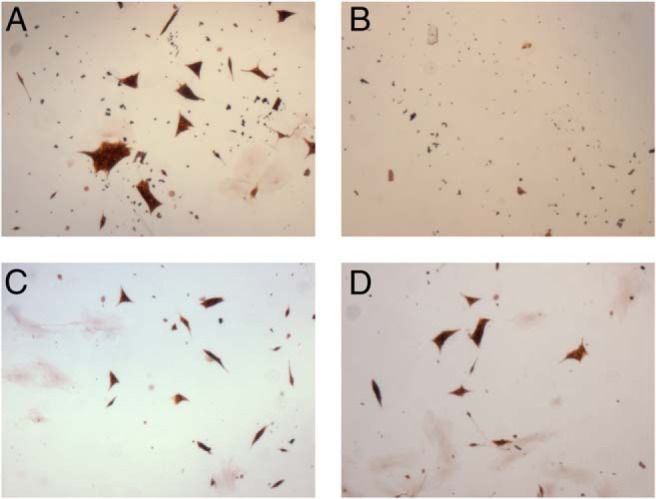
Primary cell culture growth inhibition by LMP-400. The figure shows the effect of LMP-400 on a primary cell culture, with decreasing concentrations of the drug: control (A), 1 μM (B), 0.1 μM (C), and 0.01 μM (D). The primary culture was derived from a 10- × 9- × 8-cm PHEO in a 53-year-old patient of Greek descent with typical clinical symptoms. Genetic testing for succinate dehydrogenase subunits B, C, and D was negative; other testing was not performed.
LMP-400 decreases Top1 and increases γ-H2AX levels in MTT cells
Pharmacodynamic assays for Top1 and phospho γ-H2AX were previously developed for clinical trials with LMP-400 (36, 43). Thus, we evaluated the effect of LMP-400 on these potential biomarkers in MTT cells, measuring target proteins after 8 hours of treatment with several drug concentrations in normoxic and hypoxic conditions. The levels of Top1 in MTT cells decreased in a concentration-dependent fashion in both hypoxic and normoxic conditions. On the other hand, the levels of phospho γ-H2AX, which were almost not present in control cells, peaked upon treatment with LMP-400. MTT cells showed stable levels of the HIF-1α protein in both hypoxic and normoxic conditions. This expression was decreased after 8 hours of treatment with LMP-400 (Figure 3B).
LMP-400 affected expression of HIF-1 targets in MTT cells
To determine the effect of LMP-400 treatment on Hif1a gene expression in MTT cells, we treated these cells with increasing LMP-400 concentrations. To limit the potential effect of cell apoptosis on mRNA expression, we used concentrations that did not affect cell proliferation in more than 30% of the cells. mRNA was extracted after 8 and 24 hours of LMP-400 treatment in both normoxic and hypoxic conditions (Supplemental Figure 2).
After an 8-hour treatment, we did not observe a significant decrease in Hif1a expression, but a decrease in Hif1a expression was apparent after 24 hours of treatment in both hypoxia and normoxia (Supplemental Figure 3). Because HIFs serve as transcription factors, determining the significance of changes in their expression levels is best measured by the evaluation of the expression levels of their target genes. We found a significant decrease in two very well-established preferential HIF1 targets at the highest concentration tested, irrespective of time or oxygen conditions: solute carrier family 2, facilitated glucose transporter member 1 (Slc2a1), better known as glucose transporter type 1 (Glut1), and hexokinase 2 (Hk2) (Figure 4 and Supplemental Figure 4).
Figure 4.

Glut1 (Slc2a1) mRNA level changes. The figure depicts changes in the mRNA levels of Glut1 (Slc2a1) upon 8- and 24-hour treatments with LMP-400 in hypoxic (1% O2, red column) and normoxic conditions (21% O2, blue column).
LMP-400 reduced tumor growth and metastatic potential in vivo
To elucidate the effects of LMP-400 in an in vivo model, we used a well-established model of spontaneously metastatic PHEO, taking advantage of MTT-Luc bioluminescence imaging (47). Seven days after the sc implantation of MTT-Luc cells, we started 5 continuous days of 12 mg/kg LMP-400 iv application, which led to an overall significant decrease in tumor growth (P = .0005) when measured by bioluminescence. The sc injection of MTT-Luc cells allowed us to measure tumor growth externally by caliper, and this measurement also confirmed a significant effect of LMP-400 on growth in vivo (P = .003 for wk 3; P = .015 for wk 4). Over time, the implanted cells started to migrate and metastases developed. After the mice had been euthanized, the lungs and liver were harvested and bioluminescence measurements were performed, showing that LMP-400 decreased the development of metastases (in lungs P = .002; in liver P = .091). A consecutive study with an alternative dosing schedule of 20 mg/kg once a week also showed a significant difference in tumor growth (P = .044, bioluminescence measurement) when compared with a group that received only a vehicle. The significance of the bioluminescence measurement was confirmed by external caliper measurements (wk 5 P = .037) (Figure 5).
Figure 5.

In vivo study of LMP-400. The figure shows the effect of LMP-400 on tumor growth when dosed for 5 consecutive days (A) or once a week (B).
LMP-400 as a part of combination treatment
CVD treatment represents one of the best available chemotherapeutic regimens for patients with metastatic PHEO/PGL, although it can be modified, as mentioned earlier. Because this combination cannot be tested in vitro (eg, dacarbazine, which is an essential part of this combination is a prodrug that needs to be activated by the liver), we attempted to closely simulate CVD treatment by combining cisplatin and vincristine. We aimed to show the potential of LMP-400 by adding it to this blend. Testing this combination in concentrations of original single drugs (cisplatin, vincristine, LMP-400) below and above their IC50 showed high synergism at lower concentrations, which turned into an additive effect at the second-highest concentration. At the highest concentration, in which the effect of a single drug alone was already very potent, the synergism was not present. Table 1 shows CI and DRI values when the synergism was evaluated between LMP-400, vincristine, and cisplatin.
Table 1.
Combinational Testing
| Drug Dose, μM |
Fa | CI | DRI |
||||
|---|---|---|---|---|---|---|---|
| LMP-400 | VIN | CIS | LMP-400 | VIN | CIS | ||
| 0.001 | 0.001 | 0.1 | 0.173 937 | 0.653 | 9.752 | 17.008 | 2.034 |
| 0.005 | 0.005 | 0.5 | 0.345 735 | 0.48 | 13.447 | 9.032 | 3.394 |
| 0.01 | 0.01 | 1 | 0.456 195 | 0.395 | 17.732 | 7.376 | 4.925 |
| 0.05 | 0.05 | 5 | 0.777 163 | 0.202 | 70.506 | 6.693 | 26.315 |
| 0.1 | 0.1 | 10 | 0.823 222 | 0.274 | 64.664 | 4.549 | 25.626 |
| 0.5 | 0.5 | 50 | 0.871 295 | 0.857 | 28.355 | 1.353 | 12.143 |
| 1 | 1 | 100 | 0.886 617 | 1.439 | 19.186 | 0.788 | 8.466 |
Abbreviations: CIS, cisplatin; Fa, fractions of affected cells; VIN, vincristine. Shown are the Fa with different doses of tested drugs on MTT cell survival after 48 hours of treatment with corresponding values of the CI and DRI.
Discussion
In the present study, we evaluated the complex effect exerted by LMP-400 on PHEO/PGL in vivo and in vitro. LMP-400 distinctly inhibited the growth of human and animal PHEO/PGL cells. The effect of the agent on both the pharmacodynamic markers evaluated (Top1 and γ-H2AX) was found to be significant in MTT cells. The HIF-1α protein and HIF-1 transcriptional targets were also significantly affected by LMP-400 in MTT cells.
The Food and Drug Administration approved Top1 inhibitors, topotecan and irinotecan, are derivatives of camptothecins. Although they both target Top1, their clinical use is different. Whereas topotecan is used to treat ovarian and lung cancers, irinotecan has been shown to be effective in the treatment of colon cancer (21). Pharmacodynamic and clinical limitations of camptothecin derivatives led to the development of noncamptothecin Top1 inhibitors, including LMP-400 (23). Top1 inhibitors were described as interfacial inhibitors that prevent Top1 functions, leading to double-strand DNA breaks, which, if not repaired, lead to cell death (23, 49). It was proposed that the pretreatment levels of Top1 might be an important factor in determining the effectiveness of the Top1 inhibitors and thus predicting the treatment response (36). Our evaluation of Top1 protein levels in several types of PHEO/PGL showed the highest levels of this protein (compared with normal adrenal medulla) in SDHB-mutated tumors. This is of interest because Top1 could represent a therapeutic target in this disease and could lead to the development of more effective drugs for this population.
First, we tested the efficacy of LMP-400 in vitro using MPC, MTT, and PC12 cell lines. MPC cells were derived from an Nf1 knockout mouse that developed PHEO; the MTT cell line was derived from a liver metastasis of an MPC tumor and is thus considered to be the most aggressive available PHEO cell line, which guided our decision to use this cell line for most parts of this complex drug evaluation study. The PC12 cell line is of rat origin and is used as a model cell line for neuroendocrine tumors (50, 51). LMP-400 showed efficacy in all animal PHEO cell lines, with IC50 in the tens of nanomolar concentration range. There is no available human cell line for PHEO/PGL, which would tremendously enhance the possibilities in the search for new therapeutic options. Despite extensive ongoing research, none of the attempts has been successful. This makes the primary cell cultures prepared from tumor tissue obtained from NIH patient surgeries the best option for current drug testing, although animal and human pheochromocytoma cells are known to have a different growth rate. As we previously mentioned in Results, LMP-400 substantially inhibited the growth of these primary cell cultures.
These studies suggest that LMP-400 might be a promising therapeutic avenue for PHEO/PGL. Because in vivo studies are essential for introducing a new drug into clinical practice, we took advantage of a metastatic PHEO/PGL animal model that has been used in our previous studies and allows for noninvasive, repeatable, and reproducible in vivo tumor measurement (47). In the initial animal study, when the drug was applied constitutively for 5 days, a statistically significant effect was reached, established by the measurement of bioluminescence as well as external tumor measurement by caliper. LMP-400 also showed an effect on the development of metastases. The same 5-day dosing schedule was previously tested in a model study of LMP-400 using mice bearing A375 tumor xenografts (human malignant melanoma) (43). An alternative for this monthly dosing plan is a weekly schedule using a higher single dose of 20 mg/kg. We in fact implemented this scheme of dosing into our study, and tumor growth inhibition was also significant. However, there was an obvious difference between the two approaches. Monthly dosing, with the cumulative dose applied in the early stage of MTT-Luc cell tumor development, led to very significant efficacy at the beginning of the study, which then went down gradually. In contrast, weekly dosing required a longer time to exert a significant effect, resulting in decreased formation of MTT-Luc cell tumors. We believe that the observed difference between the two dosing schemes can be caused by the aggressiveness of MTT cells.
The use of combined chemotherapeutic approaches can be beneficial for patients if the drugs show a synergistic effect, eventually leading to a reduction in drug dose with the mitigation of some side effects and reduction in the development of resistance, in contrast to full dosing of the drug (46). Despite its weaknesses, CVD chemotherapy has been found to be the best available chemotherapy regimen for PHEO/PGL (12, 16–18). We attempted to imitate the CVD regimen in in vitro conditions using vincristine and cisplatin with the addition of LMP-400 to this combination. This treatment led to a decreased cell growth in concentrations below and above the respective IC50, suggesting the possibility of adding LMP-400 to the CVD regimen. We believe that the synergism at low doses might be potentially important.
For use in clinical trials with LMP-400, two assays focused on the pharmacodynamic markers Top1 and γ-H2AX were developed (36, 43). We tested these markers when treating MTT cells with LMP-400 and observed a dramatic increase in γ-H2AX, which indicates the development of DNA damage after the treatment and can be considered as an induction of early chromatin modification after the initiation of DNA fragmentation during apoptosis (52). We also observed a decrease in Top1. Both of these effects were achieved in both normoxic and hypoxic conditions. Showing the LMP-400 effectiveness in hypoxic conditions is of great importance because hypoxic conditions are associated with tumor aggressiveness, progression, and acquired resistance to treatment (53).
It was previously shown that topoisomerase inhibitors also deliver effects beyond cytotoxicity. When LMP-400 was tested at lower concentrations (inhibiting the growth of less than 30% of cells in a given time period), a decrease in HIF-1α protein was observed. HIF-1/2α were proposed to be the mediators of hypoxic signaling in VHL- and SDHx-mutated PHEOs/PGLs (3, 54). In an unsupervised analysis of the transcriptional profile of these tumors, reduced oxidoreductase and angiogenesis/hypoxia were seen, suggesting that these tumors have similar profiles, leading to their categorization as cluster 1 tumors. Activation of receptor tyrosine kinases, possibly leading to increased transcription of HIF1α is, however, more common for cluster 2, consisting of PHEO/PGL with germline mutations in several other susceptibility genes (mainly RET, NF1, and MAX). Sporadic PHEOs/PGLs are equally distributed in both clusters (33, 55). Thus, targeting HIF-1α might be of potential interest for all PHEOs/PGLs (35, 44). Although the levels of HIF-1α and HIF-2α in PHEO/PGL, once referred to as rivaling siblings (31), were previously evaluated, no unifying pattern was found (32, 56, 57). Therefore, the balance between these two proteins in PHEO/PGL is still inconclusive but may possibly also depend on the development stage of the tumor. Changes in HIF-1α are not specific to Top1 inhibitors because similar results were obtained after treatment with Top2 inhibitors, but specific genes are likely to respond individually to topoisomerase inhibition. The response can result directly from enzyme inhibition or might be due to a secondary mechanism (58). Previous reports showed that HIF-1α changes are not transcriptional, which is not consistent with the present study because we found changes in the mRNA levels of Hif1a targets after prolonged treatment. Nevertheless, the specific pathway causing changes in Hif1a levels after topoisomerase treatment needs to be further studied. The presence of Top1 does seem to be a unifying condition for its exertion (26, 28, 29).
We have also evaluated the effects of 1 μM LMP-400 on HIF-1 target gene expression. Because this concentration is already toxic after prolonged treatment, we treated the cells with the drug for only 8 and 24 hours. The data show a consistent expressional decrease in the known HIF-1 target genes including Glut1, Hk2, and Vegfa under hypoxic conditions after both 8 and 24 hours (Figure 4 and Supplemental Figures 4 and 5). In normoxia, mRNA levels of all target genes were also significantly lower in all conditions when compared with control cells with the exception of Vegfa after an 8-hour treatment. Expression of Hif2a was increased after an 8-hour treatment, irrespective of oxygen conditions. This increase was not present after prolonged treatment (Supplemental Figure 6). Due to this observation, we were eager to evaluate its transcriptional targets, looking for a possible interplay between HIFs. It has been shown that Top1 inhibitors, rather than decreasing the expression of several genes, increase the mRNA levels of prostaglandin-endoperoxide synthase 2, also known as cyclooxygene 2 (Ptgs2 and Cox-2, respectively). This was discussed in connection with potential nuclear factor-κB activation (29). We also evaluated this gene but found a significant increase only in its mRNA levels under normoxic conditions after treatment with 1 μM of LMP-400 for 8 hours (Supplemental Figure 7).
In conclusion, LMP-400, whose effects were thoroughly evaluated on the best available PHEO/PGL models, represents a promising step in the search for new treatment options for PHEO/PGL patients.
Acknowledgments
We acknowledge Dr James Doroshow (National Cancer Institute, Bethesda, Maryland) for his support. We thank the Division of Cancer Treatment and Diagnosis, National Cancer Institute (Rockville, Maryland) for providing the LMP-400.
This work was supported the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 3880
- CI
- combination index
- CVD
- cyclophosphamide, vincristine, and dacarbazine
- DRI
- dose reduction index
- γ-H2AX
- H2A histone family, member X
- HIF
- hypoxia inducible factor
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PGL
- paraganglioma
- PHEO
- pheochromocytoma
- SDHB
- succinate dehydrogenase subunit B
- TH
- tyrosine hydroxylase
- Top1
- topoisomerase I
- VHL
- von Hippel-Lindau.
References
- 1. DeLellis RA. Pathology and genetics of tumours of endocrine organs. Lyon, France: IARC Press; 2004. [Google Scholar]
- 2. Karasek D, Shah U, Frysak Z, Stratakis C, Pacak K. An update on the genetics of pheochromocytoma. J Hum Hypertens. 2013;27:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pacak K, Jochmanova I, Prodanov T, et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013;31:1690–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Comino-Mendez I, de Cubas AA, Bernal C, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22:2169–2176. [DOI] [PubMed] [Google Scholar]
- 6. Jochmanova I, Lazurova I. A new twist in neuroendocrine tumor research: Pacak-Zhuang syndrome, HIF-2alpha as the major player in its pathogenesis and future therapeutic options. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:175–180. [DOI] [PubMed] [Google Scholar]
- 7. Vicha A, Musil Z, Pacak K. Genetics of pheochromocytoma and paraganglioma syndromes: new advances and future treatment options. Curr Opin Endocrinol Diabetes Obes. 2013;20:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adjalle R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009;41:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ayala-Ramirez M, Feng L, Habra MA, et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012;118:2804–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baudin E, Habra MA, Deschamps F, et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol. 2014;171:R111–R122. [DOI] [PubMed] [Google Scholar]
- 11. Taieb D, Kaliski A, Boedeker CC, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014;er20141026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plouin PF, Fitzgerald P, Rich T, et al. Metastatic pheochromocytoma and paraganglioma: focus on therapeutics. Horm Metab Res. 2012;44:390–399. [DOI] [PubMed] [Google Scholar]
- 13. Taieb D, Timmers HJ, Hindie E, et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imag. 2012;39:1977–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94:4757–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ilias I, Divgi C, Pacak K. Current role of metaiodobenzylguanidine in the diagnosis of pheochromocytoma and medullary thyroid cancer. Semin Nucl Med. 2011;41:364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholz T, Eisenhofer G, Pacak K, Dralle H, Lehnert H. Clinical review: current treatment of malignant pheochromocytoma. J Clin Endocrinol Metab. 2007;92:1217–1225. [DOI] [PubMed] [Google Scholar]
- 18. Matro J, Giubellino A, Pacak K. Current and future therapeutic approaches for metastatic pheochromocytoma and paraganglioma: focus on SDHB tumors. Horm Metab Res. 2013;45:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jimenez C, Rohren E, Habra MA, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep. 2013;5(4):356–371. [DOI] [PubMed] [Google Scholar]
- 20. Powers JF, Korgaonkar PG, Fliedner S, et al. Cytocidal activities of topoisomerase 1 inhibitors and 5-azacytidine against pheochromocytoma/paraganglioma cells in primary human tumor cultures and mouse cell lines. PLoS One. 2014;9:e87807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sordet O, Khan QA, Kohn KW, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anticancer Agents. 2003;3:271–290. [DOI] [PubMed] [Google Scholar]
- 23. Pommier Y, Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther. 2009;8:1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Antony S, Agama KK, Miao ZH, et al. Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer Res. 2007;67:10397–10405. [DOI] [PubMed] [Google Scholar]
- 25. Beidler DR, Cheng YC. Camptothecin induction of a time- and concentration-dependent decrease of topoisomerase I and its implication in camptothecin activity. Mol Pharmacol. 1995;47:907–914. [PubMed] [Google Scholar]
- 26. Choi YJ, Rho JK, Lee SJ, et al. HIF-1α modulation by topoisomerase inhibitors in non-small cell lung cancer cell lines. J Cancer Res Clin Oncol. 2009;135:1047–1053. [DOI] [PubMed] [Google Scholar]
- 27. Guerin E, Raffelsberger W, Pencreach E, et al. In vivo topoisomerase I inhibition attenuates the expression of hypoxia-inducible factor 1α target genes and decreases tumor angiogenesis. Mol Med. 2012;18:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64:1475–1482. [DOI] [PubMed] [Google Scholar]
- 29. Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- 30. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pollard PJ, El-Bahrawy M, Poulsom R, et al. Expression of HIF-1α, HIF-2α (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91:4593–4598. [DOI] [PubMed] [Google Scholar]
- 33. Dahia PL, Ross KN, Wright ME, et al. A HIF1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jochmanova I, Chunzhang Y, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst. 2013;105(17):1270–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfister TD, Hollingshead M, Kinders RJ, et al. Development and validation of an immunoassay for quantification of topoisomerase I in solid tumor tissues. PLoS One. 2012;7:e50494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cinelli MA, Reddy PV, Lv PC, et al. Identification, synthesis, and biological evaluation of metabolites of the experimental cancer treatment drugs indotecan (LMP400) and indimitecan (LMP776) and investigation of isomerically hydroxylated indenoisoquinoline analogues as topoisomerase I poisons. J Med Chem. 2012;55:10844–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meisenberg C, Ward SE, Schmid P, El-Khamisy SF. TDP1/TOP1 ratio as a promising indicator for the response of small cell lung cancer to topotecan. J Cancer Sci Ther. 2014;6:258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giovanella BC, Stehlin JS, Wall ME, et al. DNA topoisomerase I-targeted chemotherapy of human colon cancer in xenografts. Science. 1989;246:1046–1048. [DOI] [PubMed] [Google Scholar]
- 40. Braun MS, Richman SD, Quirke P, et al. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol. 2008;26:2690–2698. [DOI] [PubMed] [Google Scholar]
- 41. Pfister TD, Reinhold WC, Agama K, et al. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol Cancer Ther. 2009;8:1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinders RJ, Hollingshead M, Lawrence S, et al. Development of a validated immunofluorescence assay for gammaH2AX as a pharmacodynamic marker of topoisomerase I inhibitor activity. Clin Cancer Res. 2010;16:5447–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol. 2012;23:21–33. [DOI] [PubMed] [Google Scholar]
- 45. Giubellino A, Bullova P, Nolting S, et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology. 2013;154:646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. [DOI] [PubMed] [Google Scholar]
- 47. Giubellino A, Woldemichael GM, Sourbier C, et al. Characterization of two mouse models of metastatic pheochromocytoma using bioluminescence imaging. Cancer Lett. 2012;316:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amar L, Baudin E, Burnichon N, Peyrard S, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. [DOI] [PubMed] [Google Scholar]
- 49. Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov. 2012;11:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Korpershoek E, Pacak K, Martiniova L. Murine models and cell lines for the investigation of pheochromocytoma: applications for future therapies? Endocr Pathol. 2012;23:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martiniova L, Lai EW, Elkahloun AG, et al. Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin Exp Metastasis. 2009;26:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–9395. [DOI] [PubMed] [Google Scholar]
- 53. Tatum JL, Kelloff GJ, Gillies RJ, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol. 2006;82:699–757. [DOI] [PubMed] [Google Scholar]
- 54. Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. [DOI] [PubMed] [Google Scholar]
- 55. Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. [DOI] [PubMed] [Google Scholar]
- 56. Eisenhofer G, Huynh TT, Pacak K, et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer. 2004;11:897–911. [DOI] [PubMed] [Google Scholar]
- 57. Favier J, Briere JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4:e7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Collins I, Weber A, Levens D. Transcriptional consequences of topoisomerase inhibition. Mol Cell Biol. 2001;21:8437–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]