

Abstract
Prior studies demonstrated increased plasma IgE in diabetic patients, but the direct participation of IgE in diabetes or obesity remains unknown. This study found that plasma IgE levels correlated inversely with body weight, body mass index, and body fat mass among a population of randomly selected obese women. IgE receptor FcϵR1-deficient (Fcer1a−/−) mice and diet-induced obesity (DIO) mice demonstrated that FcϵR1 deficiency in DIO mice increased food intake, reduced energy expenditure, and increased body weight gain but improved glucose tolerance and glucose-induced insulin secretion. White adipose tissue from Fcer1a−/− mice showed an increased expression of phospho-AKT, CCAAT/enhancer binding protein-α, peroxisome proliferator-activated receptor-γ, glucose transporter-4 (Glut4), and B-cell lymphoma 2 (Bcl2) but reduced uncoupling protein 1 (UCP1) and phosphorylated c-Jun N-terminal kinase (JNK) expression, tissue macrophage accumulation, and apoptosis, suggesting that IgE reduces adipogenesis and glucose uptake but induces energy expenditure, adipocyte apoptosis, and white adipose tissue inflammation. In 3T3-L1 cells, IgE inhibited the expression of CCAAT/enhancer binding protein-α and peroxisome proliferator-activated receptor-γ, and preadipocyte adipogenesis and induced adipocyte apoptosis. IgE reduced the 3T3-L1 cell expression of Glut4, phospho-AKT, and glucose uptake, which concurred with improved glucose tolerance in Fcer1a−/− mice. This study established two novel pathways of IgE in reducing body weight gain in DIO mice by suppressing adipogenesis and inducing adipocyte apoptosis while worsening glucose tolerance by reducing Glut4 expression, glucose uptake, and insulin secretion.
IgE activates mast cells by binding to its high-affinity receptor Fcϵ receptor-1 (FcϵR1). This activity of IgE is essential to allergic responses (1), such as asthma. Recent studies demonstrated that IgE also activates macrophages and T cells (2, 3). All these IgE-targeting cells play detrimental roles in obesity and diabetes (4–6), suggesting the participation of IgE in these metabolic diseases. Although the direct role of IgE in obesity and diabetes remains untested, asthma associates with increased plasma IgE (7) and acts as an important risk factor of obesity and diabetes. Of 4773 subjects aged 20 years and older randomly selected from 10 348 individuals from 2005 to 2006 in the National Health and Nutrition Examination Survey in the United States, IgE concentrations correlated positively with obesity risk but not insulin resistance in asthmatic patients (8). Of 4321 children aged 2–19 years from the same population, obese and overweight children had higher plasma total IgE levels, driven largely by allergic sensitivity to foods (9). A respective study of 246 adults with asthma and other atopic disorders revealed that asthmatic patients had higher body mass indices (BMI) than nonasthmatics. Obesity associated with increased serum IgE among those patients (10). Yet in a population study of 666 patients with severe asthma, plasma IgE levels correlated negatively with BMI (11). These studies therefore do not prove the direct participation of IgE in body weight gain. Previous studies investigated IgE in patients and animals with diabetes. A linear regression analysis of a population study of 340 patients aged 55–75 years revealed a positive correlation between plasma IgE and type 2 diabetes mellitus and prediabetes status. Ordinal logistic regression demonstrated that plasma IgE correlates with the incidence of type 2 diabetes before and after adjusting for common diabetes risk factors (12, 13). In nonobese diabetic mice, anti-FcϵR1 antibody therapy activated basophils and mast cells but delayed type 1 diabetes (14). These observations from diabetic patients and mice highlight the role of IgE in diabetes.
This study design was 2-fold: to test the direct role of IgE in obesity and diabetes using FcϵR1-deficient Fcer1a−/− mice in diet-induced obese and diabetic mice and to understand the molecular and cellular mechanism by which this Ig molecule contributes to these metabolic diseases.
Materials and Methods
Patients
A random selection from a bariatric surgery program from the Institute of Cardiometabolism and Nutrition, Pitié-Salpêtrière Hospital (Paris, France) yielded a cohort of 50 Caucasian women with morbid obesity. These patients met the criteria for bariatric surgery (BMI ≥ 40 kg/m2 or ≥ 35 kg/m2 with at least one comorbidity: hypertension, type 2 diabetes, dyslipidemia, or obstructive sleep apnea syndrome) but without allergic or autoimmune diseases or antiallergy and antiautoimmunity medications that may affect plasma IgE levels. Subjects had stable weights (±3 kg) for at least 3 months before the surgery. Of the 50 patients, 18 (36%) had type 2 diabetes as defined by a fasting glycemia greater than 7 mmol/L and/or the use of an antidiabetic drug. The Ethics Committees of the CPP Ile de France 1 (number 0611351) approved the clinical investigations. All subjects gave written informed consent. A Spearman's correlation test helped test the correlation between plasma IgE concentration and clinical and biological parameters at baseline. Patient body composition was determined by dual-energy x-ray absorptiometry (DEXA; Hologic). Blood samples were obtained before the bariatric surgery after 12 hours of fasting to measure total cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides, insulin, glucose, glycated hemoglobin (HbA1c), leptin, adiponectin, inflammatory markers (highly sensitive C-reactive protein (hs-CRP) and IL-6, and IgE as previously described (12, 13, 15, 16).
Mice
We used C57BL/6 (Jackson Laboratory) and Fcer1a−/− mice (C57BL/6, N9) (2, 3). All mice used in this study were littermates. Male (or female) mice at 6 weeks of age from each group were fed a high-fat diet (HFD; D12492: 60% kilocalories from fat; Research Diets Inc) for 17 weeks. Mouse body weight was monitored weekly. After 17 weeks on a HFD, mouse total body fat and lean masses were assessed by DEXA (PIXImus). For the calorimetric analysis, these mice were placed individually in an indirect open circuit calorimeter (Oxymax System; Columbus Instruments). Oxygen and carbon dioxide concentrations by volume were monitored at the inlet and outlet parts of a partially sealed chamber, through which a known flow of ambient air was forcibly ventilated. The concentration difference measured between the parts was used to compute oxygen consumption and carbon dioxide production. The consumption and production information were presented in units of milliliters per kilogram per hour and normalized to 25°C and 760 mm Hg. Food intake was investigated by using the Oxymax feed scale device (Columbus Instruments) for 3 continuous days, and the data were presented as the average food intake per day of the last 2 days without considering the first day acclimation period. The physical activity of the mice was monitored with an OPTO-M3 activity application device (Columbus Instruments). The movements (other than scratching, grooming, digging, etc) of each animal were determined by infrared beams in the x-, y-, and z-axes. After 17 weeks on a HFD, an ip glucose tolerance test (1.5 g/kg glucose per body weight) and an insulin tolerance test (1.5 U/kg body weight) were also performed after an overnight (16 h) and a daytime 5-hour fast, respectively. The mice were killed and the fat tissue was collected. The mice were bred and maintained according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Bethesda, Maryland). The Harvard Medical School Standing Committee on Animals approved all of the animal protocols.
Cell culture
3T3-L1 cells (American Type Culture Collection; CL-173) were cultured in DMEM (Life Technologies) including 10% calf serum and L-glutamine. To induce adipogenesis, complete confluent 3T3-L1 cells were cultured in induction media containing DMEM (Life Technologies), 10% fetal bovine serum, L-glutamine, MEM sodium pyruvate, 0.0115 g/mL 3-isobutyl-1-methylxanthine (Sigma), 1 mM dexamethasone (Sigma), and 167 μM insulin (Sigma) for 2 days and for an additional 6 days without 3-isobutyl-1-methylxanthine and dexamethasone.
Islet isolation and glucose-stimulated insulin secretion by islets
Islets were isolated from 2-month-old male C57BL/6J mice (Jackson Laboratory) by the intraductal collagenase digestion method, as described previously (17). For the glucose-stimulated insulin secretion assay, after culturing for 12 hours in RPMI 1640 medium containing 5.6 mM glucose and supplemented with 10% fetal calf serum, 10 size-matched islets were incubated at 37°C for 1.5 hours in Krebs-Ringer bicarbonate buffer containing 2.8, 8.3, or 22.2 mM glucose with or without IgE (0.01, 0.1, 1, 10, or 100 μg/mL). The insulin levels in the culture media were measured using an insulin ELISA kit (Crystal Chem Inc). FcϵR1α mRNA levels were measured in total RNA extracted from 50 islets that were incubated at 37°C for 24 hours in RPMI 1640 medium containing 5.6 mM glucose with 10% fetal calf serum in the presence or absence of IgE (0.01, 0.1, 1, 10, or 100 μg/mL). Each quantitative reaction was performed in duplicate.
Quantitative real-time PCR
Total RNA was extracted from white adipose tissue (WAT), 3T3-L1 cells, bone marrow-derived macrophages, or islets using a QIAGEN RNA extraction kit (QIAGEN). Quantified total RNA using Nanodrop 2000 (Thermo Fisher Scientific Inc) was transcribed into first-strand cDNA using Superscript first-strand kit (Life Technologies). Real-time PCR (RT-PCR) was performed using SYBR green super mix (Bio-Rad Laboratories) in a Bio-Rad iCycler iQ to determine the mRNA levels of CCAAT/enhancer binding protein-α (C/EBPα), peroxisomal proliferator-activated receptor-γ (PPAR-γ), and three FcϵR1 chains (α, β, and γ) using 36B4 (acidic ribosomal protein) and β-actin as internal controls to normalize gene expression. RT-PCR data were analyzed based on ΔΔcycle threshold calculation and presented as the fold of change obtained from the value of 2̂(-ΔΔcycle threshold). All RT-PCR primer sequences are listed in Supplemental Table 1.
Immunoblotting and immunohistochemistry
WAT, brown adipose tissue (BAT), and cells were lysed in a RIPA buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, proteinase inhibitor (Roche Diagnostics Corp), and phosphatase inhibitor cocktail (Roche Diagnostics). Tissue lysates were centrifuged at 20 000 × g for 15 minutes. The supernatant was removed without interrupting the upper layer fat for protein concentration determination using the DC protein assay kit (Bio-Rad Laboratories). Tissue or cell lysate was separated by SDS-PAGE, blotted, and detected with different antibodies, including FcϵR1a, glucose transporter-4 (Glut4), B-cell lymphoma 2 (Bcl2), phosphorylated (p) c-Jun N-terminal kinase (JNK), total JNK, p-AKT, total AKT, CEBPα, PPARγ, uncoupling protein 1 (UCP1), and β-actin or glyceraldehyde 3-phosphate dehydrogenase (Table 1). WAT paraffin sections (6 μm) were prepared for immunohistochemistry with antibodies to detect macrophages (Mac-2), T cells (CD3), and FcϵR1, and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining (in situ cell death detection kit; Roche Diagnostics Corp) to detect apoptotic cells. We used AlexaFluor conjugated with different fluorochromes (Invitrogen) to show localization of FcϵR1 to inflammatory cells. All antibodies are listed in Supplemental Table 2.
Table 1.
Antibody Table
| Protein Name | Antibody Name | Manufacturer, Catalog Number, or Name of Individual Provider | Species Raised (Monoclonal or Polyclonal) | Dilution Used |
|---|---|---|---|---|
| Mac-2 | Rabbit antimouse Mac 2 | Cedarlane, 119–12296 | Rabbit polyclonal | 1:100 (IHC) |
| CD3 | Rabbit antihuman CD3 | Abcam, ab16669 | Rabbit monoclonal | 1:60 (IHC) |
| FcϵR1α | Antimouse FcϵRI-α | eBioscience, 14-5898-82 | Armenian Hamster monoclonal | 1:50 (IHC) |
| FcϵR1α | Antirabbit FcϵRI-α | Upstate, 06–725 | Rabbit polyclonal | 1:1000 (IB) |
| Bcl-2 | Bcl-2 antibody | Cell Signaling Technology, 2876S | Rabbit polyclonal | 1:1000 (IB) |
| p-JNK | Phospho-SAPK/JNK (Thr183/Tyr185) (G9) mouse mAb | Cell Signaling Technology, 5131 | Mouse monoclonal | 1:1000 (IB) |
| JNK | SAPK/JNK antibody | Cell Signaling Technology, 9252S | Rabbit polyclonal | 1:500 (IB) |
| p-AKT | Phospho-Akt (Ser473) (D9E) XP rabbit mAb | Cell Signaling Technology, 4060S | Rabbit monoclonal | 1:1000 (IB) |
| AKT | Akt antibody | Cell Signaling Technology, 9272S | Rabbit polyclonal | 1:3000 (IB) |
| Glut4 | Glut4 antibody (C-20) | Santa Cruz Biotechnology, sc-1608 | Goat polyclonal | 1:400 (IB) |
| CEBPα | C/EBPα antibody (14 AA) | Santa Cruz Biotechnology, sc-61 | Rabbit polyclonal | 1:1000 (IB) |
| PPARγ | PPARγ antibody (H-100) | Santa Cruz Biotechnology, sc-7196 | Rabbit polyclonal | 1:1000 (IB) |
| UCP1 | UCP1 antibody (C-17) | Santa Cruz Biotechnology, sc-6528 | Goat polyclonal | 1:3000 (IB) |
| β-Actin | Monoclonal anti-β-actin | Sigma, A5441 | Mouse monoclonal | 1:2000 (IB) |
Abbreviations: IB, immunoblot; IHC, immunohistochemistry; mAb, monoclonal antibody; SAPK, stress-activated protein kinase. This table is a list of antibodies used for immunoblot analysis and immunostaining.
Enzyme-linked immunosorbent assay
An ELISA determined plasma IL-6 (eBioscience), monocyte chemotactic protein-1 (MCP-1; eBioscience), IgE (BD Biosciences), insulin (Crystal Chem Inc), and serum amyloid A (Life Technologies), according to the manufacturers' instructions.
2-Deoxyglucose (2DG) uptake assay
Preadipocyte 3T3-L1 cells were differentiated to adipocytes in a 48-well plate with and without IgE (0, 1, 10, 50 μg/mL). After 2 days, glucose uptake was performed using a 2DG uptake measurement kit (Cosmo Bio Co Ltd), according to the manufacturer's instructions.
Small interfering RNA (siRNA) transfection
Both FcϵR1α and scramble control siRNAs (100 nM; Santa Cruz Biotechnology) were transfected to preadipocyte 3T3-L1 cells in a 12-well plate after electroporation with an Amaxa Cell Line Nucleofector kit (Lonza). After 24 hours, the cells were differentiated in an induction medium and cultured for 4 days followed by starvation and stimulation with 25 μg/mL IgE for 10 minutes. The cells were lysed for protein analysis.
Cell cytotoxicity assay
Preadipocyte 3T3-L1 cells were differentiated to adipocytes on an eight-well chamber slide or a 96-well plate with and without IgE (50 μg/mL) for 2–8 days before TUNEL staining (in situ cell death detection kit; Roche Diagnostics Corp), cell counting kit-8 (CCK-8), cell viability assay (Dojindo Molecular Technologies, Inc), or lactate dehydrogenase (LDH) cytotoxicity assay (LDH assay; Promega), according to the manufacturers' instructions.
Oil-red O staining
Differentiated 3T3-L1 cells with and without IgE (50 μg/mL) in a 96-well plate were fixed with 10% formalin for 1 hour, washed with 100% propylene glycol, and stained with 0.5% oil-red O for 4 hours. This procedure was followed by washing with 85% propylene glycol. For quantitative analysis, stained cell layers were extracted with isopropanol and measured at OD510 nm.
Statistical analysis
All human data are expressed as means ± SD. Correlation analyses between IgE concentration and clinical parameters were performed using Spearman's correlation. Regression plots were built after log transformation of IgE values for normalization purpose. All P values are two sided, and values of P < .05 were considered to be statistically significant. All analyses were performed using R software, version 3.0.1. All mouse data were expressed as mean ± SEM. Due to our small sample sizes and often skewed data distributions, we performed a pairwise nonparametric Mann-Whitney test followed by Bonferroni corrections to examine the statistical significance.
Results
Inverse correlation between human plasma IgE and obesity
Data obtained from the 50 obese women (aged 42 ± 11 y, BMI 50.67 ± 8.26 kg/m2) showed that serum IgE correlated negatively with BMI (P = .018, ρ = −0.33) (Figure 1A), body weight (P = .016, ρ = −0.34) (Figure 1B), and fat mass (P = .023, ρ = −0.34) (Figure 1C). Fasting glycemia, insulin, HbA1c, triglyceride, HDL, apolipoprotein A1, apolipoprotein B, aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transpeptidase, leptin, adiponectin, IL-6, and hs-CRP did not associate with IgE levels. Only total cholesterol correlated positively with IgE (P = .028, ρ = 0.31) (Supplemental Table 3). Of the 50 severely obese patients, 18 had type 2 diabetes. Diabetic obese patients were significantly older and exhibited a higher BMI, fasting glycemia, fasting insulin, and HbA1c level as expected. These patients also had a lower HDL and higher triglyceride, alanine aminotransferase, γ-glutamyl transpeptidase, IL-6 and hs-CRP levels than nondiabetic obese patients. Diabetic and nondiabetic obese patients did not exhibit significantly different plasma IgE levels, however (data not shown).
Figure 1.

Spearman's correlations between logarithmized human plasma IgE and BMI (A), body weight (B), and body fat mass (C).
FcϵR1 deficiency increases body weight gain but improves glucose tolerance in mice
This study monitored the body weight and included glucose and insulin tolerance assays in both male and female wild-type (WT) and FcϵR1-deficient Fcer1a−/− mice. Male (Figure 2A) or female (data not shown) FcϵR1-deficient Fcer1a−/− mice gained significantly more body weight than WT control mice on a HFD. Fcer1a−/− mice consumed significantly more food and gained more lean and fat mass, as determined by DEXA analysis (Figure 2B). Fcer1a−/− mice demonstrated significantly improved glucose tolerance but exhibited no difference in insulin tolerance when compared with WT control mice (Figure 2C), suggesting that Fcer1a−/− mice had improved glucose metabolism but a similar degree of insulin resistance to that of WT mice. Consistently, overnight-fasted Fcer1a−/− mice exhibited elevated glucose-induced insulin release, which showed no significant difference from WT mice at 90 minutes after the first glucose stimulation (Figure 2D). Islets from WT mice released insulin responding to glucose in a dose-dependent manner, but IgE did not affect islet insulin production at any tested doses of up to 100 μg/mL (Figure 2E). The low level expression of FcϵR1 on islets possibly triggered insignificant insulin induction responding to IgE. RT-PCR revealed about 6-fold lower FcϵR1α expression on islets than that on bone marrow-derived macrophages (Figure 2F). Nonfasted WT and Fcer1a−/− mice on a HFD showed no difference between the basal levels of plasma IgE or insulin (Figure 2G). WAT from Fcer1a−/− mice, however, had significantly lower IgE levels than WT mice (Figure 2H). Although a direct comparison remains impossible, WAT milieu may have much higher IgE concentrations (∼1600 ng/mg WAT protein from WT mice) than the plasma (about 150 ng/mL from WT mice).
Figure 2.

FcϵR1 deficiency increased obesity but improved glucose tolerance in mice. A, Body weight gain of male WT and Fcer1a−/− mice on a HFD. B, DEXA determined food intake and whole-body lean and fat masses after mice consumed a HFD for 17 weeks. Glucose and insulin tolerance test (C) and glucose-induced insulin release (D) from male WT and Fcer1a−/− mice after 17 weeks of a HFD were performed. E, Insulin production from islets treated with and without different doses of glucose and IgE. Insulin secretion is expressed as a percentage of the islet insulin content (n = 4). F, The mRNA expression levels of FcϵR1a in the islets treated with different doses of IgE (n = 4). Bone marrow-derived macrophages were used as a positive control. Plasma IgE and insulin levels (G), WAT extract IgE (H), and plasma serum amyloid A (SAA), IL-6, and MCP-1 levels (I) from male WT and Fcer1a−/− mice after 17 weeks of a HFD are shown. The number of mice per group is indicated in parentheses.
Consistent with increased body weight gain, Fcer1a−/− mice had higher plasma serum amyloid A, IL-6, and MCP-1 than WT control mice after consuming a HFD, although the difference in MCP-1 levels did not reach statistical significance (Figure 2I). Yet data showed significantly fewer Mac2-positive macrophages in both sc adipose tissue and visceral adipose tissue (VAT) from Fcer1a−/− mice than those from WT control mice (Figure 3, A and B). VAT from Fcer1a−/− mice also contained fewer CD3+ T cells than the VAT from WT mice (Figure 3C). TUNEL staining also revealed significantly fewer apoptotic cells in the WAT from Fcer1a−/− mice than in WT mice (Figure 3D), which possibly contributed to increased body weight gain and body fat mass in Fcer1a−/− mice (Figure 2, A and B). Metabolic characterization demonstrated that Fcer1a−/− mice on a HFD had lower O2 consumption and CO2 production than WT control mice (Figure 3, E and F), although the two groups did not reach statistical significance with regard to the respiratory exchange ratio (RER) and body heat (Figure 3, G and H). Although Fcer1a−/− mice showed more food intake than WT control mice (Figure 2B), their plasma leptin levels and WAT adipocyte sizes did not significantly differ (Figure 3, I and J). Reduced energy expenditure in HFD-fed Fcer1a−/− mice (Figure 3, E and F) may contribute to increased body weight gain in these mice (Figure 2A). Immunoblot analysis demonstrated much lower UCP1 expression in BAT from Fcer1a−/− mice than that from WT control mice (Figure 3K).
Figure 3.

FcϵR1 deficiency in WAT macrophage and T-cell accumulation, WAT cell apoptosis, and mouse energy expenditure. Mac2 immunostaining detected macrophages in VAT (A) and sc adipose tissue (SAT?) (B), and CD3 immunostaining detected total T cells (C) from male WT and Fcer1a−/− mice after 17 weeks of a HFD. D, TUNEL staining detected apoptotic cells in the WAT from the same groups of mice. Area under the curve (AUC) of O2 consumption (VO2) volume (E), CO2 production volume (F), RER (G), and body heat (H) from both male WT and Fcer1a−/− mice consumed a HFD for 17 weeks (n = 6–8 per group). Representative data are shown to the left (panels A–C) or right (panels D–G) panels. Plasma leptin levels as determined by ELISA (I), WAT adipocyte size (J), and BAT immunoblot analysis detected UCP1 expression (K) in both WT and Fcer1a−/− mice consumed a HFD for 17 weeks. The number of mice per group is indicated in the parentheses.
FcϵR1 deficiency in mice affects the expression of molecules involved in WAT adipogenesis, apoptosis, and glucose uptake
Immunoblot analysis (Figure 4A) and immunostaining (Figure 4B) confirmed comparable FcϵR1 expression in WAT from mice after a chow diet and a HFD. In WAT, both Mac2-positive macrophages and CD3-positive T cells all express FcϵR1 (Figure 4C). WAT from HFD-fed Fcer1a−/− mice revealed a significantly enhanced expression of p-AKT (Figure 4D), supporting the suppressive role of FcϵR1 on AKT activation (18), which requires the expression and activation of C/EBPα and PPARγ (19, 20). Increased AKT activation in WAT from Fcer1a−/− mice led to a concurrent increase of C/EBPα and PPARγ expressions (Figure 4, E and F).
Figure 4.

Immunoblot analysis of adipogenesis-associated proteins in WAT from WT and Fcer1a−/− mice after 17 weeks on a HFD. FcϵR1a immunoblot analysis (A), FcϵR1a immunostaining (B), and FcϵR1a immunofluorescent double staining together with Mac-2 or CD3 (C) of WAT from mice fed a chow diet and a HFD. Immunoblots or RT-PCR determined the expression of p-AKT and total AKT (D), C/EBPα (E), PPARγ (F), p-JNK and total JNK (G), Bcl-2 (H), and Glut4 (I) in WAT from WT and Fcer1a−/− mice that consumed a HFD. β-Actin immunoblots were used to ensure equal protein loading. Data are mean ± SEM of five independent experiments. Representative immunoblots for panels B-I are shown to the left.
Both C/EBPα and PPARγ participate in the control of adipocyte terminal differentiation and maintenance (21, 22) and macrophage apoptosis (23). Reduced macrophage and T-cell contents (Figure 3, A–C) and reduced cell apoptosis (Figure 3D) in WAT from Fcer1a−/− mice may therefore associate with increased expression of C/EBPα and PPARγ (Figure 4, E and F). Immunoblot analysis of the same WAT from WT and Fcer1a−/− mice revealed a reduced expression of the apoptosis-signaling molecule p-JNK (24) (Figure 4G) and increased expression of cell apoptosis and necrosis inhibitory molecule Bcl-2 (25) (Figure 4H), which might also contribute to increased fat mass and body weight gain in Fcer1a−/− mice (Figure 2, A and B). Improved glucose tolerance and increased release of plasma insulin after a glucose challenge in Fcer1a−/− mice (Figure 2, C and D) might also associate with increased PPARγ expression (26, 27). Immunoblot analysis revealed significantly increased Glut4 expression in WAT from Fcer1a−/− mice (Figure 4I).
IgE suppresses adipogenesis in mice
Increased fat mass in Fcer1a−/− mice (Figure 2, A and B) and increased expression of C/EBPα and PPARγ in WAT from Fcer1a−/− mice (Figure 4, E and F) suggest that IgE participates in adipogenesis. WAT from mice on a chow or HFD expressed similar levels of FcϵR1 (Figure 4, A and B). Data consistently revealed elevated levels of FcϵR1 expression (all three chains: α, β, and γ) during adipogenesis of preadipocyte 3T3-L1 (Figure 5A). Partially differentiated 3T3-L1 cells (2 d) that had increased FcϵR1 expression (compared with preadipocytes) helped treat cells with 50 μg/mL IgE, which revealed no differences in FcϵR1 (α, β, and γ) expression (Figure 5B) with and without IgE stimulation. The addition of different concentrations of IgE (10, 50, and 100 μg/mL) to 3T3-L1 cells during the first 2 days and the whole duration of 8 days of adipogenesis tested IgE participation in adipogenesis. At either treatment, 50–100 μg/mL IgE significantly blocked 3T3-L1 adipogenesis, as determined by oil-red O staining (Figure 5C). IgE inhibited increased the expression of C/EBPα and PPARγ after 2 days of 3T3-L1 differentiation, although IgE-induced PPARγ reduction did not reach statistical significance (Figure 5, D and E), likely because cells still existed at an early stage of differentiation and C/EBPα acts upstream of PPARγ (28).
Figure 5.

IgE activity in reducing 3T3-L1 cell adipogenesis. A, RT-PCR determined the mRNA levels of three FcϵR1 chains (α, β, and γ) in 3T3-L1 cells at three time points (0, 2, and 8 d) during the differentiation. B, RT-PCR determined the expression of FcϵR1 three chains (α, β, and γ) in 3T3-L1 cells differentiated for 2 days with and without 50 μg/mL IgE. C, Oil-red O staining and quantification of 3T3-L1 cells treated with different doses of IgE for the first 2 days or throughout the whole course of differentiation (8 d). Preadipocytes and fully differentiated 3T3-L1 cells without IgE treatment were used as negative and positive controls. Representative data are shown to the right. RT-PCR determined the mRNA levels of C/EBPα (D) and PPARγ (E) in 3T3-L1 cells before differentiation and after 2 days of differentiation meanwhile treated with and without 50 μg/mL IgE. Data are mean ± SEM of three to five independent experiments.
IgE activity on 3T3-L1 adipocyte apoptosis and glucose uptake
Increased fat mass in Fcer1a−/− mice (Figure 2, A and B) but reduced apoptosis in the WAT from the same mice (Figure 3D) suggest that IgE participates in inducing adipocyte apoptosis. The treatment of 3T3-L1 preadipocytes with IgE during differentiation helped test this hypothesis. IgE treatment induced adipocyte apoptosis in a time-dependent manner (Figure 6A). At both the 6- and 8-day time points, IgE completely suppressed adipogenesis, but cell apoptosis reached 7% at the 6-day time point and 45% at the 8-day time point, respectively (Figure 6, A and B), suggesting that IgE inhibited adipogenesis before inducing apoptosis. Indeed, IgE induced apoptosis of only adipocytes but not preadipocytes (Figure 6C). IgE toxicity to the cells did not engender IgE-induced adipocyte apoptosis. In 3T3-L1 cells after 2 days differentiation, when cells exhibited increased FcϵR1 expression (Figure 5, A and B) but no evident apoptosis (Figure 6A), a range of IgE concentrations did not affect 3T3-L1 cell viability or cytotoxicity as determined by an assay of CCK-8 and LDH (Figure 6, D and E).
Figure 6.

IgE activities in promoting adipocyte apoptosis and suppressing adipocyte glucose uptake. A, TUNEL staining and quantification of apoptotic cells in 3T3-L1 cells differentiated in the presence and absence of 50 μg/mL IgE for indicated days. Representative data are shown to the right. B, Oil-red O staining of the same experiment from day 6 and day 8 experiments of panel A. C, TUNEL staining of preadipocytes and fully differentiated adipocyte treated with and without IgE (50 μg/mL) to induce cell apoptosis. A CCK-8 assay determined cell viability (D) and an LDH assay determined cytotoxicity (E) of 3T3-L1 cells after 2 days of differentiation with and without different amount of IgE. F, Glucose uptake assay of 3T3-L1 cells after 2 days of differentiation with and without different amount of IgE. Preadipocytes were used as an experimental negative control. G, Glut4, p-AKT, and total AKT immunoblots of differentiated 3T3-L1 cells and treated with and without 50 μg/mL IgE for 30 minutes. H, FcϵR1α siRNA- and scramble siRNA-transfected 3T3-L1 cells and differentiated for 4 days, followed by treatment with 25 μg/mL of insulin or IgE for 10 minutes. β-Actin immunoblots were used for protein loading controls. Data are mean ± SEM of three independent experiments.
Improved glucose tolerance in Fcer1a−/− mice (Figure 2C) increased insulin secretion to the plasma from Fcer1a−/− mice after a glucose challenge (Figure 2D) and increased Glut4 expression in the WAT from Fcer1a−/− mice (Figure 4I), which suggests the participation of IgE in inhibiting glucose uptake. This study tested this hypothesis in 3T3-L1 cells after 2 days of differentiation, when 3T3-L1 increased FcϵR1 expression (Figure 5, A and B), but IgE treatment did not induce 3T--L1 apoptosis (Figure 6A) to interfere with glucose uptake. Whereas preadipocytes took up 2DG at baseline, as determined by measuring the intracellular hexokinase-phosphorylated 2DG-6-phosphate (29), differentiated 3T3-L1 cells showed a significant increase in glucose uptake. IgE treatment inhibited glucose uptake in a concentration-dependent manner (Figure 6F). A reduced expression of glucose transporters likely mediated IgE-suppressed glucose uptake. In differentiated 3T3-L1 cells, IgE reduced the expression of Glut4 and its upstream signaling molecule p-AKT (Figure 6G). In the differentiated 3T3-L1 cells, the silencing of FcϵR1 expression with its siRNA was confirmed by an FcϵR1 immunoblot analysis and increased the expression of both Glut4 and p-AKT, compared with those transfected with scrambled control siRNA (Figure 6H). This result establishes that IgE suppresses glucose uptake by reducing the expression of glucose transporters.
Discussion
This study revealed the dual participation of IgE action in obesity and diabetes, which remained consistent to observations from several human studies. Plasma IgE levels correlated negatively with BMI among patients with severe asthma (11). Data showed that plasma IgE levels also correlated negatively with body weight, BMI, and fat mass among severely obese women, although the current study observed a relatively small cohort with a power of 0.691 compared with other similar studies (11). However, this is the first correlation study linking IgE to obesity without the confounding from asthma or other allergic or autoimmune diseases. These human studies point to the role of IgE in modulating obesity. We reported previously that human plasma IgE correlated positively with type-2 diabetes (12, 13). Interruption of IgE action with an anti-FcϵR1 antibody delayed the onset of type 1 diabetes in mice (14), suggesting IgE increases plasma glucose levels.
This study also demonstrated that the interruption of IgE action in Fcer1a−/− mice increased food intake and reduced energy expenditure (O2 consumption and CO2 production), as reflected by reduced UCP1 expression in BAT from the Fcer1a−/− mice, without exhibiting significant changes in RER and body heat. These physiological changes may contribute to increased body weight gain in these mice. Although previous data suggest that high RER and body heat reduce body weight, the presence of diabetes may affect such values. In obese humans, nondiabetic patients have lower RMR than diabetic patients (30). Increased body weight but improved glucose tolerance in Fcer1a−/− mice may trigger insignificant differences in RMR and body heat between WT and Fcer1a−/− mice, although this hypothesis merits further investigation.
At the molecular and cellular levels, however, this study proposed two possible mechanisms by which IgE reduced fat mass; one is the C/EBPα and PPARγ pathway. Activation of C/EBPα is a prerequisite for PPARγ activation (28). Several signaling pathways can activate C/EBPα, including the MAPK (eg, p38 and ERK1/2) (31), the cAMP-associated protein kinase A pathway (32), and the AKT pathway (19, 20). Activation of IgE receptor FcϵR1 activates the MAPK pathway but suppresses AKT activation (18). This study found that WAT from Fcer1a−/− mice showed increased p-AKT, C/EBPα, and PPARγ, supporting a negative action of IgE on AKT activation, downstream C/EBPα and PPARγ (19, 20), and consequent adipocyte differentiation (21, 22). The second mechanism by which IgE can influence obesity involves its activity in promoting the apoptosis of adipocytes, whereas preadipocytes were fully protected. IgE can furnish survival signals to mast cells and basophils (1), death signals to macrophages and vascular cells (2), but does not affect CD4+ and CD8+ T cell survival or death (3).
This study revealed that IgE induces adipocyte apoptosis but not preadipocytes. Cell differentiation occurred before apoptosis in cultured 3T3-L1 cells. Why IgE promotes survival or apoptosis differently from one cell type to another (1–3) remains unclear. Prior studies show that PPARγ activation inhibits monocyte/macrophage migration, accumulation, and apoptosis in atherosclerotic lesions (23) and in WAT from HFD-fed mice (33). Reduced PPARγ activation in these WAT may contribute to reduced macrophages in WAT from Fcer1a−/− mice. Reduced JNK activation in WAT from Fcer1a−/− mice, however, may correlate with reduced macrophage accumulation and inflammation in WAT (34), although interrupted FcϵR1 signaling and Syk (spleen tyrosine kinase) activation may also directly suppress JNK activation. JNK activation contributes to body weight gain and glucose tolerance. The protection of JNK-deficient mice from obesity and diabetes occurred in DIO mice (35). Reduced apoptosis and p-JNK expression in WAT from Fcer1a−/− mice support the role of JNK in regulating adipocyte apoptosis (24). Although the signaling pathways that control adipocyte apoptosis have considerable complexity, IgE-mediated JNK activation, AKT/Bcl-2 suppression, and AKT-C/EBPα-PPARγ reduction may all contribute to adipocyte apoptosis, a hypothesis that merits further investigation.
In contrast to the inhibitory effect of IgE on obesity, IgE promotes experimental type 2 diabetes. IgE-suppressed p-AKT may directly impair the expression and distribution of glucose transporters, such as Glut4 (36, 37) and indirectly via reduced PPARγ expression and activation (38). Therefore, in 3T3-L1 cells, IgE reduced p-AKT and Glut4 expression and FcϵR1 knockdown by its siRNA increased p-AKT and Glut4 expression, all of which may explain concentration-dependent suppression of glucose uptake in 3T3-L1 cells by IgE. A similar pattern occurred in vivo. FcϵR1 deficiency increased WAT Glut4 expression. These observations may explain why Fcer1a−/− mice had improved glucose tolerance and glucose-induced insulin release. Figure 7 summarizes our hypothesis.
Figure 7.
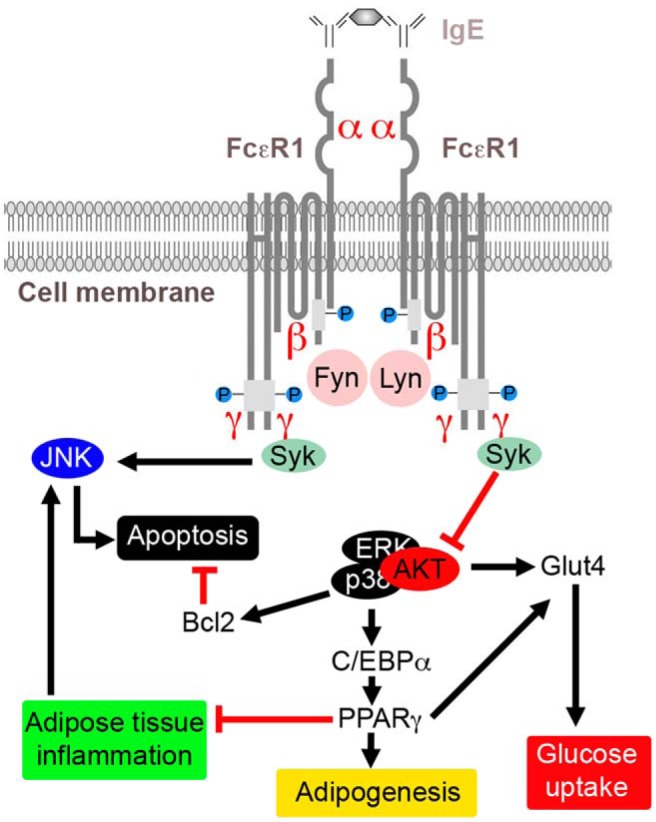
Possible mechanisms of FcϵR1-mediated IgE actions in regulating the expressions of Glut4, C/EBPα, PPARγ, and Bcl-2, and associated pathophysiological activities in obesity and diabetes.
Several questions remain unresolved. Fcer1a−/− mice consumed more food and had lower levels of energy expenditure and BAT expression of UCP1 than WT control mice. Although these observations may explain why Fcer1a−/− mice gained more body weight than WT mice, the mechanism by which IgE activity controls food uptake and energy expenditure remains unknown. Prior studies showed that leptin infusion augmented plasma IgE levels in allergen-challenged mice (39), but the absence of IgE receptor FcϵR1 did not affect plasma leptin levels. Therefore, IgE may control food uptake and energy expenditure with mechanisms other than leptin. For example, gut microbiota-associated metabolic endotoxemia and inflammation impair food uptake (40, 41). IgE may reduce food uptake by inducing gut inflammation (4, 42, 43), although this study did not test this hypothesis. We further found that Fcer1a−/− mice exhibited improved glucose tolerance and glucose-induced insulin secretion, but these mice showed similar degrees of insulin resistance and similar basal levels of plasma insulin to those from WT control mice. This study did not reveal a direct participation of IgE in suppressing glucose-induced insulin secretion in isolated islets, suggesting that elevated glucose-induced insulin secretion in Fcer1a−/− mice indirectly participated in IgE (44, 45).
All in vitro experiments in this study used IgE at concentrations (10–100 μg/mL) much higher than its physiological concentrations in human or mouse plasma (100–400 ng/mL). At 10 μg/mL, IgE showed a negligible effect in suppressing adipogenesis and weak suppression of glucose uptake. Therefore, this study used 50 μg/mL IgE to demonstrate inhibition of 3T3-L1 cell adipogenesis, promoting adipocyte apoptosis and suppressing Glut4 expression and glucose uptake. The physiological relevance of these in vitro experiments remains a key question. As described in our prior studies of atherosclerosis and abdominal aortic aneurysm (3, 4), low plasma IgE levels may not necessarily reflect the actions of tissue IgE in situ. The tissue milieu may harbor higher levels of IgE than previously surmised. This possibility remains consistent to our observation that each milligram of WAT extract from DIO mice contained about 1600 ng of IgE.
This study provides the first direct evidence of IgE participation in obesity and diabetes, although many observations still remain incompletely understood. Because atopic diseases can affect certain disadvantaged populations who remain particularly vulnerable to obesity and diabetes (8–11, 46, 47), these observations have public health implications beyond providing novel mechanistic insight. The results of this study, showing divergent actions of IgE on obesity and glucose tolerance, require consideration in the context of therapeutic targeting of IgE as well.
Acknowledgments
We thank Ms Eugenia Shvartz for her technical assistance and Ms Chelsea Swallom for her editorial assistance.
This work was supported by National Institutes of Health Grants HL60942, HL81090, HL88547 (to G.-P.S.), HL48743, HL080472 (to P.L.), and DK67536 (to R.N.K.). The clinical work performed in France was supported by Programme Hospitalier de Recherche Clinique 0276 (adiposity signals, Ethical Agreement CPP N°1, Hotel-Dieu Hospital) and by Bar-Institute of Cardiometabolism and Nutrition project as part of Investment for the Future Grant ANR-10-IAHU-05.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Bcl2
- B-cell lymphoma 2
- BMI
- body mass index
- CCK-8
- cell counting kit-8
- C/EBPα
- CCAAT/enhancer binding protein-α
- DEXA
- dual-energy x-ray absorptiometry
- 2DG
- 2-deoxyglucose
- FcϵR1
- Fcϵ receptor-1
- Glut4
- glucose transporter 4
- HbA1c
- glycated hemoglobin
- HDL
- high-density lipoprotein
- HFD
- high-fat diet
- hs-CRP
- highly sensitive C-reactive protein
- JNK
- c-Jun N-terminal kinase
- LDH
- lactate dehydrogenase
- MCP-1
- monocyte chemotactic protein-1
- p
- phosphorylated
- PPAR-γ
- peroxisomal proliferator-activated receptor-γ
- RER
- respiratory exchange ratio
- RT-PCR
- real-time PCR
- siRNA
- small interfering RNA
- TUNEL
- terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling
- UCP1
- uncoupling protein 1
- VAT
- visceral adipose tissue
- WAT
- white adipose tissue
- WT
- wild type.
References
- 1. Kawakami T, Galli SJ. Regulation of mast-cell and basophil function and survival by IgE. Nat Rev Immunol. 2002;2:773–786. [DOI] [PubMed] [Google Scholar]
- 2. Wang J, Cheng X, Xiang MX, et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe−/− mice. J Clin Invest. 2011;121:3564–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang J, Lindholt JS, Sukhova GK, et al. IgE actions on CD4+ T cells, mast cells, and macrophages participate in the pathogenesis of experimental abdominal aortic aneurysms. EMBO Mol Med. 2014;6:952–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weigmann B, Schughart N, Wiebe C, et al. Allergen-induced IgE-dependent gut inflammation in a human PBMC-engrafted murine model of allergy. J Allergy Clin Immunol. 2012;129:1126–1135. [DOI] [PubMed] [Google Scholar]
- 5. Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. [DOI] [PubMed] [Google Scholar]
- 6. Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmad Al Obaidi AH, Mohamed Al Samarai AG, Yahya Al Samarai AK, Al Janabi JM. The predictive value of IgE as biomarker in asthma. J Asthma. 2008;45:654–663. [DOI] [PubMed] [Google Scholar]
- 8. Ma J, Xiao L, Knowles SB. Obesity, insulin resistance and the prevalence of atopy and asthma in US adults. Allergy. 2010;65:1455–1463. [DOI] [PubMed] [Google Scholar]
- 9. Visness CM, London SJ, Daniels JL, et al. Association of obesity with IgE levels and allergy symptoms in children and adolescents: results from the National Health and Nutrition Examination Survey 2005–2006. J Allergy Clin Immunol. 2009;123:1163–1169, 1169.e1161–e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fitzpatrick S, Joks R, Silverberg JI. Obesity is associated with increased asthma severity and exacerbations, and increased serum immunoglobulin E in inner-city adults. Clin Exp Allergy. 2012;42:747–759. [DOI] [PubMed] [Google Scholar]
- 11. Gibeon D, Batuwita K, Osmond M, et al. Obesity-associated severe asthma represents a distinct clinical phenotype: analysis of the British Thoracic Society Difficult Asthma Registry Patient cohort according to BMI. Chest. 2013;143:406–414. [DOI] [PubMed] [Google Scholar]
- 12. Wang Z, Zhang H, Shen XH, et al. Immunoglobulin E and mast cell proteases are potential risk factors of human pre-diabetes and diabetes mellitus. PLoS One. 2011;6:e28962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Zhang H, Shen XH, et al. Immunoglobulin E and mast cell proteases are potential risk factors of impaired fasting glucose and impaired glucose tolerance in humans. Ann Med. 2013;45:220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hubner MP, Larson D, Torrero MN, et al. Anti-FcϵR1 antibody injections activate basophils and mast cells and delay type 1 diabetes onset in NOD mice. Clin Immunol. 2011;141:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aron-Wisnewsky J, Julia Z, Poitou C, et al. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women. J Clin Endocrinol Metab. 2011;96:1151–1159. [DOI] [PubMed] [Google Scholar]
- 16. Sell H, Divoux A, Poitou C, et al. Chemerin correlates with markers for fatty liver in morbidly obese patients and strongly decreases after weight loss induced by bariatric surgery. J Clin Endocrinol Metab. 2010;95:2892–2896. [DOI] [PubMed] [Google Scholar]
- 17. Kulkarni RN, Winnay JN, Daniels M, et al. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured β-cell lines. J Clin Invest. 1999;104:R69–R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feuser K, Feilhauer K, Staib L, Bischoff SC, Lorentz A. Akt cross-links IL-4 priming, stem cell factor signaling, and IgE-dependent activation in mature human mast cells. Mol Immunol. 2011;48:546–552. [DOI] [PubMed] [Google Scholar]
- 19. Peng XD, Xu PZ, Chen ML, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshiga D, Sato N, Torisu T, et al. Adaptor protein SH2-B linking receptor-tyrosine kinase and Akt promotes adipocyte differentiation by regulating peroxisome proliferator-activated receptor γ messenger ribonucleic acid levels. Mol Endocrinol. 2007;21:1120–1131. [DOI] [PubMed] [Google Scholar]
- 21. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 22. Lowell BB. PPARγ: an essential regulator of adipogenesis and modulator of fat cell function. Cell. 1999;99:239–242. [DOI] [PubMed] [Google Scholar]
- 23. Chinetti G, Griglio S, Antonucci M, et al. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273:25573–25580. [DOI] [PubMed] [Google Scholar]
- 24. Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. [DOI] [PubMed] [Google Scholar]
- 25. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. [DOI] [PubMed] [Google Scholar]
- 26. Standaert ML, Kanoh Y, Sajan MP, Bandyopadhyay G, Farese RV. Cbl, IRS-1, and IRS-2 mediate effects of rosiglitazone on PI3K, PKC-λ, and glucose transport in 3T3/L1 adipocytes. Endocrinology. 2002;143:1705–1716. [DOI] [PubMed] [Google Scholar]
- 27. Kramer D, Shapiro R, Adler A, Bush E, Rondinone CM. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism. 2001;50:1294–1300. [DOI] [PubMed] [Google Scholar]
- 28. Rosen ED, Hsu CH, Wang X, et al. C/EBPα induces adipogenesis through PPARγ: a unified pathway. Genes Dev. 2002;16:22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saito K, Lee S, Shiuchi T, et al. An enzymatic photometric assay for 2-deoxyglucose uptake in insulin-responsive tissues and 3T3-L1 adipocytes. Anal Biochem. 2011;412:9–17. [DOI] [PubMed] [Google Scholar]
- 30. Huang KC, Kormas N, Steinbeck K, Loughnan G, Caterson ID. Resting metabolic rate in severely obese diabetic and nondiabetic subjects. Obes Res. 2004;12:840–845. [DOI] [PubMed] [Google Scholar]
- 31. Bost F, Aouadi M, Caron L, Binetruy B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie. 2005;87:51–56. [DOI] [PubMed] [Google Scholar]
- 32. Liu NC, Lin WJ, Yu IC, et al. Activation of TR4 orphan nuclear receptor gene promoter by cAMP/PKA and C/EBP signaling. Endocrine. 2009;36:211–217. [DOI] [PubMed] [Google Scholar]
- 33. Foryst-Ludwig A, Hartge M, et al. PPARγ activation attenuates T-lymphocyte-dependent inflammation of adipose tissue and development of insulin resistance in obese mice. Cardiovasc Diabetol. 2010;9:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bluher M, Bashan N, Harman-Boehm I, et al. Activated Ask1-MKK4–p38MAPK/JNK stress signaling pathway in human omental fat tissue may link macrophage infiltration to whole-body Insulin sensitivity. J Clin Endocrinol Metab. 2009;94:2507–2515. [DOI] [PubMed] [Google Scholar]
- 35. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. [DOI] [PubMed] [Google Scholar]
- 36. Hernandez R, Teruel T, Lorenzo M. Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in brown adipocytes. FEBS Lett. 2001;494:225–231. [DOI] [PubMed] [Google Scholar]
- 37. Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol Biol Cell. 2006;17:4484–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR. PPARγ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J Clin Invest. 1998;101:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, Johnston RA. Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol. 2005;115:103–109. [DOI] [PubMed] [Google Scholar]
- 40. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. [DOI] [PubMed] [Google Scholar]
- 41. McHugh K, Castonguay TW, Collins SM, Weingarten HP. Characterization of suppression of food intake following acute colon inflammation in the rat. Am J Physiol. 1993;265:R1001–R1005. [DOI] [PubMed] [Google Scholar]
- 42. De Winter BY, van den Wijngaard RM, de Jonge WJ. Intestinal mast cells in gut inflammation and motility disturbances. Biochim Biophys Acta. 2012;1822:66–73. [DOI] [PubMed] [Google Scholar]
- 43. Rentzos G, Lundberg V, Stotzer PO, Pullerits T, Telemo E. Intestinal allergic inflammation in birch pollen allergic patients in relation to pollen season, IgE sensitization profile and gastrointestinal symptoms. Clin Transl Allergy. 2014;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Keane K, Newsholme P. Metabolic regulation of insulin secretion. Vitam Horm. 2014;95:1–33. [DOI] [PubMed] [Google Scholar]
- 45. Thorens B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab. 2014;16(suppl 1):87–95. [DOI] [PubMed] [Google Scholar]
- 46. Rana JS, Mittleman MA, Sheikh J, et al. Chronic obstructive pulmonary disease, asthma, and risk of type 2 diabetes in women. Diabetes Care. 2004;27:2478–2484. [DOI] [PubMed] [Google Scholar]
- 47. Stene LC, Nafstad P. Relation between occurrence of type 1 diabetes and asthma. Lancet. 2001;357:607–608. [DOI] [PubMed] [Google Scholar]