

Abstract
Ulcerative colitis (UC) belongs to inflammatory bowel disorders, a group of gastrointestinal disorders that can produce serious recurring diarrhea in affected patients. The mechanism for UC- and inflammatory bowel disorder-associated diarrhea is not well understood. The cystic fibrosis transmembrane-conductance regulator (CFTR) chloride channel plays an important role in fluid and water transport across the intestinal mucosa. CFTR channel function is regulated in a compartmentalized manner through the formation of CFTR-containing macromolecular complexes at the plasma membrane. In this study, we demonstrate the involvement of a novel macromolecular signaling pathway that causes diarrhea in UC. We found that a nitric oxide-producing enzyme, inducible nitric oxide synthase (iNOS), is overexpressed under the plasma membrane and generates compartmentalized cGMP in gut epithelia in UC. The scaffolding protein Na+/H+ exchanger regulatory factor 2 (NHERF2) bridges iNOS with CFTR, forming CFTR-NHERF2-iNOS macromolecular complexes that potentiate CFTR channel function via the nitric oxide-cGMP pathway under inflammatory conditions both in vitro and in vivo. Potential disruption of these complexes in Nherf2−/− mice may render them more resistant to CFTR-mediated secretory diarrhea than Nherf2+/+ mice in murine colitis models. Our study provides insight into the mechanism of pathophysiologic occurrence of diarrhea in UC and suggests that targeting CFTR and CFTR-containing macromolecular complexes will ameliorate diarrheal symptoms and improve conditions associated with inflammatory bowel disorders.
Inflammatory bowel disorder (IBD) represents chronic relapsing inflammatory conditions of the gastrointestinal tract and is a major cause of morbidity in the industrialized nations. IBD is broadly categorized into ulcerative colitis (UC) and Crohn disease (CD). UC extends proximally from the rectum and is restricted to the colon mucosa, whereas CD can affect any part of the gastrointestinal tract and is transmural.1 Diarrhea is the most common and distressing symptom in IBD patients, with a 50% consistency in acute flare-ups of CD and in almost 100% of UC patients, and reflects poor disease control in these patients.2–4
Cystic fibrosis transmembrane-conductance regulator (CFTR) is a cAMP-/cGMP-activated Cl−/HCO3− channel and a major epithelial apical surface transporter in the gastrointestinal tract and other secretory epithelial systems.5,6 It was reported that carriers of a loss-of-function mutation of CFTR, F508del, are eight times less likely to develop CD.7 Concerted activities of CFTR and other transporters and channels control luminal fluid levels.8–10 Regulation of CFTR activity mainly occurs in the form of macromolecular signaling complexes of CFTR assembled onto PSD-95/DLG-5/ZO-1 (PDZ) scaffolds, which enable CFTR to respond uniquely to cyclic nucleotides within specialized microdomains and to be regulated in a highly compartmentalized manner.10–12 Conclusive data on the phenotypic role of CFTR in UC-associated fluid dysregulation are lacking, and the molecular mechanism of CFTR regulation in UC was not studied previously.
The nitric oxide (NO)-producing enzyme inducible NO synthase (iNOS) is of therapeutic interest for IBD, and iNOS deficiency is correlated with reduced disease severity in animal models of experimental colitis.13–15 Colonic mucosa of IBD patients or patients in remission produce twofold to fivefold higher amounts of NO than amounts in control subjects, which correlates with the sustained expression and activity of iNOS.16,17 Data suggest that iNOS activity, although sustained, is subject to regulation in mammalian cells by protein–protein interactions.18,19 iNOS harbors a PDZ domain recognizing motif Met-Ser-Ala-Leu at its carboxyl terminal region and was reported to tether onto PDZ domain containing proteins ezrin-binding protein 50 and CFTR-associated protein 70.18,19
In this study, we unveiled a previously unknown physical and functional coupling of CFTR and iNOS that is mediated by PDZ protein Na+/H+ exchanger regulatory factor 2 (NHERF2; also known as E3KARP) at the plasma membrane of gut epithelia. This coupling potentiates CFTR chloride-channel function through generation of compartmentalized cGMP via a canonical NO-cGMP signaling pathway and leads to secretory diarrhea in UC. Our study identified a macromolecular complex of pathogenic potential and provides insights into the molecular mechanisms of the diarrheal phenotype in UC with implications for therapeutic interventions of IBD and other forms of gut inflammation.
Materials and Methods
Reagents
Zaprinast was purchased from Enzo Life Sciences (Farmingdale, NY), and dithiobis succinimidyl propionate was obtained from Pierce (Thermo Fisher, Rockford, IL). Dextran sodium sulfate (DSS; mol. wt., 36,000 to 50,000) was purchased from MP Biomedicals (Irvine, CA) and 2,4,6-trinitrobenzenesulfonic (TNBS; 1 mol/L solution in water) was obtained from Sigma-Aldrich (St. Louis, MO).
Cell Culture and Transfections
HEK293 and HT29-CL19A cells were cultured in Dulbecco’s modified Eagle’s medium-F12 and Dulbecco’s modified Eagle’s medium high glucose media (Invitrogen, Carlsbad, CA), respectively, containing 10% serum and 1% penicillin/streptomycin, and maintained in a 5% CO2 incubator at 37°C. Lipofectamine 2000 (Invitrogen) was used to express the plasmids pcDNA3-m-cygnet 2.1, pcDNA3-iNOS, pcDNA3-Flag-CFTR, pcDNA3-TM-Cl−, pCMV-hemagglutinin (HA)-NHERF2, pECFP-iNOS, pcDNA3-yellow fluorescent protein (YFP)-CFTR, and pZsYellow-NHERF2 in HEK-293 cells according to the manufacturer's instructions. pECFP-iNOS was a generous gift from Dr. Randal A. Skidgel (University of Illinois, Chicago). Stable cell lines were generated by lentiviral transfection and selected with 2 μg/mL puromycin. Knockdown of NHERF2 expression was performed with lentiviral transduction. HEK-293 cells were seeded in 12-well plates. The lentiviral particles that contained scrambled or NHERF2 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) were added to HEK-293 cells. Selection medium that contained 6 μg/mL puromycin was added to the cells at 48 hours after transduction, and knockdown-positive cells were selected for 1 week. Cells were subsequently maintained on 2 μg/mL puromycin. The expression level of NHERF2 after knockdown was detected by Western blot analysis.
DSS-Induced Colitis Murine Model
Seven- to 8-week-old C57BL/6 male mice were given 5% DSS in drinking water for 7 days, monitored regularly for total body weight, and later euthanized. Age-matched Nherf2−/− and Nherf2+/+ mice were given DSS-containing water for a period of 7 days, and control mice were given the same autoclaved drinking water without DSS.
To evaluate the contribution of CFTR in DSS-induced diarrhea, 150 μg/kg thiozolidinone CFTR inhibitor (CFTRinh-172) prepared in saline was administered intraperitoneally on the fifth day of DSS treatment after appearance of symptoms of colitis and was given twice per day for 3 days. A highly selective iNOS inhibitor, N6-(1-Iminoethyl)-l-lysine dihydrochloride (L-NIL; Cayman Chemical, Ann Arbor, MI), was administered intraperitoneally at a concentration of 10 mg/kg for 12 hours for 6, 4, and 2 days in three different mice groups treated with 5% DSS-containing water, four animals per group. Saline was administered in the control animals.
On the seventh day, mice were sacrificed, and colon tissues were removed and assessed for stool score, wet/dry weight ratio, and histologic examination. Dry/wet weight ratio was calculated by drying fecal pellets from the colon at 50°C overnight.
Induction of TNBS Colitis
Colitis was induced by a single rectal injection of 3.75 mg of TNBS dissolved in 50% ethanol–phosphate-buffered saline (PBS) solution into the descending colon of male C57BL/6 Nherf2−/− and Nherf2+/+ mice with the use of a catheter inserted 3 to 4 cm into the anus. The volume of TNBS enema was kept at 100 μL. The mice were anesthetized with intraperitoneal injection of 50 mg/kg xylazine-ketamine solution before rectal injection. The animals were observed for 3 days with regular weight measurements and afterward were euthanized for further assessment.
Determination of Clinical Scores
Stool consistency was determined on day 7 of DSS treatment and day 3 for TNBS colitis. Briefly, stool scores were determined on a scale of 0 to 5, depending on the looseness of the fecal matter: 0, well-formed pellets that did not stick to the anus; 1, semiformed stools that did not stick to the anus; 2 to 4, depending on the looseness of the fecal matter that did not stick to the anus; and 5, nonstanding liquid stool.
Histopathology Scoring for DSS Colitis
After day 7, mice were euthanized, and the entire colon was excised to measure the length of the colon. Colons were washed, fixed in formalin, and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin. Histology was scored by a pathologist in a blinded fashion (A.K.) on the basis of a combination of inflammatory cell infiltration (score 0 to 3) and tissue damage (score 0 to 3) as described previously.20 The presence of occasional inflammatory cells in the lamina propria was scored as 0, increased numbers of inflammatory cells in the lamina propria was assigned score 1, confluence of inflammatory cells that extended into the submucosa was scored as 2, and transmural extension of the infiltrate was scored as 3. For tissue damage, no mucosal damage was scored as 0, lymphoepithelial lesions were scored as 1, surface mucosal erosion or focal ulceration was scored as 2, and extensive mucosal damage and extension into deeper structures of the bowel wall was scored as 3. The combined histologic score ranged from 0 (no changes) to 6 (extensive infiltration and tissue damage).
Human Colon Sections
Sections of normal and IBD-affected, paraffin-embedded human colon tissues (5 μm thick) were used for the study. These slides were made available by the Tissue Service Core at Pathology Department (University of Tennessee Health Science Center) under the institutional review board-approved protocol 05-03854-XM. These samples and preexisting tissues from healthy and IBD individuals were de-identified.
Immunohistochemistry
Paraffin-embedded human and mouse colon sections were stained for iNOS. A few colon sections from each mouse were embedded in a Swiss roll configuration for better visualization. Slides were deparaffinized in xylene three times, 5 minutes each, followed by dehydration with ethanol. Antigen retrieval was performed with Borg DeCloaker RTU (Biocare Medical, Concord, CA) in a pressure cooker for 5 to 10 minutes. Slides were cooled for 5 minutes, and tissues were permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. Tissues were then blocked in 2.5% horse serum for 2 hours or overnight. Slides were rinsed with 1× PBS and incubated with rabbit polyclonal iNOS (dilution 1:100 to 1:150 dilution; Abcam, Cambridge, UK), rabbit polyclonal R3194 CFTR (dilution 1:100), and rabbit monoclonal NHERF2 (dilution 1:50; Cell Signaling, Danvers, MA) antibodies at 4°C overnight. Slides were incubated with Peroxidazed 1 (Biocare Medical, Concord, CA) to block endogenous peroxidase activity for 5 minutes. Biotin-conjugated rabbit horseradish peroxidase antibody (Santa Cruz Biotechnology) was used in 1:100 dilution for 30 minutes, and Vectastain ABC reagent (Vector Labs, Burlingame, CA) was used for signal amplification for another 30 minutes. Slides were developed with 3-3′-diaminobenzidine solution (Biocare Medical) according to the manufacturer's instructions. For histologic examination, slides were lightly stained with hematoxylin and eosin.
PLA
Paraffin-embedded human colon sections and formaldehyde-fixed HEK-293 cells (overexpressing iNOS and FLAG-CFTR) were used for performing proximity ligation assay (PLA; Olink Bioscience, Uppsala, Sweden). Briefly, human colon sections were deparaffinized and incubated with mouse monoclonal iNOS (dilution 1:100) and rabbit monoclonal NHERF2 (dilution 1:100) primary antibodies at 4°C overnight. For fixed cells, mouse monoclonal iNOS (dilution 1:100) and rabbit polyclonal R3194 CFTR (dilution 1:100) primary antibodies were used, and cells were incubated at 4°C overnight. The next day, PLA was performed with anti-rabbit (plus) and anti-mouse (minus) Duo link In Situ PLA probes on the samples according to the manufacturer's instructions. Images were taken with a Zeiss LSM-5 Pascal confocal microscope (Carl Zeiss Inc., Thornwood, NY).
Measurement of CFTR-Mediated I− Influx Using a YFP-Based Halide Sensor
HEK-293 cells with different protein expression profiles were cotransfected with pcDNA3-TM-Cl− sensor. After 24 hours of incubation, the transfected cells were seeded in 12- to 24-well plates and cultured for another 24 hours. The cells were washed twice with 200 μL of 136 mmol/L NaNO3 solution per well prepared in a wash buffer that contained 137 mmol/L NaCl, 4.5 mmol/L KH2PO4, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L glucose, and 5 mmol/L HEPES, pH 7.2, and 100 μL of the same buffer was added to each well. Fluorescence readings were recorded at 37°C on a FLUOstar-Omega microplate reader (BMG Labtech, Ortenberg, Germany) equipped with HQ500/20X (500 ± 10 nm) excitation and HQ535/30M (535 ± 15 nm) emission filters and two syringe pumps. Intracellular YFP fluorescence was monitored continuously for 90 to 150 intervals for 1 second each for each well. At 5 seconds after the start of fluorescence recording, 100 μL of an influx buffer (272 mmol/L NaI in the wash buffer) that contained 250 μmol/L zaprinast was added to activate CFTR-mediated I− influx.
FRET Microscopy and Data Analysis
For direct sensitized emission fluorescence resonance energy transfer (FRET), HEK-293 cells were transiently transfected with pECFP-iNOS/pZsYellow-NHERF2/pCDNA3-YFP-CFTR according to the experiment with the use of Lipofectamine 2000. Single transfected cells were used to acquire cyan fluorescent protein (CFP)- or YFP-only images for bleedthrough calculations. The corrected FRET (FRETc) was normalized with donor CFP intensity (FRETc/CFP), yielding the normalized corrected FRET (N-FRETc), and the intensity of N-FRETc images was presented in pseudocolor and monochrome mode, stretched between the low and high normalization values, according to an intensity-to-color mapped lookup table. All calculations were performed with the Channel Math and FRET modules of SlideBook software version 4.2 (Intelligent Imaging Innovations, Denver, CO) as described previously.21
For ratiometric FRET, HEK-293 cells were grown on glass-bottomed dishes (MatTek, Ashland, MA) and transfected with pcDNA3-m-cygnet 2.1 with the use of Lipofectamine 2000. Cells were washed twice with Hanks' balanced salt solution and mounted on an inverted Olympus wide-field microscope (IX51, U-Plan Fluorite 60× 1.25 NA oil-immersion objective; Olympus, Tokyo, Japan) for FRET imaging. The stage was maintained at 37°C, and the cells were maintained in the dark in Hanks' balanced salt solution. After establishing the baseline, zaprinast was added as indicated. Ratiometric FRET imaging was performed as described previously.12
Co-Immunoprecipitation Using a Cross-Linker
Protein complexes were cross-linked in HA-iNOS-HEK-293 and control HEK-293 cells with the use of 1 mmol/L cross-linker dithiobis succinimidyl propionate. Cells were then lyzed in lysis buffer (1× PBS, containing 0.2% Triton X-100, and the following protease inhibitors: 1 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL pepstatin-A, 1 μg/mL leupeptin, 1 μg/mL aprotinin), and the clear supernatant fluid was subjected to immunoprecipitation with the use of HA beads (Sigma-Aldrich). The cross-linked complex was eluted with 100 mmol/L glycine (pH 2.2) and quickly neutralized with 150 mmol/L Tris (pH 8.8). The eluted proteins were mixed with sample buffer (5×; containing 1% β-mercaptoethanol), denatured, subjected to SDS-PAGE, transferred to polyvinylidene difluoride membrane, and immunoblotted with rabbit polyclonal α-NHERF2 antibody.
PDZ Protein-Array Binding Assay
Various glutathione S-transferase (GST)-tagged PDZ proteins were immobilized on nitrocellulose membrane in various concentrations with the use of a slot-blot applicator. Purified His-S-iNOS-C-tail protein (containing the C-terminal 112 amino acids) fusion protein was added to the membrane and mixed overnight at 4°C in 3 to 5 mL of Tris-buffered saline that contained 0.1% Tween 20, 2% bovine serum albumin, and protease inhibitors. Thereafter, membrane was washed extensively with Tris-buffered saline that contained 0.1% Tween-20 and probed with S-horseradish peroxidase antibody.
Macromolecular Complex Assembly
The in vitro complex formation was performed as described previously.12 Briefly, 20 μg His-S-iNOS-C-tail protein was mixed with various amounts of GST-NHERF2 (0 to 15 μg) at 4°C for 1 hour, followed by mixing with 20 μL S-beads for another 1 hour. This step, which is called pairwise binding, was performed in 200 μL of lysis buffer (PBS that contained 0.2% Triton X-100 and protease inhibitors). The complex was washed twice with the same buffer and allowed to incubate overnight with 0.5 μg purified Flag-wild-type (WT)-CFTR at 4°C with constant mixing. The complex was washed extensively with lysis buffer, eluted with sample buffer, and immunoblotted with Flag-horseradish peroxidase antibody.
NO Assay
Colon tissue was homogenized in 1 mL of 1× PBS buffer that contained 0.2% Triton X-100, and protein concentration of the supernatant fluid was determined. The tissue supernatant fluid (100 μL) was mixed with an equal volume of Griess reagent and incubated for 20 minutes. Plate was read at 540 nm, and the nitrite levels were determined with a sodium nitrite standard curve. Nitrite levels were normalized with the total protein concentration.
cGMP Assay
Mice colons were quickly frozen in liquid nitrogen and ground to a fine powder. Tissue was dissolved in 600 μL of 6% trichloroacetic acid and spun at 8000 × g at 4°C. Pellet was lyzed in lysis buffer (Tris-Cl EDTA that contained 0.5% Triton X-100 and 0.1% SDS) for protein quantitation. Supernatant fluid was then transferred to a glass vial and washed three times with water-saturated diethyl ether (1:1). Residual ether was removed by boiling the samples briefly at 68°C. Samples were used for cGMP measurement by direct cGMP enzyme-linked immunosorbent assay from Enzo Life Sciences (Farmingdale, NY) as per the manufacturer's directions.
Statistical Analysis
Results are presented as means ± SEM for the indicated number of experiments. P < 0.05 was considered statistically significant. Most of the P values were determined with t-test unless otherwise indicated. Significance of intergroup variance was evaluated with the use of one-way analysis of variance for animal experiments.
Results
iNOS Expression Is Up-Regulated in UC Patients
To elucidate the significance of iNOS in IBD, gene microarray data on colon biopsies that were obtained mostly from the ascending colon were data-mined to assess iNOS expression levels in biopsies comprising active CD and UC disease areas.22 The diagnosis of IBD was made with established clinical, radiologic, and histologic criteria.23 The Pediatric Crohn's Disease Activity Index and Pediatric Ulcerative Colitis Clinical Activity Index were used as measures of clinical severity of IBD.22,24 We observed a significant increase in the transcript levels of iNOS in CD- and UC-affected biopsies compared with normal tissue (Figure 1).
Figure 1.
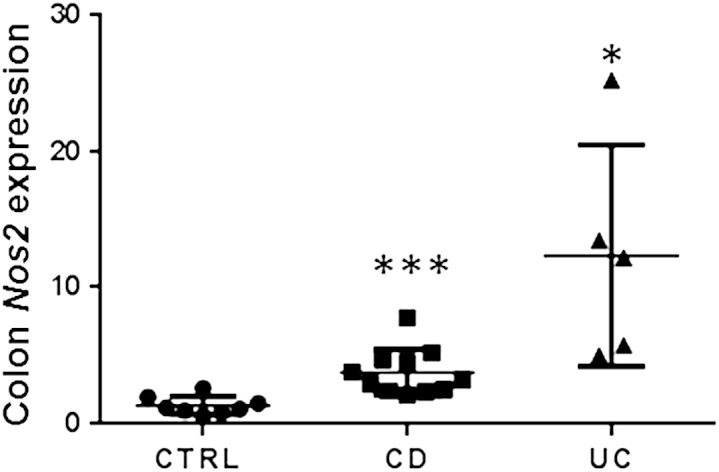
iNOS levels are up-regulated in UC. Gene expression of NOS2 (iNOS) in the CTRL, CD-, and UC-affected colon biopsies shows that iNOS levels are significantly higher in CD- and UC-affected biopsies than the control. Data are expressed as means ± SEM. ∗P < 0.05, ∗∗∗P < 0.001. Intergroup variance was evaluated with one-way analysis of variance significance test, and P values were obtained with t-test to demonstrate statistical significance between control group and CD group and control group and UC group. CD, Crohn disease; CTRL, control; iNOS, inducible nitric oxide synthase; UC, ulcerative colitis.
iNOS Interacts with NHERF2 at the Plasma Membrane in UC
Given that iNOS has a putative PDZ motif to bind to PDZ scaffolding proteins, we studied the interaction of His-S-iNOS-C-tail (amino acid 1041 to 1153) with several PDZ proteins that are expressed in gut epithelia. Our results found that NHERF2 bound the iNOS-C-tail with the highest affinity, whereas NHERF1 had weak binding and PDZK1 binding to iNOS-C-tail (Figure 2A).
Figure 2.

iNOS interacts with NHERF2. A: His-S-iNOS-C-tail protein interacts with GST-NHERF2 in a PDZ protein array binding assay. GST-tagged PDZ proteins were immobilized on a nitrocellulose membrane in a slot-blot format to generate the PDZ protein array and were used to probe their interactions with His-S-iNOS-C-tail protein. B: Representative pseudocolor images show various fluorophore-tagged proteins in HEK-293 cells: CFP-iNOS, CFP-iNOS ΔPDZ, Yellow only, Yellow-NHERF2, Yellow-PDZK1. The intensity of the N-FRETc between the respective FRET pairs was shown in a monochrome mode, according to a temperature-based look-up table. n = 3 to 5 independent experiments. CFP, cyan fluorescent protein; GST, glutathione S-transferase; HRP, horseradish perioxidase; iNOS, inducible nitric oxide synthase; N-FRETc, normalized corrected fluorescence resonance energy transfer; NHERF2, Na+/H+ exchanger regulatory factor 2; PDZ, PSD-95/DLG-5/ZO-1.
We then studied the interaction between iNOS and NHERF2 in HEK-293 cells and found that NHERF2 co-immunoprecipitated with iNOS on cross-linking the complex (Supplemental Figure S1A). That the interaction could not be detected without a cross-linker suggests that i) iNOS-NHERF2 interactions are transient inside the cell, ii) preservation of secondary/tertiary protein structure is critical for this native interaction, and iii) this interaction can also be physiologically stimulated (eg, under inflammation conditions). We could however rule out the possibility of a weak interaction between iNOS and NHERF2, because our data found the interaction to be of picomolar sensitivity (Figure 2A).
With the use of FRET, we investigated whether the interaction, quantitated as the N-FRETc value, could be detected in live HEK-293 cells. An approximate N-FRETc value of 35% ± 4% was estimated at the plasma membrane in cells that co-expressed CFP-iNOS (acts as a donor) and Yellow-NHERF2 (acts as an acceptor) (Figure 2B). Several controls were used to verify the specificity of FRET between iNOS and NHERF2. HEK-293 cells co-expressing CFP-iNOS and Yellow-fluorophore alone were used as a negative control and found little or no FRET signal (Figure 2B). We also generated a CFP iNOS-ΔPDZ in which the last three amino acids of iNOS, SALCOOH, is replaced with AAACOOH, which compromises the ability of iNOS to bind to NHERF2 in HEK-293 cells (Supplemental Figure S1B). No detectable FRET was obtained between CFP iNOS-ΔPDZ and Yellow-NHERF2 (Figure 2B). We also determined FRET between CFP-iNOS and Yellow-PDZK1 and observed considerably low FRET signals of 4% ± 0.37% (Figure 2B), consistent with our PDZ array data (Figure 2A).
Previous findings suggest iNOS activity to be largely vectorial because of its membrane localization in polarized cells.18,19 We determined the iNOS expression and localization profile in UC-affected colon tissues with the use of immunohistochemistry. We confirmed the increased iNOS expression and its predominant localization at or near the apical plasma membrane in UC-affected human colon tissues (Figure 3A) compared with non-UC colon tissues. We also determined NHERF2 expression in these biopsies and found that NHERF2 remained unchanged between UC-affected and non-UC biopsies (Figure 3A). To demonstrate the pathophysiologic relevance of iNOS-NHERF2 interactions in UC, PLA was performed in the same UC-affected and unaffected human colon tissues. Previous studies have validated the application of this assay in determining protein interactions and colocalizations in cells and tissues.25,26 We observed a significant amount of PLA signals at the plasma membrane of epithelial cells in UC-affected tissues, signifying the interaction between iNOS and NHERF2 (Figure 3B), whereas almost no PLA signal was observed in non-UC human colon tissue (Figure 3B). Thus, we determined a novel interaction of iNOS with scaffolding protein NHERF2 that may have a role in altered vectorial physiology in the IBD gut. In addition, we speculate that iNOS and the NO-cGMP pathway is predominantly operative in UC-affected colonic mucosa and influences certain critical regulators of fluid transport.
Figure 3.

iNOS interacts with NHERF2 in UC-affected colon tissues. A: Immunohistochemical staining for iNOS and NHERF2 expression in non-UC and UC-affected human colon tissue sections with the use of DAB-based method. Arrows indicate membrane localization of iNOS and NHERF2. B: Representative images show the interaction between iNOS and NHERF2 in human colon biopsies in PLA. Strong PLA signals (red; marked arrows) are observed in UC-affected human colon tissue section, showing the interaction between iNOS and NHERF2, whereas considerably low PLA signals are observed in non-UC colon. Scale bar = 100 μm. DAB, 3-3′-diaminobenzidine; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2; PLA, proximity ligation assay; UC, ulcerative colitis.
We also studied the relevance of the iNOS-NHERF2 interaction in animal models of UC. The iNOS expression was tested in a chemically induced murine colitis model by oral administration of DSS in C57BL/6 Nherf2+/+ versus Nherf2−/− mice (Supplemental Figure S2A). The DSS colitis model closely resembles UC conditions and is the most appropriate and widely used model to study UC.15 Colon tissues from the study groups were excised and processed for histologic staining. The microscopic sections of the DSS-challenged colons from Nherf2+/+ and Nherf2−/− mice showed a range of histopathologic features of DSS-induced colitis, including mucosal edema, epithelial cells and crypt damage, and immune cell infiltration that characterize an inflamed colon (Supplemental Figure S2B). iNOS-specific immunostaining confirmed similar up-regulation of iNOS in the colon epithelia in response to DSS treatment in both Nherf2+/+ and Nherf2−/− mice compared with control mice (Supplemental Figure S2C), suggesting that the absence of NHERF2 does not affect the expression of iNOS under colitis conditions.
NHERF2 Bridges CFTR and iNOS into a Macromolecular Signaling Complex
Given that iNOS binds with high affinity to NHERF2 and localizes primarily at the plasma membrane and that CFTR interacts with NHERF2 with high affinity,11 we rationalized that a macromolecular complex might exist among CFTR, NHERF2, and iNOS with NHERF2 serving as the scaffolding protein. Abilities of NHERF proteins to assemble macromolecular complexes of CFTR and to act as conduits of regulatory influences on CFTR channel activity were reported previously.10,27–29 To explore the pathophysiologic relevance of NHERF2-mediated complex formation of CFTR and iNOS, we first performed a macromolecular formation assay with purified His-S-iNOS-C-tail, GST-NHERF2, and FLAG-WT-CFTR proteins (Figure 4A). We found that a macromolecular complex between CFTR, and iNOS could only be formed in the presence of NHERF2 and that the formation of this complex increased in a NHERF2-dependent manner (Figure 4A).
Figure 4.

NHERF2 bridges CFTR and iNOS into a macromolecular signaling complex. A: A pictorial representation of the macromolecular complex assay. A macromolecular complex is detected with three purified proteins (His-S-iNOS-C-tail, GST-NHERF2, and FLAG-WT-CFTR). B: Representative images of the PLA assay in HEK-293 cells transfected with FLAG-WT-CFTR and iNOS with or without NHERF2 knockdown. PLA signal was quantified as fluorescence intensity and is represented in the form of a bar graph. C: Representative direct sensitized FRET depicts the interaction between CFTR and iNOS at the plasma membrane in HEK-293 cells. The cells were single or cotransfected with CFP-iNOS and YFP-CFTR, and the pseudocolor images show the expression. HA-NHERF2 was cotransfected to test its effect on the N-FRETc, which was represented in a monochrome mode, according to a temperature-based look-up table. Bar graph quantifies the increase in N-FRETc between CFTR and iNOS with increasing amounts of HA-NHERF2. Data are expressed as means ± SEM. n = 8 to 49 regions of interest (B); n = 3 independent experiments, 8 to 10 regions of interest per experiment (C). ∗∗∗∗P < 0.00001 determined with t-test. Scale bar = 10 μm. CFP, cyan fluorescent protein; CFTR, cystic fibrosis transmembrane-conductance regulator; GST, glutathione S-transferase; HA, hemagglutinin; iNOS, inducible nitric oxide synthase; N-FRETc, normalized corrected fluorescence resonance energy transfer; NHERF2, Na+/H+ exchanger regulatory factor 2; PLA, proximity ligation assay; WT, wild-type; YFP, yellow fluorescent protein.
We proceeded to determine NHERF2-mediated CFTR-iNOS interaction with the use of PLA in HEK-293 cells. Briefly, iNOS and FLAG-WT-CFTR were overexpressed in HEK-293 cells with or without NHERF2 knockdown with the use of siRNA (Supplemental Figure S3). The knockdown did not impair the ability of the cells to overexpress proteins, and equal levels of iNOS and FLAG-WT-CFTR were detected in scrambled or NHERF2 siRNA knockdown cells (data not shown). We found that NHERF2 knockdown reduced the amounts of PLA signals of iNOS and FLAG-WT-CFTR interactions by 50% (Figure 4B).
Previous studies have determined the capacity of NHERF proteins to modulate CFTR protein assemblies.30 We used direct sensitized FRET to test whether iNOS and CFTR interaction can be altered by controlling the expression level of NHERF2 in live cells. HEK-293 cells were cotransfected with CFP-iNOS and YFP-WT-CFTR and subjected to FRET imaging. N-FRETc signal between CFP-iNOS and YFP-WT-CFTR at the plasma membrane could be visualized (Figure 4C). Increasing the expression of NHERF2 by transfecting the cells with different amounts of pCMV-HA-NHERF2 cDNA (0, 0.5, and 1 μg) enhanced the N-FRETc signals by twofold to 2.5-fold compared with the HA-NHERF2 untransfected cells (Figure 4C). Transfection with >1 μg of HA-NHERF2 did not further increase the FRET signal, implying that the complex was already saturated (data not shown).
Because our data showed the multifold up-regulation of iNOS expression with no change in NHERF2 levels in UC-affected colon tissues compared with non-UC samples (Figure 3, A and B), we next determined CFTR levels in the same colon samples. We found that CFTR localized predominantly at the apical plasma membrane, and its expression level remained unchanged in both normal and UC colons (Supplemental Figure S4). Provided that CFTR has a pathophysiologic role in UC conditions, we speculate that it is the regulation of CFTR-governed macromolecular complexes that is altered in IBD rather than the CFTR expression level.
A Functional NO-cGMP Pathway Exists Underneath the Plasma Membrane in iNOS-Saturated Systems and Forms the Basis of Potentiation of CFTR Channel Function
It was reported that iNOS-governed microdomains involve an active NO-cGMP pathway in the presence of soluble guanylate cyclase (sGC). Despite that sGC is cytosolic, studies have reported that a functional NOS-NO-sGC-cGMP pathway can exist in microdomains and that cellular membranes can serve as a predominant site of NO synthesis on calcium flux.31,32 To study NO-cGMP signaling, we used the cGMP modulator zaprinast, which is a phosphodiesterase-5 inhibitor, in various cell culture studies.
To test whether iNOS contributes to cGMP production at the plasma membrane, ratiometric FRET was performed to monitor cGMP dynamics in HEK-293 cells that expressed a membrane-bound cGMP sensor, m-cygnet 2.1, which was generated by inserting the myristoylation-palmitoylation site into the N-terminus of cygnet 2.1 backbone sequence to anchor the protein to the plasma membrane (Supplemental Figure S5). Although a complete cellular cGMP sensor can also be used to determine the cGMP levels associated with the plasma membrane, the rationale of using a membrane-bound cGMP sensor is to provide an improved and more precise resolution of measurement of the membrane-localized fraction of cGMP. Cells that overexpressed iNOS showed a higher baseline and zaprinast-induced CFP/FRET ratio at the plasma membrane (Figure 5A), suggesting that iNOS communicates with sGC to generate localized cGMP at the plasma membrane. We next explored functional iNOS coupling with CFTR via the NO-cGMP pathway. CFTR-mediated iodide influx was determined in HEK-293 cells with (referred to as HEK-293-N) or without (referred to as HEK-293-S) NHERF2 knockdown and overexpression of FLAG-WT-CFTR, iNOS, and a YFP-based halide sensor, pCDNA3-TM-Cl−.33 CFTR-dependent halide influx in response to zaprinast was reduced by 28% in cells having the partially compromised macromolecular complex of CFTR and iNOS due to NHERF2 knockdown (103.07 fluorescence units/second) compared with that in cells with intact NHERF2 (131.3033 fluorescence units/second) (Figure 5B). CFTR function was not significantly different in HEK-293-S-FLAG-WT-CFTR and HEK-293-N-FLAG-WT-CFTR cells (Supplemental Figure S6).
Figure 5.

Signaling via a NO-cGMP pathway in the microdomains of CFTR-iNOS protein complex potentiates CFTR channel function. A: Representative pseudocolor images of CFP/FRET emission ratio before (time, 0) and after adding 250 μmol/L zaprinast (time, 10 minutes) in HEK-293 cells transiently expressing membrane-bound cGMP sensor m-cygnet 2.1 and transfected with pCDNA3 empty vector or pcDNA3-iNOS. Representative line graph shows the changes of CFP/FRET emission ratio over time on adding the agonist. B: Averaged representative traces of CFTR-mediated I− influx in HEK-293-S (scrambled siRNA) and HEK-293-N (NHERF2 siRNA) cells that overexpress FLAG-WT-CFTR and iNOS in response to 250 μmol/L zaprinast. Bar graph represents baseline and zaprinast-induced CFP/FRET emission ratio in empty vector versus iNOS-transfected cells (A) and the rate of change in YFP fluorescence intensity indicative of the rate of chloride influx (B). Data are expressed as means ± SEM. n = 3 (A); n = 6 (B). ∗P < 0.05, ∗∗P < 0.01 determined with t-test. CFP, cyan fluorescent protein; CFTR, cystic fibrosis transmembrane-conductance regulator; FRET, fluorescence resonance energy transfer; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2; NO, nitric oxide; PAR, cells with no FLAG-WT-CFTR and iNOS; WT, wild-type; YFP, yellow fluorescent protein.
Inhibition of CFTR and iNOS Activity Attenuates Diarrhea in DSS-Induced Colitis
Because CFTR plays an important role in regulating fluid homeostasis across the intestinal epithelia and in the pathogenesis of secretory diarrheas, it is important to conclusively determine the role of CFTR in the pathogenesis of diarrhea in UC.10 Ma et al34 developed a highly specific thiozolidinone CFTR inhibitor (CFTRinh-172), which significantly attenuated (>50%) cholera toxin-induced diarrhea in mice when administered intraperitoneally (150 μg/kg) or orogavaged (250 μg/kg). In our study, colitis was induced in C57BL/6 mice with the use of 5% DSS with or without intraperitoneal administration of 150 μg/kg CFTRinh-172. Mice administered with CFTRinh-172 exhibited reduced weight loss, gross fluid secretion, stool score, and wet/dry weight ratio as indicators of diarrhea compared with the untreated animals in the DSS group (Figure 6A). Interestingly, the histology score was not significantly altered with the inhibitor compared with DSS-alone treatment (Supplemental Figure S7A). To the best of our knowledge, this is the first study to evaluate the role of CFTR in DSS-induced colitis, and our data found that CFTR contributes significantly to DSS-induced diarrhea. These observations have important clinical implications for the use of CFTR inhibitors in therapeutic intervention of UC-associated diarrhea.
Figure 6.

Inhibition of CFTR and iNOS activity ameliorates diarrhea in colitis models. A: The experimental design to test the effect of a CFTR channel blocker CFTRinh-172 on fluid secretion in a mouse DSS colitis model. Line graph shows the total body weight measurements over a period of 7 days of mice in experimental groups: saline ± DSS treatment and CFTRinh-172 ± DSS. Representative isolated mouse colons with no DSS ± CFTRinh-172 and DSS ± CFTRinh-172 treatment. Arrows show the extent of fluid secretion in the colon. Middle and right bar graphs show the assessments of various disease variables of the DSS-induced colitis, including stool score and wet/dry weight ratio of the fecal pellets in the mice not receiving or receiving intraperitoneal injection of CFTRinh-172. B: The experimental design to test the effect of L-NIL on fluid secretion in a mouse DSS colitis model. Representative isolated mouse colons of the treatment group: DSS ± L-NIL to visualize gross fluid accumulation in the colon corresponding to day 1 L-NIL treatment. Arrows show the extent of fluid accumulation in the colon. Line graph shows the total body weight measurements over a period of 7 days for the experimental groups as follows: control + saline, control + L-NIL (day 1), DSS + saline, DSS + L-NIL (day 1 to 3). Middle and right bar graphs show the assessments of various disease variables of the DSS-induced colitis, including stool score and wet/dry weight ratio of the fecal pellets in the mice not receiving or receiving intraperitoneal injection L-NIL (cumulative data of DSS + L-NIL treatment group). C: Bar graph represents nitrite (left) and cGMP levels (right) in colon tissues in response to DSS and DSS + LNIL treatment. Data are expressed as means ± SEM. n = 4 experimental groups (A); n = 3 mice each in no DSS treatment group and control group (A); n = 5 to 8 mice in DSS treatment group (A); n = 7 to 8 mice per group for CFTRinh-172 treatment (A); n = 5 experimental groups (B); n = 4 to 5 mice per DSS treatment group (B); n = 3 colon tissue samples (C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 determined with t-test. Significance of intergroup variance was evaluated with one-way analysis of variance for CFTR and iNOS inhibition animal experiments. CFTR, cystic fibrosis transmembrane-conductance regulator; CFTRinh-172, thiozolidinone CFTR inhibitor; DSS, dextran sodium sulfate; iNOS, inducible nitric oxide synthase; L-NIL, N6-(1-Iminoethyl)-l-lysine dihydrochloride.
Previous studies have consistently found that the loss of iNOS causes reduced severity of DSS-induced colitis.13,35,36 However, studies that involve the use of iNOS inhibitors have generated mixed information, with the inconsistencies largely stemming from the narrow window of selectivity and specificity toward iNOS of these inhibitors.14,15 Exogenous NO-donors stimulate electrolyte secretion in guinea pig mucosa, but with the role of endogenous NO largely undetermined.37 A study documented the curative effects of iNOS inhibition with the use of L-NIL on smoke-induced pulmonary emphysema and reversal of the deterioration associated with emphysema in the presence of the drug.38 We tested the effects of intraperitoneally administered L-NIL in the murine DSS colitis model. Animals treated with L-NIL 1 day after DSS treatment appeared healthier and had significantly less weight loss and fluid secretion than mice that received saline (Figure 6, B and C). Interestingly, mice that were treated with L-NIL 3 or 5 days after DSS treatment did show improvement in body weights compared with the no-inhibitor treatment group (Figure 6C). However, it was less significant compared with the animals treated with L-NIL 1 day after colitis induction because of the enhanced severity of inflammation at day 3 compared with day 1. Overall disease activity index that involved stool score, wet/dry weight ratio, and histology score was improved in L-NIL–treated animals compared with the untreated group (presented as a cumulative data) (Figure 6B and Supplemental Figure S7B). DSS-exposed colon tissues were enriched with high amounts of NO (quantified as nitrite) and cGMP compared with the untreated tissue (Figure 6C). The amount of nitrite and cGMP was substantially reduced in the L-NIL–treated animal group (Figure 6C). These data demonstrate that CFTR and iNOS play important roles in DSS-induced diarrhea and suggest that the diarrhea-causing mechanism might be through CFTR-iNOS macromolecular complexes in DSS-induced colitis. In addition, the results signify that the NO-cGMP pathway is actively operating in DSS colitis and driven by up-regulated levels of iNOS.
Diarrhea Is Attenuated in Nherf2−/− Mice in Both DSS- and TNBS-Induced Colitis
On the basis of our in vitro data, we determined whether CFTR-NHERF2-iNOS macromolecular signaling complexes may have pathophysiologic relevance to diarrhea in UC. We tested the differences in intestinal fluid secretion and weight loss, two indicators for secretory diarrhea, between Nherf2−/− and Nherf2+/+ mice in the DSS and TNBS colitis models. TNBS-induced colitis simulates CD in humans.15 Symptoms of colitis (rectal bleeding, weight loss) were observed on the fourth day and after DSS treatment in all DSS-treated mice irrespective of genetic background.
Histology revealed slightly better preservation of mucosal integrity in Nherf2−/− compared with Nherf2+/+ DSS-treated mice colons exteriorized on day 7; however, this was not consistently observed, and no significance score could be acquired (Supplemental Figure S3). Similarly, the pathologic scores of Nherf2−/− and Nherf2+/+ TNBS-treated mice colons were not significantly different (Supplemental Figure S8). TNBS-treated mice experienced rapid weight loss on day 2 in both Nherf2−/− and Nherf2+/+ mice (Figure 7B). Nherf2−/− mice had significantly smaller maximal weight loss than Nherf2+/+ mice in both DSS and TNBS treatment groups (Figure 7, A and B). Colons were exteriorized and wet/dry weights were calculated. Gross examination of the colons suggested relatively higher fluid secretion in Nherf2+/+ mice than in Nherf2−/− mice in both the DSS- and TNBS-treated groups (Figure 7C). DSS- and TNBS-treated Nherf2+/+ mice colon sections had higher stool score and wet/dry weight ratio than those from Nherf2−/− mice (Figure 7, D and E). These studies suggested that mice lacking NHERF2 were protected against DSS- and TNBS-induced diarrhea and that NHERF2 is required to mediate the functional coupling between CFTR and iNOS that may contribute to the diarrheal manifestation of DSS in WT mice with intact NHERF2.
Figure 7.

Nherf2−/− mice have attenuated diarrhea in DSS- and TNBS-induced colitis. A: Experimental design of DSS treatment in Nherf2+/+ and Nherf2−/− mice for a period of 7 days. Line graph shows the mouse total body weight measurements over a period of 7 days for the experimental groups: Nherf2+/+ ± DSS treatment and Nherf2−/− ± DSS treatment. B: Experimental design of DSS treatment in Nherf2+/+ and Nherf2−/− mice for a period of 7 days. Line graph shows the mouse total body weight measurements over a period of 3 days for the experimental groups: Nherf2+/+ ± TNBS treatment and Nherf2−/− ± TNBS treatment. C: Representative exteriorized mice colons of the various treatment groups. Arrows show the extent of fluid accumulation in the colon. D: Bar graphs show the assessments of various disease variables of DSS-induced colitis, including stool score and wet/dry weight ratio of the fecal pellets in Nherf2+/+ and Nherf2−/− mice. E: Bar graphs show the measured stool score and wet/dry weight ratio of the fecal pellets in TNBS-treated Nherf2+/+ and Nherf2−/− mice. F: Schematic representation of CFTR and iNOS interaction in the gut epithelia and the associated molecular machinery in inflammatory bowel disorder-affected gut epithelia. Data are expressed as means ± SEM. n = 4 experimental groups (A and B); n = 3 to 4 mice per group (A); n = 5 to 8 mice per TNBS group (B); n = 3 mice per control group with no DSS treatment (D); n = 8 to 10 mice per DSS treatment group (D); n = 8 to 10 mice per TNBS treatment group (E). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 calculated with t-test. Significance of intergroup variance was evaluated with one-way analysis of variance for animal experiments. CFTR, cystic fibrosis transmembrane-conductance regulator; DSS, dextran sodium sulfate; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2; sGC, soluble guanylate cyclase; TNBS, 2,4,6-trinitrobenzenesulfonic.
Discussion
The pathology of UC-associated diarrhea is not clearly understood, and new molecular targets for its amelioration need to be identified. Suggesting a role for compartmentalized signaling (NO-cGMP signaling) in contributing to diarrhea in a pathologic condition is a novel theme. We demonstrated that iNOS is overexpressed at or near the plasma membrane in UC-affected colon mucosa, where it actively contributes to NO-cGMP signaling and is channeled to CFTR activation in localized domains in UC-affected mucosa. The question then arises: How would this mode of CFTR function regulation by iNOS in localized domains be different from that by freely diffusing iNOS? The definitive advantage of compartmentalized regulation of CFTR is the selective and specific activation of CFTR, which also allows a major proportion of iNOS function to be channeled to CFTR regulation. Therefore, it presents a therapeutic advantage to target a compartmentalized complex rather than individual molecules because these may have independent roles than when within the complex. The rationale is that it is the complex that renders CFTR function pathophysiologic in IBD. It also needs to be emphasized that for NO-cGMP signaling to be effective and communicated to CFTR, the cGMP-synthesizing enzyme guanylate cyclase and cGMP-effector system (protein kinase A/G) also need to be recruited into the complex (Figure 7F). Determination of the presence of these additional proteins in this compartmentalized mega-complex is needed for future studies.
Selective studies document down-regulation of NHERF2 under colitis conditions, but in these studies conclusive patient data are absent, or an only moderate change was observed.39,40 Similar studies report down-regulation of NHERF1 and NHERF3 in colitis.40,41 In our observation of clinical data, NHERF2 expression remains unaltered in IBD. The biological significance of NHERF proteins in mediating highly compartmentalized regulation of CFTR was recapitulated in various NHERF-protein knockout models.42 Although the effect of NHERF1 ablation on CFTR functional regulation could be determined under basal/physiologic conditions, contribution of NHERF2 and NHERF3 surfaced only on certain stimuli or deletion of CFTR. In additional studies that used NHERF1- and NHERF2-deficient mice, NHERF1 was shown to be critical for apical membrane placement of CFTR in native intestinal epithelium and optimizing cAMP- and cGMP-dependent regulation of CFTR, whereas placing NHERF2 not being of importance for these physiologic roles.43 NHERF2 confers inhibition of CFTR activity by coupling the lysophosphatidic acid receptor to CFTR on lysophosphatidic acid stimulus.11,42 Therefore, NHERF proteins follow specific physiologic and pathophysiologic contexts. NHERF2 was reported to act as a protein kinase G-anchoring protein for cyclic GMP kinase II in the cGMP-mediated regulation of sodium–hydrogen exchanger 3 (NHE3).28 In addition, cyclic GMP kinase II cGKII is known to mediate cGMP-responsive activation of CFTR. It is possible that NHERF2 exhibits a dual role in assembling CFTR and iNOS at the plasma membrane, as well as functioning as a protein kinase G-anchoring protein in the complex to spatiotemporally regulate CFTR function.
With the use of DSS- and TNBS-induced colitis, we demonstrated in vivo that Nherf2−/− mice are protected against diarrhea as indicated by the reduction in weight loss, intestinal fluid secretion, stool score, and wet/dry weight ratio. Identifying and targeting specific scaffolding activity of NHERFs in a functional context can be a therapeutic advantage because it allows simultaneous control of multiple signaling cascades in complex diseases by hitting a single protein rather than multiple disease-related proteins.44 In addition, mapping of the PDZ domains and narrowing down to smaller binding segments would confer a high degree of specificity and flexibility in the use of such therapeutic approaches.
Our group identified a cell-permeable, small-molecule compound CO-068 that preferentially inhibits lysophosphatidic acid2–NHERF2 interaction and augments CFTR activity in Calu-3 cells and CFTR-dependent fluid secretion from pig tracheal submucosal glands.45 Recently, a NHERF1-mediated complex of the chemokine receptor CXCR2 with phospholipase C-β2 was reported to regulate neutrophil infiltration during inflammation.46 Also, addition of CXCR2-specific peptide (containing the PDZ motif) was found to suppress neutrophilic chemotaxis and transepithelial migration, having the potential for therapeutic interventions in several inflammatory disorders.46
Diarrhea can result with the occurrence of either decreased absorption (absorptive diarrhea; indirectly coupled to sodium transport) or increased secretion (secretory diarrhea; majorly by CFTR overstimulation, eg, enterotoxin-induced diarrhea).47 NHE3/Slc9a3 is a major NHE present in the intestine that plays an important role in absorption of NaCl and HCO3−.48 Slc9a3−/− mice exhibit mild diarrhea, spontaneously develop colitis, and demonstrate high susceptibility to DSS injury.49,50 Many studies suggest that decreased gene expression/epithelial surface expression/activity of NHE3 and other sodium transporters relates to the incidence of absorptive diarrhea in IBD and animal colitis models.39,51,52 However, other studies discount this to be the case.51,53
CFTR remains critical to regulate electroneutral sodium chloride absorption and NHE3 regulation by augmenting its inhibition in a cAMP-dependent fashion.54–56 It is interesting that the same intracellular cAMP, Ca2+, or cGMP that activates CFTR would inhibit NHE3.55,57 Not only do CFTR and NHE3 share the same secondary messengers for regulation but also regulatory scaffolding NHERF proteins.48 NHE3 harbors a C-terminal PDZ-binding motif, similar to CFTR that would mediate NHE3 interaction with other membrane proteins, cytoskeleton, and protein kinases through NHERFs. cAMP-driven inhibition of NHE3 can be mediated via NHERF1, NHERF2, and NHERF3 (PDZK1), whereas cGMP-mediated inhibition was found to specifically require NHERF2.28,57 NO was found to inhibit NHE3 function by the NO-cGMP pathway, but without account for the effect of inducible NO under inflammation conditions for NHE3 regulation.40,58 In another study, NHE3 expression was found to be unaltered in cytokine-induced inflammation in interleukin-10–deficient mice marked by up-regulated iNOS levels.40 Therefore, the information to comprehensively suggest a role for iNOS in regulating NHE3 function is limited. It remains unclear whether CFTR is the regulatory force for NHE3 function in IBD or whether NHE3 is the major player, or if they act together or independently. We do not currently have experimental evidence to prove co-dependence or to delineate their independent contributions. It will be of further interest to identify NHE3 as also being a component of CFTR-NHERF2-iNOS regulatory complex.
We have presented several lines of evidence to support CFTR in the form of a macromolecular complex that potentially exacerbates diarrhea in colitis. Therefore, our data can be used to rationalize a targeted approach to ameliorate UC-associated diarrhea, depending on identification of specific protein–protein interactions that become pathologic or the scaffolds that assemble such pathophysiologic protein networks in this disease.
Acknowledgments
We thank Dr. David L. Armbruster (University of Tennessee Health Science Center, Memphis, TN) for critically reading the manuscript, Dr. Randal A. Skidgel (University of Illinois, Chicago, IL) for providing the pECFP-iNOS construct, J. Denise Wetzel (Cincinnati Children’s Hospital Medical Center) for editing the manuscript, and Dr. Anand Kulkarni (Tissue Services Core, UTHSC, Memphis, TN) for his assistance with the histopathology scoring.
K.A. designed and performed the experiments, analyzed data, and wrote the manuscript. C.S., W.Z., C.S.M., A.R., and S.Y. performed the experiments, revised the manuscript, and analyzed the data. A.A., Y.H., L.A.D., and X.W. analyzed the data. W.R.D. generated the cGMP sensor. A.P.N. initiated and supervised the project, designed the experiments, analyzed data, and edited the manuscript.
Footnotes
Supported by the NIH grants HL68991 (W.R.D.) and DK080834 and DK093045 (A.P.N.) and the Totman Trust for Medical Research (W.R.D.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.06.007.
Supplemental Data
A: Co-IP of NHERF2 and HA-iNOS in HEK-293 cells in the absence or presence of cross-linker DSP. B: Immunoblot shows interaction of full-length iNOS and iNOS-ΔPDZ with GST-NHERF2 with the use of a GST-pulldown from HEK-293 cells that overexpress iNOS proteins. GST-only protein was used as a negative control. Co-IP, Co-immunoprecipitation; DSP, dithiobis succinimidyl propionate; GST, glutathione S-transferase; HA, hemagglutinin; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2; PDZ, PSD-95/DLG-5/ZO-1.
A: Immunoblot for NHERF2 expression in Nherf2+/+ and Nherf2−/− mice kidney and colon lysates. B: H&E-stained untreated and DSS-treated Nherf2+/+ and Nherf2−/− mice colon sections. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections from DSS-treated Nherf2+/+ and Nherf2−/− mice. C: Representative colon sections from iNOS-specific immunostained DSS-treated and untreated Nherf2+/+ and Nherf2−/− mice. Data are expressed as means ± SEM. n = 3 to 5 (B). Scale bar = 20 μm. Original magnification: ×5 μm (inset, D). DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2.
Western blot analysis shows NHERF2 knockdown in HEK-293 cells with the use of lentiviral siRNA particles. β-Actin was used as a control. NHERF2, Na+/H+ exchanger regulatory factor 2.
Immunostaining for CFTR expression in normal and UC-affected human colon sections depicts no change in CFTR expression between these two samples. Arrows indicate membrane localization of CFTR in the colon samples. CFTR, cystic fibrosis transmembrane-conductance regulator; UC, ulcerative colitis.
Representative pseudocolor images show expression of cytosolic and membrane-bound cGMP sensors cygnet 2.1 and m-cygnet 2.1 and the schematic to show how the membrane-bound sensor was generated.
Averaged representative traces of CFTR-mediated I− influx in HEK-293-S (scrambled siRNA) and HEK-293-N (NHERF2 siRNA) that overexpress Flag-CFTR in response to 250 μmol/L zaprinast. n = 4 to 6. CFTR, cystic fibrosis transmembrane-conductance regulator; NHERF2, Na+/H+ exchanger regulatory factor 2; YFP, yellow fluorescent protein.
A: H&E-stained colon sections of the mice from DSS ± CFTRinh-172 treatment experimental group. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. B: H&E-stained colon sections of the mice from DSS ± L-NIL treatment experimental group. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. Data are expressed as means ± SEM. n = 6 to 12 (A); n = 5 to 7 (B). ∗P < 0.05, determined by t-test. CFTR, cystic fibrosis transmembrane-conductance regulator; CFTRinh-172, thiozolidinone CFTR inhibitor; DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; L-NIL, N6-(1-Iminoethyl)-l-lysine dihydrochloride.
H&E-stained colon sections of the TNBS-treated Nherf2+/+ and Nherf2−/− mice. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. Data are expressed as means ± SEM. n = 5 to 10. H&E, hematoxylin and eosin; NHERF2, Na+/H+ exchanger regulatory factor 2; TNBS, 2,4,6-trinitrobenzenesulfonic.
References
- 1.Xavier R.J., Podolsky D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Eckmann L. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann N Y Acad Sci. 2006;1072:28–38. doi: 10.1196/annals.1326.008. [DOI] [PubMed] [Google Scholar]
- 3.Goyette P., Labbe C., Trinh T.T., Xavier R.J., Rioux J.D. Molecular pathogenesis of inflammatory bowel disease: genotypes, phenotypes and personalized medicine. Ann Med. 2007;39:177–199. doi: 10.1080/07853890701197615. [DOI] [PubMed] [Google Scholar]
- 4.Tang Y., Forsyth C.B., Keshavarzian A. New molecular insights into inflammatory bowel disease-induced diarrhea. Expert Rev Gastroenterol Hepatol. 2011;5:615–625. doi: 10.1586/egh.11.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulsen J.H., Fischer H., Illek B., Machen T.E. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1994;91:5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welsh M.J., Smith A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 7.Bresso F., Askling J., Astegiano M., Demarchi B., Sapone N., Rizzetto M., Gionchetti P., Lammers K.M., de Leone A., Riegler G., Nimmo E.R., Drummond H., Noble C., Torkvist L., Ekbom A., Zucchelli M., Lofberg R., Satsangi J., Pettersson S., D'Amato M. Potential role for the common cystic fibrosis DeltaF508 mutation in Crohn's disease. Inflamm Bowel Dis. 2007;13:531–536. doi: 10.1002/ibd.20067. [DOI] [PubMed] [Google Scholar]
- 8.Kleizen B., Braakman I., de Jonge H.R. Regulated trafficking of the CFTR chloride channel. Eur J Cell Biol. 2000;79:544–556. doi: 10.1078/0171-9335-00078. [DOI] [PubMed] [Google Scholar]
- 9.Guggino W.B., Stanton B.A. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 10.Li C., Naren A.P. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr Biol (Camb) 2010;2:161–177. doi: 10.1039/b924455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C., Dandridge K.S., Di A., Marrs K.L., Harris E.L., Roy K., Jackson J.S., Makarova N.V., Fujiwara Y., Farrar P.L., Nelson D.J., Tigyi G.J., Naren A.P. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C., Krishnamurthy P.C., Penmatsa H., Marrs K.L., Wang X.Q., Zaccolo M., Jalink K., Li M., Nelson D.J., Schuetz J.D., Naren A.P. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell. 2007;131:940–951. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zingarelli B., Szabo C., Salzman A.L. Reduced oxidative and nitrosative damage in murine experimental colitis in the absence of inducible nitric oxide synthase. Gut. 1999;45:199–209. doi: 10.1136/gut.45.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beck P.L., Xavier R., Wong J., Ezedi I., Mashimo H., Mizoguchi A., Mizoguchi E., Bhan A.K., Podolsky D.K. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am J Physiol Gastrointest Liver Physiol. 2004;286:G137–G147. doi: 10.1152/ajpgi.00309.2003. [DOI] [PubMed] [Google Scholar]
- 15.Kolios G., Valatas V., Ward S.G. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rachmilewitz D., Eliakim R., Ackerman Z., Karmeli F. Direct determination of colonic nitric oxide level–a sensitive marker of disease activity in ulcerative colitis. Am J Gastroenterol. 1998;93:409–412. doi: 10.1111/j.1572-0241.1998.00409.x. [DOI] [PubMed] [Google Scholar]
- 17.Kimura H., Hokari R., Miura S., Shigematsu T., Hirokawa M., Akiba Y., Kurose I., Higuchi H., Fujimori H., Tsuzuki Y., Serizawa H., Ishii H. Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in colonic mucosa of patients with active ulcerative colitis. Gut. 1998;42:180–187. doi: 10.1136/gut.42.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro-Lerida I., Martinez-Moreno M., Ventoso I., Alvarez-Barrientos A., Rodriguez-Crespo I. Binding of CAP70 to inducible nitric oxide synthase and implications for the vectorial release of nitric oxide in polarized cells. Mol Biol Cell. 2007;18:2768–2777. doi: 10.1091/mbc.E06-12-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glynne P.A., Darling K.E., Picot J., Evans T.J. Epithelial inducible nitric-oxide synthase is an apical EBP50-binding protein that directs vectorial nitric oxide output. J Biol Chem. 2002;277:33132–33138. doi: 10.1074/jbc.M205764200. [DOI] [PubMed] [Google Scholar]
- 20.Zaki M.H., Boyd K.L., Vogel P., Kastan M.B., Lamkanfi M., Kanneganti T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penmatsa H., Zhang W., Yarlagadda S., Li C., Conoley V.G., Yue J., Bahouth S.W., Buddington R.K., Zhang G., Nelson D.J., Sonecha M.D., Manganiello V., Wine J.J., Naren A.P. Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell. 2010;21:1097–1110. doi: 10.1091/mbc.E09-08-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carey R., Jurickova I., Ballard E., Bonkowski E., Han X., Xu H., Denson L.A. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:446–457. doi: 10.1002/ibd.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner D., Otley A.R., Mack D., Hyams J., de Bruijne J., Uusoue K., Walters T.D., Zachos M., Mamula P., Beaton D.E., Steinhart A.H., Griffiths A.M. Development, validation, and evaluation of a pediatric ulcerative colitis activity index: a prospective multicenter study. Gastroenterology. 2007;133:423–432. doi: 10.1053/j.gastro.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 24.Haberman Y., Tickle T.L., Dexheimer P.J., Kim M.O., Tang D., Karns R., Baldassano R.N., Noe J.D., Rosh J., Markowitz J., Heyman M.B., Griffiths A.M., Crandall W.V., Mack D.R., Baker S.S., Huttenhower C., Keljo D.J., Hyams J.S., Kugathasan S., Walters T.D., Aronow B., Xavier R.J., Gevers D., Denson L.A. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 26.Li H., Vahlquist A., Torma H. Interactions between FATP4 and ichthyin in epidermal lipid processing may provide clues to the pathogenesis of autosomal recessive congenital ichthyosis. J Dermatol Sci. 2013;69:195–201. doi: 10.1016/j.jdermsci.2012.11.593. [DOI] [PubMed] [Google Scholar]
- 27.Cha B., Donowitz M. The epithelial brush border Na+/H+ exchanger NHE3 associates with the actin cytoskeleton by binding to ezrin directly and via PDZ domain-containing Na+/H+ exchanger regulatory factor (NHERF) proteins. Clin Exp Pharmacol Physiol. 2008;35:863–871. doi: 10.1111/j.1440-1681.2008.04931.x. [DOI] [PubMed] [Google Scholar]
- 28.Cha B., Kim J.H., Hut H., Hogema B.M., Nadarja J., Zizak M., Cavet M., Lee-Kwon W., Lohmann S.M., Smolenski A., Tse C.M., Yun C., de Jonge H.R., Donowitz M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem. 2005;280:16642–16650. doi: 10.1074/jbc.M500505200. [DOI] [PubMed] [Google Scholar]
- 29.Naren A.P., Cobb B., Li C., Roy K., Nelson D., Heda G.D., Liao J., Kirk K.L., Sorscher E.J., Hanrahan J., Clancy J.P. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci U S A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raghuram V., Mak D.O., Foskett J.K. Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain-mediated interaction. Proc Natl Acad Sci U S A. 2001;98:1300–1305. doi: 10.1073/pnas.031538898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linder A.E., McCluskey L.P., Cole K.R., 3rd, Lanning K.M., Webb R.C. Dynamic association of nitric oxide downstream signaling molecules with endothelial caveolin-1 in rat aorta. J Pharmacol Exp Ther. 2005;314:9–15. doi: 10.1124/jpet.105.083634. [DOI] [PubMed] [Google Scholar]
- 32.Zabel U., Kleinschnitz C., Oh P., Nedvetsky P., Smolenski A., Muller H., Kronich P., Kugler P., Walter U., Schnitzer J.E., Schmidt H.H. Calcium-dependent membrane association sensitizes soluble guanylyl cyclase to nitric oxide. Nat Cell Biol. 2002;4:307–311. doi: 10.1038/ncb775. [DOI] [PubMed] [Google Scholar]
- 33.Galietta L.V., Jayaraman S., Verkman A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol. 2001;281:C1734–C1742. doi: 10.1152/ajpcell.2001.281.5.C1734. [DOI] [PubMed] [Google Scholar]
- 34.Ma T., Thiagarajah J.R., Yang H., Sonawane N.D., Folli C., Galietta L.J., Verkman A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krieglstein C.F., Cerwinka W.H., Laroux F.S., Salter J.W., Russell J.M., Schuermann G., Grisham M.B., Ross C.R., Granger D.N. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med. 2001;194:1207–1218. doi: 10.1084/jem.194.9.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCafferty D.M., Miampamba M., Sihota E., Sharkey K.A., Kubes P. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut. 1999;45:864–873. doi: 10.1136/gut.45.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacNaughton W.K. Nitric oxide-donating compounds stimulate electrolyte transport in the guinea pig intestine in vitro. Life Sci. 1993;53:585–593. doi: 10.1016/0024-3205(93)90716-g. [DOI] [PubMed] [Google Scholar]
- 38.Seimetz M., Parajuli N., Pichl A., Veit F., Kwapiszewska G., Weisel F.C., Milger K., Egemnazarov B., Turowska A., Fuchs B., Nikam S., Roth M., Sydykov A., Medebach T., Klepetko W., Jaksch P., Dumitrascu R., Garn H., Voswinckel R., Kostin S., Seeger W., Schermuly R.T., Grimminger F., Ghofrani H.A., Weissmann N. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147:293–305. doi: 10.1016/j.cell.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan S., Alex P., Dassopoulos T., Zachos N.C., Iacobuzio-Donahue C., Donowitz M., Brant S.R., Cuffari C., Harris M.L., Datta L.W., Conklin L., Chen Y., Li X. Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm Bowel Dis. 2009;15:261–274. doi: 10.1002/ibd.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lenzen H., Lunnemann M., Bleich A., Manns M.P., Seidler U., Jorns A. Downregulation of the NHE3-binding PDZ-adaptor protein PDZK1 expression during cytokine-induced inflammation in interleukin-10-deficient mice. PLoS One. 2012;7:e40657. doi: 10.1371/journal.pone.0040657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeruva S., Chodisetti G., Luo M., Chen M., Cinar A., Ludolph L., Lunnemann M., Goldstein J., Singh A.K., Riederer B., Bachmann O., Bleich A., Gereke M., Bruder D., Hagen S., He P., Yun C., Seidler U. Evidence for a causal link between adaptor protein PDZK1 downregulation and Na(+)/H(+) exchanger NHE3 dysfunction in human and murine colitis. Pflugers Arch. 2015;467:1795–1807. doi: 10.1007/s00424-014-1608-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh A.K., Riederer B., Krabbenhoft A., Rausch B., Bonhagen J., Lehmann U., de Jonge H.R., Donowitz M., Yun C., Weinman E.J., Kocher O., Hogema B.M., Seidler U. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J Clin Invest. 2009;119:540–550. doi: 10.1172/JCI35541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broere N., Hillesheim J., Tuo B., Jorna H., Houtsmuller A.B., Shenolikar S., Weinman E.J., Donowitz M., Seidler U., de Jonge H.R., Hogema B.M. Cystic fibrosis transmembrane conductance regulator activation is reduced in the small intestine of Na+/H+ exchanger 3 regulatory factor 1 (NHERF-1)- but not NHERF-2-deficient mice. J Biol Chem. 2007;282:37575–37584. doi: 10.1074/jbc.M704878200. [DOI] [PubMed] [Google Scholar]
- 44.Dev K.K. Making protein interactions druggable: targeting PDZ domains. Nat Rev Drug Discov. 2004;3:1047–1056. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W., Penmatsa H., Ren A., Punchihewa C., Lemoff A., Yan B., Fujii N., Naren A.P. Functional regulation of cystic fibrosis transmembrane conductance regulator-containing macromolecular complexes: a small-molecule inhibitor approach. Biochem J. 2011;435:451–462. doi: 10.1042/BJ20101725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y., Wang S., Farooq S.M., Castelvetere M.P., Hou Y., Gao J.L., Navarro J.V., Oupicky D., Sun F., Li C. A chemokine receptor CXCR2 macromolecular complex regulates neutrophil functions in inflammatory diseases. J Biol Chem. 2012;287:5744–5755. doi: 10.1074/jbc.M111.315762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931–943. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donowitz M., Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825–872. doi: 10.1152/physrev.00030.2006. [DOI] [PubMed] [Google Scholar]
- 49.Schultheis P.J., Clarke L.L., Meneton P., Miller M.L., Soleimani M., Gawenis L.R., Riddle T.M., Duffy J.J., Doetschman T., Wang T., Giebisch G., Aronson P.S., Lorenz J.N., Shull G.E. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 50.Larmonier C.B., Laubitz D., Thurston R.D., Bucknam A.L., Hill F.M., Midura-Kiela M., Ramalingam R., Kiela P.R., Ghishan F.K. NHE3 modulates the severity of colitis in IL-10-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G998–G1009. doi: 10.1152/ajpgi.00073.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeruva S., Farkas K., Hubricht J., Rode K., Riederer B., Bachmann O., Cinar A., Rakonczay Z., Molnar T., Nagy F., Wedemeyer J., Manns M., Raddatz D., Musch M.W., Chang E.B., Hegyi P., Seidler U. Preserved Na(+)/H(+) exchanger isoform 3 expression and localization, but decreased NHE3 function indicate regulatory sodium transport defect in ulcerative colitis. Inflamm Bowel Dis. 2010;16:1149–1161. doi: 10.1002/ibd.21183. [DOI] [PubMed] [Google Scholar]
- 52.Clayburgh D.R., Musch M.W., Leitges M., Fu Y.X., Turner J.R. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–2694. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lohi H., Makela S., Pulkkinen K., Hoglund P., Karjalainen-Lindsberg M.L., Puolakkainen P., Kere J. Upregulation of CFTR expression but not SLC26A3 and SLC9A3 in ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2002;283:G567–G575. doi: 10.1152/ajpgi.00356.2001. [DOI] [PubMed] [Google Scholar]
- 54.Clarke L.L., Harline M.C. CFTR is required for cAMP inhibition of intestinal Na+ absorption in a cystic fibrosis mouse model. Am J Physiol. 1996;270:G259–G267. doi: 10.1152/ajpgi.1996.270.2.G259. [DOI] [PubMed] [Google Scholar]
- 55.Kato A., Romero M.F. Regulation of electroneutral NaCl absorption by the small intestine. Annu Rev Physiol. 2011;73:261–281. doi: 10.1146/annurev-physiol-012110-142244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahn W., Kim K.H., Lee J.A., Kim J.Y., Choi J.Y., Moe O.W., Milgram S.L., Muallem S., Lee M.G. Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3- salvage mechanisms in model systems and the mouse pancreatic duct. J Biol Chem. 2001;276:17236–17243. doi: 10.1074/jbc.M011763200. [DOI] [PubMed] [Google Scholar]
- 57.Yun C.H., Oh S., Zizak M., Steplock D., Tsao S., Tse C.M., Weinman E.J., Donowitz M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci U S A. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gill R.K., Saksena S., Syed I.A., Tyagi S., Alrefai W.A., Malakooti J., Ramaswamy K., Dudeja P.K. Regulation of NHE3 by nitric oxide in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G747–G756. doi: 10.1152/ajpgi.00294.2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Co-IP of NHERF2 and HA-iNOS in HEK-293 cells in the absence or presence of cross-linker DSP. B: Immunoblot shows interaction of full-length iNOS and iNOS-ΔPDZ with GST-NHERF2 with the use of a GST-pulldown from HEK-293 cells that overexpress iNOS proteins. GST-only protein was used as a negative control. Co-IP, Co-immunoprecipitation; DSP, dithiobis succinimidyl propionate; GST, glutathione S-transferase; HA, hemagglutinin; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2; PDZ, PSD-95/DLG-5/ZO-1.
A: Immunoblot for NHERF2 expression in Nherf2+/+ and Nherf2−/− mice kidney and colon lysates. B: H&E-stained untreated and DSS-treated Nherf2+/+ and Nherf2−/− mice colon sections. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections from DSS-treated Nherf2+/+ and Nherf2−/− mice. C: Representative colon sections from iNOS-specific immunostained DSS-treated and untreated Nherf2+/+ and Nherf2−/− mice. Data are expressed as means ± SEM. n = 3 to 5 (B). Scale bar = 20 μm. Original magnification: ×5 μm (inset, D). DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; iNOS, inducible nitric oxide synthase; NHERF2, Na+/H+ exchanger regulatory factor 2.
Western blot analysis shows NHERF2 knockdown in HEK-293 cells with the use of lentiviral siRNA particles. β-Actin was used as a control. NHERF2, Na+/H+ exchanger regulatory factor 2.
Immunostaining for CFTR expression in normal and UC-affected human colon sections depicts no change in CFTR expression between these two samples. Arrows indicate membrane localization of CFTR in the colon samples. CFTR, cystic fibrosis transmembrane-conductance regulator; UC, ulcerative colitis.
Representative pseudocolor images show expression of cytosolic and membrane-bound cGMP sensors cygnet 2.1 and m-cygnet 2.1 and the schematic to show how the membrane-bound sensor was generated.
Averaged representative traces of CFTR-mediated I− influx in HEK-293-S (scrambled siRNA) and HEK-293-N (NHERF2 siRNA) that overexpress Flag-CFTR in response to 250 μmol/L zaprinast. n = 4 to 6. CFTR, cystic fibrosis transmembrane-conductance regulator; NHERF2, Na+/H+ exchanger regulatory factor 2; YFP, yellow fluorescent protein.
A: H&E-stained colon sections of the mice from DSS ± CFTRinh-172 treatment experimental group. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. B: H&E-stained colon sections of the mice from DSS ± L-NIL treatment experimental group. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. Data are expressed as means ± SEM. n = 6 to 12 (A); n = 5 to 7 (B). ∗P < 0.05, determined by t-test. CFTR, cystic fibrosis transmembrane-conductance regulator; CFTRinh-172, thiozolidinone CFTR inhibitor; DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; L-NIL, N6-(1-Iminoethyl)-l-lysine dihydrochloride.
H&E-stained colon sections of the TNBS-treated Nherf2+/+ and Nherf2−/− mice. Bar graph represents the histology score (mucosal damage score and infiltration of immune cells) of the colon sections. Data are expressed as means ± SEM. n = 5 to 10. H&E, hematoxylin and eosin; NHERF2, Na+/H+ exchanger regulatory factor 2; TNBS, 2,4,6-trinitrobenzenesulfonic.