

Abstract
A regio-, diastereo- and enantioselective [4+3] cycloaddition between vinylcarbenes and dienes has been achieved using the dirhodium tetracarboxylate catalyst Rh2(S-BTPCP)4. This methodology provides facile access to 1,4-cycloheptadienes that are regioisomers of those formed from the tandem cyclopropanation/Cope rearrangement reaction of vinylcarbenes with dienes.
Keywords: Carbenoid, cycloaddition, rhodium, cycloheptadienes, vinyldiazoacetate
Cycloaddition reactions play a pivotal role in the synthetic design of complex natural products. The venerable Diels-Alder reaction is a notable example of the synthetic utility of cycloaddition strategies.[1] Excellent stereocontrol is routinely achieved, and with appropriate electronic bias in the diene 1 and dienophile 2, high levels of regioselectivity are also obtained to form cyclohexene 3 (Scheme 1).[2] The defined regiocontrol is a great advantage for the predictable use of the Diels-Alder reaction but it also presents a limitation as the reverse regioisomer 4 is not readily accessed. Limited methods have been developed to address this longstanding problem but they involve multistep synthetic sequences.[3] Relatedly, our laboratory has developed a rhodium-catalyzed formal [4+3] cycloaddition between vinyldiazoacetates and dienes.[4,5] This reaction is also highly regioselective, as illustrated by the reaction of diene 1 with rhodium vinylcarbene intermediate 5 to generate the cycloheptadiene 6, because it proceeds by a tandem cyclopropanation/Cope rearrangement (CPCR). In this paper we describe an alternative and mechanistically distinct [4+3] cycloaddition caused by initial attack of the diene at the vinylogous position of the vinylcarbene instead of at the carbene center. In this way, we achieve a regiochemical switch of the [4+3] cycloaddition, leading to the formation of cycloheptadiene 7.
Scheme 1.

Different cycloaddition approaches.
A representative example of the regular formal [4+3] cycloaddition of vinyldiazoacetates is the Rh2(S-PTAD)4-catalyzed reaction of 2-siloxyvinyldiazoacetate 9 with various dienes 8 (Scheme 2).[5a] Some of the most significant chiral catalysts for the reactions of vinyldiazoacetates are illustrated in Figure 1.[6] The Rh2(S-PTAD)4-catalyzed reaction proceeds with high asymmetric induction and has been used as a key reaction for the synthesis of several natural products.[5a,b] In all of the published examples to date, the reactions are highly regioselective, proceeding by an initial cyclopropanation of the electronically most favorable and sterically most accessible double bond.
Scheme 2.
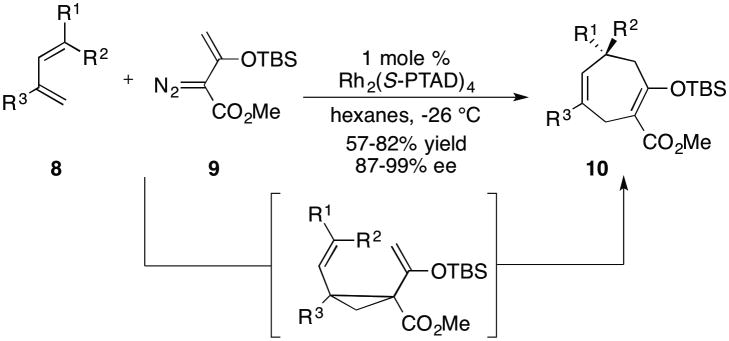
Rh2(S-PTAD)4-catalyzed tandem cyclopropanation/Cope rearrangement.
Figure 1.
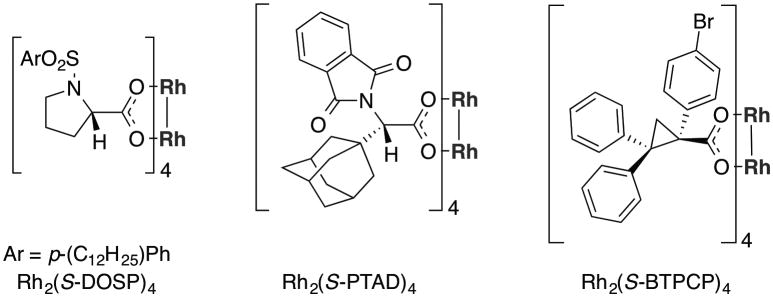
Representative chiral dirhodium catalysts.
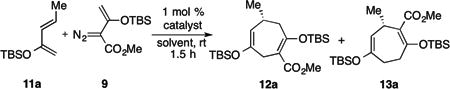
The possibility of reversing the regiochemistry of the CPCR [4+3] cycloaddition was discovered during a study of the reaction of siloxyvinyldiazoacetate 9 with 2-tert-butyldimethylsiloxybutadiene 11a catalyzed by Rh2(S-PTAD)4 (Table 1, entry 1). The major product was the typical CPCR cycloadduct 12a but a small amount of the regiosiomeric [4+3] cycloadduct 13a was also formed (12a:13a ratio, 94:6). We rationalized that the formation of the regioisomeric [4+3] cycloadduct 13a was most likely caused by a competing reaction of the diene occurring at the vinylogous position of the carbenoid, generating a zwitterionic intermediate, which then cyclizes to 13a.[7-9] Previous studies have shown that vinylogous reactivity is favored in polar solvents.[7] Indeed, when the reaction was repeated using dichloromethane as solvent, the ratio of 12a to 13a improved to 87:13, and the regiosiomeric [4+3] cycloadduct 13a was produced in 71% ee. Another major chiral catalyst for vinyldiazoacetate reactions is the proline derived dirhodium catalyst, Rh2(S-DOSP)4 (see Figure 1). The Rh2(S-DOSP)4-catalyzed reaction of 9 with 11a increased the amount of the regioisomeric [4+3] cycloadduct 13a formed. In the reaction conducted in pentane, the ratio of 12a to 13a was 79:21, whereas when dichloromethane was used as solvent, the ratio improved to 30:70 (entries 3 and 4), but with poor enantiocontrol (5% ee).
Table 1.
First observation of [4+3] cycloadduct 13a.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | solvent | catalyst | ratio[a] 12a : 13a | yield, % | ee of 13a, %[d] |
| 1 | pentane | Rh2(S-PTAD)4 | 94 :6 | 55[b] | -73 |
| 2 | CH2Cl2 | Rh2(S-PTAD)4 | 87 :13 | 43[c] | -71 |
| 3 | pentane | Rh2(S-DOSP)4 | 79 : 21 | 62[c] | 33 |
| 4 | CH2Cl2 | Rh2(S-DOSP)4 | 30 : 70 | 61[c] | 5 |
Determined by 1H NMR of crude reaction mixture,
isolated yield of 12a,
combined yield of 12a and 13a.
negative sign indicates the opposite enantiomer of 13a.
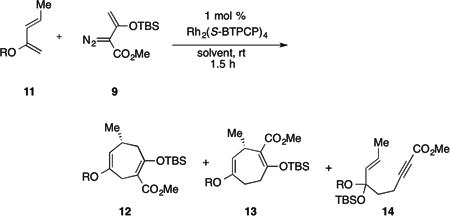
Recently, we have discovered that sterically crowded tetrakis(triarylcyclopropanecarboxylate) dirhodium catalysts are very effective at enhancing vinylogous reactivity of rhodium vinylcarbenes.[8j] Therefore, we explored the effect of Rh2(S-BTPCP)4 on the reaction of 9 with 2-siloxydienes 11 (Table 2). The Rh2(S-BTPCP)4-catalyzed reaction resulted in the formation of a third product, alkynoate 14a, in addition to the two [4+3] cycloadducts 12a and 13a. Compounds related to alkynoate 14a had been observed in the reaction of vinylcarbenes with vinyl ethers and were shown to be derived from vinylogous attack on the vinylcarbenoid, followed by a siloxy group transfer.[8c] The Rh2(S-BTPCP)4-catalyzed reaction, however, was promising because the amount of the standard cycloadduct 12a was considerably reduced (entries 1 and 2). The desired cycloadduct 13a was the dominant product when dichloromethane was used as solvent (entry 1) but the enantioinduction (87% ee vs. 54% ee) was higher when pentane was used as solvent (entry 2). Further optimization studies revealed that the siloxy group migration to form the alkynoate 14 was sensitive to the size of the siloxy group on the diene. The OTMS derivative 11b gave more of the alkynoate product 14b, but when the more sterically demanding OTIPS derivative 11c was used, only traces of the alkynoate 14c was observed. Furthermore, the size of the siloxy group also influenced carbenoid versus vinylogous reactivity, as the ratio of 12c to the desired regioisomer 13c improved to 5:95. Furthermore, the bulkier silyl groups resulted in improved levels of enantioselectivity for the reaction (70% ee for the TMS derivative 13b, 87% ee for the TBS derivative 13a, and 96% ee for the TIPS derivative 13c).
Table 2.
Optimization studies for the formation of 13.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | compound | R | solvent | ratio[a] 12 : 13 : 14 | yield, % [e] | % ee[c] |
| 1 | a | TBS | CH2Cl2 | 9 : 70 : 26 | 37[b], 16[d] | 54 |
| 2 | a | TBS | pentane | 4 : 29 : 62 | 23[b], 42[d] | 87 |
| 3 | b | TMS | pentane | 14 : 18 : 68 | 16[b], 45[d] | 70 |
| 4 | c | TIPS | pentane | 5 : 95 : trace | 59[c] | 96 |
Determined by 1H NMR of crude reaction mixture,
combined yield of 12 and 13,
adduct 13.
isolated yield of 14.
isolated yield.
Having established optimized conditions for the formation of the regiosiomeric [4+3] cycloadducts, we explored the generality of this reaction with representative 2-siloxydienes 15 (Table 3). Both 4-substituted and 3,4-disubstituted 2-OTIPS-1,3-dienes afforded the [4+3] cycloadducts 16 with good regiocontrol and moderate yields. In general, the products 16 were formed in higher yields at elevated temperatures with the diene as the limiting reagent (38-50% yield versus 65-78% yield), but the levels of asymmetric induction were generally higher at ambient temperatures with the vinyldiazoacetate 9 as the limiting agent (90-94% ee versus 82-95% ee). The [4+3] cycloaddition is restricted to moderately electron-rich dienes. Highly electron-rich dienes such as the triisopropylsilyl variant of the Danishefsky's diene results in the formation of a complex mixture of products, whereas less electron-rich dienes such as the p-nitro derivative of 15a fail to react. The absolute configuration of 16a was determined by X-ray crystallography of a derivative prepared by DIBAL reduction followed by hydrolysis. The absolute configuration of the other cycloadducts are tentatively assigned by analogy. [10]
Table 3. Reactions of 2-OTIPS-1,3-dienes 15 with 9.
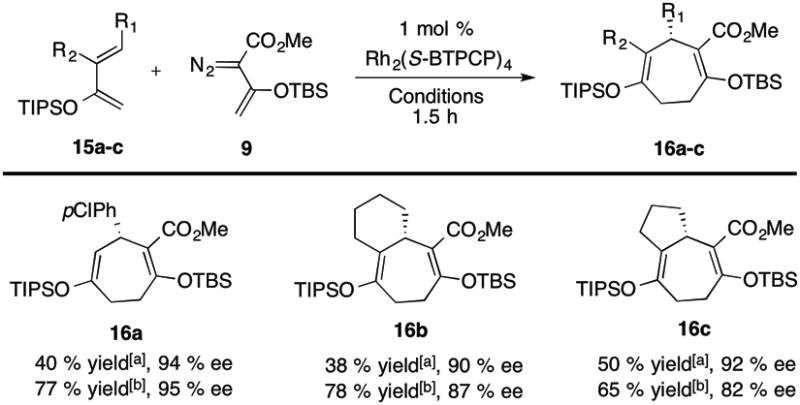
|
5 equiv. of diene, pentane, rt.
2 equiv. of 9, hexanes, reflux. Yield refers to isolated yield after silica gel chromatography.
In general, vinylogous reactivity under rhodium(II) catalysis is most common when the vinyl terminus of the carbenoid is unsubstituted.[8] Rh2(S-BTPCP)4, however, is capable of inducing vinylogous reactivity on more highly substituted vinylcarbenes.[8j] Therefore, the Rh2(S-BTPCP)4-reaction of the methyl-substituted vinyldiazoacetate 17 was examined (Table 4). Mono- and bicyclic cycloadducts 18 containing stereogenic centers at C-3 and C-7 were formed with high levels of enantiocontrol (92-99% ee). Furthermore, even though the vinyldiazoacetate 17 consists of a mixture of (E,Z) isomers, only the cis diastereomers of 18 were formed. These results suggest that only one geometrical isomer of the rhodium vinylcarbene is capable of undergoing the [4+3] cycloaddition.
Table 4.
Diastereoselective Formal [4+3] Cycloaddition.
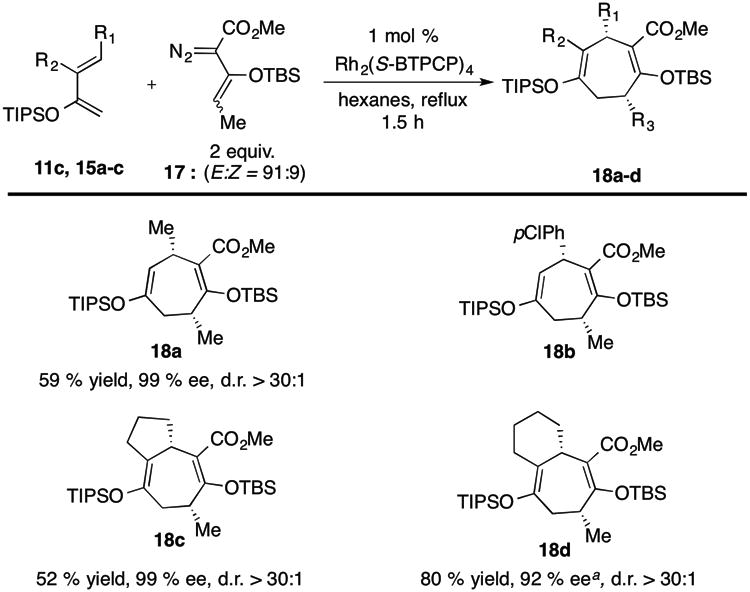
|
%ee of corresponding allylic alcohol. Yield refers to isolated yield after silica gel chromatography. Enantiomeric excess was determined by chiral HPLC.
The study was then extended to the reaction of vinyldiazoacetate 9 with 1,4-disubstituted diene 19, which would be expected to generate [4+3] cycloadducts 20 containing stereogenic centers at C-4 and C-7 (Table 5). The additional terminal substituent on the diene was expected to be a challenge to the [4+3] cycloaddition because it would add steric interference at the position of initial bond-formation and dienes 19 consisted of ∼9:1 mixture of (Z,E):(E,E) isomers. Consequently, we were pleasantly surprised to find that cycloadducts 20 were produced as single diastereoisomers with high levels of asymmetric induction (96% ee). The high diastereoselectivity is presumable caused by preferred reaction of the vinylcarbenoid on (Z,E)-19 over (E,E)-19.
Table 5.
Diastereoselective Formal [4+3] Cycloaddition.
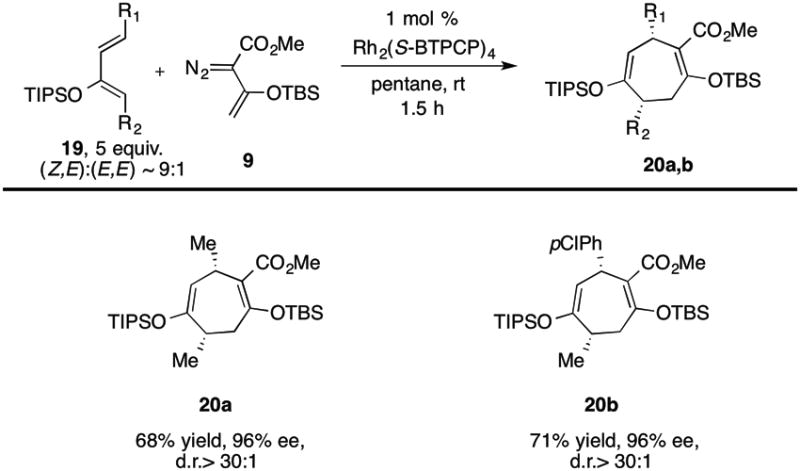
|
Yield refers to isolated yield after silica gel chromatography. Enantiomeric excess was determined by chiral HPLC
A final reaction was conducted between vinyldiazoacetate 17 and diene 19a. Even though both 17 and 19 are composed of mixtures of E/Z isomers the Rh2(S-BTPCP)4,-catalyzed reaction smoothly formed cycloadduct 21, containing three new stereogenic centers in 72% yield as a single diastereomer. Analysis of 21 by chiral HPLC was unsuccessful, but after treatment with DIBAL, the resulting cycloheptene 22 was determined to be 85% ee (reduction yield is unoptimized).
A reasonable mechanism for the [4+3] cycloadditions is shown in Scheme 4. Rh2(S-BTPCP)4 has been shown to be a sterically hindered catalyst that blocks the re-face of the carbene,[6c] and preferentially reacts at the vinylogous position when electron rich trapping agents are used. The regioisomeric [4+3] cycloaddition involves initial attack at the vinylogous position of the vinylcarbene as illustrated in 23 to generate zwitterionic intermediate 24. A similar type of zwitterionic intermediate has been proposed for the [3+2] cycloaddition between vinylcarbenes and dienes.[8d] The high diastereoselectivity of this reaction suggests that the subsequent cyclization to the cycloheptadiene 25 is faster than bond rotation. Zwitterionic intermediates have been shown to be likely in several types of stereoselective reactions of rhodium vinylcarbenes,[8] so the concept of rapid cyclization of zwitterionic intermediates without epimerization is well established. Both the diene and the vinylcarbenoid would need to react through an s-cis conformation for ring closure to occur without bond rotation. Rhodium 2-siloxyvinylcarbenes have been shown to adopt preferentially an s-cis conformation[11] and the 2-siloxy group would likely favor the s-cis conformation of the diene. The face selectivity of attack of the diene on the rhodium carbene determines the absolute configuration of the final product, even when no stereocenters are present in the zwitterionic intermediate 27, as illustrated in the conversion 26 –27 – 28. This would be the case for the examples shown in Table 3. Even though the exact trajectory of approach of the dienes is not known, the proposed mechanism can be used to rationalize why highly diastereoselective reactions are possible even when the vinylcarbene and the diene are mixtures of geometrical isomers. An (E,E) diene would have R1 in an unfavorable position pointing towards the vinylcarbene (see structure 29), whereas a Z-vinylcarbene would unlikely form because R3 would be pointing towards the catalyst surface (see structure 30).
Scheme 4.

Proposed mechanism for the [4+3] cycloaddition: a: General model; b: Model of the reaction of the usubstituted vinyldiazoacetate 9 with dienes 15a-c; c: Explanation for high diastereocontrol when the diene or vinyldiazoacetates are not pure geomertrical isomers.
In summary, an asymmetric [4+3] cycloaddition between rhodium vinylcarbenes and dienes has been developed. The reaction proceeds with the opposite regiochemistry to the traditional tandem cyclopropanation/Cope rearrangement. An efficient asymmetric cycloaddition was achieved when Rh2(S-BTPCP)4 was used as catalyst in hydrocarbon solvents. By an appropriate choice of diene and vinyldiazoacetate, cloheptadienes with up to three new stereogenic centers can be generated with excellent stereocontrol.
Experimental Section
Representative procedure for the synthesis of 13c: To a round-bottom flask was added (E)-triisopropyl(penta-1,3-dien-2-yloxy)silane (1.51 mmol, 5.00 equiv), pentane (3.5 mL), and Rh2(S-BTPCP)4 (5.3 mg, 0.0030 mmol, 0.010 equiv). A solution of methyl 3-((tert-butyldimethylsilyl)oxy)-2-diazobut-3-enoate (77.0 mg, 0.30, mmol, 1.00 equiv) in pentane (3.5 mL) was added by syringe pump over 1 h. Once the addition was complete, the reaction was allowed to stir at 23 °C for 0.5 h. The reaction was stopped by concentration under reduced pressure and purified by flash chromatography on silica gel to provide pure products.
Supplementary Material
Scheme 3.

Reaction of 17 with 19a.
Acknowledgments
**This work was supported by the National Institutes of Health (GM-099142-02). We would like to thank Dr. John Bacsa, Emory, University, for the X-Ray crystallographic analysis and Changming, Qin for the supply of Rh2(S-BTPCP)4 catalyst.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Diels O, Alder K. Justus Liebigs Ann Chem. 1928;460:98. [Google Scholar]
- 2.For selected review of intermolecular and intramolecular Diels-Alder reactions, see: Oppolzer W. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Pergamon Press; New York: 1991. pp. 315–399.Roush WR. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Pergamon Press; New York: 1991. pp. 513–550.Corey EJ. Angew Chem. 2002;114:1724.Angew Chem, Int Ed. 2002;41:1650.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis GE. Angew Chem. 2002;114:1742. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z.Angew Chem, Int Ed. 2002;41:1668.
- 3.a) Shea KJ, Staab J, Zandi KS. Tetrahedron Lett. 1991;24:2715. [Google Scholar]; b) Sammis GM, Flamme EM, Xie H, Ho DM, Sorenson EJ. J Am Chem Soc. 2005;127:8612. doi: 10.1021/ja052383c. [DOI] [PubMed] [Google Scholar]
- 4.Davies HML, Clark DM, Smith TK. Tetrahedron Lett. 1985;26:5659.Davies HML, Ahmed G, Churchill MR. J Am Chem Soc. 1996;118:10774.Davies HML, Stafford DG, Doan BD, Houser JH. J Am Chem Soc. 1998;120:3326.d) For an early review on the tandem cyclopropanation/Cope rearrangement, see: Davies HML. Tetrahedron. 1993;49:5203.
- 5.For recent examples of the use of the tandem cyclopropanation/Cope rearrangement in total synthesis, see Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J Am Chem Soc. 2009;131:8329. doi: 10.1021/ja9019484.Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J Am Chem Soc. 2010;132:12422. doi: 10.1021/ja103916t.Olson JP, Davies HML. Org Lett. 2008;10:573. doi: 10.1021/ol702844g.Jackson KL, Henderson JA, Motoyoshi H, Phillips AJ. Angew Chem. 2009;121:2382. doi: 10.1002/anie.200806111.Angew Chem, Int Ed. 2009;48:2346.Xu J, Caro-Diaz EJE, Theodorakis EA. Org Lett. 2010;12:3708. doi: 10.1021/ol1015652.
- 6.a) Davies HML, Ahmed G, Churchill MR. J Am Chem Soc. 1996;118:10774. [Google Scholar]; b) Reddy RP, Lee GH, Davies HML. Org Lett. 2006;8:3437. doi: 10.1021/ol060893l. [DOI] [PubMed] [Google Scholar]; c) Qin C, Boyarskikh V, Hansen JH, Hardcastle KI, Musaev DG, Davies HML. J Am Chem Soc. 2011;133:19198. doi: 10.1021/ja2074104. [DOI] [PubMed] [Google Scholar]
- 7.For early examples of vinylogous reactivity of rhodium vinylcarbenes, see: Davies HML, Saikali E, Clark TJ, Chee EH. Tetrahedron Lett. 1990;31:6299.Davies HML, Hu B, Saikali E, Bruzinski PR. J Org Chem. 1994;59:4535.
- 8.For recent applications of vinylogous reactivity of rhodium vinylcarbenes in synthesis, see: Lian Y, Davies HML. Org Lett. 2010;12:924. doi: 10.1021/ol9028385.Lian Y, Davies HML. Org Lett. 2012;14:1934. doi: 10.1021/ol300632p.Valette D, Lian Y, Haydek JP, Hardcastle KI, Davies HML. Angew Chem. 2012;124:8764. doi: 10.1002/anie.201204047.Angew Chem, Int Ed. 2012;51:8636.Smith AG, Davies HML. J Am Chem Soc. 2012;134:18241. doi: 10.1021/ja3092399.Wang X, Xu X, Zavalji PY, Doyle MP. J Am Chem Soc. 2011;133:16402. doi: 10.1021/ja207664r.Xu X, Zavalij PY, Hu W, Doyle MP. J Org Chem. 2013;78:1583. doi: 10.1021/jo302696y.Qian Y, Zavalij PJ, Hu W, Doyle MP. Org Lett. 2013;15:1564. doi: 10.1021/ol400339c.Wang X, Abrahams QM, Zavalij PY, Doyle MP. Angew Chem. 2012;124:6009. doi: 10.1002/anie.201201917.Angew Chem Int Ed. 2012;51:5907.Qian Y, Xu X, Wang X, Zavalij PJ, Hu W, Doyle MP. Angew Chem. 2012;124:6002. doi: 10.1002/anie.201202525.Angew Chem Int Ed. 2012;51:5900.Qin C, Davies HML. J Am Chem Soc. 2013;135:14516. doi: 10.1021/ja4069003.
- 9.For a recent review on the vinylogous transformations of 9, see: Xu X, Doyle MP. Acc Chem Res. 2014;47:1396. doi: 10.1021/ar5000055.
- 10.For x-ray crystallographic data for the derivative of 16a, see the Supporting Information. The crystal structure has been deposited at the Cambridge Crystallographic Data Centre under deposition number CCDC 979611.
- 11.Hansen JH, Gregg TM, Ovalles SR, Lian Y, Autschbach J, Davies HML. J Am Chem Soc. 2011;133:5076. doi: 10.1021/ja111408v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.