

Abstract
Introduction
When establishing the physiological roles of specific receptors in normal and disease states, it is critical to have selective antagonist ligands for each receptor in a receptor system with several subtypes. The melanocortin receptors have five subtypes referred to as the melanocortin 1 receptor, melanocortin 2 receptor, melanocortin 3 receptor, melanocortin 4 receptor and melanocortin 5 receptor, and they are of critical importance for many aspects of human health and disease.
Areas covered
This article reviews the current efforts to design selective antagonistic ligands for the five human melanocortin receptors summarizing the currently published orthosteric and allosteric antagonists for each of these receptors.
Expert opinion
Though there has been progress, there are still few drugs available that address the many significant biological activities and diseases that are associated with these receptors, which is possibly due to the lack of receptor selectivity that these designed ligands are currently showing. The authors believe that further studies into the antagonists’ 3D conformational and topographical properties in addition to future mutagenesis studies will provide greater insight into these ligands which could play a role in the treatment of various diseases in the future.
Keywords: allosteric antagonist, melanocortin receptors, orthosteric antagonists, receptor antagonists
1. Introduction
The melanocortin receptors (MCRs) are a class of five 7-transmembrane GPCRs that are critically involved in many important aspects of human health and disease including pigmentation, feeding behavior, response to stress, immune response, sexual motivation and behavior, pain, energy utilization, learning, sebaceous gland secretion and many others [1–4]. Though the pigmentary hormone α-melanocyte stimulating hormone (α-MSH) and the stress hormone, adrenal corticotropic hormone (ACTH), were among the early peptide hormones and neurotransmitters whose structures were determined [5], the cloning of the five receptors was only accomplished in the early 1990s [2,3]. The cloning of these receptors and the availability of the biological potent, stable and bioavailable α-MSH analogs NDP-α-MSH (Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2, MT-I) [6] and MT-II (Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys)-NH2) [7,8] and the potent melanocortin 3 receptor (MC3R) and melanocortin 4 receptor (MC4R) antagonist SHU-9119 [Ac-Nle-c[Asp-His-D-Nal (2′)-Arg-Trp-Lys]-NH2 [9] (it is a potent agonist at the melanocortin 1 receptor (MC1R) and melanocortin 5 receptor (MC5R)) led to an explosion of scientific interest. Though there were earlier reports of α-MSH derivatives and analogs that had weak antagonist activity in pigmentary assays (MC1R) [10,11], none of these compounds turned out to be useful in vivo nor did they provide critical insights into the structural features that could be utilized for the design of more potent analogs. Similarly for ACTH, though ACTH-11 to -24 was reported to be a weak antagonist to the steroidogenic receptor for ACTH [12], it provided little insight into designing potent antagonists for the ACTH receptor (melanocortin 2 receptor (MC2R)). Despite these early shortcomings, in 1977, Schwyzer, who made many early outstanding contributions to this area, stated: ‘I personally believe that we have about reached the limits of insight that can be reasonably provided by structure-activity studies – except in the realm perhaps of stimulus-effect coupling’ [13]. Since then, it has become very clear that given the complexity of hormone and neurotransmitter biological activity of these hormone/neurotransmitter systems, the development of structural and conformational considerations that can lead to biologically potent, stable, bio-available and receptor selective antagonist analogs is essential for sorting out and understanding the biological activities of these hormones and neurotransmitters and their specific receptors. Furthermore, it is clear that highly receptor selective agonist and antagonist ligands are essential for determining the specific contributions of each specific hormone/neurotransmitter and its receptor to a specific biological effect in health and disease states that may involve the specific endogenous ligand and receptor. Indeed, systematic approaches to developing such ligands including antagonist ligands have been developed (for antagonist design reviews see for example [14–18]). In this paper, we review the approaches which have been taken for the design of selective antagonist ligands, including allosteric antagonists, for the MCRs. Though there is still much to be developed, we hope this review will aid in the needed future developments.
2. Design of melanocortin receptor selective antagonist ligands
A major focus in designing new melanotropin analogs and mimetics is to gain potency, selectivity and improved pharmacological properties that will be suitable for biological and medical applications. Ligand structure-based drug design has been used for many years in peptide and peptide mimetic-based drug design [18] and has played a critical role in the development of melanocortin ligands [19]. Moreover, the development of sophisticated biophysical and computational methodologies provides additional important tools for designing constrained analogs that can provide critical insights for antagonist ligand design.
This review, due to space limitations, will primarily examine efforts that have been made to modify the natural peptide ligands for the human MCRs (hMCRs). We have previously reviewed the strategies that can be used to establish a peptide pharmacophore (e.g., [16,18–20]). Further design, especially conformational and topographical constraint, (β-turn mimetics and so on are needed, and many of these methods have been applied to develop stable and selective melanotropins. In addition, 3D structural analysis (NMR, X-ray) of selected ligands, combined with computational studies have aided in the design of selective small molecule hMCR ligands, and chimeric or hybrid structures also have been used more recently. N-methylation of the amide bonds also has been introduced into melanocortin drug design [21]. Substituting amide protons with methyl groups can result in receptor subtype selectivity, and in addition other pharmacological properties can be improved, such as metabolic stability, lipophilicity, enhanced potency, enhanced bioavailability and BBB and gut permeability. All of these approaches may also turn an agonist into an antagonist.
3. hMC1R antagonists
MC1R is mainly present in the skin of most animals where it is involved in the control of the relative amounts of pheomelanin and photoprotective eumelanin. Appropriate MC1R agonist ligands may offer protection against damaging and mutagenic effects of UV radiation or be useful in the diagnosis and treatment of certain types of skin cancer, such as melanoma [l]. MC1R is also present on the surface of various immune cells (e.g., macrophages, fibroblasts, monocytes, mast cells, neutrophils) and is a potential target for the development of ligands for the treatment of acute and chronic inflammatory diseases, neurodegenerative diseases and systemic host reactions [22]. In the CNS, the MC1R is present only on neurons in the periaqueductal grey matter of the midbrain [23], where it is thought to have a role in pain control [24,25].
The first selective human MC1R (hMC1R) antagonist with nanomolar potency was Ac-Nle-Asp-Trp-D-Phe-Nle-Trp-Lys-NH2 [10] (pA2 = 8.4 in frog skin) but with agonist activities at the MC3R, MC4R and MC5R (IC50 = 260, 60 and 910 nM, respectively) [24]. All efforts to develop a structure–activity relationship (SAR) were unsuccessful as modified analogs were either agonists or inactive. However, the analog was used to demonstrate that the hMC1R is important in modulating pain by female specific mechanisms of analgesia [24].
A promising early antagonist of the hMC1R was c[Gly-Cpg-D-Nal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 (1, Table 1) [26] which was highly selective versus the human MC4R (hMC4R) (1, 110-fold, Table 1) though only modestly selective versus the MC3R. Again, efforts to establish a SAR around this α-MSH/deltorphin chimera were unsuccessful, but it did lead us to explore other cyclic melanotropin derivatives which have been more successful.
Table 1.
Ligands selective for the hMC1R as antagonists.
| Peptides* | hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | EC50 (nM) | Act % | IC50 (nM) | EC50 (nM) | Act % | IC50 (nM) | EC50 (nM) | Act % | IC50 (nM) | EC50 (nM) | Act % | |
| 1 | 12 | > 10,000 | 0 | 44 | > 10,000 | 0 | 1300 | > 10,000 | 0 | ND | ND | ND |
| 2 | 4 | > 10,000 | 0 | 124 | > 1000 | 25 | 105 | > 5000 | 6 | 672 | 205 | 55 |
| 3 | 7 | > 10,000 | 0 | 158 | > 1000 | 17 | 144 | > 10,000 | 7 | 546 | 105 | 63 |
| 4 | 17 | > 10,000 | 0 | 3900 | > 103 | NA | > 10,000 | NA | NA | 1300 | 1300 | NA |
IC50 = Concentration of peptide at 50% specific binding (n = 4). NB = 0% of 125I-NDP-α-MSH displacement observed at 10 (μM. Percent binding efficiency = maximal % of [125I]-NDP-α-MSH displacement observed at 10 (μM; NA: No activity at 10−4 M; ND: Not Determined.
Peptides: 1. c[Gly-Cpg-D-Nal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 (Cpg = cyclopentylglycine); 2. c[CO-p-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2; 3. c[CO-m-C6H4-CO-His-D-Nal(2′)-Arg-Trp-Lys]-NH2; 4. c[(CH2)3 CO-Gly-His-D-Phe-Arg-D-Trp-Cys(S)]-Asp-Arg-Phe-Gly-NH2.
The cyclic peptides 2 and 3 (Table 1) by Mayorov et al. [27] show the effects of the linker arm rigidity and ring size on the antagonist selectivity of MCRs for the hMC1R. This study explored the use of a novel cyclic lactam α-MSH template. A variety of dicarboxylic acid linkers were introduced between the α-amino group of His6 and the ε-amino group of Lys10 which led to high-affinity, selective hMCR-1 and -5 (hMC1R and human MC5R (hMC5R)) antagonists [27]. The incorporation of more hydrophilic groups into the linker arm was found to be unfavorable for both binding potency and receptor selectivity. Peptides 2 and 3 containing highly conformationally constrained hydrophobic linkers (m- and p-phthalic acids) were found to be selective nanomolar range hMC1R antagonists (IC50 = 4 and 7 nM, respectively), whereas the employment of a small conformationally constrained linker (maleic acid) resulted in a high-affinity (IC50 =19 nM) and selective hMC5R antagonist (not shown). The newly developed melanotropins have served as critical biochemical tools for elucidating the full spectrum of functions performed by the physiologically important MC1R and MC5Rs.
Another successful approach has been the design of cyclic sulfide analogs (manuscript in preparation) of which compound 4 is an example. As shown in Table 1, this compound is a highly potent (IC50 = 17 nM) antagonist at the hMC1R and has excellent selectivity against the human MC3R (hMC3R), MC4R and MC5R (230, > 600, 76). It was used to demonstrate the importance of the hMC1R in mediating opioid-induced hyperalgesia [28]. A structure–activity study is being prepared for publication.
4. Human MC2R antagonists
The MC2R, also known as the ACTH receptor, requires a different pharmacophore than the other subtypes of MCRs. ACTH is the endogenous ligand for the MC2R. ACTH is a 39-residue peptide whose first 13 amino acids are identical to the sequence of α-MSH. However, unlike other MCRs, the C-terminal cationic portion of the POMC product in ACTH is necessary for MC2R binding. Binding of ACTH to the MC2R increases the adenylate cyclase activity and thus the activation of protein kinase A. Shorter peptides ACTH1-24 and ACTH1-23 were shown to have agonist properties similar to that of the full length ACTH, whereas a 13-residue peptide ACTH11-24 was shown to have antagonist properties (Table 2) [29]. It was also shown that a peptide with an N-methyltryptophan instead of Trp at the 9 position in ACTH1-24 had antagonist activity [30]. In a recent study by Kovalitskaya et al., it was shown that the cationic C-terminal sequence -Lys-Lys-Arg-Arg- (ACTH15-18) had antagonist activity [31]. This tetrapeptide binds to membranes of the rat adrenal cortex with high affinity (Table 2), but does not show any adenylate cyclase activity even at 1000 nM concentration. Therefore, they concluded that the tetrapeptide KKRR (ACTH15-18) is an antagonist for the ACTH receptor. The same research group has also found in a separate study that a synthetic peptide GKVLKKRR corresponding to the fragments 81 – 88 of human pro-IL-1α protein acts as an antagonist at the MC2R [32]. They chose this peptide because it has 80% sequence similarity with fragments 10 – 18 of ACTH. Clearly, much additional work is needed to develop useful and selective MC2R antagonist ligands.
Table 2.
ACTH antagonists and their inhibition of [3H] ACTH11-24 in rat adrenal cortex membranes by unlabeled peptides.
| Compound | Peptide sequence | IC50 (nM) | Ki (nM) |
|---|---|---|---|
| ACTH1-24 | SYSMEHFRWGKPVGKKRRPVKVYP | ||
| ACTH11-24 | KPVGKKRRPVKVYP | 11.3 | 1.7 |
| ACTH15-18 | KKRR | 15.1 | 2.3 |
| pIL-1α81-88 | GKPVGKKRR | 7.6 | 2 |
5. hMC3R antagonists
Pharmacological and genetic studies have shown that MC3R is involved in the control of energy homeostasis, host inflammatory responses, thermoregulation and cardiovascular function. Some observations have also suggested that this receptor might be an inhibitory autoreceptor which suppresses release of α-MSH on POMC neurons. In the CNS, MC3R is usually colocalized with MC4R, and selective MC3R ligands are needed to understand and separate physiological actions of these two receptors. There are suggestions that such compounds could be useful in the treatment of disorders of nutrient imbalance and have protective effects in myocardial ischemia, reperfusion-induced heart arrhythmias and/or in some inflammatory conditions [33–36].
A very great effort has been made to develop highly potent hMC4R-selective agonists and antagonists due to the proposed involvement of this receptor in the regulation of feeding behavior, sexual behavior and other biological activities. On the other hand, comparatively little attention has been given to obtaining selective ligands for the hMC3R especially considering the dearth of specific evidence of its physiological functions. The availability of highly selective antagonists for the hMC3R relative to the hMC4R should greatly enhance this understanding. Studies of MC3R-deficient mice show increased fat mass, reduced lean mass and higher feed efficiency than their wild-type littermates [37]. Furthermore, peripheral injections of an hMC3R selective agonist [D-Trp8]γ-MSH can stimulate food intake in mice suggesting an important role of this receptor subtype in the regulation of feeding and energy partitioning [38]. In addition, possible involvement of the hMC3R in the regulation of inflammatory responses and cardiovascular function have also been proposed (e.g., [39,40]). Finally, emerging evidence points to a potential role of the hMC3R in the regulation of erectile function and sexual behavior [41], which provides further impetus for the development of highly selective hMC3R agonists and antagonists.
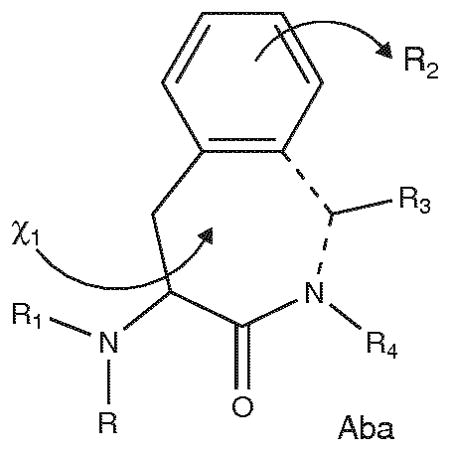
Several approaches to the design of hMC3R-selective agonists and antagonists have been described in the literature. Among the natural MSHs, γ-MSH exhibits substantial hMC3R agonist selectivity, whereas α-MSH and β-MSH show little selectivity for any specific receptor subtype. For selective antagonists for the MC3R the D-Nal(2′)7 substitution for D-Phe7 as used in SHU-9119 [9], but in a novel cyclic structure (5, Table 3), resulted in good binding affinities at all four receptor subtypes and converted a full hMC3R agonist to a good affinity somewhat selective hMC3R antagonist [42]. Interestingly, when His6 was substituted with a proline residue a similarly selective hMC3R antagonist analog was obtained with particularly good selectivity versus the MC1R (6, Table 3). Proline plays an important role to stabilize the secondary structure. The structurally similar α-MSH analog 7 also was reported to be a highly potent hMC3R antagonist (7, Table 3) with good selectivity against the hMC4R and the hMC5R [43]. This peptide shows an improved antagonist potency and selectivity toward the hMC3R compared to the parent peptide, the nonselective hMC3R/hMC4R antagonist SHU9119 [9]. A further improvement for the MC3R in addition to the cis olefin bridged compound 7 [43] was the bridged heterocyclic 2,3-pyrazine-containing compound (data not shown [43]). The cyclic lactam analog containing the special Aba residue (compound 8, Table 3) had > 200-fold selectivity against the MC1R and the MC4R, and to a lesser extent the MC5R, and was a potent antagonist at the MC3R [44]. Interestingly, the Aba-D-Nal(2′) analog (data not shown, [44]) displayed high affinity hMC3R and hMC5R antagonist properties (IC50 = 43 and 87 nM, respectively), but weak binding affinity for the hMC4R (IC50 = 1.7 μM). The observed lack of agonist activity at the hMC3R points to possible steric interference from Aba. Analog 9 [45] was designed as a hybrid α-MSH-AGRP structure. This hybrid showed high antagonist binding affinity at the hMC3R (1.7 nM) and good selectivities against the hMC1R and hMC4R, but also is a potent antagonist at the hMC5R
Table 3.
Selective hMC3R antagonists.
| No. | Sequence | hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50, nM |
EC50, nM |
% Max effect |
IC50, nM |
EC50, nM |
% Max effect |
IC50, nM |
EC50, nM |
% Max effect | IC50, nM |
EC50, nM |
% Max effect |
||
| MT-II | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 0.2 | 0.3 | 100 | 1.25 | 1.85 | 100 | 1.07 | 2.87 | 100 | 7.47 | 3.3 | 100 |
| SHU-9119 | Ac-Nle-c[Asp-His-D-NAL(2′)-Arg-Trp-Lys]-NH2 | 0.2 | 0.3 | 100 | 2.3 | NA | 0 | 0.6 | NA | 0 | 0.9 | 1.2 | 97 |
| 5 | (o)C-(CH2)3-C(O)-c[His-D-Nal(2′)-Arg-Trp-Lys]NH2 | ND | ND | ND | Ki 5.9 | NA | 0 | Ki 220 | 1200 | 22.8 | Ki 26 | 1 | 81 |
| 6 | c[CO-(CH2)3-CO-Pro-D-Nal(2′)-Arg-Trp-Lys]-NH2 (AVM133) | > 10,000 | NA | 0 | 11 pA2 = 9.8 | NA | 0 | 330 | NA | 0 | 27 | 23 | 40 |
| 7 | c[CO-cis-CH = CH-CO-Pro-D-Nal(2′)-Arg-Trp-Lys]-NH2 | > 10,000 | NA | 0 | 20 | NA | 0 | 520 | NA | 0 | 120 | NA | 0 |
| 8 | Ac-Nle-c[Asp-Aba-D-Phe-Arg-Trp-Lys]-NH2 | > 10,000 | NA | 0 | 50 | NA | 0 | > 10,000 | NA | 0 | 2900 | NA | 0 |
| 9 | c[Nle-Val-D-Nal(2′)-Arg-Trp-Glu]-NH2 (AVM127) | 4000 | > 10,000 | 0 | 1.7 | > 10,000 | 0 | 180 | 16 | 13 | 2.2 | > 10,000 | 0 |
| 10 | Ac-Nle-c[Asp-β-Ala-D-Nal(2′)-Arg-Trp-Lys]-NH2 | ND | ND | ND | 210 | NA | 0 | NB | 9900 | 0 | NB | NA | 0 |
| 11 | Ac-Nle-c[Asp-Mamb-D-Nal(2′)-Arg-Trp-Lys]-NH2 | ND | ND | ND | 67 | NA | 0 | > 3000 | NA | 0 | 2500 | NA | 0 |
| 12 | Ac-Nle-c[Asp-Acpc-D-Nal(2′) Arg-Trp-Lys]-NH2 | ND | ND | ND | 2.5 | NA | 0 | 270 | NA | 0 | 4.1 | 390 | 50 |
| 13 | H-Tyr-Val-Nle-Gly-His-D-Phe-Arg-D-Nal(2′)-Asp-Arg-Phe-Gly-NH2 | 0.7 | 2 | 87 | 6 | NA | 0 | 6 | 36 | 100 | 5 | 200 | 67 |
| 14 | Ac-c[Cys-Arg-D-Phe-Cys]-Trp-NH2 | NB | NA | 0 | 420 | 2300 | 28 | NB | NA | 0 | NB | NA | 0 |
Structure of Aba:
D-Nal(2′), NH2; ND: Not determined; NA: No activity at 10−4 M; NB: No binding at 10−4 M
Analog 10 (Table 3) [46], a cyclic analog related to SHU-9119, was a weak but selective antagonist at the hMC3R and completely inactive as a ligand at the hMC4R and hMC5R. These results indicated that a simple, unconstrained amino-acid residue ((β-Ala) in the 6 position of MT-II can lead to selective ligands for the hMC3R. Interestingly, analog 11 (Table 1), which differs from 10 by having a Mamb (3-aminomethylbenzoic acid) residue in position 5, was a more potent and highly selective antagonist for the hMC3R [46]. These data suggest that the Mamb residue has a considerable impact in the formation of ligand–receptor complex but only for antagonists at the hMC3R. It is possible that the same residue could destabilize the ligand–receptor interactions for other MCRs.
Replacement of His6 with Acpc (1-aminocyclo-propane-1-carboxylic acid), a highly side chain constrained amino acid in SHU-9119 [47], leads to a potent antagonist at the hMC3R (IC50 = 2.5 nM) (12, Table 3). Interestingly, the compound is a potent partial agonist at the hMC5R (Table 3).
Substitution of Trp8 with D-Nal(2′)8 and Phe6 with D-Phe6 in [Nle3]-γ-MSH-NH2 forms a unique selective antagonist for the hMC3R, 13. In this group of hybrid melanotropin peptides, analog 13 was found to be a hMC3R selective antagonist (IC50 = 6 nM), but with potent agonist activity at the MC1R, MC4R and MC5R In this analog, the sterically bulky D-Nal(2′)8 was substituted for the D-Trp8, which converts a hMC3R selective agonist to a hMC3R selective antagonist [48]. The side chains of residue 8 in peptide analog 13 [D-Nal(2′)] favored the gauche(+) conformation. It was proposed that the gauche(+) conformation of the side chain of the residue 8 may be important for selective hMC3R antagonist activity.
Analog 14 is a noncompetitive allosteric antagonist of the hMC3R which was designed as a hybrid AGRP/MSH pharmacophore. Compared to the previous cyclized melanotropins with a 23 membered cyclic β-turn pharmacophore, this peptide has only a 17 membered ring and there is no β-turn moiety involved in the pharmacophore. The orientation of the aromatic groups of Phe and Trp and of the positive charged Arg, which are critically important for binding to the hMC3R, is changed in 14. This could explain why this peptide is a noncompetitive allosteric ligand for the hMC3R [49].
6. hMC4R antagonists
The MC4R is mainly expressed in the CNS where it is found in virtually all areas [50] and also is present in sensory neurons in the periphery [51,52]. It has a major role in control of feeding behavior [53,54]. hMC4R agonists were shown to increase metabolic rate and reduce appetite in various models of rodent obesity [54], while hMC4R antagonists reversed these effects [54]. Consequently, hMC4R peptide and non-peptide agonists were sought which could be developed as potential therapeutic agents for the treatment of obesity and other feeding disorders. In addition, hMC4R antagonists also have been designed and evaluated for the potential treatment of anorexia, wasting in the elderly, or cachexia which accompanies many chronic diseases [55]. Additionally, the hMC4R has been recognized as an important target for the development of compounds which could alleviate sexual dysfunction [41,56,57].
As shown in Table 4, many peptide and non-peptide small ligands have been designed and found to be highly selective antagonist ligands for the hMC4R. Perhaps this is not surprising due to the great interest in both academia and the pharmaceutical industry in obesity, sexual function and other bioactivities associated with the hMC4R.
Table 4.
Selective hMC4R antagonists.
| No. | Sequence | hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50, nM |
EC50, nM |
% Max effect |
IC50, nM |
EC50, nM |
% Max effect |
IC50, nM |
EC50, nM |
% Max effect |
IC50, nM |
EC50, nM |
% Max effect |
||
| MT-II | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 0.2 | 0.3 | 100 | 1.25 | 1.85 | 100 | 1.07 | 2.87 | 100 | 7.47 | 3.3 | 100 |
| SHU-9119 | Ac-Nle-c[Asp-His-D-Nal(2′)-Arg-Trp-Lys]-NH2 | 0.2 | 0.3 | 100 | 1.9 | NA | 0 | 1.07 | 2 | 0 | 3 | 3.3 | 97 |
| 15 | Cyclo(6β-10(ε)-(succinyl6-D-Nal(2′)-Arg-Trp-Lys) NH2 | ND | ND | ND | Ki 150 | NA | 0 | 0.5 | NA | 0 | Ki 540 | 530 | ND |
| 16 | Ac-c[Cys-Nle-Arg-His-D-Nal(2prime;)-Arg-Trp-Gly-Cys-NH2 | Ki 18.6 | NA | NA | Ki 5.45 | NA | NA | Ki 0.29 | NA | NA | Ki 3.29 | NA | NA |
| 17 | H-Tyr-Val-Nle-Glu-His-Phe-Arg-D-Nal(2′)-Asp-Arg-Phe-Gly-NH2 | ND | ND | ND | Ki 15 | 2400 | 31 | Ki 1.5 | > 10,000 | NA | Ki 17 | 28 | 80 |
| 18 | H-Tyr-Val-c[Pen-Gly-His-Phe-Arg-Trp-Cys]-Arg-Phe-Gly-NH2 | ND | ND | ND | Ki 150 | 400 | 77 | Ki 12 | > 10,000 | NA | Ki 1700 | > 10,000 | NA |
| 19 | c[Nle-Asp-D-Phe-Arg-Trp-Glu]-NH2 | > 10,000 | NA | 0 | > 10,000 | NA | 0 | 150 | NA | 0 | > 10,000 | NA | 0 |
| 20 | Ac-c[Cys-Glu-His-D-Nal(2′)-Arg-Trp-Gly-Cys]-Pro-Pro-Lys-Asp-NH2 | Ki 108 | ND | ND | Ki 54 | - | 0 | Ki 3.16 | - | 0 | Ki 694 | - | - |
| 21 | Ac-c[Cys-Glu-His-diCl-D-Phe-Arg-Trp-Gly-Cys]-Pro-Pro-Lys-Asp-NH2 | 60.1 | NA | NA | 73.7 | NA | NA | 0.95 | NA | NA | 211 | NA | NA |
| 22 | Ac-Nle-c[Asp-Aic-D-Nal(2′)-Arg-Trp-Lys]-NH2 | ND | ND | ND | 190 | NA | 0 | 1.3 | NA | 0 | 12 | 17 | 73 |
| 23 | Ac-Nle-c[Asp-Cpe-D-Nal(2′)-Arg-Trp-Lys]-NH2 | ND | ND | ND | 110 | NA | 0 | 0.51 | NA | 0 | 16 | 45 | 79 |
| 24 | Ac-DNal(2′)-Arg-Nal(2′)-NH2 | NB | - | - | NB | - | - | 11.6 | NA | 0 | NB | - | - |
| 25 | Ac-c[Cys-His-D-Phe-Nα-guanidinylbutyl-Cys]-Trp-NH2 | NB | - | - | NB | - | - | 1.8 | NA | NB | - | - | |
| 26 | Ac-c[Cys-His-D-Phe-Cys]-Nα-guanidinylbutyl-D-Trp-NH2 | NB | - | - | > 1000 | - | - | 0.7 | > 1000 | 18 | NB | - | - |
| MT-II | Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 | 0.2 ± 0.01 | 0.3 ± 0.04 | 100 | 1.25 ± 0.2 | 1.85 ± 0.2 | 100 | 1.07 ± 0.3 | 2.87 ± 0.52 | 100 | 7.47 ± 0.23 | 3.3 ± 0.7 | 100 |
| SHU-9119 | Ac-Nle-c[Asp-His-N-NAL(2′)-Arg-Trp-Lys]-NH2 | 0.2 ± 0.01 | 0.3 ± 0.04 | 100 | 1.9 ± 0.2 | NA | 0 | 1.07 ± 0.3 | 2 ± 0.2 | 0 | 3 ± 0.3 | 3.3 ± 0.7 | 97 |
| 27 | 1-[(S)-2-(4-fluorophenyl)-2-(4-isopropylpiper adin-1-yl)ethyl]-4-[4(2-methoxynaphthalen-1-yl) butyl]piperazine | NB | - | - | NB | - | - | Ki 7.6 | NA | 0 | ND | ND | ND |
| 28 | (1-[2-(4-Fluorophenyl)-2-(4-methylpiperazin-1-yl)ethyl]-4-[4-(1-naphthyl)butyl]piperazine | > 10,000 | - | - | > 10,000 | - | - | 124 | 5100 | 0 | > 10,000 | - | - |
| 29 | Pyrrolidine derivatives, Figure 1 | 140 | ND | ND | 1400 | ND | ND | 8.6 | NA | 0 | 380 | ND | ND |
| 30 | Structure Figure 1 | ND | ND | ND | ND | ND | ND | 330 | NA | 0 | ND | ND | ND |
| 31 | Structure Figure 1 | ND | ND | ND | ND | ND | ND | 160 | NA | 0 | ND | ND | ND |
| 32 | Structure Figure 1 | 111 | NA | - | 46 | NA | - | 1 | NA | 0 | 26 | NA | - |
| 33 | Structure Figure 1 | 5700 | NA | NA | 1100 | NA | NA | 4.2/2.8* | NA | NA | 540 | NA | NA |
| 34 | Structure Figure 1 | NA | NA | NA | Ki 640 | NA | NA | Ki 1.8 | NA | NA | NA | NA | NA |
| 35 | Structure Figure 1 | ND | ND | ND | 790 | NA | - | Ki 3.2 | NA | 0 | ND | ND | ND |
IC50 = Concentration of peptide at 50% specific binding (n = 4). EC50 = Effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (n = 4). % Max effect = % of cAMP produced at 10 μM ligand concentration in relation to MT-II. NA = 0% cAMP accumulation observed at 10 μM. The peptides were tested at a range of concentration from 10−10 to 10−5 M. ND: Not determined.
The antagonist properties of the lead compounds were evaluated by their ability to competitively displace the MT-II agonist in a dose-dependent manner, at up to 10 μM. The pA2 values were obtained using the Schild analysis method.
Two radioisotopes were used ([125I]NDP-α-MSH and [125I]AGRP).
NA: No activity at 10−5 M; NB: No binding at 10−5 M.
Among the first selective antagonist ligands for the MC4R were the peptides 15 and 16 in Table 4. Compound 15 ([58], known as MBP10), is a cyclic tetrapeptide derivative based on the antagonist pharmacophore of SHU-9119. It was found to be quite selective versus the MC3R and MC5R (330- and 1100-fold), respectively. It was not tested against the MC1R. The cyclic disulfide peptide 16 [59] also based on the SHU-9119 pharmacophore was not very selective for the MC4R (~ 10- to 60-fold) but was very potent.
The linear γ-MSH analog 17, H-Tyr-Val-Nle-Gly-His-Phe-Arg-D-Nal(2′)-Asp-Arg-Phe-Gly-NH2 (Table 4), was found to be a potent and selective antagonist at the hMC4R [60] while the cyclic Pen3-Cys9 analog 18 also is a potent and selective MC4 antagonist [60], a potent partial agonist at the MC1R and a potent full agonist at the MC5R. The D-Nal(2′)-containing analog 18, on the other hand, is a potent and selective antagonist at the hMC4R, and a moderate potent agonist at the hMC3R (IC50 = 150 nM) and a weak antagonist (IC50 = 1700 nM) at the MC5R [60]. Compound 19 (Table 4) is an interesting cyclic analog related to γ-MSH [45] which, though a modest binder, has highly selective antagonist activity at the MC4R with no binding to the MC1R, MC3R and MC5R.
The cyclic disulfide-containing analog 20 (Table 4) has been found to be a selective MC4R antagonist, which enhances feeding behavior [61]. Another cyclic disulfide analog 21 (Table 4) showed high affinity (Ki = 0.95 nM) and high selectivity (80-fold) for the MC4R over the MC3R [62]. Thus, 21 shows both higher affinity and higher selectivity for the MC4R compared to the earlier described MC4R selective ligand 20 (Table 4).
We found some time ago that replacement of the His5 residue in ligands related to MT-II and SHU-9119 with proline analogs such as 2-aminoindone-2-carboxylic acid (Aic) and 1-amino-1-cyclopentane carboxylic (Cpe) gave analogs 22 and 23 (Table 4) [63]. They had high subnanomolar binding affinities at the MC4R and were antagonists with good selectivity as well, with 22 being 150-fold selective for the MC4R versus MC3R, and 23 being 220-fold selective for the MC4R versus MC3R [63]. Interestingly, as discussed earlier, the analog which contains a 1-aminocyclopropane-1-carboxylic acid in the 5 position to give Ac-Nle-c[Asp-Acpc-D-Nal(2)-Arg-Trp-Lys]-NH2 [47] is a highly potent (IC50 = 2.5 nM) and selective (110-fold) ligand for the MC3R rather than the MC4R [63]. These results suggest that proper substitution in the 5 position replacing the hydrophilic His residue with various proline analogs leads to a potent and MCR selective agent.
A tripeptide analog 24 with D-Nal(2′) and Nal(2′) residues was found to be a potent antagonist at the MC4R [64] with no binding at the MC1R, MC3R and MC5R.
There is increasing evidence that allosteric ligands (agonists and antagonists) for GPCRs such as the MCRs might provide an advantage in modulating their biological responses especially in disease states. Recently, we have discovered allosteric ligands for the hMC4R (25 and 26, Table 4) [65]. These ligands were designed as structural analogs with the arginine side chain moiety in different relationships to the other pharmacophore side chain groups for the MCRs [65]. As discussed for peptide 14, these two peptides are cyclized with 17 membrane rings, and the critical aromatic amino acids Phe, Trp and the charged amino acid Arg, which are important for binding to the hMC4R, are re-orientated. These changes made them noncompetitive hMC4R antagonists. In addition, compared to the peptide 14 the N-guanidinylbutyl and D-Trp might contribute to selectivity between the hMC3R and hMC4R. The unique pharmacological properties of 25 and 26 indicate that they may modulate the hMC4R in novel ways. Thus, they may provide a unique specific tool to examine the functions of the MC4R in animals.
Recently, a series of substituted piperazinebenzylamines exemplified by analogs 27 and 28 have been reported as potent and selective MC4R antagonists [66,67]. Interesting are the MC4R peptide antagonists such as (1-[(S)-2-(4-fluorophenyl)-2-(4-isopropylpiperadin-1-yl)ethyl]-4-[4-(2-methoxynapthalen-1-yl)butyl]piperazine (MCL 0129), analog 27 and the MC4R antagonist 28 which also had serotonin transport inhibitory activity and has the structure (1-[2-(4-fluorophenyl)-2-(4-methylpiperazin-1-yl)ethyl]-4-[4-(1-naphthyl)butyl]piperazine (MCL0042) (28) [67]. It was suggested that the MC4R antagonists may be useful for the treatment of stress-related disorders such as depression and anxiety [67].
A wide variety of complex heterocyclic ligands have been reported that are claimed to be potent and selective antagonists ligands for the MC4R (Table 4 and Figure 1). As can be seen from the table and the figure, though it is claimed that these ligands are selective antagonist ligands for the hMC4R, a complete evaluation of the ligands at the hMC1R, hMC3R, hMC4R and hMC5R have not been reported in many cases. Compounds 29, 32 and 33 ([68–70], respectively) are the only compounds which have been fully evaluated for their selectivity against the hMC1R, hMC3R, hMC4R and hMC5R. It is interesting to note that compound 29 which is shown as two different isomers in Figure 1 has totally different biological activities. The S,S,R-isomer has agonist activity, but the R,S,S-isomer has antagonist activities. We list the others and leave it to the individual reader to evaluate the extent to which claims of receptor selectivity can be accepted and utilized to plan future work (30 [71], 31 [72], 34 [73], 35 [73]).
Figure 1.


Structures of compound in Table 4.
A general question which arises is whether the non-peptide antagonists for the MC4R are actual peptide mimetics, that is, do they utilize the same structure features as peptide antagonists to bind to the hMC4R in its antagonist conformation or do they bind by some other mode to the receptor that maintains the receptor structure in an inactive state. The studies of Yang and co-workers for the native orthosteric agonist ligand α-MSH (MTI) indicate that the conserved amino acids of the hMCRs are involved in the binding, whereas for a small molecule agonist, such as THIQ, in addition to the conserved amino acids of the hMCRs, other non-conserved amino acids of the hMCRs are involved to the binding [49,74]. It is proposed that for the non-peptide antagonists, the non-conserved amino acids can provide the extra stability to keep the receptor structure in an inactive state. However, a much more careful analysis is needed to more clearly evaluate the similarities and differences between peptide and non-peptide antagonists. In addition, computational aided molecular docking of antagonists towards the MC4R may also provide useful insights.
7. hMC5R antagonists
MC5R is expressed in many peripheral tissues [75], in particular the secretory epithelia of many exocrine glands where it affords secretory and trophic controls. The MC5R may also be present in the CNS, but its function in this location is unknown. In rodents, this receptor has been shown to play a role in pheromone and lipid production in the exocrine glands [76]. Together with the other MCRs, the MC5R might be involved in the control of some immune effects [77]. It has been suggested that MC5R agonists may alleviate conditions such as dry eyes and mouth [22], and MC5R antagonists might perhaps be useful in the treatment of acne [78]. We report here on hMC5R antagonists that have been published. In vivo evaluation of these ligands thus far has not been done.
A number of synthetic antagonist ligands for the hMC5R have been reported, but most do not have appreciable selectivity. In this review, we discuss only those ligands which have appreciable selectivity for the hMC5R.
The first highly selective hMC5R ligand was a cyclic disulfide-containing derivative of an α-MSH–β-MSH hybrid where the cyclic part was α-MSH related and the C-terminal β-MSH related (36, Table 5) [79]. As shown in Table 5, 36 is a potent (IC50 =10 nM) hMC5R antagonist that does not bind to the hMC1R, and hMC4R.
Table 5.
Selective hMC5R antagonists.
| Compound | hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) |
EC50 (nM) |
%Act. at 10 μM |
IC50 (nM) |
EC50 (nM) |
%Act. at 10 μM |
IC50 (nM) |
EC50 (nM) |
%Act. at 10 μM |
IC50 (nM) |
EC50 (nM) |
%Act. at 10 μM |
||
| 36 | Ac-cyclo(Cys-Glu-His-D-Phe-Arg-Trp-D-Cys)-Pro-Pro-Lys-Asp-NH2 | NB | NA | NA | 5600 | > 10,000 | 0 | > 10,000 | NA | 0 | 10 | 10,000 | 0 |
| 37 | x-His-Nal(2′)-Arg-Trp-y* | >2000 | NA | 0 | > 1000 | 50 | 21 | > 5000 | NA | 0 | 130 | NA (pA2) 8.3) | 0 |
| 38 | x-Cha-DPhe-Arg-Trp-y* | >2000 | 807 | 92 | > 1000 | 4600 | 179 | NB | NA | 0 | 38 | NA | 0 |
| 39 | x-Pro-DPhe-Arg-Trp-y* | > 1000 | NA | 0 | 380 | > 1000 | 190 | > 1000 | NA | 0 | 58 | NA | 0 |
| 40 | HMC013‡ | NB | NA | 0 | NB | NA | 0 | NB | NA | 0 | 0.50 | NA | 0 |
| 41 | HMC014‡ | NB | NA | 0 | NB | NA | 0 | NB | NA | 0 | 0.10 | NA | 0 |
| 42 | HMC021‡ | NB | NA | 0 | NB | NA | 0 | NB | NA | 0 | 0.40 | NA | 0 |
| 43 | HMC025‡ | NB | NA | 0 | NB | NA | 0 | NB | NA | 0 | 0.10 | NA | 0 |
NB: No binding at 10−5 M; NA: No activity at 10−5 M.
Potential allosteric ligand for hMC5R based on the partial binding efficacy [81].
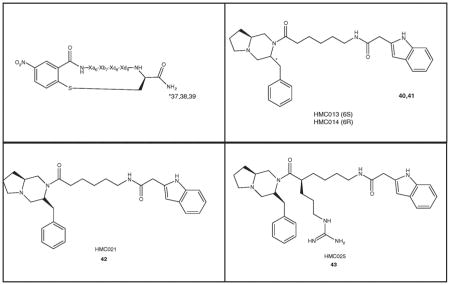
Grieco et al. [80] developed an alkylthioaryl-bridged macrocyclic peptide template related to the MT-II and SHU-9119 and the analogs obtained produced several cyclic ligands of varying potencies and MC5R antagonist selectivities (37 – 39, Table 5) [81]. These 20-membered macrocycles were synthesized by a tandem combination using solid phase peptide synthesis and microwave-assisted reactions. Biological assays for binding affinities and adenylate cyclase activities for the hMC1R, hMC3R, hMC4R and hMC5R showed that the three compounds 37, 38 and 39 (Table 5) are selective antagonists at the hMC5R. In particular, compounds 37 and 38 are selective and competitive hMC5R antagonists with IC50 values of 130 (pA2 = 8.3) and 38 nM, respectively. They can provide important tools for further in vivo biological investigations of the hMC5R.
A non-peptide class of hMC5R selective antagonist ligands has been reported by Cain et al. [80]. These are bicyclic heterocyclic compounds 40 - 43 in Table 5. All four of these compounds appear to bind only to the hMC5R [80], though some related analogs bind to other MCRs (data not shown). Based on binding profiles all of these four compounds act as allosteric antagonists at the hMC5R.
8. Expert opinion
It has become clear, though there is still much to be learned, that ligands can be designed that are potent and selective antagonists for the various hMCRs. These ligands have been and will continue to be valuable tools for helping to sort out the complex pharmacology and physiology of the melanotropin peptides and their receptors. However, there still are not many orthosteric antagonists for the hMCRs that have > 1000-fold selectivity, and so greater effort is needed to obtain more selective ligands. In addition, the specific structural and 3D conformational and topographical properties that determine antagonist versus agonist activity, and most importantly, the 3D structural correlates of ligand antagonist selectivity for the five different MCRs are still to be determined. We have investigated some aspects of this structural pharmacology for antagonists, but there is still much to be learned in this regard as it can provide critical insights. Similarly, though mutagenesis studies have provided important insights into melanocortin agonist ligand–MCR interactions [49,74], virtually nothing is known about the differences associated with agonist versus antagonist ligand–MCR interactions. Ultimately, these studies and related studies using novel biophysical and biochemical methods will be invaluable for the development of a wide variety of melanotropin ligands for the treatment of various diseases, including cancer, obesity, anorexia, sexual dysfunction, immune response, diabetes, pigmentary disorders and many others in which the MCRs play a critical role.
Article highlights.
Obtaining selective antagonist ligands for the five melanocortin receptor is outlined.
Obtaining receptor selective antagonists for the five receptor subtypes has been difficult.
Structural modifications of pharmacophore elements in sequence space, conformational space and topographical space have been used to obtain receptor selective antagonists.
Antagonist SAR is different than agonist SAR for the melanocortin receptors.
Receptor selective antagonists for all five melanocortin receptors have been identified but selectivities of 1000-fold for one receptor are rare.
This box summarizes key points contained in the article.
Acknowledgments
The authors thank M Collie for her help in preparing this review.
Footnotes
Declaration of interest
The research reported from our laboratory has primarily been supported by a grant from the National Institutes of Health (DK17420).
Bibliography
- 1.Hadley ME, editor. The Melanotropic peptides. I, II and III. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 2.Cone RD, editor. The melanocortin system. Ann NY Acad Sci. 2003;994:387. [Google Scholar]
- 3.Cone RD, editor. The melanocortin receptors. Humana Press, Inc; Totowa NJ: 2000. p. 551. [Google Scholar]
- 4.Vandry H, Eberle AN, editors. The melanotropic peptides. Ann NY Acad Sci. 1993;680:687. [Google Scholar]
- 5.Kreiger DT, Ganong WF, editors. ACTH and related peptides: structure, regulation and action. Ann NY Acad Sci. 1977;297:664. [PubMed] [Google Scholar]
- 6.Sawyer TK, Sanfilippo PJ, Hruby VJ, et al. [Nle4, D-Phe7]-alpha-melanocyte stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci USA. 1980;77:5754–8. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Obeidi F, Hadley ME, Pettitt BM, et al. Design of a new class of superpotent cyclic alpha-melanotropins based on quenched dynamic simulations. J Amer Chem Soc. 1989;111:3413–16. [Google Scholar]
- 8.Al-Obeidi F, de Lauro Castrucci AM, Hadley ME, et al. Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: design based on molecular dynamics. J Med Chem. 1989;32:2555–61. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 9.Hruby VJ, Lu D, Sharma SD, et al. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-c[Asp5,D-Phe7, Lys10]alpha-MSH(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–61. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 10.Al-Obeidi F, Hruby VJ, Hadley ME, et al. Design, synthesis and biological activities of a potent and selective alpha-melanotropin antagonist. Int J Peptide Protein Res. 1990;35:228–34. doi: 10.1111/j.1399-3011.1990.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 11.Medzihradszky K. The bio-organic chemistry of alpha-melanotropin. Medical Res Rev. 1982;2:247–70. doi: 10.1002/med.2610020303. [DOI] [PubMed] [Google Scholar]
- 12.Seelig S, Lindley BD, Sayers G. A new approach to the structure-activity relationship for ACTH analogs using isolated adrenal cortex cells. Methods Enzymol. 1975;39D:347–59. doi: 10.1016/s0076-6879(75)39031-9. [DOI] [PubMed] [Google Scholar]
- 13.Schwyzer R. ACTH: a short introduction review. In: Kreiger DT, Ganong WF, editors. ACTH and related peptides: structure, regulation and action Ann NY Acad Sci. Vol. 297. 1977. pp. 3–26. [Google Scholar]
- 14.Hruby VJ. Conformational restrictions of biologically active peptides via amino acid side chain groups. Life Sci. 1982;31:189–99. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 15.Hruby VJ. Design of peptide superagonists and antagonists: conformational and dynamic considerations. In: Vida JA, Gordon M, editors. Conformationally directed drug design peptides and nucleic acids as templates or targets. ACS Symposium Series No. 251. Washington, DC: 1984. pp. 9–27. [Google Scholar]
- 16.Hruby VJ, Al-Obeidi F, Kazmierski WM. Emerging approaches in the molecular design of receptor selective peptide ligands: conformational, topographical and dynamic considerations. Biochemical J. 1990;268:249–62. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowell SM, Balse-Srinivasan PM, Ahn JM, et al. Design and synthesis of peptide antagonists and inverse agonists for G-protein coupled receptors. In: Iyengar R, Hildebrandt JD, editors. Methods in Enzymology. Vol. 343. Academic Press; San Diego, CA: 2002. pp. 49–72. [DOI] [PubMed] [Google Scholar]
- 18.Hruby VJ. Designing peptide receptor agonists and antagonists. Nat Rev Drug Discov. 2002;1:847–58. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 19.Hruby VJ, Wilkes BC, Cody WL, et al. Melanotropins: structural, conformational, and biological considerations in the development of superpotent and superprolonged analogs. Peptide Protein Rev. 1984;3:1–64. [Google Scholar]
- 20.Hruby VJ, Li G, Haskell-Luevano C, et al. Design of peptides, proteins, and peptidomimetics in chi space. Biopolymers. 1997;43:219–66. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Doedens L, Opperer F, Cai M, et al. Multiple N-Methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. J Amer Chem Soc. 2010;132:8115–28. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golz S, Bruggermeier U, Geerts A. US20070037226. Diagnostics, drug screening and therapeutics for diseases associated with human melanocortin 1 receptor (MC1R) 2007
- 23.Xia Y, Wikberg J, Chhajlani V. Expression of melanocortin 1 receptors in periaqueductal gray matter. Neuroreport. 1995;6:2193–6. doi: 10.1097/00001756-199511000-00022. [DOI] [PubMed] [Google Scholar]
- 24.Mogil JS, Wilson SG, Chesler EJ, et al. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci USA. 2003;100:4867–72. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liem E, Joinen T, Tsueda K, et al. Increased sensitivity to thermal pain and reduced subcutaneous lidocaine efficacy in redheads. Anesthesiology. 2005;102:509–14. doi: 10.1097/00000542-200503000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han G, Quillan JM, Carlson K, et al. Design of novel chimeric melanotropin-deltorphin analogues. Discovery of the first potent human melanocortin 1 receptor antagonist. J Med Chem. 2003;46:810–19. doi: 10.1021/jm020355o. [DOI] [PubMed] [Google Scholar]
- 27.Mayorov AV, Han SY, Cai M, et al. Effects of macrocycle size and rigidity on melanocortin receptor-1 and -5 selectivity in cyclic lactam alpha-melanocyte-stimulating hormone analogs. Chem Biol Drug Design. 2006;67:329–35. doi: 10.1111/j.1747-0285.2006.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juni A, Cai M, Stankova M, et al. Sex-specific mediation of opioid-induced hyperalgesia by the melanocortin-1 receptor. Anesthesiology. 2010;112:181–8. doi: 10.1097/ALN.0b013e3181c53849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapas S, Cammas F, Hinson J, et al. Agonist and receptor binding properties of adrenocorticotropin peptides using the cloned mouse adrenocorticotropin receptor expressed in a stably transfected HeLa cell line. Endocrinology. 1996;137:3291–4. doi: 10.1210/endo.137.8.8754753. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann K, Montibeller JA, Finn FM. ACTH antagonists. Proc Natl Acad Sci USA. 1974;71:80–3. doi: 10.1073/pnas.71.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovalitskaya IA, Kolobov AA, Kampe-Nemm EA, et al. Synthetic peptide KKRR corresponding to the human ACTH fragment 15-18 is an antagonist of the ACTH receptor. Russ J Bioorg Chem. 2008;34:24–9. [PubMed] [Google Scholar]
- 32.Navolotskaya EV, Kovalitskaya YA, Sadovnikov VB, et al. Synthetic ACTH-Like peptide GKVLKKRR, corresponding to the fragment 81–88 of human pro-interleukin-1 alpha, acts as an antagonist of ACTH receptor. Int J Pept Res Ther. 2008;14:10–15. [Google Scholar]
- 33.Butler AA, Kesterson RA, Khong K, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–21. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 34.Ellacott KLJ, Murphy JG, Marks DL, et al. Obesity-induced inflammation in white adipose tissue is attenuated by loss of melanocortin-3 receptor signaling. Endocrinology. 2007;148(12):6186–94. doi: 10.1210/en.2007-0699. [DOI] [PubMed] [Google Scholar]
- 35.Sutton GM, Perez-Tilve D, Nogueiras R, et al. The melanocortin-3 receptor is required for entrainment to meal intake. J Neurosci. 2008;28(48):12946–55. doi: 10.1523/JNEUROSCI.3615-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Getting SJ, Di Filippo C, Christian HC. MC-3 receptor and the inflammatory mechanisms activated in acute myocardial infarct. J Leukoc Biol. 2004;76:1–9. doi: 10.1189/jlb.0306175. [DOI] [PubMed] [Google Scholar]
- 37.Santoro N, Perrone L, Cirillo G, et al. Effect of the melanocortin-3 receptor C17A and G241A variants on weight loss in childhood obesity. Am J Clin Nutr. 2007;85(4):950–3. doi: 10.1093/ajcn/85.4.950. [DOI] [PubMed] [Google Scholar]
- 38.Marks DL, Hruby VJ, Brookhart G, et al. The regulation of food intake by selective stimulation of the type 3 melanocortin receptor (MC3R) Peptides. 2006;27:259–64. doi: 10.1016/j.peptides.2005.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ni X-P, Butler AA, Cone RD, et al. Central receptors mediating the cardiovascular actions of melanocyte stimulating hormones. J Hypertens. 2006;24(11):2239–46. doi: 10.1097/01.hjh.0000249702.49854.fa. [DOI] [PubMed] [Google Scholar]
- 40.Li SJ, Varga K, Archer P, et al. Melanocortin antagonists define two distinct pathways of cardiovascular control by alpha- and gamma-melanocyte-stimulating hormones. J Neurosci. 1996;16:5182–8. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King SH, Mayorov AV, Balse-Srinivasan P, et al. Melanocortin receptors, melanotroic peptides and penile erection. Curr Topics Med Chem. 2007;7:1111–19. [PMC free article] [PubMed] [Google Scholar]
- 42.Kavarana MJ, Trivedi D, Cai M, et al. Novel cyclic templates of alpha-MSH give highly selective and potent antagonists/agonists for human melanocortin-3/4 receptors. J Med Chem. 2002;45:2644–50. doi: 10.1021/jm020021z. [DOI] [PubMed] [Google Scholar]
- 43.Mayorov AV, Cai M, Palmer ES, et al. Structure-activity relationships of cyclic lactam analogues of alpha-melanocyte-stimulating hormone (alpha-MSH) targeting the human melanocortin-3 receptor. J Med Chem. 2008;51:187–95. doi: 10.1021/jm070461w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ballet S, Mayorov AV, Cai M, et al. Novel selective human melanocortin-3 receptor ligands: use of the 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one (Aba) scaffold. Bioorg Med Chem Lett. 2007;17:2492–8. doi: 10.1016/j.bmcl.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayorov AV, Cai M, Chandler KB, et al. Development of cyclic gamma-MSH analogues with selective hMC3R agonist and hMC3R/hMC5R antagonist activities. J Med Chem. 2006;49:1946–52. doi: 10.1021/jm0510326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grieco P, Cai M, Han G, et al. Further structure-activity studies of lactam derivatives of MT-II and SHU-9119: their activity and selectivity at human melanocortin receptors 3, 4 and 5. Peptides. 2007;28:1191–6. doi: 10.1016/j.peptides.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grieco P, Lavecchia A, Cai M, et al. Structure-activity studies of the melanocortin peptides: discovery of potent and selective affinity antagonists for the hMC3 and hMC4 receptors. J Med Chem. 2002;45:5287–94. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 48.Cai M, Mayorov AV, Cabello C, et al. Novel 3D pharmacophore of alpha-MSH/gamma-MSH hybrids leads to selective human MC1R and MC3R analogues. J Med Chem. 2005;48:1839–48. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 49.Chen M, Cai M, Aprahamian CJ, et al. Contribution of the conserved amino acids of the melanocortin-4 receptor in Nle4,D-Phe7]-alpha-melanocyte-stimulating hormone binding and signaling. J Biol Chem. 2007;282:21712–19. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mountjoy K, Mortrud M, Low M, et al. Localization of the melanocortin-4 receptor (MC4R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 51.Starowicz K, Bilecki W, Sieja A, et al. Melanocortin 4 receptor is expressed in the dorsal root ganglions and down-regulated in neuropathic rats. Neurosci Lett. 2004;358:79–82. doi: 10.1016/j.neulet.2003.12.096. [DOI] [PubMed] [Google Scholar]
- 52.Tanabe K, Gamo K, Aoki S, et al. Melanocortin receptor 4 is induced in nerve-injured motor and sensory neurons of mouse. J Neurochem. 2007;101:1145–52. doi: 10.1111/j.1471-4159.2006.04432.x. [DOI] [PubMed] [Google Scholar]
- 53.Fan W, Boston BA, Kesterson RA, et al. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 54.Cone RD. Studies of the physiological functions of the melanocortin system. Endocr Rev. 2006;27:736–49. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- 55.Jean-Christophe P, Hofbauer K. Monoclonal antibodies directed to the melanocortin-4 receptor (MC4R) and their use in the treatment of cachexia and related conditions. Can Pat Appl. 2009 [Google Scholar]
- 56.Wessells H, Fuciarelli K, Hansen J, et al. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. J Urol. 1998;160:389–93. [PubMed] [Google Scholar]
- 57.Wessells H, Gralnek D, Dorr R, et al. Effect of an alpha-melanocyte stimulating hormone analog on penile erection and sexual desire in men with organic erectile dysfunction. Urology. 2000;56:641–6. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 58.Bednarek MA, MacNeil T, Kalyani RN, et al. Selective, high affinity peptide antagonists of alpha-melanotropin action at human melanocortin receptor 4: their synthesis and biological evaluation in vitro. J Med Chem. 2001;44:3665–72. doi: 10.1021/jm010165y. [DOI] [PubMed] [Google Scholar]
- 59.Kask A, Mutulis F, Muceniece R, et al. Discovery of a novel superpotent and selective melanocortin-4 receptor antagonist (HS024): evaluation in vitro and in vivo. Endocrinology. 1998;139:5006–14. doi: 10.1210/endo.139.12.6352. [DOI] [PubMed] [Google Scholar]
- 60.Balse-Srinivasan P, Grieco P, Cai M, et al. Structure-activity relationships of gamma-melanocortin MC3, MC4, and MC5 receptors. Discovery of highly selective hMC3R, hMC4R, and hMC5R analogues. J Med Chem. 2003;46(23):4965–73. doi: 10.1021/jm030119t. [DOI] [PubMed] [Google Scholar]
- 61.Schioth HB, Mutulis F, Muceniece R, et al. Discovery of novel melanocortin4 receptor selective MSH analogues. Br J Pharmacol. 1998;124:75–82. doi: 10.1038/sj.bjp.0701804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skuladottir GV, Jonsson L, Skarhedinsson JO, et al. Long term orexigenic effect of a novel melanocortin 4 receptor selective antagonist. Br J Pharmacol. 1999;126:27–34. doi: 10.1038/sj.bjp.0702264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grieco P, Lavecchia A, Cai M, et al. Structure-activity studies of the melanocortin peptides: discovery of potent and selective affinity antagonists for the hMC3 and hMC4 receptors. J Med Chem. 2002;45(24):5287–94. doi: 10.1021/jm0202526. [DOI] [PubMed] [Google Scholar]
- 64.Chakia S, Ogawab S, Todaa Y, et al. Involvement of the melanocortin MC4 receptor in stress-related behavior in rodents. Eur J Pharmacol. 2003;474:95–101. doi: 10.1016/s0014-2999(03)02033-8. [DOI] [PubMed] [Google Scholar]
- 65.Ying J, Gu X, Cai M, et al. Design, synthesis, and biological evaluation of new cyclic melanotropin peptide analogues selective for the human melanocortin-4 receptor. J Med Chem. 2006;49(23):6888–96. doi: 10.1021/jm060768f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chaki S, Hirota S, Funakoshi T, et al. Anxiolytic-like and antidepressant-like activities of MCL0129 (1-[(S)-2-(4-fluorophenyl)-2-(4-isopropylpiperadin-1-yl)ethyl]-4-[4-(2- methoxynaphthalen-1-yl)butyl] piperazine), a novel and potent nonpeptide antagonist of the melanocortin-4 receptor. J Pharmacol Exp Therap. 2003;304 (2):818–26. doi: 10.1124/jpet.102.044826. [DOI] [PubMed] [Google Scholar]
- 67.Chaki S, Oshida Y, Ogawa S, et al. MCL0042: a nonpeptidic MC4 receptor antagonist and serotonin reuptake inhibitor with anxiolytic- and antidepressant-like activity. Pharmacol Biochem Behav. 2005;82(4):621–6. doi: 10.1016/j.pbb.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Jiang W, Tran JA, Tucci FC, et al. Synthesis and characterization of pyrrolidine derivatives as potent agonists of the human melanocortin-4 receptor. Bioorg Med Chem Lett. 2007;17(23):6546–52. doi: 10.1016/j.bmcl.2007.09.079. [DOI] [PubMed] [Google Scholar]
- 69.Sharma S, Shi Y-Q, Wu Z, et al. US 2004/0152134A1. Bicyclic melanocortin-specific compounds. 2004
- 70.Pontillo J, Arellano M, Tran IA, et al. Structure-activity relationship of a series of cyclohexylpiperidines bearing an amide side chain as antagonists of the human melanocortin-4 receptor. Bioorg Med Chem Lett. 2005;15:3434–8. doi: 10.1016/j.bmcl.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 71.Vos TJ, Balani S, Blackburn V, et al. Identification and structure-activity relationships of a new series of melanocortin-4 receptor antagonists. Bioorg Med Chem Lett. 2006;16:2302–5. doi: 10.1016/j.bmcl.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 72.Nakazato A, Ishii T, Nozawa H. JP2005035983. Use of piperazine derivatives as MC4 receptor antagonists and therapeutic agents containing them for treatment anxiety neurosis or depression. 2005
- 73.Pontillo J, Tran JA, Markison S, et al. A potent and selective nonpeptide antagonist of the melanocortin-4 receptor induces food intake in satiated mice. Bioorg Med Chem Lett. 2005;15:2541–6. doi: 10.1016/j.bmcl.2005.03.053. [DOI] [PubMed] [Google Scholar]
- 74.Yang Y, Cai M, Chen M, et al. Key amino acid residues in the melanocortin-4 receptor for nonpeptide THIQ specific binding and signaling. Regul Peptides. 2009;155(1–3):46–54. doi: 10.1016/j.regpep.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Labbe O, Desarnaud F, Eggerickx D, et al. Molecular cloning of a mouse melanocortin 5 receptor gene widely expressed in peripheral tissues. Biochemistry. 1994;33(15):4543–9. doi: 10.1021/bi00181a015. [DOI] [PubMed] [Google Scholar]
- 76.Chen W, Kelly MA, Opitz-Araya X, et al. Exocrine gland dysfunction in MC5 -R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell. 1997;91(6):789–98. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 77.Hiramoto K, Hashimoto M, Orita K, et al. alpha-Melanocyte-Stimulating hormone plays an important role in the onset of pollinosis in a pollen allergy mouse model. Int Arch Allergy Immunol. 2010;153(1):13–18. doi: 10.1159/000301574. [DOI] [PubMed] [Google Scholar]
- 78.Blaskovich MAT, Cassidy PJ. Preparation of 1,4-diazepan-2-one peptidomimetics as MC5R melanocortin receptor modulators for treating conditions related to this receptor. U.S. Patent Appl Publ Chemical Indexing Equivalent to 151:313916 US. PCT Int Appl. 2009:282. [Google Scholar]
- 79.Balse-Srinivasan PM, Grieco P, Cai M, et al. Structure-activity relationships of novel cyclic alpha-MSH/beta-MSH hybrid analogues that lead to potent and selective ligands for the human MC3R and human MC5R. J Med Chem. 2003;46:3728–33. doi: 10.1021/jm030111j. [DOI] [PubMed] [Google Scholar]
- 80.Grieco P, Cai M, Liu L, et al. Design and microwave assisted synthesis of novel macrocyclic peptides active at melanocortin receptors: discovery of potent and selective hMC5R receptor antagonists. J Med Chem. 2008;51(9):2701–7. doi: 10.1021/jm701181n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cain JP, Mayorov AV, Cai M, et al. Design, synthesis, and biological evaluation of a new class of small molecule peptide mimetics targeting the melanocortin receptors. Bioorg Med Chem Lett. 2006;16(20):5462–7. doi: 10.1016/j.bmcl.2006.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]