

Abstract
Motivation: A rapid progression of esophageal squamous cell carcinoma (ESCC) causes a high mortality rate because of the propensity for metastasis driven by genetic and epigenetic alterations. The identification of prognostic biomarkers would help prevent or control metastatic progression. Expression analyses have been used to find such markers, but do not always validate in separate cohorts. Epigenetic marks, such as DNA methylation, are a potential source of more reliable and stable biomarkers. Importantly, the integration of both expression and epigenetic alterations is more likely to identify relevant biomarkers.
Results: We present a new analysis framework, using ESCC progression-associated gene regulatory network (GRNescc), to identify differentially methylated CpG sites prognostic of ESCC progression. From the CpG loci differentially methylated in 50 tumor–normal pairs, we selected 44 CpG loci most highly associated with survival and located in the promoters of genes more likely to belong to GRNescc. Using an independent ESCC cohort, we confirmed that 8/10 of CpG loci in the promoter of GRNescc genes significantly correlated with patient survival. In contrast, 0/10 CpG loci in the promoter genes outside the GRNescc were correlated with patient survival. We further characterized the GRNescc network topology and observed that the genes with methylated CpG loci associated with survival deviated from the center of mass and were less likely to be hubs in the GRNescc. We postulate that our analysis framework improves the identification of bona fide prognostic biomarkers from DNA methylation studies, especially with partial genome coverage.
Contact: tsengsm@mail.ncku.edu.tw or ycw5798@mail.ncku.edu.tw
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Recently, systematic biological approaches to study cancer have provided unprecedented views of molecular changes in many cancers. For example, the mutagenesis within a network of general human cancer signaling genes (Cui et al., 2007) and the protein expression within a protein–protein interaction network (Ostlund et al., 2010) have led to the discovery of subnetworks involving cancer-related genes. The combination of protein–protein networks with gene expression microarray datasets has also been used to distinguish metastatic from non-metastatic tumor samples (Chuang et al., 2007; Garcia et al., 2012) or to identify biomarkers correlated with patient survival (Li et al., 2012). More recently, Sun and Wang (2013) used a genetic network as a reference to estimate the penalty score of a conditional logistic regression model and applied it on a matched tumor–normal analysis of DNA methylation array data to identify a list of candidate CpG sites associated with hepatocellular cancer development. Kim et al. (2012) attempted to integrate more biological resources like the epigenomic, transcriptomic and protein interactome data to identify glioblastoma prognostic biomarkers using gene expression and DNA methylation-based networks. Although DNA methylation can be used as a powerful and promising prognostic indicator alone (Laird, 2003), none of the aforementioned network-based studies, integrating DNA methylation, gene expression or protein expression information performed experimental validation of the identified biomarkers. This can be because of the large number of candidate biomarkers within networks, making their validation and use in the clinic more difficult.
Affecting >450 000 patients annually, esophageal carcinoma with squamous cell carcinoma (ESCC) as the predominant histological subtype worldwide is the sixth leading cause of cancer-related mortality, with >400 000 deaths per year (Pennathur et al., 2013; van Hagen et al., 2012). Late presentation with already existing lymph node metastasis (LNM) followed by rapid progression explains the poor outcome of the disease (Bollschweiler et al., 2006). Metastasis requires certain steps like primary tumor initialization and proliferation, blood vessel/lymphatic channel intravasation, cell arrest and extravasation and proliferation at secondary target sites/organs (Hunter et al., 2008). Metastasis can arise from tumor cells that have undergone phenotypic changes called epithelial-to-mesenchymal transition (EMT), gaining plasticity and circulating and seeding ability. There is no observable change in DNA methylation during the transforming growth factor beta-mediated EMT in AML12 mouse hepatocyte (McDonald et al., 2011). But such DNA methylation is able to be involved in gene regulation during the EMT of prostate cell line, EP156T (Ke et al., 2010). Identifying epigenetic alterations occurring during ESCC progression is therefore not only essential for a detailed understanding of the molecular biology underlying the disease progression but also to improve clinical prognosis and develop more sophisticated treatment strategies.
Until now, only few genomic regions, such as the retrotransposon-related long interspersed element 1 (Iwagami et al., 2013) or the gene regulatory elements, showing methylation alterations have been identified as possible biomarkers for LNM and/or patient survival in ESCC. Of the gene annotation-based studies, hyper-methylation at CpG islands (CGIs) in the vicinities of PAX6 (paired box 6) and RN7SKP211 (RNA, 7SK small nuclear pseudogene 211) were significantly associated with LNM and disease-free survival in 96 patients (Gyobu et al., 2011). Hyper-methylated CGIs located within UCHL1 (ubiquitin carboxyl-terminal esterase L1) (Mandelker et al., 2005), FHIT (fragile histidine triad) (Lee et al., 2006), GRIN2B (glutamate receptor, ionotropic, N-methyl D-aspartate 2B) (Kim et al., 2007) and GADD45G (growth arrest and DNA-damage-inducible, gamma) (Guo et al., 2013) promoter regions were associated with poor survival. However, these studies only validated CpG sites in a small set of candidate genes, therefore limiting the scope of the findings. A more comprehensive analysis is likely to reveal new DNA methylation as biomarkers associated with LNM and survival.
In this study, we use a new comprehensive approach to efficiently identify and validate DNA methylation sites as putative prognostic biomarkers of ESCC progression. We propose an intuitive framework, and demonstrate its ability to identify CpG sites of prognostic value. The framework leverages an ESCC progression-associated gene regulatory network (GRNescc) to identify methylated sites with significant prognostic value. By taking into account differentially methylated CpG sites whose corresponding gene promoters are ranked, the ranked CpG sites are purified/selected via a top-k precision test in network. We validate the results on a selection of 20 CpG loci in a separate cohort of ESCC patients and demonstrate that this framework is capable of identifying novel sites of DNA methylation with prognostic impact that had not been discovered by previous approaches.
2 METHODS
2.1 Patients and biopsy specimens
This study was verified and qualified by the institutional review board of National Cheng Kung University Hospital from May 1, 2010 to July 31, 2011 under contract number ‘HR-99-021’. The ethics committee specifically waived the need for informed consent forms because the data were publicly obtained from an observational study and analyzed anonymously. We enrolled 100 ESCC patients admitted to the Cancer Center and Pathology department, National Cheng Kung University Hospital (N = 80) and the Cancer Center, China Medical University Hospital (N = 20). Primary ESCC specimens and matched normal tissues, located >10 cm from the primary site, were collected through surgical resection. Pathologic examination of the resected surgical specimens was performed following a standardized protocol, and the specimens were classified according to the sixth edition of the UICC TNM (Union for International Cancer Control, TNM Classification of Malignant Tumours) system and the WHO classification. Although surgically resected tumor tissue and corresponding normal tissue samples were collected from two separate hospitals, the samples were processed by the same laboratory, using the same protocol, therefore limiting potential batch effects. Follow-up of enrolled patients was performed at 6 months interval, with the last follow-up performed at least 12 months and up to 104 months after diagnosis for living patients. The enrolled patients were randomly split between screening (50 patients) and validation (50 patients) cohorts. The general clinicopathological characteristics of the enrolled patients are shown in Supplementary Table S1.
2.2 Construction of an ESCC-related gene regulatory network
We built a general gene regulatory network (GRNg) using three publicly available networks: Pathway Commons (11/2011 version; Cerami et al., 2011), BioGRID 3.1.79 (Stark et al., 2006) and KEGG (Kyoto Encyclopedia of Genes and Genomes) (09/2011 version; Ogata et al., 1999). The consolidated network features 1 294 769 gene regulations (edges) and 12 803 genes (nodes). In this study, we focused more on direct gene regulations because the DNA methylation is a major epigenetic event that blocks binding of transcription factors to promoters of target genes, or modifies chromatin structure, which in turn blocks transcription factor binding (Suzuki and Bird, 2008). Therefore, only interactions derived from transcriptional regulation were considered, excluding protein–protein and protein compound interactions. We then generated GRNescc. We selected 186 genes by curating the literature and identified genes whose expression pattern is associated with ESCC progression (Supplementary Table S2) before January 2012. The progression refers to the cancer metastasis, proliferation, arrest, invasion and patient survival. We excluded genes showing differential expression between tumor and normal but that could not be associated with ESCC progression. We then generated the GRNescc as a subnetwork of GRNg using the following steps: (i) Initiate an empty distance matrix with a length equal to the number of literature-curated genes, (186 × 186), (ii) calculate the shortest distance between each pair of genes projected on GRNg using the Dijkstra's algorithm and (iii) calculate the shortest paths of each pair of genes on GRNescc using a breadth-first search algorithm. The resulting GRNescc contained 1 013 604 interactions between 4636 genes. A non-ESCC progression-associated gene regulatory network (GRNg-escc) was also derived from the complement of GRNescc in GRNg. More precisely, after generating the GRNescc via the above three steps, every gene (node) of GRNescc and its connected gene–gene regulations (edges) in GRNg were removed from GRNg. We therefore called the rest part GRNg-escc as a negative control used in this study.
2.3 Generation and analysis of DNA methylation microarray data
2.3.1 Microarray data generation
The genomic DNA from primary ESCC and normal esophagus specimen was extracted using proteinase K digestion and phenol–chloroform extraction. One microgram of DNA was then converted using bisulfite following the directions from the EpiTect Bisulfite kit (Qiagen, Duesseldorf, Germany), converting unmethylated cytosines to uracil and then to thymidine in the subsequent PCR step. We used the Illumina’s GoldenGate Methylation Assay Cancer Panel I (1505 CpG dinucleotides located in the promoter of 807 genes; Illumina, San Diego, CA, USA) following the manufacturer’s instructions. The data are available at the NCBI/GEO database (GSE51287).
2.3.2 Microarray data analysis
The ratio of fluorescent signals was computed from the two alleles beta = (max(M, 0))/(|U| + |M| + 100), where U is the green fluorescent signal (Cy3) from an unmethylated allele and M is the red signal (Cy5) from a methylated allele, generated by the Illumina's proprietary software (BeadStudio). The beta-value reflects the methylation level of each CpG site (Bibikova et al., 2006), and their distribution is shown in Supplementary Figure S1A. To allow further statistical analyses able to be applicable to these values across different samples, the beta-values were then normalized using the function of normalize.loess implemented in Bioconductor affy package with four parameters including epsilon (0.01), log.it (F), span (0.4) and maxit (5). Then we kept all normalized values positive by adding an absolute (the minimum value). Their distribution is shown in Supplementary Figure S1B. We identified significantly differentially methylated CpG sites between tumor and normal using a two-tailed Student’s t-test (P < 0.05). CpG loci had a significant increase (respectively decrease) in methylation when methylation is increased by N-fold or greater (respectively −N-fold) in the tumor compared with normal, with N corresponding to the median of absolute fold changes between tumor and normal.
2.3.3 Identification of CpG sites associated with progression
We constructed a contingency table for each significantly different CpG site, counting the number of patients with or without LNM and for which the probe significantly increased or decreased methylation CpG. This table can be used to analyze the relationships between two categorical variables: methylation change in tumor (increase/decrease) and metastasis status (N0/N1). We then calculated, for each CpG site, the following six correlation metrics for each CpG site: PhiCoefficient (Cramer, 1946), OddsRatio (Edwards, 1963), PiatetskyShapiroMeasure (Piatetsky-Shapiro, 1991), LiftMeasure (Tufféry, 2011), AddedValue (Sahar and Mansour, 1999) and KlosgenMeasure (Klösgen, 1992). The detailed equations are given as Supplementary Method S1. For each of these metrics, a positive value indicates a positive correlation between the direction of the methylation change and the LNM status, at each CpG site. This resulted in the identification of 130 progression-associated CpG sites.
2.3.4 Identification of CpG sites associated with survival
The last follow-up of enrolled patients was performed at least 12 months after diagnosis for living patients. The first group (‘Good’ survival) included patients who were still alive after 12 months following tumor resection and the second group (‘Bad’ survival) consisted of patients who died within 12 months post-resection. Coincidentally, the two groups have the identical number of patients. As a consequence, a perfect classifier would separate the cohort into two groups of equal size. For this reason, we imposed the comparisons with groups of patients of equal size, and grouped them according to the methylation change of the tested CpG: we ranked patients according to their fold methylation change [FC = log2 (tumor/normal)] of the probe, and automatically selected the FC threshold (FCt) leading to an equal number of patients with FC < −FCt (decreased-methylation) and FC > FCt (increased-methylation). The association with survival was determined by preforming a logrank-test.
2.4 Network analysis
The top-k precision (TP) is an ubiquitous correlation metric (Fagin et al., 2003). To test whether the top-ranked CpG sites are prevalent in a network, the measurement is given by the following two equations.
where GRN represents the processed network, and Gi represents the currently indexed gene promoter probe containing a CpG site within a list of ranked CpG sites.
2.5 Pyrosequencing validation and survival analysis
The bisulfite-converted DNA was pyrosequenced using the PyroMark Q24 (Qiagen). We designed specific pyrosequencing primer and PCR primer using the specialized software (PyroMark Assay Design 2.0) to target the CpG sites in the promoter region of selected gene (Supplementary Table S3). Bisulfite-modified DNA was dissolved in 20 μl H2O, and 1 µl of DNA template was used for PCR amplification. Hot-start PCR was performed with PyroMark PCR Kit (Qiagen), and pyrosequencing was carried out according to the manufacturer’s protocol (Qiagen). The target CpG sites were evaluated by converting the resulting pyrograms to numerical values for peak heights. The percentage of methylation was calculated as the mean of all CpG analyzed (Vaissiere et al., 2009). We finally performed a survival analysis by using these methylation percentages to validate the screening cohort-derived candidate probes containing CpG sites.
2.6 Quantitative RT-PCR
For the mRNA quantifications, we performed SYBR Green and TaqMan® Gene Expression Assay (Life Technologies Corporation) qRT-PCR methods to detect mRNAs in the same validation cohort. If genes were not suitable for the primer design of SYBR Green qRT-PCR, we alternatively performed the TaqMan® method using its commercial primers (Supplementary Table S4). We analyzed the results using the cycle threshold method (Ct). Only strong signals with high expressions (Ct < 35) were used for a further correlation analysis with promoter methylation.
3 RESULTS AND DISCUSSION
3.1 Overall framework and ESCC network construction
Cancer metastatic progression may be associated with multiple gene regulatory changes, some of them mediated by aberrant promoter CpG methylation. To identify the CpG probes where methylation is the most associated with disease progression, we propose the following framework comprising five different steps. (i) We construct a disease-specific gene regulatory network. This is done by extracting the minimal path subnetwork containing genes important for the disease progression, as identified through manually curated references (Fig. 1 panel I). The complement of this subnetwork in the global network is extracted and used as a negative control (see also Section 2.2). (ii) We identify cancer-specific CpG methylation events (Fig. 1 panel II), then (iii) we select CpG sites where methylation change is the most associated with progression (Fig. 1 panel III). (iv) We then rank these candidate CpG sites by the association of the methylation change with patient’s survival (Fig. 1 panel IV), and finally (v) we use the disease-specific network to select the top candidate CpG sites associated with disease progression and patient’s survival (Fig. 1 panel V). Applying this approach to ESCC progression, we first built a GRNg using publicly available networks (Section 2). This GRNg features 1 294 769 gene regulations (edges) and 12 803 genes (nodes). We then extracted the minimal-path subnetwork featuring 186 genes associated with ESCC progression (Section 2 and Supplementary Table S2). This GRNescc contains 1 013 604 interactions between 4 636 genes. Finally, we derived a non-ESCC-associated gene regulatory network (GRNg-escc) from the complement of GRNescc in GRNg. A Gene Ontology (GO) analysis (Dennis et al., 2003) revealed that the genes in GRNescc were enriched in biological processes such as chemotaxis, cell adhesion, cell migration and angiogenesis, compared with the GRNg-escc (Fig. 2). These processes are important for metastasis and cancer progression. This observation indicates that the genes in GRNescc are related to cancer progression, extending our initial gene list and likely accounting for unsuspected regulatory patterns important for disease progression and metastasis.
Fig. 1.
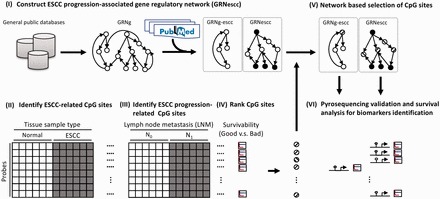
Schematic overview of data processing steps. (I) Development of literature-guided gene regulatory networks. The circles and arrows represent the regulatory genes and regulations, respectively. (II) Identification of differentially methylated CpG sites associated with ESCC. (III) Selection of differentially methylated CpG sites associated with ESCC progression. (IV) Ranking of CpG sites based on the association with patient survival. (V) Selection of the ranked CpG sites using a network-based approach. (VI) Validation of network-selected top-ranked CpG sites in a new patient cohort. The circles indicate increased methylation (upward diagonal), decreased methylation (downward diagonal) in the tumor or literature curated (filled)
Fig. 2.
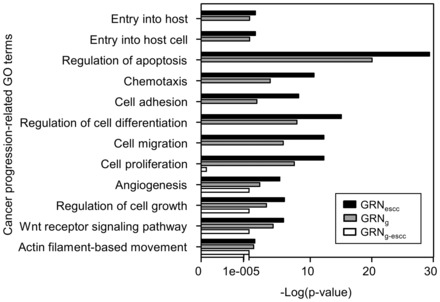
Enrichment analysis of biological processes GO terms for the three different GRNs studied. P-value as a function of GO terms
3.2 Identification of candidate CpG sites associated with ESCC progression
Using a microarray, we measured the methylation status of 1505 CpG sites located in the promoter of 807 cancer-related genes in 50 ESCC tumors and matched normal esophageal tissue. We identified 309 differentially methylated sites between tumor and normal (t-test nominal P < 0.05), of which 108 and 201 had decreased and increased methylation in the tumor, respectively. We then determined which CpG sites were associated with cancer progression, e.g. lymph node metastasis, in our cohort. Using a compendium of six correlation metrics, we looked for CpG site with significant changes in methylation in the tumor of metastatic patients (LNM classification N1). Using this approach, we were able to identify 130 CpG sites in the promoter of 109 genes, which are associated with ESCC lymph node metastasis. To characterize these sites and extract a specific ESCC progression epigenetic signature, we further analyzed them using the global regulatory network.
3.3 Network-based selection of Candidate CpG sites associated with survival
To increase our confidence in the biological significance of the CpG sites identified above, we calculated their association with patient survival. Using a dynamic classification of patients with increased and decreased CpG methylations to compare groups of the same size (section 2.3.4), we ranked the 130 CpG sites by decreasing association of their methylation status and patient survival (log-rank test P-value). To further select the CpG sites where methylation status is the most likely to be associated with disease progression, we examined the genes associated with them and how well they map to the GRNescc network. We noticed that the top-ranked genes were prominent in the GRNescc network, compared with random (Top-k precision, Fig. 3A). In contrast, the top-ranked genes were depleted in the GRNg-escc network compared with random (Fig. 3B). This observation therefore suggests that our methodology enriches for CpG sites located in the promoters of genes important for progression and survival.
Fig. 3.
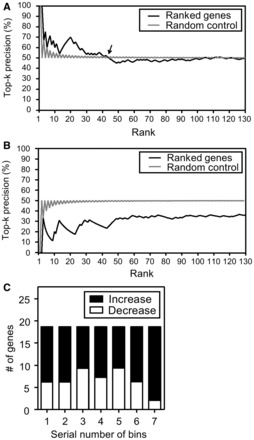
Characterization of genes in GRNs ranked by association with survival. (A) Top-k precision as a function of gene rank for GRNescc. The arrow points to the greatest rank where the precision is above random control (k = 44). (B) Top-k precision as a function of gene rank for GRNg-escc. (C) Relative proportion of CpGs with decreasing and increasing methylation from decreasing rank bins. Each bin contains 18 genes. Increase: methylation increased in tumor; Decrease: methylation decreased in tumor
To further distinguish the relative importance of increase and decrease in methylation at the CpG sites in the promoter of the genes in the networks, we split the ranked list of 130 CpG sites in equal size bins of decreasing association with survival (Fig. 3C). There was no significant bias between increased and decreased methylation at CpG in the promoters of these genes, indicating that both repression and activation of genes in the network may contribute to ESCC progression.
We finally selected 44 best candidate CpG sites (Fig. 3A –arrow) for further analysis (Supplementary Table S5). Of the 44 CpG sites, 22 were located in the promoters of genes belonging to GRNescc (referred to as In-CpG sites) and 22 where in promoters of genes outside GRNescc (referred to as Out-CpG sites). This enrichment of CpG with changing methylation is significantly different from what can be expected by chance (χ2 test P < 0.0001). Moreover, only 5 of the 22 In-CpGs and none of the 11 Out-CpGs were located in the promoters of the 186 genes that seeded the GRNescc network, suggesting that the In-CpG methylation changes were likely to be associated with progression and close to the 186 genes in network. To confirm this possibility, we compared the average distance between the 186 seed genes and the genes whose promoters have CpG methylation changes associated with progression. Of the 22 Out-CpG sites, 11 were located in the promoters of genes belonging to GRNg-escc (referred to as Out-CpGg-escc sites), and 11 were in the promoters of genes not represented in the GRNg network. The genes with In-CpG were significantly closer to a seed gene than the genes with Out-CpGg-escc (average distance of 2.7 versus 3.2, t-test P-value < 1E-30). The average distance of genes with In-CpG to a seed gene is in fact similar to the average distance of the seed genes between themselves (2.7 versus 2.6, t-test P-value = 3.6E-09). This suggests that network approach enables the identification of CpG methylation changes in the promoter of genes not previously associated with progression and, therefore, increases the number of potential prognostic biomarkers that can be tested. Moreover, although both inside and outside the GRNescc had the identical number (22) of genes, the 22 genes in GRNescc were originally from top-ranked genes (Fig. 3A and B and Supplementary Table S5—the median rank of In-CpG sites versus the median rank of Out-CpG sites = 18 versus 27). The 22 In-CpG sites in genes from GRNescc are therefore more likely to have a methylation status associated with ESCC metastatic progression and are good candidates to test their prognostic value.
3.4 Validation of the findings
To validate the association between these candidate CpG sites methylation and survival (Fig. 1 panel VI), we decided to measure the association in a new cohort (validation cohort) of 50 patients with ESCC and matched normal esophageal tissues. We were able to design specific primers for 10 In-CpGs as well as 10 control Out-CpGs (Section 2 and Supplementary Table S3).
We first checked the validity of the methodology by determining the methylation level of these 20 CpG sites in the screening cohort (N = 50 patients). This analysis showed that the methylation level determined by pyrosequencing was highly correlated with the one obtained from the microarray (Supplementary Fig. S2; r = 0.78), therefore demonstrating the technical validity of the approach. We then examined the methylation of these 20 CpGs in the validation cohort (N = 50 patients). We first noticed that, the methylation change is significant in 12/20 CpG (P < 10−4), either through increased (N = 7) or decreased methylation (N = 5) in the tumor. Additionally, the methylation changes of 8/10 In-CpGs and 0/10 Out-CpG were associated with patient survival (Table 1—log-rank test P < 0.05). Of eight validated In-CpGs, only one is located in the promoter of a seed gene (MAPK4, Supplementary Table S2), suggesting the increased sensitivity provided by the network approach. To further confirm the association between methylation changes and survival, we performed a Cox regression analysis using SPSS v17 on the methylation changes in addition to other clinical variable (Supplementary Table S6). We calculated the hazard ratio (HR) of cancer death risk of variables including promoter methylation change, TNM stage, local LNM status, distant metastases status, age and drinking status. This analysis showed that methylation changes at five of eight validated In-CpGs and distant metastasis were associated with a significantly increased risk (HR > 1) or decreased risk (HR < 1) of cancer-related death, and three In-CpGs showed a borderline significance, while none of the methylation changes at Out-CpGs (negative control group) was associated with the risk of cancer-related death. A multivariate analysis further showed that 5/8 validated In-CpGs remain significant association with prognosis even after accounting for the presence of distant metastasis.
Table 1.
Pyrosequencing-based validation of methylated In-CpG and Out-CpG sites
| CpGs | Distance from TSS | Gene | Fold change (P-valuea) | Association with survival P-valueb | Survival correlation directionc |
|---|---|---|---|---|---|
| In-CpG | +64 | JAK3 | 1.8 (<0.0001***) | 0.033* | − |
| In-CpG | −1,121 | PAX6 | 1.3 (0.314) | 0.041* | − |
| In-CpG | −115 | CFTR | 1.3 (0.047*) | 0.188 | NA |
| In-CpG | −516 | E2F5 | 1.6 (<0.0001***) | 0.024* | − |
| In-CpG | −272 | CD81 | 1.1 (0.921) | 0.031* | − |
| In-CpG | +53 | CCL3 | −1.4 (<0.0001***) | 0.015* | + |
| In-CpG | −8 | CSF3R | −1.1 (0.429*) | 0.154 | NA |
| In-CpG | −804 | INS | −1.2 (<0.0001***) | 0.040* | − |
| In-CpG | +273 | MAPK4 | −1.2 (<0.0001***) | 0.001** | − |
| In-CpG | −456 | PGR | −1.4 (<0.0001***) | 0.023* | + |
| Out-CpG | −38 | SLC5A8 | 2.0 (0.008*) | 0.153 | NA |
| Out-CpG | +26 | PENK | 2.0 (<0.0001***) | 0.079 | NA |
| Out-CpG | −546 | HS3ST2 | 1.9 (<0.0001***) | 0.323 | NA |
| Out-CpG | +3 | KCNK4 | 1.7 (<0.0001***) | 0.091 | NA |
| Out-CpG | −299 | SEZ6L | 1.3 (0.142) | 0.252 | NA |
| Out-CpG | −22 | ZIM2 | 1.2 (<0.0001***) | 0.206 | NA |
| Out-CpG | −455 | ADCYAP1 | 2.7 (<0.0001***) | 0.536 | NA |
| Out-CpG | −1,394 | PI3 | −1.1 (0.248) | 0.245 | NA |
| Out-CpG | +340 | SFTPA1 | −1.4 (<0.0001***) | 0.400 | NA |
| Out-CpG | −721 | TRPM5 | −1.1 (0.023*) | 0.324 | NA |
Note: TSS, transcription start site; NA, not applicable; Fold change, pyrosequencing values between matched ESCC and normal adjacent tissue. aP-value of t-test. bP-value of log-rank test. cThe direction of correlation was considered as ‘+’ (respectively ‘−’) when the methylation increase in tumor led to a good (respectively poor) survival rate. *P < 0.05; **P < 0.001; ***P < 0.0001.
The validation results (Table 1), including the methylation changes between tumor and normal tissues and the direction of survival associations, of all of the eight validated CpGs were consistent with the results in the screening cohort (Supplementary Table S7 and Supplementary Fig. S3). These results suggest that, despite the limitation of the cohort size to identify significant methylation changes in the tumors, the network-based framework was able to enrich for CpG sites significantly associated with survival.
Promoter CpG methylation usually results in transcriptional repression. In Supplementary Figure S4, we measured the expression changes between normal esophagus and ESCC for six genes with strong reliable signals (Section 2) in the GRNescc and whose promoters contain CpG associated with survival. We can identify trends for negative correlation for four increased methylation CpGs or positive correlation for two decreased methylation CpGs at least 10 patients in the validation cohort. Referring to previous literature, a hyper-methylated CpG island located 5300 bp upstream from the transcriptional start site of PAX6 was found to date as the only biomarker to be associated with LNM (Gyobu et al., 2011). However, the authors claimed that although this CpG island was unlikely to be associated with repression of PAX6, it was quantified in four ESCC cell lines in three of which PAX6 was expressed in spite of CpG island methylation. Their results suggested that the methylation status would not always correlate with gene expression. Therefore, in agreement with this study, our results indicate that methylation changes at selected CpG sites can be good prognostic markers even in absence of a clear effect on transcriptional regulation.
A role for inflammation in tumorigenesis is emerging. Inflammatory responses play pivotal roles at tumor progression including tumor initiation, promotion, invasion and metastasis. Tumors are frequently surrounded by an inflammatory microenvironment rich in cytokines, chemokines and immune cells infiltration, which promote malignant cellular growth. These factors are produced by the tumor cells or its surrounding tissue and contribute to malignant progression (Grivennikov et al., 2010). Interestingly, we also validated several genes associated with inflammation. For example, CCL3 is a cytokine in the TNF inflammation pathway (Wang et al., 2013), and JAK3 is predominantly expressed in immune cells and transduces a signal in response to its activation via tyrosine phosphorylation by interleukin receptors (Krejsgaard et al., 2011). Different cytokines can either promote or inhibit tumor development and progression (Lin and Karin, 2007). Previous studies indicated that interleukin 6 (IL-6), a pro-inflammatory cytokine that mediates chronic inflammation, may play an import role in inflammation-driven oral carcinogenesis. Notably, Jacqueline and associates recently found that IL-6 induces hyper-methylation and gene silencing mediated by DNMTs (mammalian DNA methyltransferases) (Gasche et al., 2011). In this study, the changes of global DNA methylation and gene-specific promoter methylation patterns by IL-6 treatment in oral cancer cells were examined. The increased promoter methylation changes were identified in several tumor suppressor genes, including CHFR, GATA5 and PAX6 (Gasche et al., 2011). Of these, we confirmed PAX6 with increased methylation in ESCC. Together, the role of inflammation in relation of promoter methylation of PAX6, CCL3 and JAK3 in ESCC tumor microenvironment is worthy of further investigation.
3.5 GRNescc network topology
In an effort to understand better the importance of the network in identifying significant associations with cancer progression and survival, we characterized further the network topology. Previous studies have shown that the network topology of certain genes might have functional implications in a cell. For example, an enrichment of genes having lethal knockout phenotypes possessed a high-degree (hub) property in a Saccharomyces cerivisiae gene co-expression network (Carter et al., 2004). Therefore, it is plausible that the CpG methylation changes of promoters of genes in GRNescc might have certain interesting distributions. We examined whether the gene promoters contain differentially methylated CpG possessed specific characteristics in the GRNescc network. We ranked these genes based on their decreasing association with patient survival. We noticed that both the barycenter score (White and Smyth, 2003) and the closeness centrality (Opsahl et al., 2010) showed a negative correlation with significance (Fig 4A and B). This observation suggests that the genes associated with survival tend to deviate from the center of mass of the GRNescc and to be more located at the periphery of the network. This is similar to a recent work that age-associated epigenetic drift occurs preferentially in genes that occupy peripheral network positions (West et al., 2013). In another analysis shown in Figure 4C and D, the significance for survival was negatively correlated with the Hyperlink-Induced Topic Search (HITS) hub and the HITS authority (Kleinberg, 1999). Derived for algorithms used to rate web pages based on topic significance, this observation again suggests that the ESCC progression genes are not the most connected nodes but rather stem away from them.
Fig. 4.
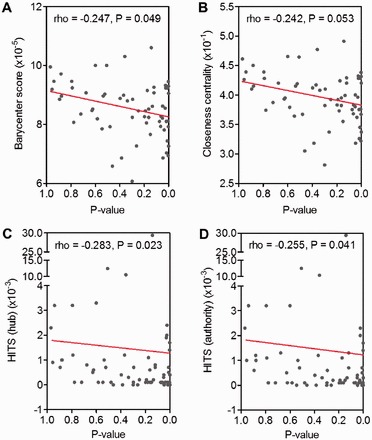
Node topological measurements in GRNescc. (A) Barycenter scores, (B) Closeness centrality, (C) HITS hub and (D) HITS authority as a function of log-rank test P-values of ranked genes. The further right along x-axis indicates the greater propensity for ESCC progression. Red line: A linear regression. P: P-value is calculated by testing Spearman’s rank-order correlation coefficient (rho) with an F-distribution
4 CONCLUSIONS
In this study, we proposed a new framework that uses literature-guided GRN to enhance the results and interpretation of DNA methylation microarray experiments. Specifically, the framework helps prioritize differentially methylated genes for their impact on cancer progression and survival. We validated the results in an independent cohort and confirmed that the selected CpG sites were significantly associated with patient survival, even in absence of a direct correlation with the gene expression. Eight of 10 validated CpG sites significantly correlated with patient survival. These were located in the promoters of JAK3, PAX6, E2F5 and CD81 (increased methylation), and in the promoters of CCL3, INS, MAPK4 and PGR (decreased methylation). Interestingly, the position of the survival-associated genes in the GRNescc network significantly deviated from the center of mass. We postulate that the topology of progression-associated network could help identify progression-associated genes before any data collection. Our results demonstrate that the use of regulatory networks and prior expression studies can help identify bona fide DNA-methylation prognostic biomarkers. Although our focus is the identification of biomarkers for a clinical use via a methodological innovation, the functional exploration of these biomarkers is also worthy of further investigation.
Funding: This research was partially supported by the National Science Council, Taiwan [Research Project (NSC102-2627-B-006-011, NSC101-2627-B-006-003); Overseas Project for Post Graduate Research (NSC102-2917-I-006-023)]; Ministry of Health and Welfare, Taiwan [MOHW103-TDU-PB-211-133005]; the Top University Program by the Ministry of Education, Taiwan. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of interest: none declared.
Supplementary Material
REFERENCES
- Bibikova M, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollschweiler E, et al. Staging of esophageal carcinoma: length of tumor and number of involved regional lymph nodes. Are these independent prognostic factors? J. Surg. Oncol. 2006;94:355–363. doi: 10.1002/jso.20569. [DOI] [PubMed] [Google Scholar]
- Carter SL, et al. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics. 2004;20:2242–2250. doi: 10.1093/bioinformatics/bth234. [DOI] [PubMed] [Google Scholar]
- Cerami EG, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HY, et al. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer H. Mathematical Methods of Statistics. Princeton, NJ: Princeton University Press; 1946. [Google Scholar]
- Cui Q, et al. A map of human cancer signaling. Mol. Syst. Biol. 2007;3:152. doi: 10.1038/msb4100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Edwards AWF. The measure of association in a 2 × 2 table. J. R. Stat. Soc. 1963;126:109–114. [Google Scholar]
- Fagin R, et al. Proceedings of the fourteenth annual ACM-SIAM symposium on Discrete algorithms. 2003. Comparing top k lists. Society for Industrial and Applied Mathematics, Baltimore, MD, pp. 28–36. [Google Scholar]
- Garcia M, et al. Interactome-transcriptome integration for predicting distant metastasis in breast cancer. Bioinformatics. 2012;28:672–678. doi: 10.1093/bioinformatics/bts025. [DOI] [PubMed] [Google Scholar]
- Gasche JA, et al. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer. 2011;129:1053–1063. doi: 10.1002/ijc.25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, et al. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, et al. Decreased expression and aberrant methylation of Gadd45G is associated with tumor progression and poor prognosis in esophageal squamous cell carcinoma. Clin. Exp. Metastasis. 2013;30:977–992. doi: 10.1007/s10585-013-9597-2. [DOI] [PubMed] [Google Scholar]
- Gyobu K, et al. Identification and validation of DNA methylation markers to predict lymph node metastasis of esophageal squamous cell carcinomas. Ann. Surg. Oncol. 2011;18:1185–1194. doi: 10.1245/s10434-010-1393-5. [DOI] [PubMed] [Google Scholar]
- Hunter KW, et al. Mechanisms of metastasis. Breast Cancer Res. 2008;10(Suppl. 1):S2. doi: 10.1186/bcr1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwagami S, et al. LINE-1 hypomethylation is associated with a poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Ann. Surg. 2013;257:449–455. doi: 10.1097/SLA.0b013e31826d8602. [DOI] [PubMed] [Google Scholar]
- Ke XS, et al. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. doi: 10.1186/1471-2164-11-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. Multi-analyte network markers for tumor prognosis. PloS One. 2012;7:e52973. doi: 10.1371/journal.pone.0052973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, et al. A promoter methylation pattern in the N-methyl-D-aspartate receptor 2B gene predicts poor prognosis in esophageal squamous cell carcinoma. Clin. Cancer Res. 2007;13:6658–6665. doi: 10.1158/1078-0432.CCR-07-1178. [DOI] [PubMed] [Google Scholar]
- Klösgen W. Problems for knowledge discovery in databases and their treatment in the statistics interpreter explora. Int. J. Intell. Syst. 1992;7:649–673. [Google Scholar]
- Kleinberg JM. Authoritative sources in a hyperlinked environment. J. ACM. 1999;46:604–632. [Google Scholar]
- Krejsgaard T, et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J. Invest. Dermatol. 2011;131:1331–1338. doi: 10.1038/jid.2011.27. [DOI] [PubMed] [Google Scholar]
- Laird PW. The power and the promise of DNA methylation markers. Nat. Rev. Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- Lee EJ, et al. Aberrant methylation of fragile histidine triad gene is associated with poor prognosis in early stage esophageal squamous cell carcinoma. Eur. J. Cancer. 2006;42:972–980. doi: 10.1016/j.ejca.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Li J, et al. SurvNet: a web server for identifying network-based biomarkers that most correlate with patient survival data. Nucleic Acids Res. 2012;40:W123–W126. doi: 10.1093/nar/gks386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelker DL, et al. PGP9.5 promoter methylation is an independent prognostic factor for esophageal squamous cell carcinoma. Cancer Res. 2005;65:4963–4968. doi: 10.1158/0008-5472.CAN-04-3923. [DOI] [PubMed] [Google Scholar]
- McDonald OG, et al. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011;18:867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, et al. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opsahl T, et al. Node centrality in weighted networks: generalizing degree and shortest paths. Soc. Netw. 2010;32:245–251. [Google Scholar]
- Ostlund G, et al. Network-based Identification of novel cancer genes. Mol. Cell Proteomics. 2010;9:648–655. doi: 10.1074/mcp.M900227-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennathur A, et al. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- Piatetsky-Shapiro G. Discovery, Analysis, and Presentation of Strong Rules. Knowledge Discovery in Databases. Cambridge, MA: AAAI/MIT Press; 1991. [Google Scholar]
- Sahar S, Mansour Y. An empirical evaluation of interest-level criteria. In: Dasarathy BV, editor. Data Mining and Knowledge Discovery: Theory, Tools, and Technology. Orlando, FL, USA; 1999. [Google Scholar]
- Stark C, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Wang S. Network-based regularization for matched case-control analysis of high-dimensional DNA methylation data. Stat. Med. 2013;32:2127–2139. doi: 10.1002/sim.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Tufféry S. Data Mining and Statistics for Decision Making. 2011. translated from the French Data Mining et statistique décisionnelle. John Wiley & Sons, Chichester, GB. [Google Scholar]
- Vaissiere T, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243–252. doi: 10.1158/0008-5472.CAN-08-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hagen P, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 2012;366:2074–2084. doi: 10.1056/NEJMoa1112088. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. Tumor necrosis factor alpha- and interleukin-1beta-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013;65:832–842. doi: 10.1002/art.37819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J, et al. Distinctive topology of age-associated epigenetic drift in the human interactome. Proc. Natl Acad. Sci. USA. 2013;110:14138–14143. doi: 10.1073/pnas.1307242110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S, Smyth P. Proceedings of the ninth ACM SIGKDD international conference on Knowledge discovery and data mining. Washington, DC: ACM; 2003. Algorithms for estimating relative importance in networks; pp. 266–275. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.