

Abstract
Cilia are thought to harbour a membrane diffusion barrier within their transition zone (TZ) that compartmentalises signalling proteins. How this “ciliary gate” assembles and functions remains largely unknown. Contrary to current models, we present evidence that Caenorhabditis elegans MKS-5 (orthologue of mammalian Mks5/Rpgrip1L/Nphp8 and Rpgrip1) may not be a simple structural scaffold for anchoring > 10 different proteins at the TZ, but instead, functions as an assembly factor. This activity is needed to form TZ ultrastructure, which comprises Y-shaped axoneme-to-membrane connectors. Coiled-coil and C2 domains within MKS-5 enable TZ localisation and functional interactions with two TZ modules, consisting of Meckel syndrome (MKS) and nephronophthisis (NPHP) proteins. Discrete roles for these modules at basal body-associated transition fibres and TZ explain their redundant functions in making essential membrane connections and thus sealing the ciliary compartment. Furthermore, MKS-5 establishes a ciliary zone of exclusion (CIZE) at the TZ that confines signalling proteins, including GPCRs and NPHP-2/inversin, to distal ciliary subdomains. The TZ/CIZE, potentially acting as a lipid gate, limits the abundance of the phosphoinositide PIP2 within cilia and is required for cell signalling. Together, our findings suggest a new model for Mks5/Rpgrip1L in TZ assembly and function that is essential for establishing the ciliary signalling compartment.
Keywords: cilia, ciliary gate, MKS5, PIP2, transition zone
Introduction
Cilia are organelles that grow from a modified centriole (basal body) and possess diverse functions employed by most animal cell types (Satir & Christensen, 2007; Pedersen & Rosenbaum, 2008; Gerdes et al, 2009; Drummond, 2012). Motile cilia propel gametes, generate flow across cell surfaces, and have sensory functions. Non-motile (primary) cilia play key roles in chemosensation/olfaction, mechanosensation, and photosensation. Several signalling pathways, orchestrated by chemoreceptors, G-protein-coupled receptors (GPCRs), Hedgehog signalling components, and cyclic nucleotide-producing guanylate/adenylate cyclases, are known to be cilium-associated (Wong & Reiter, 2008; Veland et al, 2009; Johnson & Leroux, 2010; Goetz and Anderson, 2010; Wallingford & Mitchell, 2011; Christensen et al, 2012). Hence, cilia are critical for vertebrate physiology and development, and their disruption cause human disorders (ciliopathies) that collectively affect nearly all organs (Baker & Beales, 2009; Nigg & Raff, 2009; Bettencourt-Dias et al, 2011; Lee & Gleeson, 2011; Oh & Katsanis, 2012).
Primary cilia are believed to function as cellular antennae by compartmentalising signal transduction molecules within a membrane-bound compartment that is compositionally distinct from the cell body (Pazour & Bloodgood, 2008; Emmer et al, 2010). Evidence of a membrane diffusion barrier or “gate” at the ciliary base is starting to emerge—in particular with the proximal-most region of the cilium, termed transition zone (TZ), playing important roles (Craige et al, 2010; Garcia-Gonzalo et al, 2011; Williams et al, 2011; Chih et al, 2012; Garcia-Gonzalo & Reiter, 2012; Reiter et al, 2012; Awata et al, 2014). The TZ is characterised by Y-shaped ultrastructures that make microtubule axoneme-to-ciliary membrane connections and likely compose/organise a “ciliary necklace” consisting of a series of annular intramembrane particles of unknown function (Gilula & Satir, 1972; Fisch & Dupuis-Williams, 2011; Reiter et al, 2012).
Recently, eleven Caenorhabditis elegans proteins were localised to the TZ, and most were attributed roles in restricting inappropriate entry of periciliary membrane-associated protein (TRAM-1) or retinitis pigmentosa 2 (RPI-2) into cilia (Williams et al, 2008, 2011; Huang et al, 2011; Roberson et al, 2015). Genetic and cell biology analyses suggest that nematode TZ proteins group into two functional modules: an “MKS” module containing MKS-1 (orthologue of mammalian Mks1), MKSR-1 (B9d1), MKSR-2 (B9d2), MKS-2 (Tmem216), MKS-3 (Tmem67), MKS-6 (Cc2d2a), and JBTS-14 (Tmem237); and, an “NPHP” module consisting of NPHP-1 and NPHP-4 (orthologues of Nphp1 and Nphp4, respectively; Huang et al, 2011; Williams et al, 2011). Abrogating individual TZ-localised MKS or NPHP module proteins, or multiple proteins within one module, has little apparent impact on nematode ciliogenesis. However, disrupting any MKS module protein in combination with NPHP-1 or NPHP-4 results in synergistic defects, including loss of basal body–TZ membrane anchoring, loss of TZ ultrastructure, and axoneme malformations (Huang et al, 2011; Williams et al, 2011). Localisation experiments corroborate the notion of two independent modules; specifically, some MKS module components mislocalise in MKS mutants, and NPHP-1 is mislocalised in the absence of NPHP-4; conversely, disruption of MKS proteins does not affect NPHP proteins, and vice versa. The molecular basis or reason for the existence of two modules is not understood. Intriguingly, MKS-5 (Rpgrip1/Rpgrip1L orthologue) appears to hold a more “central” position at the TZ, potentially bridging both modules (Huang et al, 2011; Williams et al, 2011; Roberson et al, 2015). How the coiled-coil and C2-domain containing MKS-5 protein performs this role is unknown, but could conceivably be as a “scaffold” that links the various TZ proteins to the axoneme and potentially the membrane at the TZ.
Most mammalian counterparts of the above-mentioned proteins are also TZ-localised and modulate the ciliary entry/exit of various proteins; notably, their abrogation is associated with compromised Hedgehog signalling (Garcia-Gonzalo et al, 2011; Sang et al, 2011; Chih et al, 2012; Roberson et al, 2015) and linked to one or more related ciliopathies, including Meckel syndrome (MKS), Joubert syndrome (JBTS), nephronophthisis (NPHP), Bardet–Biedl syndrome (BBS), Leber's congenital amaurosis (LCA), and oral-facial-digital syndrome (OFD). These ciliopathies are characterised by overlapping clinical ailments encompassing brain and skeletal malformations, kidney disease, and retinal degeneration (Baker & Beales, 2009; Hildebrandt et al, 2011; Lee & Gleeson, 2011; Novarino et al, 2011; Oh & Katsanis, 2012).
Given the evolutionary conservation, ultrastructural complexity, and essential signalling functions of the TZ, we strive to shed light on its molecular composition and organisation, as well as mechanism of formation and function, using C. elegans as a model system. Our findings suggest a novel role for MKS-5 in the biogenesis and function of the TZ. First, we provide evidence that MKS-5 (Rpgrip1L/Rpgrip1) acts as an assembly factor for building the TZ rather than a simple scaffold for all TZ proteins, including the C. elegans orthologues of the mammalian TZ proteins, Tmem17 and Tmem138 (Chih et al, 2012; Lee et al, 2012). This function is required for the formation of characteristic TZ ultrastructure, and correct cellular signalling. We dissect the coiled-coil and C2 domains of MKS-5 to reveal their functional interactions with MKS and NPHP module components, and illuminate how the MKS/NPHP modules perform redundant roles in sealing the ciliary compartment at the transition fibres and TZ. Importantly, we demonstrate that MKS-5 establishes a ciliary zone of exclusion (CIZE), which excludes signal transduction proteins and compartmentalises them within the ciliary compartment. Finally, we show that phosphatidylinositol 4,5-bisphosphate (PIP2) is enriched at the periciliary membrane compartment and excluded from the cilium in an MKS-5-dependent manner. Collectively, our findings provide mechanistic insights into the role of MKS-5 in the formation and function of an important subciliary domain required for the compartmentalisation of signalling proteins, and cellular signalling.
Results
Identification of TMEM-17 and TMEM-138 as C. elegans transition zone proteins
In genome-wide screens for candidate C. elegans ciliary genes, we identified ZK418.3/tmem-17 and F10B5.9/tmem-138 as possessing X-box motifs, suggesting regulation by the ciliogenic RFX transcription factor DAF-19 (Blacque et al, 2005; Chen et al, 2006). Tmem17 belongs to a protein superfamily with two additional members in mammals, Tmem80 and Tmem216, while C. elegans (like protists and other “basal” metazoans) encodes only two members, tmem-17/Tmem17 and mks-2/Tmem216; notably, Mks2/Tmem216 is associated with MKS and JBTS (Valente et al, 2010). To shed light on the possible ciliary functions of C. elegans TMEM-17 and TMEM-138, we created transgenic lines bearing GFP translational fusion constructs driven by their endogenous promoters.
Consistent with having ciliary roles, both genes are expressed exclusively in most or all ciliated sensory neurons (data not shown). Furthermore, both TMEM-17 and TMEM-138 are highly enriched in a ∼0.8-μm-long region immediately distal to the basal body-associated transition fibres (Fig1A and B; transition fibres and axonemes are marked with the intraflagellar transport (IFT) protein, XBX-1/DLIC) (see also Roberson et al, 2015). This localisation pattern is precisely like other TZ-localised proteins (Williams et al, 2008, 2011; Huang et al, 2011). Our findings corroborate the recent discovery that mammalian Tmem17 is a TZ protein, as is Tmem138; notably, the latter protein is implicated in JBTS (Chih et al, 2012; Lee et al, 2012). In the sections below, we integrate these proteins into an expanded TZ functional interaction network.
Figure 1.

- Schematic depicting phasmid (tail) cilia at dendritic tips (DTs), including basal body-associated transition fibres (TFs) and transition zone (TZ).
- TMEM-17::EGFP and TMEM-138::EGFP localise to the TZ in some, but not all, TZ mutants indicated; lack of localisation is denoted by a dotted ellipse. The co-marker tdTomato-tagged XBX-1 marks TFs and the ciliary axoneme.
- MKS and NPHP protein localisation in mks-2 and tmem-17 mutants. In mks-2 animals, MKS-1, MKSR-1, MKSR-2, MKS-3, MKS-6, and TMEM-17 are delocalised (dotted ellipse) but NPHP-1, NPHP-4, and MKS-5 remain at the TZ. All tested TZ proteins remain at the TZ of the tmem-17 mutant.
- NPHP-4 and NPHP-1 localise to both the TZ and TFs in wild-type animals, but are only found at TFs in the mks-5 mutant. The CHE-13 reporter shown marks TFs and the entire axoneme. Note the specific overlap (see merge) between NPHP-1 and NPHP-4 with the TF in the mks-5 mutant.
Data information: (B–D) Proteins are shown in green (tagged with EGFP), red (tdTomato), orange (YFP), or blue (CFP). Scale bar: 4 μm.
Transition zone formation and sealing of ciliary compartment requires TMEM-17 and MKS-2 working together with NPHP-4
We acquired likely null mutants of tmem-17,tmem-138, and mks-2 and tested these for uptake of the lipophilic fluorescent dye, DiI, which permeates sensory neurons through intact, environmentally exposed cilia (Inglis et al, 2009). Whereas IFT/BBS mutants with compromised ciliary structures have strong dye-filling (Dyf) defects (Perkins et al, 1986; Starich et al, 1995; Blacque et al, 2004), tmem-17 and tmem-138 mutants, similar to mks-2 mutants (Huang et al, 2011), take up dye normally (FigEV1A and unpublished observations). Moreover, tmem-17 mutants correctly avoid a high-osmolarity solution (FigEV1B), unlike the chemosensory-defective IFT/BBS mutants (Perkins et al, 1986; Starich et al, 1995; Blacque et al, 2004; Inglis et al, 2009). Using dye-filling and behavioural (chemosensory) assays, as well as transmission electron microscopy (TEM), we tested these mutants for genetic interactions amongst themselves and other TZ-associated mutants—anticipating potential synthetic (functional) interactions, as observed previously between MKS and NPHP module mutants (Williams et al, 2008, 2010, 2011; Huang et al, 2011). tmem-17;nphp-4 double mutants display synthetic dye-filling (Dyf) and chemosensory phenotypes not seen in tmem-17;mks-3,tmem-17;mksr-1, or tmem-17;mks-2 animals (FigEV1B), indicating a likely structural defect in the tmem-17;nphp-4 double mutant, as well as assigning TMEM-17 to the MKS module of proteins. Importantly, the Dyf phenotype of both tmem-17;nphp-4 (FigEV1A) and mks-2;nphp-4 (Huang et al, 2011) was rescued with their respective TZ-localised GFP translational fusion reporters (FigEV2A). Intriguingly, tmem-138 did not present a synthetic Dyf phenotype with nphp-4,mksr-1, or mks-5 (data not shown); thus, a clear functional association with either MKS or NPHP module remains uncertain for this evolutionarily conserved TZ protein (see also below).
Figure EV1.
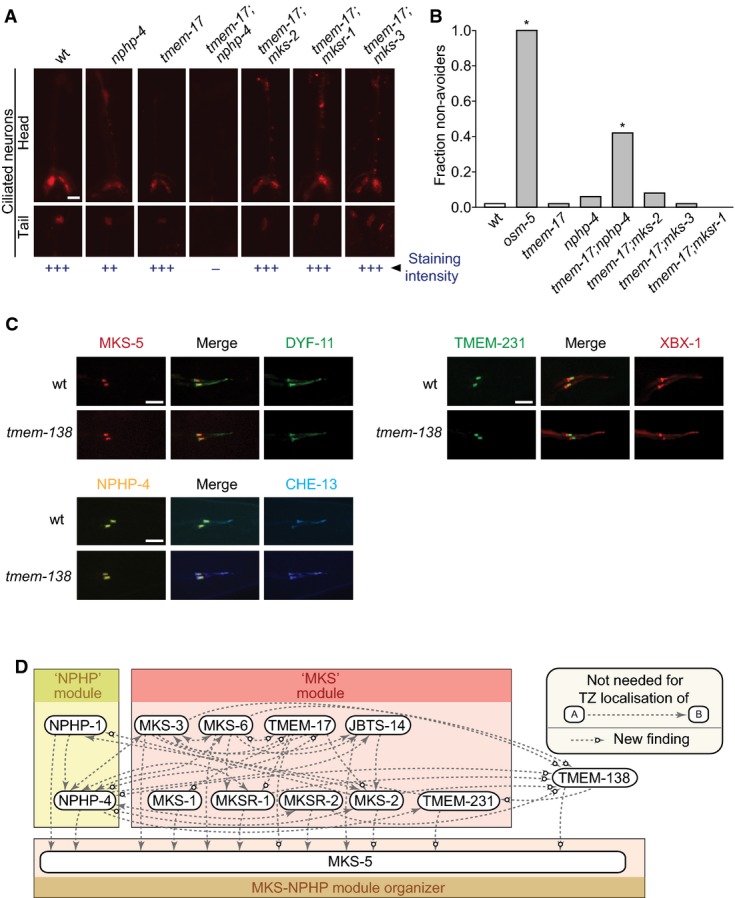
- Disruption of tmem-17 alone does not cause overt ciliary defects as measured by uptake of fluorescent dye through environmentally exposed cilia. tmem-17;nphp-4 double mutants are defective in dye uptake while tmem-17;mks-2,tmem-17;mksr-1 and tmem-17;mks-3 take up the dye similar to tmem-17 alone. Scale bar: 20 μm.
- tmem-17 mutants avoid high-osmolarity solutions, as do tmem-17;mks-2,tmem-17;mksr-1, and tmem-17;mks-3 double mutants. tmem-17;nphp-4 double mutants are partially defective in sensing osmolarity, though not as severely as osm-5 (IFT mutant) animals, which are completely oblivious of the hyperosmotic barrier (*P < 0.0001, chi-squared test).
- TZ markers expressed in wild-type and the tmem-138 mutants. MKS-5, TMEM-231 (MKS module), and NPHP-4 (NPHP module) do not require TMEM-138 for TZ localisation. Scale bars: 4 μm.
- Model showing all of the negative interaction amongst MKS/NPHP proteins, where a particular MKS/NPHP protein (at the arrowhead) is localised properly in a specific TZ mutant; a white circle on the arrowhead indicates new data obtained in this study.
Figure EV2.
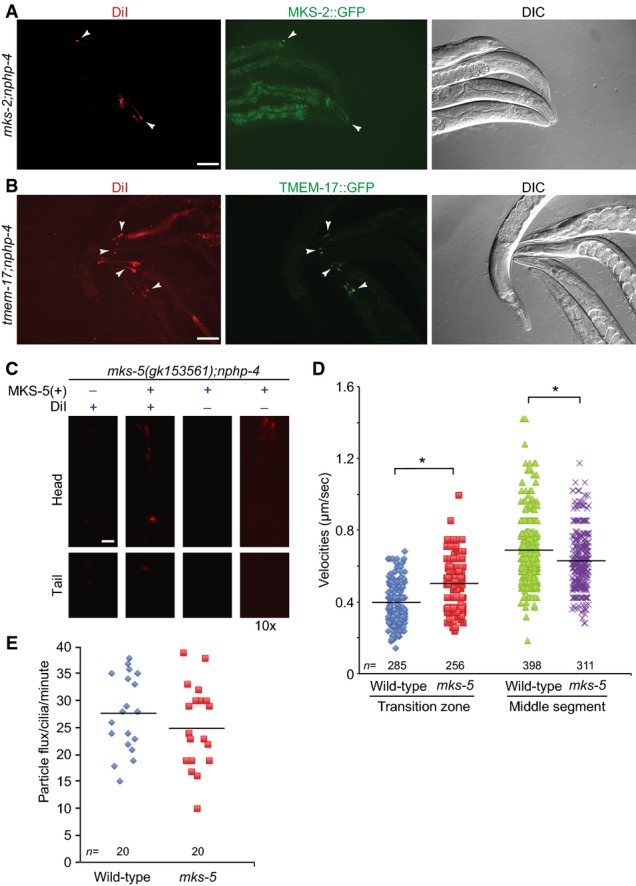
- mks-2;nphp-4 mutants display a failure to uptake the lipophilic dye DiI which can be rescued by the wild-type MKS-2::GFP reporter. Shown are transgenic animals (arrowheads) that express the reporter and uptake of the dye, while their nontransgenic siblings cannot uptake the dye. Scale bar: 100 μm.
- tmem-17;nphp-4 mutants display a failure to uptake the lipophilic dye DiI which can be rescued by the wild-type TMEM-17::GFP translational reporter. Shown are transgenic animals (arrowheads) that express the reporter and uptake of the dye, while their nontransgenic siblings cannot uptake the dye. Scale bar: 100 μm.
- The mks-5(gk153561);nphp-4 mutants display a Dyf phenotype, which can be rescued with expression of the wild-type MKS-5::tdTomato translational reporter. In the third set of head and tail panels, transgenic worms are shown at the same exposure time, but the signal is not detectable. In the right-hand set of panels, a tenfold longer exposure shows detectable signal for the MKS-5::tdTomato reporter, indicating that the signal for the transgenic worms is coming from the dye. Scale bar: 20 μm.
- Scatter plots of measured IFT velocities for wild-type and mks-5 mutants using the CHE-13::YFP translational reporter. In the transition zone (TZ; proximal ˜0.8 μm of the cilium), the mean IFT velocity is 0.39 μm/s in wild-type animals, but increases to 0.49 μm/s in the mks-5 mutant, which lacks a detectable TZ. In the middle segment, mean IFT velocity is 0.69 μm/s, while in the mks-5 mutant, it is significantly slower at 0.63 μm/s. *P < 0.001, t-test.
- IFT flux (in particles per minute) is shown for wild-type and mks-5 mutants. No difference is observed between the flux of IFT particles in wild-type and mks-5 animals (P > 0.05, t-test).
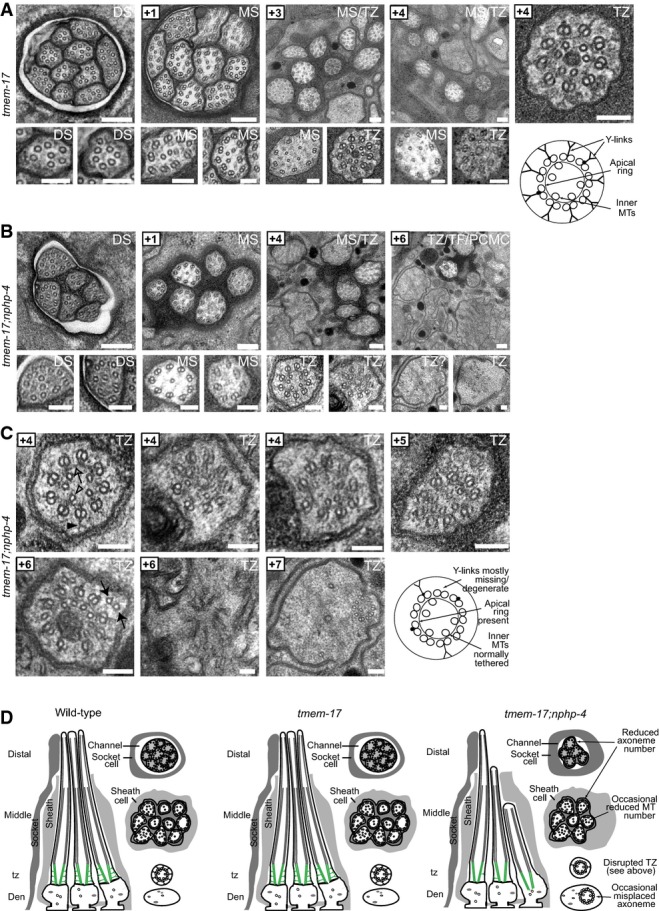
We then sought to observe the ciliary ultrastructure of the tmem-17 and mks-2 mutants by TEM. In wild-type animals, TEM cross-sections of amphid channel ciliary bundles reveal ∼3-μm-long distal segments consisting of singlet microtubules, followed by ∼3-μm-long middle segments containing nine doublet microtubules with internal singlets, followed by ∼0.8-μm-long TZs exhibiting Y-links connecting doublet microtubules to the membrane, and finally, in the most proximal positions, basal body-associated transition fibres (Perkins et al, 1986) (Fig2). The mks-2 and tmem-17 single mutant amphid (head) channel cilia display essentially identical ultrastructures, with no obvious anomalies except one of the 10 axonemes is frequently missing in mks-2 worms. In striking contrast, mks-2;nphp-4 and tmem-17;nphp-4 double mutants exhibit abrogated basal body–TZ attachment to the ciliary membrane, missing Y-links, and many shorter axonemes (Figs EV3 and EV4). Importantly, the mks-2;nphp-4 double mutant displays TZ defects that are significantly more severe than that of the nphp-4 single mutant, which presents with fewer or reduced Y-links; the TZ defects present in the tmem-17;nphp-4 double mutant appear more severe than the nphp-4 mutant, but to a lesser extent (FigEV5). Together, our results suggest that similar to mks-1,mksr-1,mksr-2,mks-3,mks-6, and jbts-14 (Huang et al, 2011; Williams et al, 2011), both tmem-17 and mks-2 are MKS module components that genetically/functionally interact with the NPHP module. This function is needed to establish correct TZ ultrastructure and “seal” the ciliary compartment by creating essential transition fibre and TZ membrane attachments.
Figure 2.
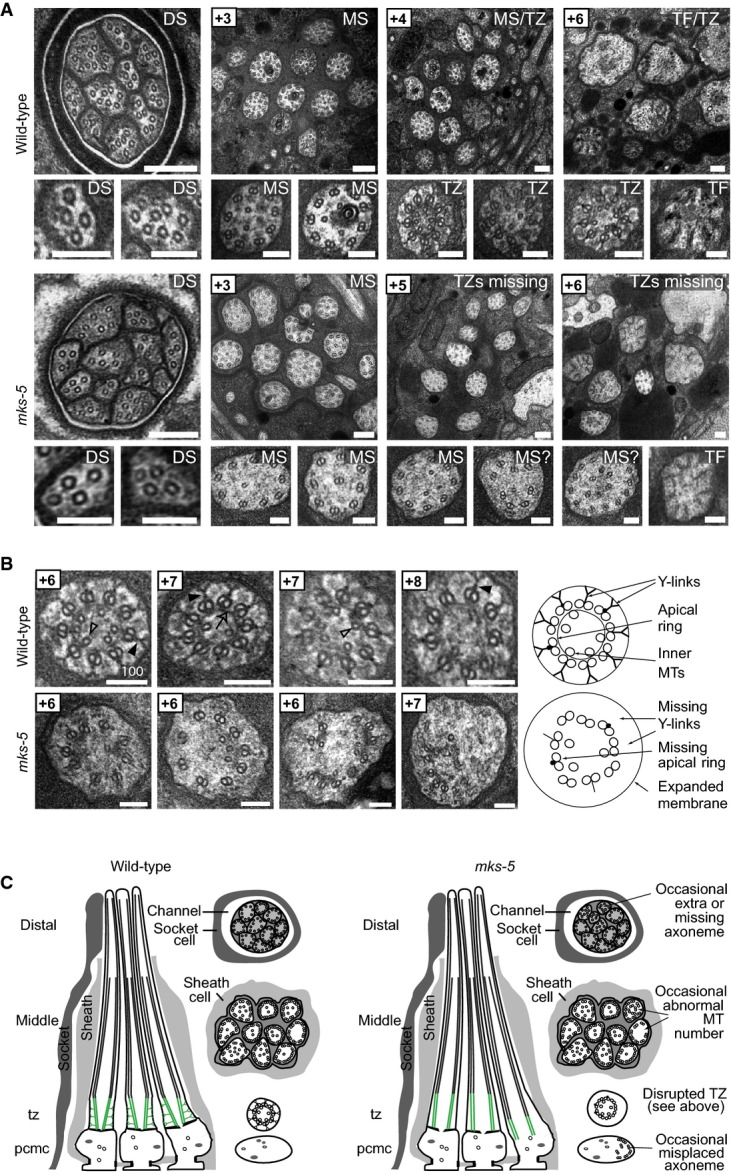
- Transmission electron microscopy (TEM) serial cross-section images of wild-type and mks-5(tm3100) amphid channel cilia. Boxed numbers denote proximal positioning of section relative to left-most (anterior) section of the animal. Wild-type amphid channels contain 10 axonemes, each possessing the following: a distal segment (DS) with singlet microtubules (MTs); a middle segment (MS) with doublet MTs and inner singlet MTs; a transition zone (TZ) with Y-link connectors, and doublet MT-associated apical ring; and a basal body/transition fibre (TF) region. mks-5 worms possess grossly normal DSs (although one axoneme is frequently missing), and MSs that frequently display MT singlets and doublets reduced in number and occasionally displaced (see high-magnification images). TZs are not observed in mks-5 worms, replaced instead by abnormal middle segments (labelled “MS?”). Scale bars: 200 nm (low-magnification images); 100 nm (high-magnification images).
- The TZ region of mks-5 mutants lacks all distinguishing features, including the apical ring (arrow), Y-links (closed arrowhead), and tight connection to the ciliary membrane. Doublet MTs of mks-5 mutant TZs are often displaced. Open arrowhead: inner singlet MT. Scale bars: 100 nm.
- Schematics summarising ultrastructures of wild-type and mks-5 mutants. PCMC, periciliary membrane compartment.
Figure EV3.
Transmission electron microscopy of tmem-17 and tmem-17;nphp-4
- A tmem-17 mutant cilium ultrastructure is similar to wild-type worms, possessing all characteristic features, including singlet microtubules (MTs) in the distal segments (DSs), an ordered ring of doublet MTs surrounding internal singlet MTs in the middle segments (MSs), and transition zones (TZ) possessing Y-links and an apical ring tethering inner singlet MTs to outer doublet MTs. Scale bars: 200 nm (low-mag. images); 100 nm (high-mag. images).
- B, C In tmem-17;nphp-4 double mutants, 3–4 middle and distal segments are missing and remaining axonemes sometimes possess missing doublet MTs. Most TZs lack some Y-link structure (closed arrowhead), and frequently other defects are observed, such as ectopic singlet MTs (closed arrow), reduced MT numbers (+7 image), and detachment from the ciliary/dendritic membrane (+7 image) indicative of an axoneme misplaced within the dendrite (see schematic). The apical membrane (open arrow) tethering inner singlet MTs (open arrowhead) to outer doublet MTs remains largely intact. Together, these ultrastructural defects are characteristic of mutants that carry a mutation in a member of both the NPHP module (nphp-4) and the MKS module (tmem-17). Scale bars: 200 nm (low-mag. images in B); 100 nm (high-mag. images in B and in C).
- D Schematics summarising the ultrastructure phenotypes.
Figure EV4.
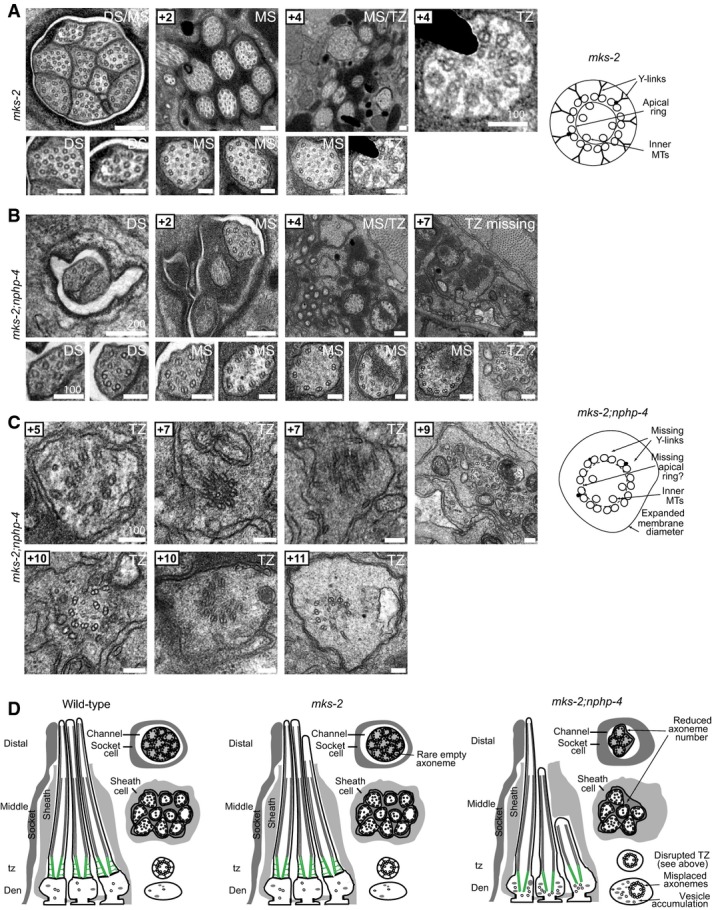
Transmission electron microscopy of mks-2 and mks-2;nphp-4
- A mks-2 mutant cilium ultrastructure is mostly similar to wild-type worms, possessing all characteristic features, including singlet microtubules (MTs) in the distal segments (DSs), an ordered ring of doublet MTs surrounding internal singlet MTs in the middle segments (MSs), and transition zones (TZ) possessing Y-links and an apical ring tethering inner singlet MTs to outer doublet MTs. However, occasional axonemes possess abnormal numbers of singlet or doublet MTs (bottom-left image). Scale bar: 200 nm (low-mag. images); 100 nm (high-mag. images).
- B, C In mks-2;nphp-4 double mutants, many middle and distal segments are missing. TZs completely lack Y-links and are detached from the ciliary/dendritic membrane indicative of axonemes anchored within the dendrite (i.e. misplaced; see schematic). The apical membrane that tethers inner singlet MTs to outer doublet MTs is difficult to discern, and doublet MTs are frequently disorganised (+10 image; left). Misplaced TZs are often surrounded by abnormal membrane (vesicle-like) accumulations (+5, +9, +10 images). These ultrastructural defects are characteristic of mutants that carry a mutation in both a member of the NPHP module (nphp-4) and the MKS module (mks-2). Scale bars: 200 nm (low-mag. images in B); 100 nm (high-mag. images in B and in C).
- D Schematics summarising the ultrastructure phenotypes.
Figure 5.

- EGFP-tagged SRBC-66 and ODR-10 both localise to the entire axonemes of ASK and AWB neuron cilia, respectively, and are excluded from the TZ in wild type (wt); these markers occupy the entire cilium, including the TZ, in mks-5 mutants. TAX-4 and NPHP-2, orthologues of the cyclic nucleotide-gated channel CNGA1 and NPHP2/inversin respectively, which are found in the inversin (Inv) compartment in wt, also localise to the TZ in mks-5 animals. NPHP-2::EGFP was co-localised with XBX-1::tdTomato and are shown in the phasmid cilia while TAX-4::EGFP is shown in amphid cilia. den, dendrite; TZ, transition zone; BB, basal body; arrowheads indicate gap is present; * indicates no gap. Scale bars: 4 μm.
- Quantification of cilia observed to have a clear gap in signal (i.e. CIZE) at the TZ in wt animals compared to mks-5 mutants for the ODR-10, SRBC-66, and NPHP-2 protein markers. *P < 0.001, chi-squared test.
- Schematic depicting the CIZE, which excludes signalling proteins from the TZ in wt animals but not mks-5 animals, which lack TZ ultrastructure.
- NPHP-2 signal length is longer in mks-5 animals compared to wt, consistent with the presence of an axonemal region that lacks TZ ultrastructure. *P < 0.001, t-test, error bars are standard deviation.
- The mks-5 mutant displays a dauer formation phenotype compared to wild-type when presented with synthetic ASCR#2 dauer pheromone. ASCR#2 is detected by the SRBC-66 G-protein-coupled receptor, and downstream signalling includes the cGMP-dependent channel, TAX-4. *P < 0.001, chi-square test.
Figure EV5.
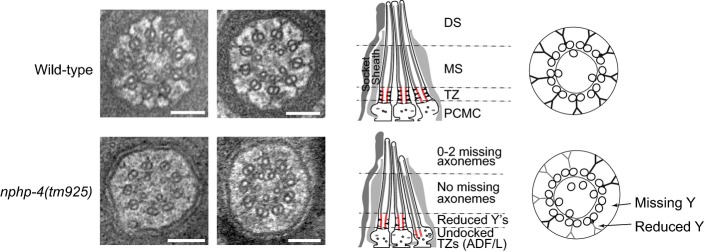
Transmission electron microscopy of nphp-4
Shown are cross-section images of amphid channel cilia at the transition zone. In nphp-4 mutants, transition zone Y-links are often missing and when present have lower electron density (light grey) compared to wild type. Schematics on the right summarise the ultrastructure phenotypes. Scale bars: 100 nm.
Mapping TMEM-17, TMEM-138, and MKS-2 to a TZ hierarchical organisation
To establish functional relationships between TMEM-17, TMEM-138, and other TZ proteins, we assayed for potential changes in TZ protein localisation in various TZ gene mutants. In wild-type animals and mks-3,mks-6, and nphp-4 mutants, TMEM-17 is tightly associated with the TZ (Fig1B). In contrast, TMEM-17 localisation is perturbed in mks-2,mksr-1,mksr-2, and mks-5 mutants, consistent with a functional association with the MKS module (Fig1B). Interestingly, TMEM-138 localises normally to the TZ in wild-type, mks-1,mksr-1,mksr-2, mks-2, mks-3,mks-6, and nphp-4 animals, suggesting that it is either a “core” rather than “peripheral” TZ component or, potentially, functionally/physically separate from the NPHP or MKS modules. Yet, TMEM-138 is mislocalised in mks-5 animals (Fig1B)—consistent with it being associated with an established “core” TZ protein.
Reciprocal experiments were performed, assessing the localisation of TZ proteins in available tmem-17,tmem-138, and mks-2 mutants. We previously reported that the novel TZ protein JBTS-14 mislocalises in mks-2 animals (Huang et al, 2011); similarly, TMEM-17, MKS-1, MKSR-1, MKSR-2, MKS-3, and MKS-6 all require MKS-2 for proper TZ localisation, while NPHP-4 and MKS-5 do not (Fig1C). None of the tested TZ proteins require TMEM-17 or TMEM-138 for localisation (Figs1C and EV1C). These results, together with our extensive “negative interaction” data (i.e. no effect of the mutants on TZ protein localisation; FigEV1D), suggest a hierarchical organisation network for the TZ proteins. Specifically, our data suggests the following assignments: MKS-2 represents a “core” MKS module component, similar to B9 domain proteins (MKS-1, MKSR-1, and MKSR-2), while TMEM-17 appears more “peripheral,” like JBTS-14, MKS-3, and MKS-6 (Huang et al, 2011; Williams et al, 2011); TMEM-138 is either a core TZ protein “close” to MKS-5 in its hierarchy and/or belongs to a different module that is also MKS-5-associated. Together, the properties of MKS-5 are unique, suggesting it is the major scaffold/assembly factor for all TZ proteins (summarised in Fig5C model figure, below).
NPHP module proteins require MKS-5 for TZ localisation but not basal body–transition fibre localisation
Since TMEM-17, TMEM-138, and previously reported MKS module proteins all require MKS-5 for TZ localisation, we questioned whether NPHP module proteins are truly localised within a “smaller” TZ region in mks-5 mutants, as we previously suggested (Williams et al, 2011), rather than absent from the TZ. In our revised experimental approach, we visualised fluorescently tagged NPHP-1 or NPHP-4 together with the IFT protein CHE-13/IFT57, which marks the basal body–transition fibre region and ciliary axoneme. We discovered that unlike other MKS module TZ proteins, NPHP-1 and NPHP-4 have a dual localisation, not only at the TZ, but also at transition fibres (Fig1D). This secondary localisation is most apparent in the mks-5 mutant, where NPHP-1 and NPHP-4 are clearly absent from the TZ but remain at the transition fibres, co-localising with the IFT protein CHE-13 (Fig1D). Hence, every identified TZ protein in C. elegans requires MKS-5 for correct TZ localisation, implicating it as central organiser or assembly factor of the MKS-NPHP TZ network. Furthermore, the dual localisation of NPHP module components (at transition fibres and TZ) is also a key finding: it is relevant to the redundant roles of MKS and NPHP module components in sealing the ciliary compartment, which we discuss later.
MKS-5 is required for the formation of TZ ultrastructure
Given that MKS-5 is needed for the correct localisation of all TZ proteins, we investigated by TEM analysis whether its absence abrogates TZ ultrastructure. Similar to wild-type animals, we found that mks-5 mutants possess a full complement of amphid channel cilia with grossly normal middle and distal segments, and transition fibres; however, the Y-link and apical ring structures characteristic of the TZ are conspicuously absent (Fig2). One seemingly apparent result from this structural defect is a partial disconnection or “loosening” between the axoneme and the proximal-most region of the ciliary membrane. The disconnection, however, is not as severe as what is observed in MKS-NPHP module double mutants (e.g. mks-2;nphp-4; Fig EV4 and Williams et al, 2011). Critically, this unique mks-5 mutant phenotype, where the TZ is absent, is exacerbated in the nphp-4 mutant background; indeed, not only does the mks-5;nphp-4 double mutant lacks Y-links, but the association between the basal body/TZ region with the ciliary membrane is completely lost, similar to MKS-NPHP double mutants (FigEV4; Williams et al, 2011). Together, our TEM findings indicate that MKS-5 is required for TZ ultrastructure formation in and of itself and that anchoring of the basal body and proximal axoneme region to the ciliary membrane also requires the NPHP module component, NPHP-4. These data help clarify the redundant functions of the MKS and NPHP modules in sealing the ciliary compartment, an important finding we discuss later.
Localisation to two distinct foci spanning the TZ suggests an assembly role for MKS-5
Having established that MKS-5 appears to be a key “anchor” or assembly factor for TZ proteins, we wondered whether MKS-5 localisation itself depends on other MKS/NPHP module components. Although MKS-5 localisation appears superficially wild-type in TZ mutants (Williams et al, 2011) (Fig3A), further observations, and with additional TZ mutants, reveal altered localisation patterns. In all MKS module mutants tested, MKS-5 localisation unexpectedly appears as two distinct, roughly symmetrical spots that span the TZ region (Fig3A). We quantified the prevalence of this dual localisation in two MKS module mutants, mks-2 and mks-3 (Fig3C). Not only does the occurrence of two distinct loci correlate with the severity of the mutant (mks-2 mutants show loss of MKS module proteins; Fig1C), but it also correlates with the overall length of the MKS-5 signal (Fig3B). Based on these results, we hypothesised that our ability to observe two foci in mutants was because of the presence of a slightly longer TZ in the various mutants; in wild-type animals, the two MKS-5 signals could not be distinguished due to the resolution limits of confocal microscopy.
Figure 3.

- In wild-type worms, tdTomato-tagged MKS-5 localises to the transition zone (TZ) at a single obloid punctum. In the MKS module mutants mksr-1,mksr-2,mks-2,tmem-17,jbts-14,mks-3, and mks-6, as well as the double mutant mks-2;mksr-1, MKS-5 localises to two puncta at the TZ. MKS-5 localisation in the NPHP module mutants nphp-1 and nphp-4 and the double mutant nphp-1;nphp-4 displays another small point of localisation, distal to the TZ along the axoneme (arrowheads). Scale bar: 4 μm.
- Mean length of the MKS-5 signal in the wild-type (wt), as well as mks-3 and mks-2 mutant strains. mks-2 mutants have disrupted MKS module protein localisation and are therefore more severe than mks-3, which do not affect localisation of any TZ proteins. The TZ is significantly longer in mks-3 mutants compared to wt, while in mks-2 it is significantly longer compared to either mks-3 or N2. *P < 0.001, t-test, error bars are 95% confidence interval.
- Fraction of transition zones displaying two peaks of MKS-5 fluorescence. In wt, none of the TZ confocal images display two peaks. In mks-3 and mks-2 mutants, the proportion with two distinct fluorescence signals was significantly increased compared to wt. mks-2 mutants also showed significantly more TZs with two peaks compared to mks-3. *P < 0.001, chi-square test.
- In double mutants with mutations in genes from each of the NPHP and MKS modules, mks-2;nphp-1 and mks-2;nphp-4, most cilia show MKS-5 localised not only to the TZ but often dispersing distally along some cilia. Additional phenotypes observed in these latter double mutants (short dendrite, long cilium, mis-oriented cilium) are shown. cb, cell body. Scale bar: 4 μm.
In nphp-1 or nphp-4 single mutants, and nphp-1;nphp-4 double mutants, MKS-5 also displays two foci, as well as an additional small punctum distal to the TZ (Fig3A). Since disrupting both modules has synthetic ciliogenesis phenotypes (this study and references Huang et al, 2011; Williams et al, 2008, 2010, 2011), we also introduced the MKS-5 reporter into the mks-2;nphp-1 and mks-2;nphp-4 strains. In both double mutants, MKS-5 is TZ-localised, but displays dual signals and often disperses distally along the axoneme (Fig3B). Taken together, our findings point to interdependent associations between MKS-5 and MKS/NPHP module components, such that removing MKS-5 prevents MKS/NPHP proteins from localising to the TZ, whereas disrupting any other TZ protein influences MKS-5 localisation. They also suggest that MKS-5 acts as an assembly factor rather than a scaffold for TZ proteins, a possibility we consider in greater detail in the discussion section.
The MKS-5 coiled-coil domain is sufficient for ciliary localisation and MKS module TZ localisation while NPHP module TZ localisation requires the MKS-5 C2-N domain
Given the central role of MKS-5 in establishing the TZ, we sought to determine which of its domains are needed for its own localisation, and the localisation of MKS/NPHP module components, to the TZ. MKS-5 contains at its N-terminus a large predicted coiled-coil region, followed by two C2 domains and an Rpgr interaction domain (RID) (Arts et al, 2007; Delous et al, 2007; Williams et al, 2011) (Fig4A). Our truncation studies with MKS-5 reveal that removing ∼1/2 of the (N-terminal-most) coiled-coil region results in weaker TZ localisation, with accumulation at the periciliary membrane (Fig4B). In contrast, MKS-5 lacking the entire coiled-coil region shows no distinct TZ localisation or trafficking to cilia (Fig4B). Consistent with these findings, the coiled-coils alone are sufficient for the TZ localisation of MKS-5 (Fig4B; bottom panels).
Figure 4.

- Schematic of predicted MKS-5 functional domains depicts 8 tandem coiled-coils, two C2 domains (C2-N and C2-C), and an Rpgr interaction domain (RID). Also shown are the missense alleles screened for synthetic dye-filling phenotypes (Dyf).
- Schematics depict the localisation of the MKS-5 FL and truncation proteins. Top three panels: full-length (FL) MKS-5::tdTomato localises to the TZ, adjacent to the TF region marked by the IFT markers (DYF-11::EGFP or CHE-13::CFP). Loss of the first half of the coiled-coil region results in substantially reduced MKS-5 localisation to the TZ and accumulation at the dendritic tip. Removal of the entire coiled-coil region results in complete loss of TZ localisation. Bottom four panels: truncations of the C-terminal half, retaining the TZ-localising coiled-coils, were co-expressed in the mks-5 null with MKS-2 or NPHP-1. The MKS-5 full-length and truncation constructs lacking the RID both rescue NPHP-1 and MKS-2 TZ localisation. Removing the C2-C or both C2 domains rescues MKS-2 localisation but results in loss of NPHP-1 at the TZ (with NPHP-1 still retained at TFs). Schematics depict the localisation of the MKS-5 FL and truncation proteins. Scale bar: 4 μm.
- Fluorescence images of dye-filling experiments show that one of the eight tested mks-5 alleles (gk153561), resulting in a D772N alteration, results in a synthetic Dyf in combination with nphp-4. None of the alleles, including gk153561, were Dyf when genetically crossed with mksr-1. A model mapping the position of D772 relative to the synaptotagmin-7 C2 domain crystal structure suggests that the aspartic acid residue lies within the β-sandwich interface, potentially altering the stability of the domain and disrupting calcium binding (shown as spheres) or lipid binding (not in the structure). Amino acid sequence alignment of the Caenorhabditis elegansMKS-5 C2-C domain with both H. sapiens (Hs) Rpgrip1 and Rpgrip1L shows conservation at D772. Scale bar: 20 μm.
- No change in the localisation of NPHP-1::CFP (TF and TZ), NPHP-2::EGFP (inversin compartment), or MKS-2::EGFP (TZ) is detected in the mks-5(gk153561) mutants. Scale bar: 4 μm.
We then tested which MKS-5 domains permit TZ localisation of representative MKS and NPHP module components (MKS-2 and NPHP-1, respectively). C-terminal truncations of MKS-5 were expressed in the mks-5 mutant (leaving intact coiled-coils since they confer TZ localisation) and tested for “rescue” of MKS-2 and NPHP-1 localisation. As expected, full-length MKS-5 rescues the localisation of both TZ proteins (Fig4B). A truncation construct lacking the RID domain also fully rescues the localisation of both MKS-2 and NPHP-1 (Fig4B). Interestingly, a smaller MKS-5 truncation product (lacking C-terminal C2-C and RID domains) rescues the localisation of MKS-2, but not NPHP-1 (Fig4B). This latter finding is consistent with a previous report showing physical interaction of the C2-C domain of both Rpgrip1L and Rpgrip1 with Nphp4, which itself is required for Nphp1 localisation in both mammalian cells and C. elegans (Roepman et al, 2005; Winkelbauer et al, 2005; Arts et al, 2007). An even smaller MKS-5 truncation product, lacking the two C2 and the RID domains, also rescues the localisation of MKS-2, but not NPHP-1 (Fig4B). This construct contains the coiled-coils, indicating that in addition to localising MKS-5 to the TZ, these are sufficient for anchoring/assembling a “core” component of the MKS module.
Mutation of a conserved residue in the C2-C domain of MKS-5 impairs its function
To further explore MKS/NPHP module functional interactions with each MKS-5 domain, we obtained mks-5 missense alleles from the Million Mutation Project (http://genome.sfu.ca/mmp/about.html). Eight alleles were selected based on amino acid change and location to a predicted domain (Fig4A). Strains containing the point mutations were crossed with mksr-1 or nphp-4 mutants, and the resulting double mutants were tested for synthetic dye-filling (Dyf) defects indicative of disrupted ciliary structures. While none of the eight mks-5 mutants showed a Dyf phenotype, one was synthetic Dyf with nphp-4 but not mksr-1 (Fig4C); the synthetic Dyf phenotype is rescued by the wild-type MKS-5::tdTomato reporter (Fig EV2C). This latter mks-5 mutant (gk153561) differs from the mks-5 single null mutant, which is synthetic Dyf with both nphp-4 and mksr-1 (Williams et al, 2011). Interestingly, the mks-5(gk153561) allele did not perturb NPHP-1, NPHP-2, or MKS-2 localisation, suggesting a functional rather than a direct protein–protein interaction defect in MKS-5 (Fig4D). The mutation in the MKS-5 protein, D772N, occurs ina conserved residue within the second C2 domain (Fig4C). Modelling its position relative to the synaptotagmin-7 C2 domain crystal structure suggests that the alteration may impair the stability of the β-sandwich, potentially disrupting the presumed Ca2+ and/or lipid-binding functions of the C2 domain (Nalefski & Falke, 1996) (Fig4C).
MKS-5 establishes a CIZE at the TZ that is required for compartmentalisation of signalling proteins
Given our discovery that MKS-5 is essential for forming the TZ, but not the ciliary compartment per se (Fig2), we realised that we could use this to investigate the properties of the TZ and role in creating a membrane diffusion barrier (“ciliary gate”) in the context of an essentially intact ciliary axoneme. We first compared in wild-type and mks-5 mutant animals the localisation of two G-protein-coupled receptor (GPCR) proteins, ODR-10 and SRBC-66, present in AWB and ASK neuronal cilia, respectively (Sengupta et al, 1996; Kim et al, 2009) (Fig5A). We observed one clear difference: both proteins are conspicuously absent from the TZ in wild-type animals—but not in mks-5 mutants, where their localisation extends to the basal body/transition fibres (Fig5A). To reinforce this observation, we analysed two additional cilium-localised signalling proteins, namely NPHP-2 (orthologue of NPHP2/inversin) (Warburton-Pitt et al, 2012) and TAX-4 (cGMP-gated channel related to CNGA1) (Coburn & Bargmann, 1996; Komatsu et al, 1996) (Fig5A). Strikingly, these two proteins are also excluded from the TZ in wild-type, but not mks-5 mutant, animals (Fig5A–C). Quantification reveals that the ciliary region occupied by NPHP-2 is longer in mks-5 mutants compared to wild type (3.85 μm versus 2.67 μm, respectively; Fig5D), suggesting that the proximal ciliary region (axoneme/membrane) normally harbouring the TZ may still be present, but as we show above by TEM (Fig2) lacks ultrastructure (including Y-links). Together, these findings implicate the presence of what we term the CIZE, a TZ functionality that precludes the localisation of signalling proteins.
To confirm that impaired compartmentalisation of signalling proteins in mks-5 animals has functional consequences on cellular signalling, we focussed on the insulin-, cGMP-, and TGF-β-dependent dauer development signalling pathway. Notably, two proteins we show are localised incorrectly to the TZ in mks-5 animals (Fig5A), namely TAX-4 (cGMP-dependent channel) and SRBC-66 (dauer pheromone-specific GPCR), are required for cilia-dependent dauer pheromone sensation/signal transduction (Kim et al, 2009; Macosko et al, 2009). Indeed, we observe a previously unreported dauer pheromone sensory defect in mks-5 mutants (Fig5E). Our finding is consistent with impaired ciliary GPCR and cGMP signalling, which dauer pheromone olfactory signalling depends on. It also supports the reported chemotaxis defect of mks-5 animals to several odours (Liu et al, 2011), which also signal through GPCR and cGMP pathways. Importantly, our results parallel those reported for the abrogated Hedgehog signalling defects present upon disruption of TZ proteins in mammals (Garcia-Gonzalo et al, 2011; Chih et al, 2012; Thomas et al, 2012).
Loss of MKS-5 alters IFT velocity at the TZ
Given that the mks-5 mutant lacks a TZ but contains a largely intact axoneme, we sought to determine what effect this might have on IFT machinery, which normally transits from the basal body–transition fibres through the TZ to the tip of the cilium, and back. Using a CHE-13::YFP reporter, we analysed the IFT velocity and flux in the TZ and middle segment of cilia in both wild-type and mks-5 mutants. We show for the first time that in wild-type cilia, IFT exhibits an average velocity of 0.39 μm/s within the TZ (first 0.8 μm of the ciliary axoneme; FigEV2D) and accelerates to the previously reported ∼0.7 μm/s in the middle segment. These findings suggest an effect on the IFT machinery by the TZ. Consistent with this possibility, IFT velocity shows a statistically significant increase at the TZ region of the mks-5 mutant (to 0.49 μm/s) and a decrease at the middle segment (to 0.63 μm/s), possibly indicating a change in the TZ/ciliary environment (FigEV2D). Despite the difference in observed velocity, no difference in IFT flux (particles/minute) was observed between wild-type and mks-5 mutant cilia (FigEV2E). Hence, MKS-5 and/or other TZ proteins influence IFT behaviour (velocities), possibly through interaction with TZ proteins (Zhao & Malicki, 2011), and/or a difference in motility within a specific lipid micro-environment (see below). Yet, IFT flux remains the same, which may explain why there are no gross defects in ciliary axoneme structure in mks-5 mutants.
PIP2 is enriched at the periciliary membrane compartment and excluded from the cilium in an MKS-5-dependent manner
The presence of a CIZE, representing a membrane diffusion (exclusion) barrier at the TZ, suggests two possibilities. One is that there is a proteinaceous gate, which restricts the free diffusion of lipid-associated and transmembrane proteins across the TZ. Such a gate could function in a manner similar (or complementary) to that proposed for the septin-based gate at the base of cilia (Hu et al, 2010; Reiter et al, 2012). The other possibility, not mutually exclusive, is a differential lipid composition at the TZ that preferentially excludes membrane-associated proteins (Trimble & Grinstein, 2015). We chose to probe the latter possibility, given that virtually all TZ proteins are membrane associated—containing transmembrane domains (Mks2/Tmem216, Mks3/Tmem67, Tmem138, Jbts14/Tmem237), or lipid-binding C2 (Mks5, Mks6, Nphp1, Nphp4) and related B9 (Mks1, Mksr1/B9d1, Mksr2/B9d2) domains (Zhang & Aravind, 2012).
To study such a TZ “lipid gate” function, we sought to identify a lipid marker that was largely excluded from the TZ in wild-type animals and potentially free to enter this region in the absence of a functional TZ. We first tested the fluorescent lipophilic dye, DiI, which enters via exposed ciliated sensory neuron endings (Hedgecock et al, 1985). Interestingly, DiI staining was restricted to the proximal region of cilia, encompassing the TZ and middle segment (Fig6A). Like the lipidated membrane proteins ARL-13 and NPHP-2/inversin (Cevik et al, 2013), DiI is not detectable in the distal segment (Fig6A). Hence, there is a lipid barrier or gate at the interface between the middle and distal ciliary segments that may restrict membrane proteins/lipids from entering or being retained within the distal segment.
Figure 6.

- Co-localisation of the red fluorescent lipophilic dye DiI and OSM-5, a component of the IFT-B complex. GFP-tagged OSM-5 marks the basal body (BB), transition zone (TZ), and entire length of the amphid cilia axoneme. DiI co-localises with only the proximal or middle segment (MS) portion of the OSM-5 localisation, showing an exclusion of the dye from the distal segment (DS). This indicates that there is a barrier to DiI accumulation in the distal segment. Scale bar: 4 μm.
- Co-localisation of GFP-tagged PLCδ-PH with tdTomato-tagged XBX-1 in the male tail rays shows enrichment, at the PCMC in wild-type animals (top panels, differential interference contrast (DIC) microscopy of male tail rays 8 and 9). In mks-5 mutants, most male tail cilia display PIP2 probe signal extending distally into the cilium. Scale bar: 4 μm.
- Co-localisation of GFP-tagged PLC-δ-PH with MKSR-2 and TRAM-1 within phasmid cilia. MKSR-2 marks the TZ, and TRAM-1 marks the PCMC. In wild-type animals, the majority of the PIP2 probe signal localises to the PCMC and decreases in signal intensity distally into the cilium across the TZ. In mks-5 mutants, which lack an identifiable TZ, the PIP2 probe signal extends distally into the cilium. Shown in the graphs are the quantification of ten cilia for each strain indicated; error bars are median absolute deviation and dots indicate P-values (small: P < 0.05, medium: P < 0.01, large: P < 0.001, Kruskal–Wallis test). In the mks-5 mutant, PIP2 ectopic ciliary localisation is observed simultaneously with TRAM-1 leaking into the cilium, as well as MKSR-2 delocalisation, as previously reported (Williams et al, 2011). Scale bars: 4 μm.
Since DiI could not be used to probe the gating function of the TZ, we chose to test a reporter for the phosphoinositide PI(4,5)P2 (abbreviated PIP2). In Trypanosoma brucei, PIP2 is found in the flagellar pocket region and excluded from flagella (Demmel et al, 2014); in C. elegans, PIP2 is implicated in ciliary targeting of receptor proteins (Bae et al, 2009). We used a reporter specific for PIP2 (PH domain of PLCδ1 fused to GFP), expressed in different cell types: one, under the control of a pan-ciliated-neuron promoter (bbs-8), and the other, under control of a male-cilia-specific promoter (pkd-2) (Bednarek et al, 2007; Bae et al, 2009). We found PLCδ1-PH-GFP to be highly concentrated near the ciliary base (Fig6B and C). To assess the specific localisation of this PIP2-enriched membrane region, we co-expressed MKSR-2::tdTomato (TZ-localised), XBX-1::tdTomato (basal body- and whole cilium-localised), or TRAM-1::tdTomato (periciliary membrane-localised) together with the PIP2 reporter.
In wild-type animals, PLCδ1-PH-GFP is most strongly localised at the periciliary membrane compartment (PCMC) and displays some localisation along distal dendrites. The PCMC in C. elegans is analogous to the ciliary pocket in mammals and the flagellar pocket in T. brucei (Molla-Herman et al, 2010; Demmel et al, 2014). Using ciliary co-markers, we observe that PIP2 extends into the basal body/transition fibre region, but decreases through the TZ, and is largely excluded from the cilium (Fig6B and C). In contrast, disruption of the TZ in the mks-5 mutant results in prominent PIP2 enrichment along the entire length of the cilium, in addition to its localisation at the PCMC and basal body (Fig6B and C). Together, these findings reveal a specific lipid enrichment of PIP2 in the PCMC and basal body region, which decreases progressively at the TZ and is largely excluded from the more distal ciliary region; MKS-5 and perhaps more generally, the TZ, is essential in maintaining this PIP2 distribution.
Discussion
In this study, we demonstrate that C. elegans MKS-5 (mammalian Rpgrip1L/Rpgrip1 orthologue) plays an essential role in building a ciliary transition zone (TZ) that compartmentalises signalling proteins and acts to exclude the phosphoinositide PIP2 from the cilium. MKS-5 is required for the correct localisation of all known C. elegans TZ proteins—including NPHP-1, NPHP-4, and two others not previously characterised in the nematode (TMEM-17 and TMEM-138); moreover, MKS-5 is essential for the formation of TZ ultrastructure, including Y-shaped axoneme-to-membrane connectors (Fig7A). We also show that MKS-5, possibly via other TZ components, establishes a CIZE that compartmentalises signalling proteins distally within the axoneme and excludes the PIP2 from the cilium, which is normally enriched in the periciliary membrane (Fig7A and D).
Figure 7.

- Summary depicting role of MKS-5 in forming a TZ that helps limit ciliary PIP2 concentration and compartmentalises signalling to the distal ciliary region. MKS and NPHP module components are shown in yellow and green, respectively, and MKS-5 in red. Inset shows organisational hierarchy of TZ proteins, with MKS-5 playing a central role in assembling two distinct modules (MKS and NPHP); arrows indicate requirement for particular TZ proteins in localising other(s) to the TZ (e.g. MKS-5 is required for correct NPHP-4 localisation), and white circles on arrowheads denote new functional interactions uncovered in this study.
- Model to explain the redundant functions of the MKS and NPHP modules in basal body docking, formation of the TZ, and sealing of the ciliary compartment. MKS module components operate at the TZ, whereas NPHP module proteins act at the TZ and basal body–transition fibres. Loss of either module impairs the ciliary gate (Williams et al, 2011) but does not severely impact TZ formation or ciliogenesis; disruption of both affects the “seal” formed between the transition fibres/TZ and ciliary membrane.
- Two models for the role of MKS-5 in TZ assembly during ciliogenesis. Different stages shown are separated by arrows and lead to a mature (fully formed) TZ at the bottom. Left: MKS-5 acts as a structural anchor (“scaffold”) for MKS/NPHP module components to build TZ ultrastructure (including Y-links) constructed from MKS and NPHP module components. In this model, MKS-5 is a scaffold at each TZ Y-link structure (green). Right: MKS-5 acts as an assembly factor (“chaperone”) in assembling MKS/NPHP module components. In this model, MKS-5 borders the forming TZ as it assists in the assembly of the Y-links using MKS/NPHP module components.
- Distinct lipid domains help establish a CIZE at the TZ, and compartmentalise signalling proteins within the ciliary organelle. The phosphoinositide PI(4,5)P2 (PIP2) is enriched at the periciliary membrane and displays much reduced concentrations towards the distal ciliary region. Likely diffusion barriers are shown as orange double-headed arrows. The lipophilic dye DiI is excluded from the distal segment (DS) consisting of singlet microtubules (MTs), defining another lipid environment. The so-called inversin (inv) compartment, which contains inversin and other signalling proteins, represents the region between the two likely different lipid domains, at the TZ and DS.
- Potential role of PIP2 in the trafficking of ciliary GPCRs. Interactions between GPCRs and IFT machinery (via TULP3; Mukhopadhyay et al, 2010) are stabilised by the high concentration of PIP2 at the periciliary membrane–basal body region (the site of IFT machinery assembly), and weakened distal to this domain, causing cargo release and retention within the cilium by the CIZE. The ciliary inositol polyphosphate 5-phosphatases INPP5E and OCRL1 may participate in reducing PIP2 concentrations by hydrolysing PIP2 to PI4P within the cilium. The anterograde IFT machinery is shown moving from its site of docking/assembly through the TZ and into the distal regions of the cilium (retrograde transport is not shown).
Importantly, the CIZE likely exists in mammalian cilia. For example, the signalling proteins adenylate cyclase 3, polycystin-2/PKD2 (TRP cation channel), Hedgehog-associated smoothened GPCR, and somatostatin receptor 3 all appear to localise distally to the TZ (Garcia-Gonzalo et al, 2011; Chih et al, 2012). The localisation of another mammalian ciliary protein, Arl13b, was recently shown to not overlap with Rpgrip1L (Cevik et al, 2013). The same holds true for rhodopsin, which is excluded from the photoreceptor TZ (i.e. “connecting cilium”) and concentrated in the distal region of the axoneme (outer segment; see, e.g. Keady et al, 2011).
Assessing whether abrogation of mammalian Mks5/Rpgrip1L disrupts the TZ and causes leakage of signalling proteins into the TZ (CIZE), as well as in and/or out of cilia, will be important; however, one will need to bear in mind that mammals possess two related proteins, Rpgrip1 and Rpgrip1L, with likely redundant/overlapping roles (Arts et al, 2007; Delous et al, 2007; Coene et al, 2011). Loss of Rpgrip1 and Rpgrip1L both result in retinal ciliopathies, although disrupting Rpgrip1L causes more severe, multi-systemic ciliopathies (Khanna et al, 2009; Coene et al, 2011). Nevertheless, loss of Rpgrip1 results in “ruffling” of the membrane connections at the photoreceptor connecting cilium (TZ), as well as partial loss of Y-links (Patil et al, 2012)—both phenotypes being reminiscent of what we observe in the C. elegans mks-5 mutant (partial disconnection of the membrane and axoneme) and MKS-NPHP double TZ mutants (complete disconnection) (Figs2, EV3 and EV4; Williams et al, 2011). Further evidence of a functional redundancy between Rpgrip1L and Rpgrip1 is that both proteins bind NPHP4 at their C2-C domain (Roepman et al, 2005; Arts et al, 2007); the same domain we show in C. elegans MKS-5 is required for proper TZ localisation of NPHP-1, whose localisation itself depends on NPHP-4 (Fig4B). Furthermore, just as C. elegans NPHP-4 is missing from the TZ in mks-5 mutants (Fig1D); Patil et al (2012) observed loss of NPHP4 from Rpgrip1 mutant photoreceptor cilia. Finally, Rpgrip1L also has many ciliopathy-modifying alleles, including a heterozygous allele in a patient with Leber's congenital amaurosis homozygous for a premature stop in Rpgrip1 (Khanna et al, 2009). Hence, we hypothesise that removing the sole Rpgrip1/Rpgrip1L-related protein in C. elegans (MKS-5) models the disruption of both Rpgrip1 and Rpgrip1L, where we would predict a complete loss of TZ ultrastructure and function in mammalian cilia.
It is unclear how the TZ establishes a CIZE that acts as a membrane diffusion barrier (“ciliary gate”) which regulates the exit, and entry, of ciliary signalling proteins (Craige et al, 2010; Williams et al, 2011; Garcia-Gonzalo et al, 2011; Chih et al, 2012; Reiter et al, 2012; Garcia-Gonzalo & Reiter, 2012; Roberson et al, 2015; Fig7A and D). Our finding that the phosphoinositide PIP2 is largely excluded from cilia through the function of MKS-5 provides clues regarding a potential functional mechanism. Specifically, the TZ may harbour a specialised lipid domain that restricts the free diffusion of membrane-associated proteins and lipids such as PIP2. This could explain our observation of a CIZE devoid of transmembrane proteins (GPCRs, CNG channel) as well as a lipidated protein, ARL-13, observed by Cevik et al (2013) to be excluded from the TZ in both C. elegans and mammalian cilia. Indeed, ARL-13 freely diffuses within the distal part of the axoneme, as expected, but not across the TZ and into the periciliary membrane—unless TZ ultrastructure is missing (i.e. in the mks-5 and mks-2;nphp-4 mutants) (Cevik et al, 2013). Such a membrane/lipid diffusion barrier could work hand-in-hand with the septin barrier or gate that exists at the base of cilia, in proximity to the TZ (Hu et al, 2010). Indeed, Chih et al (2012) presented evidence for such a possibility.
Previous studies have implicated PIP2 phosphoinositides in association with cilia. These include deflagellation in Chlamydomonas (Quarmby et al, 1992), localisation of two PIP2 5′ phosphatases (OCRL1 and INPP5E/JBTS1) to cilia (Jacoby et al, 2009; Coon et al, 2012), and detection of PIP2 in Paramecium as well as scallop cilia (Stommel & Stephens, 1985; Suchard et al, 1989). Using MKSR-2, TRAM-1, and XBX-1 as co-markers, we show that in C. elegans, PIP2 is enriched at the periciliary membrane and basal body region (Fig6B and C), similar to that observed in trypanosomes (Demmel et al, 2014). The function of PIP2 may be structural, for instance by creating a specific membrane composition and binding a specific C2 domain (Evans et al, 2006), and/or part of a signalling cascade as was shown for Chlamydomonas deflagellation where PIP2 is catalysed to IP3 and DAG in the promotion of deflagellation just distal to the TZ (Quarmby et al, 1992).
Another intriguing possibility is that PIP2 functions specifically at the ciliary base in membrane protein transport. This hypothesis is based on the recent finding that TULP3, which is associated with the intraflagellar transport (IFT) subcomplex A, requires PIP2 to transport GPCRs to cilia (Mukhopadhyay et al, 2010). Given that PIP2 is enriched near the basal body, where the IFT complex is assembled (Deane et al, 2001; Williams et al, 2011), PIP2-associated cargo within the IFT “train” would be stabilised/retained and then, upon encountering the more distal region of the axoneme lacking (or with reduced) PIP2, would be released (Fig7E). Although Mukhopadhyay and colleagues speculate that PIP2 is hydrolysed to IP3 and DAG in this process, it is also possible that the cilium-localised INPP5E 5-phosphatase—which catalyses PIP2 to PI4P and whose loss of function phenocopies mutations in other TZ genes (causative in Joubert syndrome and related disorders)—could be part of the mechanism regulating PIP2 abundance within the cilium. Another cilium-localised 5-phosphatase ciliopathy protein, OCRL1 (and its paralogue INPP5B), could play a role similar or complementary to that of INPP5E in breaking down PIP2 (Coon et al, 2012; Luo et al, 2012, 2013). The mobilisation of cargo across the TZ by IFT might be further regulated by direct interactions between IFT proteins and specific TZ components (Boldt et al, 2011; Zhao & Malicki, 2011; Reiter et al, 2012). Our finding of an MKS-5-dependent reduced IFT velocity within the TZ relative to the more distal portion of the axoneme (Fig EV2) may be consistent with such TZ–IFT protein interactions. Alternatively, or in addition, a different lipid composition coincident with the CIZE within the TZ may help explain this effect on the membrane-associated IFT particles.
Intriguingly, there appears to be a strong connection between regulation of PIP2 and Joubert syndrome. As discussed above, loss of any one of many TZ proteins (including MKS5/RPGRIP1L) results in Joubert syndrome, as does loss of the PIP2 phosphatase INPP5E or ARL13B/arl-13, which is required for proper ciliary localisation of INPP5E (Cantagrel et al, 2008; Bielas et al, 2009; Humbert et al, 2012). Furthermore, INPP5E ciliary localisation itself depends on at least one TZ protein, TMEM231 (Roberson et al, 2015). Given the importance of PIP2 in ciliary receptor trafficking (Mukhopadhyay et al, 2010), ciliary signalling (Quarmby et al, 1992), and roles in membrane shape/structure/connection to the cytoskeleton (Czech, 2000), it seems likely that loss in the regulation/distribution of PIP2 plays a central role in ciliopathies such as Joubert syndrome.
In addition to extending the functional localisation/interaction network or “hierarchy” of TZ proteins (Fig7C) and providing insights into possible ciliary transport mechanisms, our findings also help elucidate the role of MKS/NPHP-associated proteins in ciliogenesis. Our previous model ascribed redundant roles for MKS and NPHP module proteins in basal body docking at the membrane, prior to or coincident with IFT-dependent axoneme extension (Williams et al, 2011; Reiter et al, 2012). Our new findings provide a plausible molecular explanation for this functional redundancy, first described by Williams et al (2008) and supported in Williams et al (2011) (Fig7B).
In brief, we show that NPHP-1 and NPHP-4 localise to and function at two distinct regions that make membrane connections: the basal body-associated transition fibres and TZ (Fig1E). Disrupting the NPHP module does not impair basal body docking to the membrane (Jauregui et al, 2008), likely because the TZ (still containing MKS module proteins) provides a sufficiently strong membrane anchor that is known to form (Reiter et al, 2012) in early ciliogenesis. Disrupting the MKS module (i.e. in single TZ mutants) also permits basal body anchoring (Williams et al, 2011) (Figs EV3 and EV4), ostensibly because the NPHP module acts at the transition fibres and TZ to stabilise basal body–membrane connections. Complete removal of the TZ (as in the mks-5 mutant) corroborates these findings; although both MKS and NPHP modules are lost from the TZ, NPHP-1/NPHP-4 remains at transition fibres, likely promoting the basal body anchoring to the membrane we observe in the mks-5 mutant (Fig7B). It is notable that a comparable scenario is described for the mammalian Rpgrip1 mutant, where the Y-links appear to be missing from the distal TZ/connecting cilium, causing disconnection or “ruffling” of the TZ membrane, but the basal body is properly anchored (Patil et al, 2012). Thus, our model describing the functional redundancy between MKS and NPHP module proteins (Fig7B), organised by MKS-5 (Rpgrip1L/Rpgrip1), is likely relevant to mammalian ciliogenesis, and ciliary gate formation.
A surprising discovery made during the course of our studies is that in all TZ mutants, the localisation of MKS-5 is altered from the obloid shape in wild type of ∼0.7 μm in length to two distinct puncta spanning a length of ∼1 μm (Fig3), something not observed for numerous other TZ proteins (Fig1). This finding, which implies that MKS-5 does not necessarily co-localise with other TZ proteins, leads us to question whether MKS-5 is strictly a scaffold that is part of the Y-links, and hence acts as a structural anchor for other TZ proteins. The loss of Y-links we observe in an mks-2;nphp-4 double mutant (Fig EV4), which occurs despite MKS-5 still localising to the TZ (Fig3A), is also consistent with the notion that MKS-5 is not a major structural component of the Y-links. The dual localisation pattern of MKS-5 may not be observed by conventional confocal microscopy in wild-type animals due to the lack of resolving power, but can be gleaned in a TZ mutant where the TZ is significantly longer (Fig3A). Immunogold staining of Rpgrip1 supports this hypothesis, since the protein localises not along the length but to both ends of the connecting cilium (TZ) in photoreceptor cilia (Patil et al, 2012). Based on these findings, we propose a potential new model for MKS-5 protein function, namely that it acts not as a scaffold for TZ proteins, but rather, as an assembly factor (Fig7). Whether MKS-5 operates as a “molecular chaperone” (meaning it is not part of the final structure, e.g. Y-links) will be difficult to ascertain biochemically, although additional localisation experiments by immunogold staining and super-resolution microscopy should help clarify the question of whether MKS-5/Rpgrip1/Rpgrip1L is part of the Y-link assembly or not.
In conclusion, our work provides novel insights into the role of C. elegans MKS-5 in forming a functional TZ membrane diffusion barrier that compartmentalises signalling proteins within cilia. Further molecular analyses of Rpgrip1L and Rpgrip1 should help to reveal comparable roles for the mammalian proteins in TZ biogenesis and function, with important implications for understanding signal transduction pathways (e.g. Hedgehog signalling) and vertebrate development. Our studies also bring into focus the function of MKS-5 and the TZ in excluding PIP2 from cilia, potentially acting as a lipid “gate” that likely has important implications for the trafficking of ciliary proteins. Our findings also help link, in a general manner, the TZ and PIP2 distribution to ciliopathies such as Joubert syndrome.
Materials and Methods
Caenorhabditis elegans strain construction and imaging
All strains were maintained and cultured as previously described (Brenner, 1974). Strains with the following mutations were obtained from the C. elegans Gene Knockout Consortium (http://celeganskoconsortium.omrf.org/), the National Bioresource Project (http://shigen.lab.nig.ac.jp/c.elegans/index.jsp) and the Caenorhabditis Genetics Center (www.cbs.umn.edu/CGC/) and outcrossed to wild type (N2) a minimum of four times: tmem-17(gk177766) (Q23stop), tmem-138(tm5624) (331-bp deletion plus 12-bp insertion, which deletes the first 45 codons and part of the upstream regulatory region), mksr-1(ok2092), mksr-2(tm2452),mks-5(tm3100),mks-3(tm2547), mks-6(gk674), jbts-14(gk287997),nphp-1(ok500), and nphp-4(tm925). mks-2(nx111) was previously reported as a deletion of the entire coding region (Huang et al, 2011). GFP-tagged protein constructs were introduced into listed genetic backgrounds by standard C. elegans mating methods. Genotyping of the strains generated was performed by single-worm PCR. GFP-tagged TMEM-17, and TMEM-138 translational reporters were constructed by including the native promoter, all the introns and exons fused in-frame to EGFP. The ppkd-2::PLCδ-PH::GFP was previously reported (Bae et al, 2009), and the pbbs-8::PLCδ-PH::GFP was generated by adding the 341-bp upstream sequence of bbs-8 to the coding sequence of the PLCδ-PH::GFP and UTR previously reported (Bednarek et al, 2007). Transgenic C. elegans strains were created for the reporters, and subcellular localisation by fluorescence microscopy was ascertained as described (Inglis et al, 2009). A minimum of 50 individual worms were analysed for each strain displaying a mislocalisation phenotype. Strains were anaesthetised with 10 mM levamisole in M9 buffer, mounted slides with 2% agarose pads, and observed by epifluorescence or spinning-disc (WaveFX; Quorum Technologies) confocal microscopy performed on an Zeiss Axio Observer Z1 with a Hamamatsu 9100 EMCCD camera. Image capture and visualisation were performed on Volocity (PerkinElmer).
Caenorhabditis elegans phenotypic assays for ciliary structure, chemosensation, and dauer pheromone sensation
Defective osmotic avoidance was assayed as described using five individuals per ring of 8 M glycerol for 10 min. Externally exposed ciliated sensory neurons were filled with DiI as described previously (Inglis et al, 2009). Pheromone sensation assays were performed as described using ASCR#2 (Kim et al, 2009). The length of NPHP-2::GFP signals in wild-type and mks-5 mutants was measured using Volocity software.
Transmission electron microscopy (TEM)
TEM analyses were performed on young adult worms as previously described (Williams et al, 2011).
mks-5 missense allele screen
Specific missense mutations were selected based on the amino acid change (non-conservative) and location to predicted protein domains. The alleles selected, namely mks-5(gk153546),mks-5(gk153548),mks-5(gk153549),mks-5(gk153554),mks-5(gk153556),mks-5(gk153560),mks-5(gk153561), and mks-5(gk153565), were obtained from the C. elegans Gene Knockout Consortium. All eight alleles were crossed to nphp-4(tm925) heterozygous males and mksr-1(ok2092) hemizygous males. F1 progeny were singly cloned, allowed to lay eggs, and PCR-tested for either the nphp-4 or mksr-1 mutations. Those heterozygous for nphp-4 or mksr-1 were selected, and their progeny were dye-filled with DiI as noted above. F2 progeny that were Dye-filling (Dyf) defective were singly cloned, and their progeny (F3) were tested for Dyf phenotypes. Only those that gave 100% Dyf progeny in the F3 and F4 generations were kept and genotyped. Only mks-5(gk153561) crossed with nphp-4 gave inherited Dyf phenotypes, and four separate F2 progeny were found to have a persistent Dyf phenotype in the F3 and F4 generations. All four proved to be homozygous for both mks-5(gk153561) and nphp-4(tm925) by single-worm PCR. The rescue strain was created by transforming the mks-5(gk153561);nphp-4(tm925) double mutant with the MKS-5::tdTomato translational reporter.
MKS-5 truncation analyses
Truncated, fluorescently tagged MKS-5 constructs were created by PCR stitch on the previously reported MKS-5::tdTomato construct (Williams et al, 2011), connecting the pre-truncated PCR product, including the native mks-5 promoter, to the post-truncation sequences, including tdTomato and unc-54 3′ UTR. Truncations include deletion of the following amino acids: 2–241 (removes the first four predicted coiled-coils), 2–566 (removes all eight coiled-coils), 878–1306 (removes the RID), 697–1306 (removes the C2-C and RID), and 565–1306 (removes C2-N, C2-C, and the RID). Transgenic worms were created using the truncation constructs co-transformed with the specified ciliary co-markers.
Fluorescence intensity, transition zone length, and dual localisation measurements
Fluorescence intensity and length of the MKS-5::tdTomato signals were measured using the program Volocity and drawing a line longitudinally through the TZ or periciliary membrane compartment (PCMC) to MS for PIP2 comparisons. Images were excluded if any pixels were saturated. Length was calculated from the proximal to the distal pixels that were higher than 50% of the maximum intensity for each individual TZ. To determine the number of TZs with dual localisation, the intensity profiles were inspected for number of local maxima.
Acknowledgments
We thank Don Moerman (University of British Columbia), Robert Waterston (University of Washington), and the Caenorhabditis Genetics Center (CGC) for providing Million Mutation Project (MMP) and other valuable strains. This work was funded by the Canadian Institutes of Health Research (CIHR grant MOP142243; to MRL) and March of Dimes (to MRL), Seventh Framework Programme FP7/2009 (SYSCILIA grant agreement 241955 to OEB), and Science Foundation Ireland (Principal Investigator award 11/PI/1037 to OEB). MRL and VLJ hold Michael Smith Foundation for Health Research (MSFHR) senior scholar and postdoctoral awards, respectively. VLJ also holds a KRESCENT postdoctoral award.
Author contributions
VLJ performed experiments and co-wrote the manuscript. CL, RVB, LC, and SM performed experiments. OEB and MRL supervised and co-wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Expanded View Figures PDF
Review Process File
References
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Awata J, Takada S, Standley C, Lechtreck KF, Bellvé KD, Pazour GJ, Fogarty KE, Witman GB. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci. 2014;127:4714–4727. doi: 10.1242/jcs.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YK, Kim E, L'Hernault SW, Barr MM. The CIL-1 PI 5-phosphatase localizes TRP Polycystins to cilia and activates sperm in C. elegans. Curr Biol. 2009;19:1599–1607. doi: 10.1016/j.cub.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- Bednarek EM, Schaheen L, Gaubatz J, Jorgensen EM, Fares H. The plasma membrane calcium ATPase MCA-3 is required for clathrin-mediated endocytosis in scavenger cells of Caenorhabditis elegans. Traffic. 2007;8:543–553. doi: 10.1111/j.1600-0854.2007.00547.x. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. Centrosomes and cilia in human disease. Trends Genet. 2011;27:307–315. doi: 10.1016/j.tig.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, Badano JL, Mah AK, Beales PL, Davidson WS, Johnsen RC, Audeh M, Plasterk RH, Baillie DL, Katsanis N, Quarmby LM, Wicks SR, Leroux MR. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004;18:1630–1642. doi: 10.1101/gad.1194004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Perens EA, Boroevich KA, Inglis PN, Li C, Warner A, Khattra J, Holt RA, Ou G, Mah AK, McKay SJ, Huang P, Swoboda P, Jones SJ, Marra MA, Baillie DL, Moerman DG, Shaham S, Leroux MR. Functional genomics of the cilium, a sensory organelle. Curr Biol. 2005;15:935–941. doi: 10.1016/j.cub.2005.04.059. [DOI] [PubMed] [Google Scholar]
- Boldt K, Mans DA, Won J, van Reeuwijk J, Vogt A, Kinkl N, Letteboer SJ, Hicks WL, Hurd RE, Naggert JK, Texier Y, den Hollander AI, Koenekoop RK, Bennett J, Cremers FP, Gloeckner CJ, Nishina PM, Roepman R, Ueffing M. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest. 2011;121:2169–2180. doi: 10.1172/JCI45627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik S, Sanders AAWM, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, Hori Y, Horn N, Hetterschijt L, Wdowicz A, Mullins A, Kida K, Kaplan OI, van Beersum SEC, Wu KM, Letteboer SJF, Mans DA, Katada T, Kontani K, Ueffing M, et al. Active transport and diffusion barriers restrict joubert syndrome-associated ARL13B/ARL-13 to an inv-like ciliary membrane subdomain. PLoS Genet. 2013;9:e1003977. doi: 10.1371/journal.pgen.1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Mah A, Blacque OE, Chu J, Phgora K, Bakhoum MW, Newbury CR, Khattra J, Chan S, Go A, Efimenko E, Johnsen R, Phirke P, Swoboda P, Marra M, Moerman DG, Leroux MR, Baillie DL, Stein LD. Identification of ciliary and ciliopathy genes in Caenorhabditis elegans through comparative genomics. Genome Biol. 2006;7:R126. doi: 10.1186/gb-2006-7-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol. 2012;226:172–184. doi: 10.1002/path.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn CM, Bargmann CI. A putative cyclic nucleotide-gated channel is required for sensory development and function in C. elegans. Neuron. 1996;17:695–706. doi: 10.1016/s0896-6273(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Coene KL, Mans DA, Boldt K, Gloeckner CJ, van Reeuwijk J, Bolat E, Roosing S, Letteboer SJ, Peters TA, Cremers FP, Ueffing M, Roepman R. The ciliopathy-associated protein homologs RPGRIP1 and RPGRIP1L are linked to cilium integrity through interaction with Nek4 serine/threonine kinase. Hum Mol Genet. 2011;20:3592–3605. doi: 10.1093/hmg/ddr280. [DOI] [PubMed] [Google Scholar]
- Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, Lowe M, Beales PL, Aguilar RC. The lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet. 2012;21:1835–1847. doi: 10.1093/hmg/ddr615. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol. 2001;11:1586–1590. doi: 10.1016/s0960-9822(01)00484-5. [DOI] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Demmel L, Schmidt K, Lucast L, Havlicek K, Zankel A, Koestler T, Reithofer V, de Camilli P, Warren G. The endocytic activity of the flagellar pocket in Trypanosoma brucei is regulated by an adjacent phosphatidylinositol phosphate kinase. J Cell Sci. 2014;127:2351–2364. doi: 10.1242/jcs.146894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond IA. Cilia functions in development. Curr Opin Cell Biol. 2012;24:24–30. doi: 10.1016/j.ceb.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmer BT, Maric D, Engman DM. Molecular mechanisms of protein and lipid targeting to ciliary membranes. J Cell Sci. 2010;123(Pt 4):529–536. doi: 10.1242/jcs.062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JH, Murray D, Leslie CC, Falke JJ. Specific translocation of protein kinase Calpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol Biol Cell. 2006;17:56–66. [Google Scholar]
- Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella – back to the future! Biol Cell. 2011;103:249–270. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol. 2012;197:697–709. doi: 10.1083/jcb.201111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. J Cell Biol. 1972;53:494–509. doi: 10.1083/jcb.53.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Thomson JN, Perkins LA. Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev Biol. 1985;111:158–170. doi: 10.1016/0012-1606(85)90443-9. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, Greenberg CR, McLeod DR, Bernier FP, Chudley AE, Muller T, Shboul M, Logan CV, Loucks CM, Beaulieu CL, Bowie RV, et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci USA. 2012;109:19691–19696. doi: 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis PN, Blacque OE, Leroux MR. Functional genomics of intraflagellar transport-associated proteins in C. elegans. Methods Cell Biol. 2009;93:267–304. doi: 10.1016/S0091-679X(08)93014-4. [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, Gergely F, Riley JH, Pérez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- Jauregui AR, Nguyen KC, Hall DH, Barr MM. The Caenorhabditis elegans nephrocystins act as global modifiers of cilium structure. J Cell Biol. 2008;180:973–988. doi: 10.1083/jcb.200707090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Leroux MR. cAMP and cGMP signaling: sensory systems with prokaryotic roots adopted by eukaryotic cilia. Trends Cell Biol. 2010;20:435–444. doi: 10.1016/j.tcb.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Keady BT, Le YZ, Pazour GJ. IFT20 is required for opsin trafficking and photoreceptor outer segment development. Mol Biol Cell. 2011;22:921–930. doi: 10.1091/mbc.E10-09-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, Macdonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Sato K, Shibuya M, Zeiger DM, Butcher RA, Ragains JR, Clardy J, Touhara K, Sengupta P. Two chemoreceptors mediate developmental effects of dauer pheromone in C. elegans. Science. 2009;326:994–998. doi: 10.1126/science.1176331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H, Mori I, Rhee JS, Akaike N, Ohshima Y. Mutations in a cyclic nucleotide-gated channel lead to abnormal thermosensation and chemosensation in C. elegans. Neuron. 1996;17:707–718. doi: 10.1016/s0896-6273(00)80202-0. [DOI] [PubMed] [Google Scholar]
- Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Silhavy JL, Lee JE, Al-Gazali L, Thomas S, Davis EE, Bielas SL, Hill KJ, Iannicelli M, Brancati F, Gabriel SB, Russ C, Logan CV, Sharif SM, Bennett CP, Abe M, Hildebrandt F, Diplas BH, Attie-Bitach T, Katsanis N, et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science. 2012;335:966–969. doi: 10.1126/science.1213506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang M, Xia Z, Xu P, Chen L, Xu T. Caenorhabditis elegans ciliary protein NPHP-8, the homologue of human RPGRIP1L, is required for ciliogenesis and chemosensation. Biochem Biophys Res Commun. 2011;410:626–631. doi: 10.1016/j.bbrc.2011.06.041. [DOI] [PubMed] [Google Scholar]
- Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD, Weinreb RN, Travers JB, Khanna H, Sun Y. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet. 2012;21:3333–3344. doi: 10.1093/hmg/dds163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N, Kumar A, Conwell M, Weinreb RN, Anderson R, Sun Y. Compensatory role of inositol 5-phosphatase INPP5B to OCRL in primary cilia formation in oculocerebrorenal syndrome of Lowe. PLoS ONE. 2013;8:e66727. doi: 10.1371/journal.pone.0066727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Pokala N, Feinberg EH, Chalasani SH, Butcher RA, Clardy J, Bargmann CI. A hub-and-spoke circuit drives pheromone attraction and social behaviour in C. elegans. Nature. 2009;458:1171–1175. doi: 10.1038/nature07886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, Emmerson C, Romeo K, Bourdoncle P, Schmitt A, Saunier S, Spassky N, Bastin P, Benmerah A. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci. 2010;123:1785–1795. doi: 10.1242/jcs.059519. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24:2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Novarino G, Akizu N, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011;147:70–79. doi: 10.1016/j.cell.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh EC, Katsanis N. Cilia in vertebrate development and disease. Development. 2012;139:443–448. doi: 10.1242/dev.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil H, Tserentsoodol N, Saha A, Hao Y, Webb M, Ferreira PA. Selective loss of RPGRIP1-dependent ciliary targeting of NPHP4, RPGR and SDCCAG8 underlies the degeneration of photoreceptor neurons. Cell Death Dis. 2012;3:e355. doi: 10.1038/cddis.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Bloodgood RA. Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008;85:115–149. doi: 10.1016/S0070-2153(08)00805-3. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- Perkins LA, Hedgecock EM, Thomson JN, Culotti JG. Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev Biol. 1986;117:456–487. doi: 10.1016/0012-1606(86)90314-3. [DOI] [PubMed] [Google Scholar]
- Quarmby LM, Yueh YG, Cheshire JL, Keller LR, Snell WJ, Crain RC. Inositol phospholipid metabolism may trigger flagellar excision in Chlamydomonas reinhardtii. J Cell Biol. 1992;116:737–744. doi: 10.1083/jcb.116.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson EC, Dowdle WE, Ozanturk A, Garcia-Gonzalo FR, Li C, Halbritter J, Elkhartoufi N, Porath JD, Cope H, Ashley-Koch A, Gregory S, Thomas S, Sayer JA, Saunier S, Otto EA, Katsanis N, Davis EE, Attié-Bitach T, Hildebrandt F, Leroux MR, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209:129–142. doi: 10.1083/jcb.201411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman R, Letteboer SJ, Arts HH, van Beersum SE, Lu X, Krieger E, Ferreira PA, Cremers FP. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc Natl Acad Sci USA. 2005;102:18520–18525. doi: 10.1073/pnas.0505774102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O'Toole JF, Held S, Reutter HM, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Chou JH, Bargmann CI. odr-10 encodes a seven transmembrane domain olfactory receptor required for responses to the odorant diacetyl. Cell. 1996;84:899–909. doi: 10.1016/s0092-8674(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Starich TA, Herman RK, Kari CK, Yeh WH, Schackwitz WS, Schuyler MW, Collet J, Thomas JH, Riddle DL. Mutations affecting the chemosensory neurons of Caenorhabditis elegans. Genetics. 1995;139:171–188. doi: 10.1093/genetics/139.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel EW, Stephens RE. Calcium-dependent phosphatidylinositol phosphorylation in lamellibranch gill lateral cilia. J Comp Physiol. 1985;157:441–449. doi: 10.1007/BF00615144. [DOI] [PubMed] [Google Scholar]
- Suchard SJ, Rhoads DE, Kaneshiro ES. The inositol lipids of Paramecium tetraurelia and preliminary characterizations of phosphoinositide kinase activity in the ciliary membrane. J Protozool. 1989;36:185–190. doi: 10.1111/j.1550-7408.1989.tb01072.x. [DOI] [PubMed] [Google Scholar]
- Thomas S, Legendre M, Saunier S, Bessières B, Alby C, Bonnière M, Toutain A, Loeuillet L, Szymanska K, Jossic F, Gaillard D, Yacoubi MT, Mougou-Zerelli S, David A, Barthez MA, Ville Y, Bole-Feysot C, Nitschke P, Lyonnet S, Munnich A, et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am J Hum Genet. 2012;91:372–378. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimble WS, Grinstein S. Barriers to the free diffusion of proteins and lipids in the plasma membrane. J Cell Biol. 2015;208:259–271. doi: 10.1083/jcb.201410071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, Adams M, Szymanska K, Mazzotta A, Lee JE, Tolentino JC, Swistun D, Salpietro CD, Fede C, Gabriel S, Russ C, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton-Pitt SR, Jauregui AR, Li C, Wang J, Leroux MR, Barr MM. Ciliogenesis in Caenorhabditis elegans requires genetic interactions between ciliary middle segment localized NPHP-2 (inversin) and transition zone-associated proteins. J Cell Sci. 2012;125:2592–2603. doi: 10.1242/jcs.095539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK. Functional redundancy of the B9 proteins and nephrocystins in C. elegans ciliogenesis. Mol Biol Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Masyukova SV, Yoder BK. Normal ciliogenesis requires synergy between the cystic kidney disease genes MKS-3 and NPHP-4. J Am Soc Nephrol. 2010;21:782–793. doi: 10.1681/ASN.2009060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, Yoder BK, Leroux MR. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelbauer ME, Schafer JC, Haycraft CJ, Swoboda P, Yoder BK. The C. elegans homologs of nephrocystin-1 and nephrocystin-4 are cilia transition zone proteins involved in chemosensory perception. J Cell Sci. 2005;118:5575–5587. doi: 10.1242/jcs.02665. [DOI] [PubMed] [Google Scholar]
- Wong SY, Reiter JF. The primary cilium at the crossroads of Mammalian hedgehog signaling. Curr Top Dev Biol. 2008;85:225–260. doi: 10.1016/S0070-2153(08)00809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Aravind L. Novel transglutaminase-like peptidase and C2 domains elucidate the structure, biogenesis and evolution of the ciliary compartment. Cell Cycle. 2012;11:3861–3875. doi: 10.4161/cc.22068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Malicki J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J. 2011;30:2532–2544. doi: 10.1038/emboj.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File