

Abstract
Background
Allopolyploid genome needs wide structural variation to deal with genomic shock. The introgression line, generated via asymmetric somatic hybridization, is introgressed with a minimum of exogenous chromatin, which also leads to genomic shock to induce genetic variation. However, the extent of its genomic variation and its difference from allopolyploidies remains unknown.
Methods
Here, we explored this issue using the bread wheat cultivar SR3, a derivative of an asymmetric somatic hybrid between the cultivar JN177 and an accession of tall wheatgrass (Thinopyrum elongatum). The ESTs (expressed sequence taqs) were large-scale sequenced using the cDNA library constructed in each of SR3 and JN177. Point mutations and indels (insertions and deletions) of SR3 were calculated, and their difference from the genetic variation of bread wheat and its ancestors were compared, with aim to analyze the extent and pattern of sequence variation induced by somatic hybridization.
Results
Both point mutations and indels (insertions and deletions) were frequently induced by somatic hybridization in the coding sequences. While the genomic shock caused by allopolyploidization tends to favor deletion over insertion, there was no evidence for such a preference following asymmetric somatic hybridization. The GC content of sequence adjacent to indel sites was also distinct from what has been observed in allopolyploids.
Conclusions
This study demonstrates that asymmetric somatic hybridization induces high frequency of genetic variation in a manner partially different from allopolipoidization. Asymmetric somatic hybridization provides appropriate material to comprehensively explore the nature of the genetic variation induced by genomic shock.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1974-6) contains supplementary material, which is available to authorized users.
Keywords: Introgression line, Allopolyploidy, Single nucleotide polymorphism, Insertion, Deletion, Genetic variation
Background
Anthropogenic selection applied during the domestication and improvement of a crop species inevitably reduces its genetic base, while its non-domesticated wild relatives retain genetic diversity. The wild relatives thus represent a valuable genetic resource for breeders. Introgressing genetic material from a wild species is conventionally attempted via sexual hybridization [1, 2], but somatic hybridization (the induced fusion of somatic cells, followed by their in vitro regeneration) can offer an alternative route, especially where viable sexual hybrids have proven difficult or impossible to establish [3]. Unlike in an allopolyploid, where the two (or more) parental nuclear genomes are combined with a uniparental cytoplasm (that of the female parent of the hybrid), in a somatic hybrid, both the nucleus and the cytoplasm are biparental. In a refinement of the approach, termed asymmetric hybridization, the donor cells are irradiated prior to the fusion process, which has the effect of reducing the contribution of the donor’s genome to the somatic hybrid, thereby promoting the introgression of smaller donor segments [4, 5].
Bread wheat (Triticum aestivum) is an allohexaploid species which has evolved through two successive natural hybridizations. The first brought together the genomes of an as yet unidentified (possibly extinct) member of the Sitopsis section of the genus Aegilops (B genome) and T. urartu (A genome) to form the allotetraploid BA genome species T. turgidum [6]. The second event involved a domesticated form of T. turgidum and Ae. tauschii (D genome) to form the bread wheat BAD genome [6]. Allopolyploidy, in forcing a pair of non-identical genomes to co-exist within a single nucleus, has been associated with a range of genomic rearrangements and DNA modifications [7, 8], encapsulated by the term “genomic shock” [9]. Some of these changes affect gene expression, either by altering gene sequences and/or by altering their epigenetic regulation [9–20], thereby providing the polyploid genome with extensive potential for novelty and plasticity [20–23]. Once a wide hybrid (whether sexual or somatic) has been established, introgression from one parental genome to the other can occur via recombination or meiosis-driven chromosome breakage and reunion. In an asymmetric somatic hybrid, the pre-hybridization irradiation treatment of the donor cells has the effect of fragmenting its genome, so introgression can occur via end-joining of fragments, most easily during the course of mitosis [24]. This event also leads to a strong genomic shock and therefore induces genomic variation. However, in asymmetric somatic fused cells, most donor chromatin is eliminated, and very small amount of chromatin fragments are introgressed into the recipient genome. Thus, chromosomal rearrangement and large fragment deletion, the common events during the process of diploidization of allopolyploidies, seldom happen in asymmetric somatic hybrid cells [3]. This difference between the chromosomal behaviors in asymmetric somatic hybrids and allopolyploidies suggest that their patterns of genomic variation may be distinct from each other.
We previously generated an asymmetric somatic hybrid between the bread wheat cultivar JN177 (with modest salt tolerance) and tall wheatgrass (Thinopyrum elongatum) (wheat’s close relative, one of the monocots with highest salt tolerance) via fusing the wheat protoplast and UV-irradiated tall wheatgrass protoplast (chromatin was fragmented), and a number of derivatives of this hybrid have been selected based on the expression of a favorable phenotype [3, 5, 25, 26]. Some of these lines have proven to be stably introgressed with several (5 ~ 7) observed chromatin fragments of tall wheatgrass by cytogenetic analysis [27], and be sufficiently phenotypically stable to allow their release as a novel cultivar [25, 26]. One of these is the cultivar SR3 whose genome possesses five observed fragments of tall wheatgrass [27]. It has demonstrated improved levels of tolerance to both salinity and drought, and higher yield in saline land than its parent JN177 [28, 29]; the tolerance is partially due to the high tolerance to oxidative stress and superior ability of redox homeostasis maintenance [28, 30]. We found that the genome of SR3 and other derivatives occurred high frequency of genetic variation using a set of molecular biomarker assays [31], and had numerous single nucleotide polymorphisms (SNPs) and insertion/deletion (indels) when comparing genomic sequences of glutenin gene family [32]. Moreover, such genetic variation was majorly induced by asymmetric somatic hybridization, while the contribution of other factors such as protoplast formation, UV radiation, callus induction and plant regeneration was quite slight [31, 32]. However, the characteristics of sequence variation such as nucleotide substitution and indel at the genome-scale level as well as its difference from polyploidization have not been addressed. Although contrasting allelic sequences have been identified between SR3 and JN177 associated mostly with genes involved in the response to abiotic stress [30, 33, 34], a more global picture of the alterations induced by asymmetric somatic hybridization awaits the application of genome-wide re-sequencing and transcriptomic analysis. While global genomic re-sequencing remains overambitious given the large size of the wheat genome [35] along with the high content of repeat sequences (>80 %) [36], current DNA sequencing platforms are well capable of uncovering sequence variation in the transcriptome. Here, the sequencing of a large number of cDNAs isolated from both SR3 and JN177 is described and used to show the extent of the sequence variation induced by asymmetric somatic hybridization.
Results
cDNA sequencing
A total of 19,045 SR3 and 10,327 JN177 clones were sequenced, resulting in the acquisition of, respectively, 18,192 and 9770 usable sequences (Additional file 1: Table S1). The sequences resolved into 9634 unigenes (2097 contigs and 7537 singletons) from SR3, and 7107 unigenes (1207 contigs and 5900 singletons) from JN177, of which full length cDNAs were 4825 and 2975, respectively (Additional file 1: Table S1). The length of most of the unigenes laid in the range 700–1000 nt (Additional file 2: Figure S1), and their mean GC content was 53.85 % (SR3) and 55.46 % (JN177). The BLASTn analysis of the unigenes revealed that 2581 were shared (>96 % identity) between SR3 and JN177 (Table 1). In all, 5072 (71.4 %) of the JN177 and 7284 (75.6 %) of the SR3 unigenes shared >96 % identity with sequences represented in the wheat EST database (Table 1).
Table 1.
BLASTn-based homology comparisons of unigene sequences
| Alignment | Total unigenes | Matched unigenes | ||
|---|---|---|---|---|
| Total | >96 % identity | >96 % identity coverage (%) | ||
| SR3-JN177 | 9635 | 4677 | 2581 | 75.25 |
| SR3-Ta | 9635 | 9097 | 7284 | 85.22 |
| JN177-Ta | 7107 | 6466 | 5072 | 84.14 |
SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank
Frequency of single nucleotide polymorphisms (SNPs) in the unigene sequences
Based on the unigene sequences sharing >96 % identity, 15,226 SNPs were identified within the unigene sequence shared between SR3 and JN177, equivalent to a SNP frequency of 11.33 per 1000 nt of coding sequence (Table 2). The transition and transversion frequencies were, respectively, 6.70 and 4.63 per 1000 nt. The SNP frequency between JN177 and the sequences represented in the wheat EST database (JN177 vs Ta comparison) was only about one half of this level (5.77 per 1000 nt) (Table 2), demonstrating that the somatic hybridization process was effective in inducing point mutations. A comparison based on the sequences of the unigenes shared between the BA progenitor tetraploid (T. turgidum) and the A genome carrier T. monococcum revealed a SNP frequency of 15.48 per 1000 nt, while that between T. turgidum and Ae. speltoides (related to the B genome progenitor) was 18.51, indicating that a high frequency of mutation was induced during the formation of allotetraploid wheat. Similarly, the estimated SNP frequencies between bread wheat and T. monococcum, Ae. speltoides, T. turgidum and Ae. tauschii (D genome progenitor) were, respectively, 12.02, 16.24, 12.13 and 5.40 per 1000 nt (Table 3). Thus the mutation frequency induced by the somatic hybridization process appeared to be similar in extent to that induced by allopolyploidization. The frequency of SNPs between the unigene sequences of bread wheat and those of either T. monococcum or Ae. speltoides was less than that between T. turgidum and either T. monococcum or Ae. speltoides unigenes (Table 3). This coincided with the finding that the SNP frequency of SR3 and wheat database EST (SR3 vs Ta alignment) was lower than those of the SR3 vs JN177 alignment (Table 2). The SNP frequency between SR3 unigene sequences and those of the A, B, BA and D genome species was similar to those between JN177 unigenes and those of the A, B, BA and D genome species (Table 3).
Table 2.
The SNP frequencies in SR3 and JN177
| SNPs | Type | SNPs (per 1000 nt) | ||
|---|---|---|---|---|
| SR3-JN177 | SR3-Ta | JN177-Ta | ||
| Transitions | C → T | 2.065 | 0.788 | 0.727 |
| T → C | 1.776 | 1.016 | 1.031 | |
| G → A | 1.542 | 1.168 | 0.843 | |
| A → G | 1.318 | 0.641 | 0.602 | |
| Total | 6.701 | 3.613 | 3.203 | |
| Transversions | A → T | 0.267 | 0.154 | 0.139 |
| T → A | 0.252 | 0.208 | 0.180 | |
| C → G | 1.142 | 0.399 | 0.412 | |
| G → C | 1.084 | 0.680 | 0.671 | |
| G → T | 0.455 | 0.320 | 0.297 | |
| T → G | 0.386 | 0.273 | 0.281 | |
| A → C | 0.507 | 0.243 | 0.239 | |
| C → A | 0.533 | 0.349 | 0.342 | |
| Total | 4.627 | 2.625 | 2.561 | |
| Total SNPs | 11.328 | 6.238 | 5.764 | |
SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank
Table 3.
SNP and indel frequencies among wheat and its ancestors
| Query | Subject | SNPs (per 1000 nt) | Indels (per 1000 nt) | ||||
|---|---|---|---|---|---|---|---|
| Transition | Transversion | Total | Insertion | Deletion | Total | ||
| Triticum turgidum | Triticum monococcum | 9.436 | 6.048 | 15.484 | 0.622 | 1.282 | 1.904 |
| Aegilops speltoides | 10.415 | 8.091 | 18.506 | 0.565 | 0.717 | 1.282 | |
| Triticum aestivum | Triticum monococcum | 7.258 | 4.762 | 12.020 | 0.688 | 0.769 | 1.457 |
| Aegilops speltoides | 9.709 | 6.531 | 16.240 | 0.810 | 0.606 | 1.417 | |
| Triticum turgidum | 7.096 | 5.036 | 12.131 | 1.259 | 0.755 | 2.014 | |
| Aegilops tauschii | 4.368 | 1.028 | 5.396 | 0.257 | 2.055 | 2.312 | |
| JN177 | Triticum monococcum | 9.848 | 6.395 | 16.243 | 0.910 | 0.473 | 1.383 |
| Aegilops speltoides | 9.631 | 7.060 | 16.692 | 0.725 | 0.526 | 1.251 | |
| Triticum turgidum | 7.299 | 6.113 | 13.412 | 1.252 | 0.348 | 1.601 | |
| Aegilops tauschii | 5.010 | 1.474 | 6.484 | 0.000 | 4.421 | 4.421 | |
| SR3 | Triticum monococcum | 10.053 | 6.358 | 16.411 | 1.319 | 0.554 | 1.873 |
| Aegilops speltoides | 10.654 | 6.931 | 17.585 | 1.141 | 0.581 | 1.722 | |
| Triticum turgidum | 7.709 | 5.352 | 13.060 | 1.480 | 0.469 | 1.949 | |
| Aegilops tauschii | 9.123 | 1.610 | 10.733 | 0.716 | 4.651 | 5.367 | |
| Triticum aestivum | 3.613 | 2.625 | 6.238 | 1.047 | 0.307 | 1.354 | |
The size distribution of indels in the unigene sequences
The indels ranged from 1 nt to 574 nt, and a majority of the indels involved only 1 nt. A significant number of indels was revealed by aligning the matched unigene sequences, with the frequency of larger indels (>23 nt) being clearly less than that of the small ones (1–10 nt) (Table 4). There had 82.14 % unigenes possessing small indels when compared between SR3 and JN177, higher than those from SR3 vs Ta and JN177 vs Ta comparisons. On the contrary, 6.70 % unigenes had large indels in the comparison between SR3 and JN177, lower than those of other two comparisons. There had more unigenes with small insertions than those with small deletions, and the difference was stronger in the SR3 vs Ta and JN177 vs Ta comparisons. Unigenes with large insertions were similar to those with large deletions in the SR3 vs JN177 comparison, but unigenes with large deletions were more abundant than those with large insertions in the other two comparisons. The comparison between the JN177 (and similarly SR3) unigene sequences with those represented in the wheat EST database showed that for small indels, the ratio of insertion to deletion frequency was negatively correlated to indel length (R2 = 0.62 and 0.72, respectively) (Fig. 1a); the ratio was >1 for indels shorter than 6 nt, and <1 for indels longer than 6 nt. However, for the larger indels, the insertion to deletion ratio was positively correlated to indel length (R2 = 0.59 and 0.65, respectively) (Fig. 1b); in indels ranging in length from 20 to 70 nt, the ratio was just 0.01–0.06, rising to 0.28–0.86 for indels of length 71–200 nt, and to ~1.5 for indels longer than 200 nt (Fig. 1b). The SR3 vs JN177 comparison revealed an insertion to deletion ratio of ~1 irrespective of indel length (Fig. 1a, b).
Table 4.
Indel variation in SR3 and JN177 unigene sequences
| Sizes | Type | SR3-177 (%) | SR3-TA (%) | 177-Ta (%) |
|---|---|---|---|---|
| Small indels | Insertion | 51.57 | 58.02 | 37.66 |
| (1–10 nt) | Deletion | 30.57 | 17.02 | 14.57 |
| Indel | 82.14 | 75.04 | 52.23 | |
| Large indels | Insertion | 3.45 | 1.75 | 1.32 |
| (23–574 nt) | Deletion | 3.25 | 9.8 | 8.94 |
| Indel | 6.70 | 11.55 | 10.26 |
“Small” indels were defined as those lying within an aligned sequence, while “large” indels split matched sequence into two separate alignments. The table shows the percentage of unigenes affected by insertion/deletions. SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank
Fig. 1.
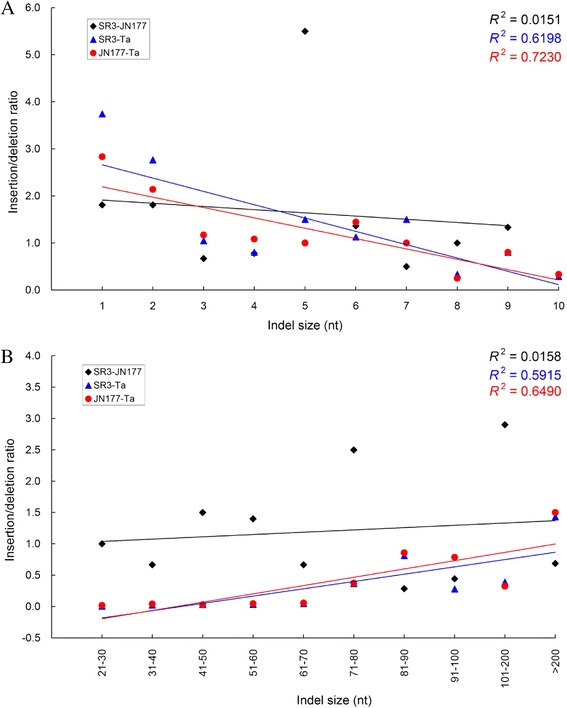
The distribution of indel lengths. a: small indels. b: large indels. The insertion/deletion ratio was obtained by dividing the number of insertions by the number of deletions. SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank. The correlation between indel size and insertion/deletion ratio was performed using the Pearson correlation analysis
The frequency of indels in the unigene sequences
Small indels were used to calculate the indel frequency because they were markedly more abundant than large indels (indel numbers not shown). In all, the 2581 matched unigenes derived from the SR3 vs JN177 comparison revealed 2120 indels (1.58 per 1000 nt). Based on the JN177 sequences, these comprised 1331 insertions and 789 deletions in SR3, equivalent to frequencies of, respectively, 0.99 and 0.59 per 1000 nt (Table 5). In the comparison with the sequences represented in the wheat EST database, the indel frequency in SR3 was 1.36 per 1000 nt. The similar comparison between JN177 and the sequences represented in the wheat EST database revealed an indel frequency of only 0.93 per 1000 nt, implying that the asymmetric somatic hybridization process was effective in inducing indels in coding sequence.
Table 5.
The frequency of small indels in SR3 and JN177 unigene sequences
| Length (nt) | SR3-177 (per 1000 nt) | SR3-TA (per 1000 nt) | 177-Ta (per 1000 nt) |
|---|---|---|---|
| 1 | 1.319 | 1.229 | 0.823 |
| 2 | 0.109 | 0.063 | 0.048 |
| 3 | 0.076 | 0.032 | 0.031 |
| 4 | 0.031 | 0.009 | 0.009 |
| 5 | 0.010 | 0.006 | 0.004 |
| 6 | 0.019 | 0.008 | 0.008 |
| 7 | 0.004 | 0.001 | 0.002 |
| 8 | 0.003 | 0.001 | 0.004 |
| 9 | 0.005 | 0.002 | 0.003 |
| 10 | 0.001 | 0.002 | 0.001 |
| Insertions | 0.990 | 1.047 | 0.672 |
| Deletion | 0.587 | 0.307 | 0.260 |
| Indels | 1.577 | 1.354 | 0.932 |
“Small” and “large” indels defined as in Table 4. Indel frequency was expressed as the number of indels per 1000 nt of unigene sequence
To compare the induction rates of indels caused by somatic hybridization and allopolyploidization, equivalent calculations were made using the matched unigene sequences present in bread wheat and its relatives/progenitor species (Table 3). Comparing the unigenes of T. turgidum with those of T. monococcum and Ae. speltoides revealed an indel frequency of, respectively, 1.90 and 1.28 per 1000; the equivalents between bread wheat and its various related species ranged from 1.42 to 2.31 per 1000 nt, with the comparison involving Ae. tauschii producing the highest estimate. The indel frequencies estimated using the unigenes of JN177 and SR3 also lay in the range 1 to 2 per 1000 nt, with the exception of the comparison with Ae. tauschii, where the frequencies were, respectively, 4.42 and 5.37 per 1000 nt (Table 3).
In the SR3 vs JN177 comparison, the insertion frequency (0.99 per 1000 nt) was 1.69 fold to the deletion frequency (0.59 per 1000 nt) (Table 5). Based on the sequences represented in the wheat EST database, the insertion frequencies were higher by 3.41 and 2.58 fold than deletion frequencies in SR3 and JN177, respectively. Especially, the insertion frequency in the SR3 vs Ta comparison (1.05 per 1000 nt) was similar to the SR3 vs JN177 comparison (0.99 per 1000 nt), but its indel frequency was lower than the latter. Thus, the preference to small insertion was decreased in wheat asymmetric somatic hybrids in comparison with allopolyploid wheat.
The function of unigenes showing sequence polymorphism
To know whether the genetic variation is associated with their biological processes, we selected sequences participating in gene expression regulation and other processes for analysis. A selection of polymorphic unigenes represented in the JN177 and SR3 libraries (948 and 1519, respectively) as well as in the wheat EST database showed that for gene expression regulation, the frequency of SNPs differed most notably in genes involved in nucleosome assembly and chromatin assembly/disassembly (Table 6). The least polymorphic comparison was between JN177 and the wheat EST database unigenes (4.59 SNPs per 1000 nt for the genes involved in the former category and 3.72 per 100 nt in the latter). The same comparison between SR3 and JN177 produced, respectively, 18.17 and 19.21 SNPs per 1000 nt. The next most variable genes were those encoding products involved in translation and post-translational modification, where the SR3 vs JN177 comparison revealed a SNP frequency of, respectively, 13.30 and 13.97 per 1000 nt, while the JN177 vs Ta comparison showed 8.69 and 7.98, respectively. The range in SNP frequency for genes associated with metabolic processes ranged from 5.45 to 6.29 per 1000 nt in the JN177 vs Ta comparison and 9.16–14.25 per 1000 nt in the SR3 vs JN177 comparison (Additional file 3: Table S2). SNP frequencies were also higher in the SR3 vs Ta comparison than in the JN177 vs Ta comparison for genes encoding proteins involved in various metabolic processes except for glycolysis as well as nucleobase, nucleoside, nucleotide and nucleic acid metabolic process (Additional file 3: Table S2). This difference in SNP frequencies was also found in ESTs of protein fate, transport, cell redox homeostasis, and response to (oxidative) stress (Additional file 4: Table S3). With respect to unigenes varying at the level of indels, the frequency of polymorphism was lower in the JN177 vs Ta than in the SR3 vs JN177 comparison (Table 6; Additional files 3 and 4: Table S2 and S3). Indel events were noticeably rare in genes involved in nucleosome assembly and chromatin assembly/disassembly (Table 6). The respective frequencies were 0.43 and 0.38 per 1000 nt in the JN177 vs Ta comparison and 0.95 and 0.92 per 1000 nt in the SR3 vs JN177 comparison.
Table 6.
The function of unigenes affected by SNP and indels in SR3 and JN177
| Process | Unigene number | SNP (per 1000 nt) | InDel (per 1000 nt) | |||||
|---|---|---|---|---|---|---|---|---|
| JN177 | SR3 | SR3-JN177 | SR3-Ta | JN177-Ta | SR3-JN177 | SR3-Ta | JN177-Ta | |
| Chromatin assembly or disassembly | 54 | 92 | 18.17 | 7.07 | 4.59 | 0.95 | 0.85 | 0.43 |
| Nucleosome assembly | 49 | 83 | 19.21 | 6.8 | 3.72 | 0.92 | 0.75 | 0.38 |
| Regulation of transcription | 96 | 204 | 10.79 | 9.85 | 11.49 | 1.94 | 1.53 | 1.22 |
| RNA processing | 35 | 85 | 10.39 | 10.65 | 9.73 | 1.94 | 1.33 | 1.02 |
| Translation | 254 | 373 | 13.30 | 8.77 | 8.69 | 1.71 | 1.40 | 1.08 |
| Protein modification process | 266 | 440 | 11.39 | 10.61 | 10.19 | 1.87 | 1.46 | 1.32 |
| Protein phosphorylation | 181 | 230 | 10.81 | 12.08 | 11.7 | 2.09 | 1.53 | 1.41 |
| Post-translational protein modification | 13 | 12 | 13.97 | 9.92 | 7.98 | 1.28 | 1.23 | 1.00 |
SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank
Sequences flanking indels in the unigenes
The sequences flanking indels were characterized by calculating the GC content of the ten nucleotides flanking either side of the indel. There was no obvious difference in GC content between 5′ and 3′ terminal flanking sequence in any of the comparisons (JN177 or SR3 vs wheat database ESTs, SR3 vs JN177) (data not shown). The trend of GC content was similar when the second to tenth nucleotides of the flanking sequence were considered (Fig. 2). The GC content of the nucleotides positioned three, six and nine away from the indels was higher than that of the ones positioned four, five, seven or eight away in the 3′ terminal flanking sequences, but the rule was not found in the 5′ terminal flanking sequences. The GC content of the flanking sequence was higher in the SR3 vs JN177 comparison (53.47–54.04 %) than in the other two comparisons (51.88–52.88 % in JN177 vs Ta, 50.99–52.63 % in SR3 vs Ta). In the SR3 vs Ta and JN177 vs Ta comparisons, the GC content of the flanking sequence of the deletions was higher than that of the insertions, while the content of the indels was close to that of the insertions. However, in the SR3 vs JN177 comparison, the GC content of the flanking sequence of the deletions was generally lower than that of the insertions, and the difference between the deletions and insertions was weaker than that in the JN177 or SR3 vs Ta comparisons; the content of the flanking sequence of the indels did not bias toward either the deletions or insertions. In the JN177 or SR3 vs Ta comparisons, with respect to the nucleotide lying on the 5′ side of the indels, the GC content of the nucleotide adjacent to the deletions was significantly lower than that of other nucleotides in the flanking sequence, but the GC content of the nucleotide adjacent to the insertions was significantly higher than that of other nucleotides in the flanking sequence (Fig. 2b, c). On the other hand, for the nucleotides lying on the 3′ side of the indels, the GC content of the nucleotide adjacent to the deletions remained high and was comparable to that of the second and third flanking nucleotides, but the GC content of the nucleotide adjacent to the insertions was significantly lower than that of the other flanking nucleotides (Fig. 2b, c). In contrast in the SR3 vs JN177 comparison, for the nucleotides lying on the 5′ side of the indel, the GC content of the nucleotide adjacent to both the deletions and insertions was higher than that of the second to tenth nucleotides of the 5′ flanking sequence, while for the nucleotides lying on the 3′ side of the indels, the GC content of the nucleotide adjacent to both the deletions and insertions was lower than the second and third nucleotides (Fig. 2a).
Fig. 2.
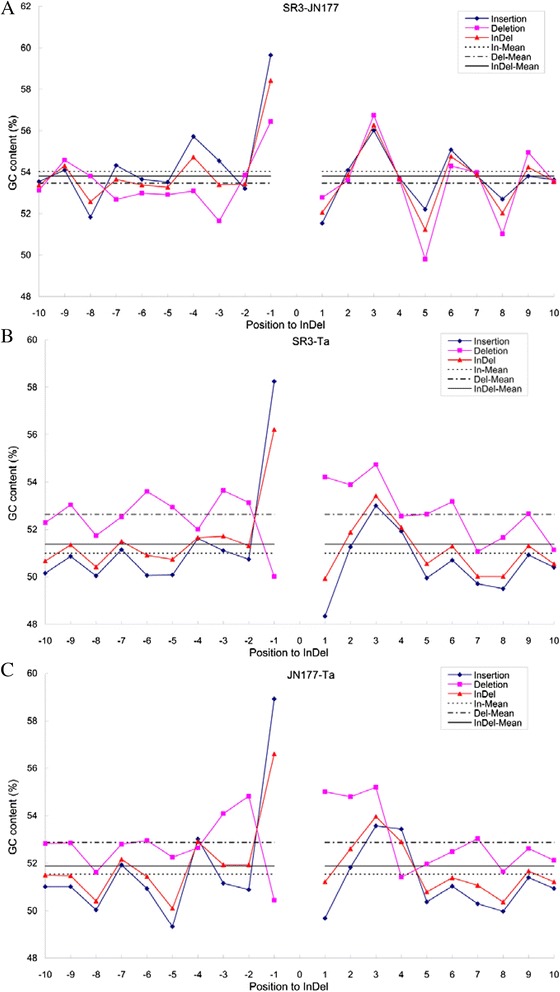
Variation in the GC content in the sequence immediately flanking indels. a: SR3-JN177, JN177 unigene sequences queried with those of SR3. b: SR3-Ta, SR3 unigene sequences queried with wheat ESTs housed in GenBank. c: JN177-Ta, JN177 unigene sequences queried with wheat ESTs housed in GenBank. -10 ~ −1: The tenth to first nucleotides on the 5′ side of the indel. 1 ~ 10: The first to tenth nucleotides on the 3′ side of the indel. In-mean, Del-mean and indel-mean: the GC content of 5′ and 3′ flanking sequences of insertions, deletions and indels, respectively
Indel classification
We further compared the characteristic of flanking sequences of large indels in the SR3 vs JN177 comparison. The flanking sequences of 45 large insertions and 39 large deletions identified in the SR3 vs JN177 comparison were identical (Fig. 3a, d). Two examples were SR3_2LCP226_E06 (474 nt insertion) and SR3_firstas1573 (34 nt deletion) (Additional file 5: Figure S2A, B). A second group had 40 insertions and 36 deletions, whose two terminals possessed repeated sequences in SR3 and JN177 (Fig. 3b, e). The flanking sequence of SR3_5V50 (179 nt insertion) harbors a run of G’s; the 3′ flanking sequence of SR3_firstas843 (55 nt deletion) includes two copies of CATCCC in JN177 but only one in SR3 (Additional file 5: Figure S2C, D). The repeat motifs present in the flanking sequence ranged in length from 1 to 51 nt (data not shown). A 1 nt motif was present in 23 of the insertions and 19 of the deletions, dominated by runs of G (data not shown). The third group, in which the flanking sequence was modified (Fig. 3c, f), is exemplified by SR3_firstas1573 (141 nt deletion), where SNPs were generated at three positions adjacent to the deletion (Additional file 5: Figure S2E). A few of the unigenes experienced multiple indel events: SR3_firstas1573 carries two deletions, one belonging to the first group and the other to the third group (Additional file 5: Figure S2B, E). Other variants included the induction of translocated and chimeric sequences. In the homologs SR3_2LCP192_G10 and JN177_firstas231, the same sequence was found in positions 1187–1367 in the former allele, but at 1–181 in the other. SR3_2LCP192_G10 also harbors a large deletion with a repeat sequence CAAGAAGGA (Additional file 6: Figure S3A). SR3_firstas716 nucleotides 88–196 do not align with JN177_LCP139_D11 nucleotides 157–278, but their terminal sequences are identical (Additional file 6: Figure S3B).
Fig. 3.
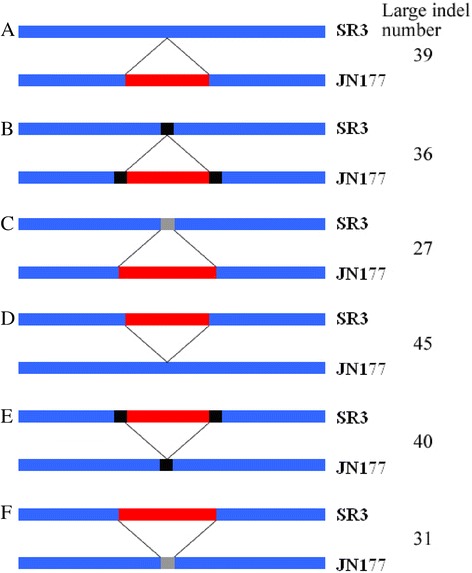
Hypothetical model for the formation of large indels induced during asymmetric somatic hybridization. Blue block: unigenes shared by SR3 and JN177. Red block: insertion and deletion fragments. Black block: repetitive sequences in the indel flanking sequence. Gray blocks: small fragments adjacent to insertions and deletions, differing sequence between SR3 and JN177. SR3-JN177: JN177 unigene sequences queried with those of SR3. SR3-Ta: SR3 unigene sequences queried with wheat ESTs housed in GenBank. JN177-Ta: JN177 unigene sequences queried with wheat ESTs housed in GenBank
Discussion
Asymmetric somatic hybridization induces sequence variation
A manifestation of genomic shock, which has been experimentally observed in de novo wide hybrids and inferred from the analysis of natural allopolyploids [37], is a widespread alteration of DNA sequence in the form of both point mutations and indels [38–40]. The alternation was found confirmed by the comparison between bread wheat and its ancestors (Table 3) as well as between SR3/JN177 and sequences of wheat EST database (Table 2,5). The present comparison between the transcriptomes of SR3 and JN177 has shown that the somatic hybridization process also perturbs the genome in this way (Table 2,5). AFLP fingerprinting has also suggested that non-genic sequence also appear to suffer mutation [31]. Cytogenetically and Phenotypically, SR3 (as well as other derivatives of wheat-based somatic hybrids) has been shown to be highly stable [5, 25, 26], which implies that genetic instability prevails only in the immediate aftermath of the wheat somatic hybridization event, at least in the short term.
The epigenetic modification of genic sequences is a common feature of allopolyploid genomes [12, 13, 41, 42]. The proportion of loci experiencing an alteration in cytosine methylation in a newly synthesized allohexaploid wheat has been estimated to be as high as 13 % [12]. As methylated cytosines, especially those present as a CpG dinucleotide, are readily converted to thymine [43], so - as demonstrated in Arabidopsis thaliana - DNA methylation can represent a major source of SNP formation [44]. In plant genomes, the bias of synonymous codon usage towards thymine and adenine is also driven by DNA methylation [45]. Both somatic hybridization and allopolyploidization result in change in cytosine methylation [12, 41, 42, 46, 47]. We recently observed that the frequency of cytosine methylation changes in wheat genome exerted by somatic hybridization was 23.6 % through the methylation-sensitive amplification polymorphism (MSAP) analysis [31], and was more intense than the frequency (13 %) exerted by allopolyploidization in a newly synthesized allohexaploid wheat [12]. This mechanism may therefore explain why C/T SNPs were more common in the SR3 vs JN177 comparison (2.07 per 1000 nt) than in the JN177 vs wheat database ESTs comparison (Table 2). Cytosine methylation polymorphisms are known to account for at least some of the differential gene transcription shown by SR3 and JN177 [48]; a further contributor may well be chromatin remodeling. The frequency of SNPs (18.17 and 19.21 per 1000 nt) and indels (0.95 and 0.92 per 1000 nt) induced SR3 involved genes participating in chromatin assembly/disassembly and nucleosome assembly (Table 6), consistent with the induction of widescale epigenetic changes resulting from somatic hybridization.
The evolutionary path followed by most polyploids is to reduce their genome size by deletion [9, 12, 42, 49–53]. After many generations of such activity, many of the sequences which have become duplicated as a result of the allopolyploidization event revert to single copy and the genome becomes increasingly diploid-like [9]. A good example of such a cryptic polyploid is maize [54, 55]. Some of the sequences lost in SR3 (compared to JN177) were of substantial length (Fig. 1b), indicating that the response to somatic hybridization also includes sequence deletion. On the other hand, the preference to large insertion was alleviated in SR3-JN177 alignment, and the preference to either insertion or deletion was not correlative to indel size (Fig. 1b). Consistently, AFLP and southern blotting outcomes found that the extent of sequence loss over generations from the products of somatic hybridization was relatively modest [31]. These point to a potential difference in the way that genomes respond to polyploidization as opposed to somatic hybridization. A major difference is that a de novo sexual hybrid harbors the full genomes of the donor and recipient, while in the asymmetric somatic hybrid, only fragments of the donor are retained from the outset, and many are presumably eliminated prior to the regeneration from tissue culture. Based on this, the suggestion is that the potential disruptive effect of donor DNA is possibly much less.
Asymmetric somatic hybridization was performed between UV-irradiated tall wheat grass protoplast and unirradiated wheat protoplast, both of which were produced from calli. During this process follows mutagenic factors including protoplast formation, UV irradiation, callus induction, and plant regeneration [3]. We found that these mutagenic factors had substantially slight effect on the genetic variation of SR3 and other other derivatives of wheat-based somatic hybrids by a set of molecular biomarker assays and comparing glutenin genomic sequences [31, 32]. Therefore, it could be concluded that the high frequency of genetic variation majorly comes from the genomic shock during asymmetric somatic hybridization.
Putative mechanisms underlying indel formation induced by somatic hybridization
Numerous indels differentiate the genomes of SR3 and JN177 (Table 5). The GC content of the indels identified in the SR3 vs JN177 comparison was comparable to that in the indels differentiating the JN177 unigenes from those present in other wheat. As a result, it is not tenable to suggest that indels induced by the somatic hybridization process reflect a particular level of GC content. The proposition that indel formation, along with the other genomic changes induced by allopolyploidization, is non-random [56] does however require that some feature of the local sequence acts as a recognition feature. The GC content of the nucleotide 5′ adjacent only to the indels differed from that of other nucleotides in flanking sequence (Fig. 2), which does suggest that GC content could be a candidate signal for indel formation. Note that the GC content of the sequences adjacent to indels differed between the SR3 vs JN177 and the JN177 vs database wheat ESTs comparisons (Fig. 2), which implies that the mechanism underlying indel formation during somatic hybridization may well be distinct from that controlling the process in a sexual hybrid. Repetitive sequences are particularly susceptible to expansion and contraction, and we found 76 indels in the SR3 vs JN177 comparison harbored repetitive sequences in their flanking sequence (Fig. 3b, e and Additional file 5: Figure S2C, D). Repetitive sequences flanking indels may serve as recombination sites to promote indel formation via homologous recombination, because double strand breaks (DSBs) appear to be common in regions enriched for repetitive sequences, and these “fragile sites” represent hotspots for recombination [57].
The possible association between sequence variation and phenotype in SR3
The genetic variation was found to affect gene expression or the activity of gene production [9–20], therefore lead to phenotypic alteration. Numerous transcriptomic and proteomic differences have been established between SR3 and JN177 [28, 29, 58]. Here, it was revealed that a substantial number of unigenes (involved in a range of plant processes) harbored SNPs and/or indels between SR3 and JN177 (Table 6, Additional file 3 and 4: Tables S2 and S3). Especially, the high salt tolerance of SR3 is closely associated with its superior ability of antioxidation and redox homeostasis maintenance [28, 30], and here the unigenes participating in cell redox homeostasis and response to stress and oxidative stress also has higher SNP and indel frequencies in SR3 (Additional file 4: Table S3). Moreover, EST sequences that absolutely matched to the probes of cDNA array with differential expression profiles [58] possessed some SNPs and indels, and some of these sequence variants could well contribute to the phenotypic difference between SR3 and JN177 (Additional file 7: Table S4). One established example is the gene TaCHP, which is more strongly transcribed in SR3 than in JN177 and is associated with SR3’s salinity tolerance [33]. The SR3 TaCHP allele differs from that present in JN177 by a deletion in its promoter sequence. A second example concerns TaSRO1, also associated with the salinity tolerance of SR3; the SR3 and JN177 alleles differ by three SNPs, one of which is responsible for the higher activity of the SR3 allele [30]. The sequences of the SR3 and JN177 alleles of TaFLS1 (likewise associated with salinity tolerance) have been shown to be differentially methylated, and this polymorphism can account for their differential transcript abundance [48]. The headline conclusion to be drawn from this analysis is that asymmetric somatic hybridization can induce a high frequency of both genetic and epigenetic variation, some of which is likely responsible for alterations in the phenotype.
During standard somatic hybridization, the genomes of biparents emerge together, which induces drastic genomic shock and therefore leads to large scale of genome rearrangement, sequence elimination and genetic variation [3]. As a result, the hybrids often exhibit development dysfunction and sterility [3]. For asymmetric somatic hybridization, the protoplasts of donor (tall wheatgrass) are irradiated and its genome is fragmented, so in the fused cells of donor and recipient (wheat), most of chromatin fragments of donor are eliminated and a very few fragments are introgressed into the genome of recipient, while the genome of recipient has almost no structural change [3]. This event induces moderate genomic shock and results in bearable genetic variation. Therefore, the hybrids are fertile and their wheat derivatives often exhibit diverse phenotypes such as high salt and drought tolerance, superior stripe rust resistance, as well as high quality and yield traits (low stem, big ear, big grain, and high yield, etc.) [3, 5, 25, 26], which can be selected to breed cultivar with improved agricultural traits.
Methods
Stress treatment and cDNA library construction
Given that SR3 has demonstrated improved levels of tolerance to both salinity and drought, we proposed to compare the sequence variation of ESTs that are produced under the control and after exposure to saline and osmotic stresses to gain the association between genetic variation and abiotic stress tolerance. Three-leaf-stage seedlings of both cv. SR3 and cv. JN177 were exposed to half strength Hoagland’s liquid medium containing either 200 mM NaCl for 0, 0.5, 1, 12, 24, 48 or 72 h, or to 18 % PEG for 0, 0.5, 1, 12, 24 or 48 h. RNA was extracted from both roots and leaves using the Trizol reagent (Invitrogen, USA), and purified using an OligotexTM-dT30 mRNA Purification Kit (TaKaRa, Japan). Equimolar amounts of the various RNA samples were mixed to obtain both an SR3 and a JN177 mRNA pool, which formed the template for the construction of two cDNA libraries using a CloneMiner™ cDNA Library Construction Kit (Invitrogen, USA), according to the manufacturer’s protocol. Given that the genetic variation induced by asymmetric somatic hybridization was remarkably higher than that induced by other mutagenic factors protoplast formation, UV radiation, callus induction and plant regeneration [31, 32], other cDNA libraries reflecting the genetic variation of these mutagenic factors were not constructed.
cDNA sequencing and sequence assembly
The two cDNA libraries were cultured on LB agar plates containing 50 mg/mL kanamycin overnight at 37 °C. The monoclones were selected randomly, and the inserts present sequenced based on the M13F primer by the Sanger sequencing method using ABI 3700 sequencer under the control of Sequencing Analysis 5.2.0 with the default parameters. The cDNAs were directionally inserted in the plastid, and the inserted sequences were sequenced from 5′-terminal using the M13F primer, which can increase the proportion of full-length cDNA to be sequenced. To ensure the quality of sequences, only 800 nt fragments from the reading start were selected for following analysis. Plastid and adapter sequences were removed to assume the sequencing quality of Q20 using the Seqman programme [59]. Low quality and short (<100 nt) sequences were removed with EGassemble (http://egassembler.hgc.jp). The quality of the cleaned sequences was further checked via randomly selecting more than 2000 sequences to analyze sequencing peak graph. The edited sequences were assembled using CAP3 software to produce a set of contigs and singletons (overlap 50 nt, identity 95 %) [60].
Local BLAST and annotation
The resulting contig and singleton sequences were subjected to a BLASTn search, applying an E-value cut-off of 1E-10 and an HSP length cut-off adjusted to 33, to the EST sequences of bread wheat, T. monococcum (modern cultivated diploid A genome carrier), Ae. tauschii (diploid D genome progenitor species), T. turgidum (BA tetraploid progenitor species) and Ae. speltoides (diploid related to the B genome ancestor) mounted in GenBank (http://www.ncbi.nlm.nih.gov/genbank/dbest) (download date: Dec 12, 2014). A BLASTx search of the non-redundant protein database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/) was used predict the full length cDNA sequences, where the matched EST sequence possessing encoding sequence of N-terminal amino acid (containing start codon ATG) was defined as full length cDNA. The SR3 and JN177 sequences were also subjected to a BLASTx search of the Arabidopsis cDNA database (ftp://ftp.arabidopsis.org/home/tair/Sequences/ATH_cDNA_EST_sequences_FASTA/ATH_cDNA_sequences_20101108.fas), applying an E-value cut-off of 1E-10 and an HSP length cut-off adjusted to 33. Matching sequences were classified by gene ontology using the TAIR GO annotation tool (http://www.arabidopsis.org/tools/bulk/go/index.jsp) and processed for GO classification and pathway enrichment using Blast2GO [61] using the entire GO-annotated collection of SR3 and JN177 ESTs as the reference.
Polymorphism calculation
The SNP pattern was defined as the nucleotide of query sequence (e.g., A) with the nucleotide of subject sequence (e.g., G) as the reference (SNP was G → A). The insertion and deletion were also defined with the subject sequence as the reference. The sequences with BLASTn identity > 96 % were selected for calculation SNP and indel frequencies to exclude the interference of paralogous genes as possible [62], which were calculated as the ratio of total SNP and indel amounts to the total length of matched regions of all ESTs with identity > 96 %, respectively. The matched coverage (>96 % identity coverage) was defined as the ratio of the total length of all matched regions of ESTs with identity >96 % to the total length of these ESTs. To analyze the possible association between genetic variation and transcriptional pattern, the sequences were subject to BLASTn against the sequences (60 nt) of differentially expressed probes of wheat cDNA array in our previous study exploring the transcriptomic difference between SR3 and JN177 seedlings under the control and after exposure to NaCl and PEG treatments [58]. The unigenes that absolutely matched probe sequences were selected for calculating SNPs and indels.
Conclusions
Asymmetric somatic hybridization induces high frequency of point mutations and indels, with a stronger intense than the genetic variation during allopolyplodization, which is the molecular basis of the phenotypic alteration of somatic hybrids. Moreover, the genetic variation induced by asymmetric somatic hybridization has different characteristics from those induced by allopolyplodization, demonstrating the specific genomic evolution in somatic hybrids. Therefore, asymmetric somatic hybridization provides appropriate material to comprehensively explore the nature of the genetic variation induced by genomic shock.
Sequences used for analysis in this work were submitted to Genbank (Accession number: JZ881292 - JZ892704).
Acknowledgments
This work was supported by the Natural Science Foundation of China (31171175) and the Major Program of the Natural Science Foundation of China (31430060).
Additional files
Details of the cDNA libraries and the contig assembly. (DOCX 11 kb)
The distribution of unigene sequences. (TIFF 943 kb)
SNP and indel frequencies in unigenes participating in metabolic processes. (DOCX 14 kb)
SNP and indel frequencies in unigenes participating in other processes. (DOCX 14 kb)
Characterization of indels. (A) An insertion (291–764) present in an SR3 unigene has flanking sequence which matches that in its JN177 homolog. (B) A deletion (490–523) present in an SR3 unigene has flanking sequence which matches that in its JN177 homolog. (C) An insertion (143–320) present in an SR3 unigene, which harbors a run of G’s (shown in green) in both flanking sequences. (D) A deletion (664–747) present in an SR3 unigene which harbors a CATCCC repeat (shown in green) in both flanking sequences. (E) A deletion (729–868) present in an SR3 unigene, in which the identity of three nucleotides (676–678) differ between the SR3 and JN177 homologs. (TIFF 2567 kb)
Translocations and sequence chimeras induced by somatic hybridization. (A) In the homologs SR3_2LCP192_G10 and JN177_firstas231, the identical sequence is found in positions 1187–1367 in the former allele, but at 1–181 in the other. SR3_2LCP192_G10 also harbors a large deletion with a repeat sequence CAAGAAGGA. (B) In SR3_firstas716, nucleotides 88–196 do not align with JN177_LCP139_D11 nucleotides 157–278, but their terminal sequences are identical. (TIFF 1660 kb)
Sequence variation for cDNAs which have been shown to be differentially transcribed in SR3 and JN177. a: The treatments were conducted in our previous transcriptomic analysis (Liu et al., Plant Mol Biol, 2012;78:159–69). b: The patterns of differential expressed probes in cDNA microarray (Liu et al., Plant Mol Biol, 2012;78:159–69). “up-regulated”: unigenes which were up-regulated in SR3, “down-regulated”:unigenes which were up-regulated in SR3. Control: half strength Hoagland’s liquid medium, PEG 0.5 h: seedlings exposed to 30 % PEG6000 for 0.5 h, NaCl 0.5 h and 24 h:seedlings exposed to 345 mM NaCl for 0.5 h and 24 h, respectively. c: The number of unigenes that absolutely matched the sequences of probes. (DOCX 14 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MW, CL and GX designed the study. MW, CL, TX and YW conducted the experiments and analyzed the data. MW and GX wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Mengcheng Wang, Email: wangmc@sdu.edu.cn.
Chun Liu, Email: liuchun@mail.sdu.edu.cn.
Guangmin Xia, Phone: 86-531-88364525, Email: xiagm@sdu.edu.cn.
References
- 1.Tanksley SD, McCouch SR. Seed banks and molecular maps: unlocking genetic potential from the wild. Science. 1997;277:1063–6. doi: 10.1126/science.277.5329.1063. [DOI] [PubMed] [Google Scholar]
- 2.Zamir D. Improving plant breeding with exotic genetic libraries. Nat Rev Genet. 2001;2:983–9. doi: 10.1038/35103590. [DOI] [PubMed] [Google Scholar]
- 3.Xia G. Progress of chromosome engineering mediated by asymmetric somatic hybridization. J Genet Genomics. 2009;36:547–56. doi: 10.1016/S1673-8527(08)60146-0. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths S, Sharp R, Foote TN, Bertin I, Wanous M, Reader S, et al. Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature. 2006;439:749–52. doi: 10.1038/nature04434. [DOI] [PubMed] [Google Scholar]
- 5.Feldman M. Origin of cultivated wheat. The World Wheat Book: A history of wheat breeding. Paris: Lavoisier Publishing; 2001. [Google Scholar]
- 6.Soltis DE, Soltis PS, Tate JA. Advances in the study of polyploidy since plant speciation. New Phytol. 2003;161:173–91. doi: 10.1046/j.1469-8137.2003.00948.x. [DOI] [Google Scholar]
- 7.Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, et al. Polyploidy and angiosperm diversification. Am J Bot. 2009;96:336–48. doi: 10.3732/ajb.0800079. [DOI] [PubMed] [Google Scholar]
- 8.Ozkan H, Levy AA, Feldman M. Alloploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell. 2001;13:1735–47. doi: 10.1105/tpc.13.8.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song K, Lu P, Tang K, Osborn TC. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc Natl Acad Sci USA. 1995;92:7719–23. doi: 10.1073/pnas.92.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieseberg LH. Polyploid evolution: keeping the peace at genomic reunions. Curr Biol. 2001;11:R925–8. doi: 10.1016/S0960-9822(01)00556-5. [DOI] [PubMed] [Google Scholar]
- 11.Shaked H, Kashkush K, Ozkan H, Feldman M, Levy AA. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell. 2001;13:1749–59. doi: 10.1105/tpc.13.8.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet. 2003;33:102–6. doi: 10.1038/ng1063. [DOI] [PubMed] [Google Scholar]
- 13.Adams KL, Wendel JF. Novel patterns of gene expression in polyploid plants. Trends Genet. 2005;21:539–43. doi: 10.1016/j.tig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Tian L, Lee HS, Wei NE, Jiang H, Watson B, et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics. 2006;172:507–17. doi: 10.1534/genetics.105.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flagel L, Udall J, Nettleton D, Wendel J. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 2008;6:16. doi: 10.1186/1741-7007-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hovav R, Udall JA, Chaudhary B, Rapp R, Flagel L, Wendel JF. Partitioned expression of duplicated genes during development and evolution of a single cell in a polyploid plant. Proc Natl Acad Sci USA. 2008;105:6191–5. doi: 10.1073/pnas.0711569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha M, Lu J, Tian L, Ramachandran V, Kasschau KD, Chapman EJ, et al. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc Natl Acad Sci USA. 2009;106:17835–40. doi: 10.1073/pnas.0907003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pang M, Woodward AW, Agarwal V, Guan X, Ha M, Ramachandran V, et al. Genome-wide analysis reveals rapid and dynamic changes in miRNA and siRNA sequence and expression during ovule and fiber development in allotetraploid cotton (Gossypium hirsutum L.) Genome Biol. 2009;10:R122. doi: 10.1186/gb-2009-10-11-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rapp RA, Udall JA, Wendel JF. Genomic expression dominance in allopolyploids. BMC Biol. 2009;7:18. doi: 10.1186/1741-7007-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comai L. The advantages and disadvantages of being polyploid. Nat Rev Genet. 2005;6:836–46. doi: 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZJ. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 2007;58:377–406. doi: 10.1146/annurev.arplant.58.032806.103835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–3. doi: 10.1126/science.1153585. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Xia G. The place of asymmetric somatic hybridization in wheat breeding. Plant Cell Rep. 2014;33:595–603. doi: 10.1007/s00299-013-1552-9. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Xiang F, Xia G, Chen H. Transfer of small chromosome fragments of Agropyron elongatum to wheat chromosome via asymmetric somatic hybridization. Science in China Series C Life Sciences. 2004;47:434–41. doi: 10.1360/03yc0150. [DOI] [PubMed] [Google Scholar]
- 25.Xia GM, Xiang FN, Zhou AF, Wang HA, Chen HM. Asymmetric somatic hybridization between wheat (Triticum aestivum L.) and Agropyron elongatum (Host) Nevishi. Theor Appl Genet. 2003;107:299–305. doi: 10.1007/s00122-003-1247-7. [DOI] [PubMed] [Google Scholar]
- 26.Chen SY, Xia GM, Quan TY, Xiang FN, Yin J, Chen HM. Introgression of salt-tolerance from somatic hybrids between common wheat and Thinopyrum ponticum. Plant Sci. 2004;167:773–9. doi: 10.1016/j.plantsci.2004.05.010. [DOI] [Google Scholar]
- 27.Wang J, Xiang FN, Xia GM. Agropyron elongatum chromatin localization on the wheat chromosomes in an introgression line. Planta. 2005;221:277–86. doi: 10.1007/s00425-004-1443-y. [DOI] [PubMed] [Google Scholar]
- 28.Peng Z, Wang M, Li F, Lv H, Li C, Xia G. A proteomic study of the response to salinity and drought stress in an introgression strain of bread wheat. Mol Cell Proteomics. 2009;8:2676–86. doi: 10.1074/mcp.M900052-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang MC, Peng ZY, Li CL, Li F, Liu C, Xia GM. Proteomic analysis on a high salt tolerance introgression strain of Triticum aestivum/Thinopyrum ponticum. Proteomics. 2008;8:1470–89. doi: 10.1002/pmic.200700569. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Liu S, Wang M, Wei T, Meng C, Wang M, et al. A wheat SIMILAR TO RCD-ONE gene enhances seedling growth and abiotic stress resistance by modulating redox homeostasis and maintaining genomic integrity. Plant Cell. 2014;26:164–80. doi: 10.1105/tpc.113.118687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S, Li F, Kong L, Sun Y, Qin L, Chen S, et al. Genetic and epigenetic changes in somatic hybrid introgression lines between wheat and tall wheatgrass. Genetics. 2015;199:1035–45. doi: 10.1534/genetics.114.174094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng DS, Xia GM, Zhao SY, Chen FG. Two quality-associated HMW glutenin subunits in a somatic hybrid line between Triticum aestivum and Agropyron elongatum. Theor Appl Genet. 2004;110:136–44. doi: 10.1007/s00122-004-1810-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Lv J, Zhao X, Ai X, Zhu X, Wang M, et al. TaCHP: A wheat zinc finger protein gene down-regulated by abscisic acid and salinity stress plays a positive role in stress tolerance. Plant Physiol. 2010;154:211–21. doi: 10.1104/pp.110.161182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Xu C, Yang Y, Xia G. Functional analysis of TaDi19A, a salt-responsive gene in wheat. Plant Cell Environ. 2010;33:117–29. doi: 10.1111/j.1365-3040.2009.02080.x. [DOI] [PubMed] [Google Scholar]
- 35.Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms. Ann Bot. 1995;76:113–76. doi: 10.1006/anbo.1995.1085. [DOI] [Google Scholar]
- 36.Bennett MD, Smith JB. Nuclear DNA amounts in angiosperms. Philos Trans R Soc Lond B Biol Sci. 1976;274:227–74. doi: 10.1098/rstb.1976.0044. [DOI] [PubMed] [Google Scholar]
- 37.McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- 38.Kawaura K, Mochida K, Enju A, Totoki Y, Toyoda A, Sakaki Y, et al. Assessment of adaptive evolution between wheat and rice as deduced from full-length common wheat cDNA sequence data and expression patterns. BMC Genomics. 2009;10:271. doi: 10.1186/1471-2164-10-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wicker T, Krattinger SG, Lagudah ES, Komatsuda T, Pourkheirandish M, Matsumoto T, et al. Analysis of intraspecies diversity in wheat and barley genomes identifies breakpoints of ancient haplotypes and provides insight into the structure of diploid and hexaploid Triticeae gene pools. Plant Physiol. 2009;149:258–70. doi: 10.1104/pp.108.129734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker GLA, Edwards KJ. A genome-wide analysis of single nucleotide polymorphism diversity in the world’s major cereal crops. Plant Biotech J. 2009;7:318–25. doi: 10.1111/j.1467-7652.2009.00412.x. [DOI] [PubMed] [Google Scholar]
- 41.Comai L. Genetic and epigenetic interactions in allopolyploid plants. Plant Mol Biol. 2000;43:387–99. doi: 10.1023/A:1006480722854. [DOI] [PubMed] [Google Scholar]
- 42.Kashkush K, Feldman M, Levy AA. Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics. 2002;160:1651–9. doi: 10.1093/genetics/160.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–4. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 45.Qin Z, Cai Z, Xia G, Wang M. Synonymous codon usage bias is correlative to intron number and shows disequilibrium among exons in plants. BMC Genomics. 2013;14:56. doi: 10.1186/1471-2164-14-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han FP, Fedak G, Ouellet T, Liu B. Rapid genomic changes in interspecific and intergeneric hybrids and allopolyploids of Triticeae. Genome. 2003;46:716–23. doi: 10.1139/g03-049. [DOI] [PubMed] [Google Scholar]
- 47.He P, Friebe BR, Gill BS, Zhou JM. Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Mol Biol. 2003;52:401–14. doi: 10.1023/A:1023965400532. [DOI] [PubMed] [Google Scholar]
- 48.Wang M, Qin L, Xie C, Li W, Yuan J, Kong L, et al. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physioly. 2014;55:1354–65. doi: 10.1093/pcp/pcu059. [DOI] [PubMed] [Google Scholar]
- 49.Feldman M, Liu B, Segal G, Abbo S, Levy AA, Vega JM. Rapid elimination of low-copy DNA sequences in polyploid wheat: A possible mechanism for differentiation of homoeologous chromosomes. Genetics. 1997;147:1381–7. doi: 10.1093/genetics/147.3.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B, Vega JM, Segal G, Abbo S, Rodova M, Feldman M. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. I. Changes in low-copy non-coding DNA sequences. Genome. 1998;41:272–7. doi: 10.1139/g98-011. [DOI] [PubMed] [Google Scholar]
- 51.Liu B, Vega MJ, Feldman M. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome. 1998;41:535–42. doi: 10.1139/g98-052. [DOI] [PubMed] [Google Scholar]
- 52.Ma XF, Fang P, Gustafson JP. Polyploidization-induced genome variation in triticale. Genome. 2004;47:839–48. doi: 10.1139/g04-051. [DOI] [PubMed] [Google Scholar]
- 53.Ma XF, Gustafson JP. Timing and rate of genome variation in triticale following allopolyploidization. Genome. 2006;49:950–8. doi: 10.1139/G06-078. [DOI] [PubMed] [Google Scholar]
- 54.Gaut BS, Doebley JF. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc Natl Acad Sci USA. 1997;94:6809–14. doi: 10.1073/pnas.94.13.6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaut BS, Le Thierry d’Ennequin M, Peek AS, Sawkins MC. Maize as a model for the evolution of plant nuclear genomes. Proc Natl Acad Sci USA. 2000;97:7008–15. doi: 10.1073/pnas.97.13.7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bento M, Gustafson P, Viegas W, Silva M. Genome merger: from sequence rearrangements in triticale to their elimination in wheat-rye addition lines. Theor Appl Genet. 2010;121:489–97. doi: 10.1007/s00122-010-1325-6. [DOI] [PubMed] [Google Scholar]
- 57.Wicker T, Buchmann JP, Keller B. Patching gaps in plant genomes results in gene movement and erosion of colinearity. Genome Res. 2010;20:1229–37. doi: 10.1101/gr.107284.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu C, Li S, Wang M, Xia G. A transcriptomic analysis reveals the nature of salinity tolerance of a wheat introgression line. Plant Mol Biol. 2012;78:159–69. doi: 10.1007/s11103-011-9854-1. [DOI] [PubMed] [Google Scholar]
- 59.Swindell SR, Plasterer TN. SEQMAN. Springer New York: Sequence data analysis guidebook; 1997. [Google Scholar]
- 60.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 62.Baldauf SL. The deep roots of eukaryotes. Science. 2003;300:1703–6. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]