

Abstract
Divalent metal-ion transporter-1 (DMT1) is a widely expressed iron-preferring membrane-transport protein that serves a critical role in erythroid iron utilization. We have investigated its role in intestinal metal absorption by studying a mouse model lacking intestinal DMT1 (i.e., DMT1int/int). DMT1int/int mice exhibited a profound hypochromic-microcytic anemia, splenomegaly, and cardiomegaly. That the anemia was due to iron deficiency was demonstrated by the following observations in DMT1int/int mice: 1) blood iron and tissue nonheme-iron stores were depleted; 2) mRNA expression of liver hepcidin (Hamp1) was depressed; and 3) intraperitoneal iron injection corrected the anemia, and reversed the changes in blood iron, nonheme-iron stores, and hepcidin expression levels. We observed decreased total iron content in multiple tissues from DMT1int/int mice compared with DMT1+/+ mice but no meaningful change in copper, manganese, or zinc. DMT1int/int mice absorbed 64Cu and 54Mn from an intragastric dose to the same extent as did DMT1+/+ mice but the absorption of 59Fe was virtually abolished in DMT1int/int mice. This study reveals a critical function for DMT1 in intestinal nonheme-iron absorption for normal growth and development. Further, this work demonstrates that intestinal DMT1 is not required for the intestinal transport of copper, manganese, or zinc.
Keywords: copper absorption, iron deficiency, iron-deficiency anemia, iron-refractive iron-deficiency anemia, manganese absorption, SLC11A2, zinc metabolism
iron deficiency is the most prevalent micronutrient deficiency worldwide (4, 50). Its deficiency leads to iron-deficiency anemia, and to neurological and developmental disorders in children (3, 4). Since there exists no regulated mechanism for the excretion of iron, regulation of the whole body iron economy is achieved by tightly controlling absorption of the metal (21). Failure to regulate iron absorption in a manner appropriate to iron status is characteristic of several hereditary iron-overload disorders and iron-refractive iron-deficiency anemia (12, 19, 21).
Divalent metal-ion transporter-1 (DMT1; reviewed in Ref. 52) is a widely expressed mammalian proton-coupled iron transporter (23, 38). Mice in which the SLC11A2 gene coding for DMT1 was globally inactivated (i.e., SLC11A2−/−) exhibited a severe hypochromic-microcytic anemia and did not survive more than 7 days (22). A critical role for DMT1 in erythroid iron acquisition was confirmed by the following observations: 1) transfusion of red blood cells, but not parenteral iron injections, improved survival of SLC11A2−/− mice; and 2) lethal dose-irradiated wild-type mice into which the investigators transplanted hematopoietic stem cells from SLC11A2−/− mice exhibited defective erythropoiesis (22).
The microcytic (mk) mouse and Belgrade (b) rat models, inbred strains that harbor a Gly185→Arg mutation in DMT1 (17, 18), also exhibited an anemia phenotype. Parenteral iron injections partially improved the condition, and tissue or vesicle preparations from the mk mouse and b/b rat revealed deficiencies in intestinal iron transport (14, 30). The mk mouse and b/b rat anemia phenotypes were, however, less severe than that of the SLC11A2−/− knockout model, a finding that may be explained by the observation that the G185R mutant retains a fraction of wild-type activity (52).
Rare mutations in human DMT1 result in severe hypochromic-microcytic anemia (52); however, that four of five probands also exhibited hepatic iron overload has raised questions as to the importance of DMT1 in intestinal iron absorption and iron homeostasis. Nevertheless, hepatic iron loading is also apparent in the b/b rat model (54). Targeted deletion of DMT1 in the mouse intestine has provided a superior model in which to test the role of DMT1 in intestinal iron absorption. Initial characterization of the intestine-specific DMT1 knockout mouse (i.e., DMT1int/int) revealed a progressive anemia that was prevented by parenteral iron injection (22), consistent with a primary defect in iron absorption; however, iron absorption has not been directly examined in that model.
In the present study, we have further examined the role of DMT1 in intestinal metal absorption by studying the DMT1int/int mouse. Our initial goal was to test a critical role for DMT1 in intestinal iron absorption and provide a detailed analysis of the DMT1int/int phenotype in male and female mice. We examined the effects of loss of intestinal DMT1, with and without parenteral iron, on iron homeostasis and the expression of iron-related genes. Second, we directly tested the contribution of DMT1 to intestinal nonheme-iron absorption by measuring 59Fe absorption from an oral dose, and localized the site of the primary lesion. Gunshin et al. (22) found that the SLC11A2+/− (global) heterozygote exhibited lower iron stores than did wild-type mice, and suggested that haploinsufficiency of intestinal absorption could explain this finding. We have more directly tested this possibility here in heterozygotes (DMT1+/int) of the intestinal knockout mouse model.
Whereas DMT1 is reactive with a broad range of divalent metal ions in vitro, it exhibits a strong preference for Fe2+ over the other physiologically relevant metal ions it can transport (27). We ranked these according to the specificity constant as follows: Fe2+ > Co2+, Mn2+ >> Zn2+. A central objective in the present study was to determine which of these metals relies upon DMT1 for their absorption. We have tested the hypothesis that intestinal DMT1 is required for the absorption of copper, manganese, and zinc by examining metal metabolism and directly measuring intestinal absorption of radiotracer metals in the DMT1int/int mouse. We did not test cobalt because the nutritional requirements for free cobalt (i.e., aside from cobalamin) are very minor. We included copper for the following reasons. 1) Previous studies demonstrated that Ctr1 is a primary high-affinity Cu+ transporter in mammals, particularly within the intestine; however, a Ctr1-independent transport activity has been detected in Ctr1−/− mouse embryonic fibroblasts (33). 2) Whereas we previously found no evidence that copper is a transported substrate of DMT1 in vitro (27), others have measured copper transport in vitro and concluded that DMT1 can transport Cu+ or Cu2+ (1, 2, 16, 29, 34). Conversely, in two of the studies just cited, investigators proposed that Ctr1 is also capable of transporting Fe2+ (16, 34), so we also examined the expression of Ctr1 in the intestinal DMT1int/int mouse.
MATERIALS AND METHODS
Reagents.
Reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Animals.
We examined iron metabolism in adult mice with intestine-specific knockout of DMT1, heterozygotes, and their wild-type littermates, following a protocol approved by the University of Cincinnati Institutional Animal Care and Use Committee. We generated the DMT1int/int model by crossing a floxed DMT1 mouse line (DMT1fl/fl) (22) on the 129S6 strain with the villin-Cre transgenic line (Vil-Cre) (15) on the C57BL/6 strain. Experimental mice (F2) were of a mixed background, 75% 129S6, 25% C57BL/6 except as follows. The Vil-Cre mouse line was also backcrossed over 9 generations onto the 129S6 strain and crossed with the DMT1fl/fl line to produce 1) DMT1int/int mice on a homogeneous 129S6 background used in experiments described in Figs. 9, I–L, and 10; and 2) wild-type and Cre-positive mice on a homogeneous 129S6 background used in the experiment described in Table 2. All Cre-positive mice were hemizygous for the Vil-Cre recombinase transgene. Animals were genotyped by obtaining DNA from tail clips and analyzed as described previously (15, 22). Mice were fed a standard chow containing 350 ppm Fe (7922 NIH-07, Harlan Laboratories, Indianapolis, IN).
Fig. 9.

Intestinal iron, copper, and manganese absorption in iron-replete male control (DMT1+/+) and intestinal DMT1 knockout (DMT1int/int) mice. Conscious mice were dosed with 0.1 μCi 59Fe or 54Mn or 1 μCi 64Cu per gram body weight via oral-intragastric gavage. Blood radiotracer data (DMT1+/+, gray symbols, lines; and DMT1int/int, black symbols, lines) were analyzed by 2-way ANOVA with repeated measures over time but, for clarity, are displayed as mean, SD. A–C: 59Fe absorption in DMT1+/+ (n = 3) and DMT1int/int (n = 7). A: blood 59Fe content as a function of time after dosing, interaction (P < 0.001) B: enterocyte 59Fe content at 4 h, analyzed by using the rank-sum test (P = 0.017). C: liver 59Fe content at 4 h (P = 0.033). D: serial hematocrit measurements for DMT1+/+ (gray symbols and lines) and DMT1int/int (black symbols and lines) mice as a function of time after the oral radiotracer dose. Each line-and-scatter plot depicts the data for an individual mouse. Two-way ANOVA with repeated measures (time) revealed a main effect of time (P < 0.001) but not genotype (P = 0.54); no interaction (P = 0.26). E–H: 64Cu absorption in DMT1+/+ (n = 7) and DMT1int/int (n = 5) mice. E: blood 64Cu content as a function of time after dosing, no interaction (P < 0.10). F: enterocyte 64Cu content at 4 h (P = 0.88). G: liver 64Cu content at 4 h (P = 0.94). H: serial hematocrit measurements for DMT1+/+ (gray) and DMT1int/int (black) mice as a function of time after the oral radiotracer dose. Each plot depicts the data for an individual mouse. Two-way ANOVA with repeated measures revealed main effects of time (P < 0.001) and genotype (P = 0.008) but no interaction (P = 0.31). I–L: 54Mn absorption in DMT1+/+ (n = 3) and DMT1int/int (n = 5) mice. I: blood 54Mn content as a function of time after dosing, no interaction (P = 0.34). J: enterocyte 54Mn content at 4 h (P = 0.56). K: liver 54Mn content at 4 h (P ≥ 0.99). L: serial hematocrit measurements for DMT1+/+ (gray) and DMT1int/int (black) mice as a function of time after the oral radiotracer dose. Each plot depicts the data for an individual mouse. Two-way ANOVA with repeated measures revealed no effects (P ≤ 0.48).
Fig. 10.

Phenotypic characterization of male mice heterozygous for intestinal DMT1. Male control (DMT1fl/+|Cre−; A–F, n = 6–11; G–J, n = 3) and intestine-specific DMT1 heterozygous (DMT1+/int; A–F, n = 13–14; G–J, n = 4) mice at ∼150 days of age. A: hematocrit. B: hemoglobin (Hgb) concentration. C: mean corpuscular volume (MCV). D: serum iron concentration. E: serum transferrin saturation. F: liver nonheme iron content. G–J: expression of iron-related intestinal and hepatic genes. qPCR data are expressed as geometric mean (and SD) relative fold expression and are unitless. Student's t-tests and the FDR procedure revealed no differences between DMT1+/+ and DMT1+/int (all comparisons, 0.058 ≤ P ≤ 0.77, except liver nonheme iron, Pi = 0.011, which was found not significant by the FDR procedure).
Table 2.
Effect of introducing the Cre transgene in 129S6 mice
| Cre− | Cre+ | Pi, Significant by FDR? | |
|---|---|---|---|
| Body weight, g | 24.9 ± 5.8 (6) | 24.7 ± 2.1 (8) | 0.91, No |
| Hematocrit, % | 45.8 ± 2.2 (6) | 45.9 ± 2.2 (8) | 0.97, No |
| Red blood cell count, × 106/μl | 9.1 ± 0.5 (5) | 9.3 ± 0.5 (7) | 0.55, No |
| Mean corpuscular volume, fl | 53.0 ± 1.7 (5) | 53.3 ± 0.5 (6) | 0.66, No |
| [Hemoglobin], g/dl | 14.7 ± 0.8 (5) | 14.6 ± 0.6 (7) | 0.81, No |
| Mean corpuscular hemoglobin, pg | 16.2 ± 0.3 (5) | 15.7 ± 0.4 (7) | 0.042, No |
| Mean corpuscular [hemoglobin], g/dl | 30.6 ± 1.3 (5) | 30.1 ± 0.7 (7) | 0.45, No |
| Serum Fe, μM | 39.9 ± 15.5 (6) | 20.8 ± 7.9 (8) | 0.011, No |
| Transferrin saturation, % | 67.9 ± 14.5 (5) | 49.9 ± 19.3 (8) | 0.10, No |
| Spleen weight, mg/g body wt | 2.1 ± 0.7 (6) | 2.2 ± 0.2 (8) | 0.86, No |
| Heart weight, mg/g body wt | 5.1 ± 0.6 (6) | 4.9 ± 0.1 (7) | 0.43, No |
| Liver nonheme Fe, μmol/g wet tissue | 5.3 ± 0.8 (6) | 4.1 ± 0.3 (8) | 0.004, No |
| Heart nonheme Fe, μmol/g wet tissue | 1.0 ± 0.3 (6) | 1.3 ± 0.1 (8) | 0.030, No |
| Spleen nonheme Fe, μmol/g wet tissue | 17.2 ± 4.1 (5) | 20.2 ± 3.7 (8) | 0.20, No |
Data are means ± SD (n). Body and tissue weights, hematological and blood-iron variables, and tissue nonheme iron were analyzed in male mice (wild type for DMT1) lacking (Cre−) or hemizygous (Cre+) for the villin-Cre transgene. Mean (SD) age of Cre− mice, 106 (35) days (n = 6), did not differ from that of Cre+ mice, 112 (7) days (n = 8) (P = 0.62). Student's t-tests and the FDR procedure revealed that no variable differed between Cre− and Cre+.
Characterization of the DMT1int/int mouse model.
We sampled peripheral blood via cardiac puncture and isolated tissues from mice of ∼120 days of age subjected to isoflurane anesthesia (by inhalation, to effect) in the following set of experiments: 1) male DMT1 wild type lacking (Cre−) or bearing (Cre+) the Cre recombinase transgene to examine any effect of introducing Cre (Table 2); 2) male experimental control DMT1fl/fl|Cre− (hereafter DMT1+/+) compared with DMT1int/int (Figs. 1A, 2–4, 6, 8, and 9); 3) female experimental control (DMT1+/+) compared with DMT1int/int (Figs. 5 and 7); and 4) male DMT1fl/+ with and without the Cre transgene, i.e., control mice compared with mice heterozygous for intestinal DMT1 (Fig. 10).
Fig. 1.
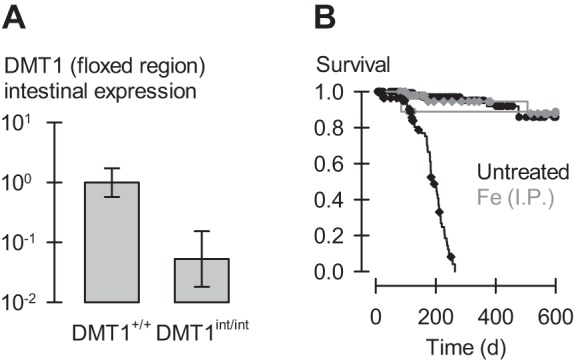
Intestinal DMT1 knockout mouse model. A: qPCR analysis of the expression of full-length DMT1 in male DMT1-floxed mice lacking (DMT1+/+, n = 3) or bearing the villin-Cre transgene (DMT1int/int, n = 4) at ∼120 days of age. We used a primer targeting the floxed region of DMT1 (exon 7–9) (Table 1). Data are geometric mean, SD relative fold expression and are unitless; P = 0.008 by Student's t-test. B: effect of ablation of intestinal DMT1 on survival in the mouse. Survival analysis (over the study period 1–600 days) of untreated DMT1+/+ (black circles, n = 215), untreated DMT1int/int (black diamonds, n = 125), as well as DMT1+/+ (gray circles, n = 11) and DMT1int/int (gray diamonds, n = 56) given Fe (ip) at age approximately 28 and 56 days. Steps denote events; symbols denote censored observations. Log-rank statistic, P < 0.001; Holm-Šidák pairwise comparisons revealed that untreated DMT1int/int differed from untreated DMT1+/+ and Fe-treated DMT1int/int (P < 0.001) but that no other groups differed from one another (0.071 > P > 0.97).
Fig. 2.
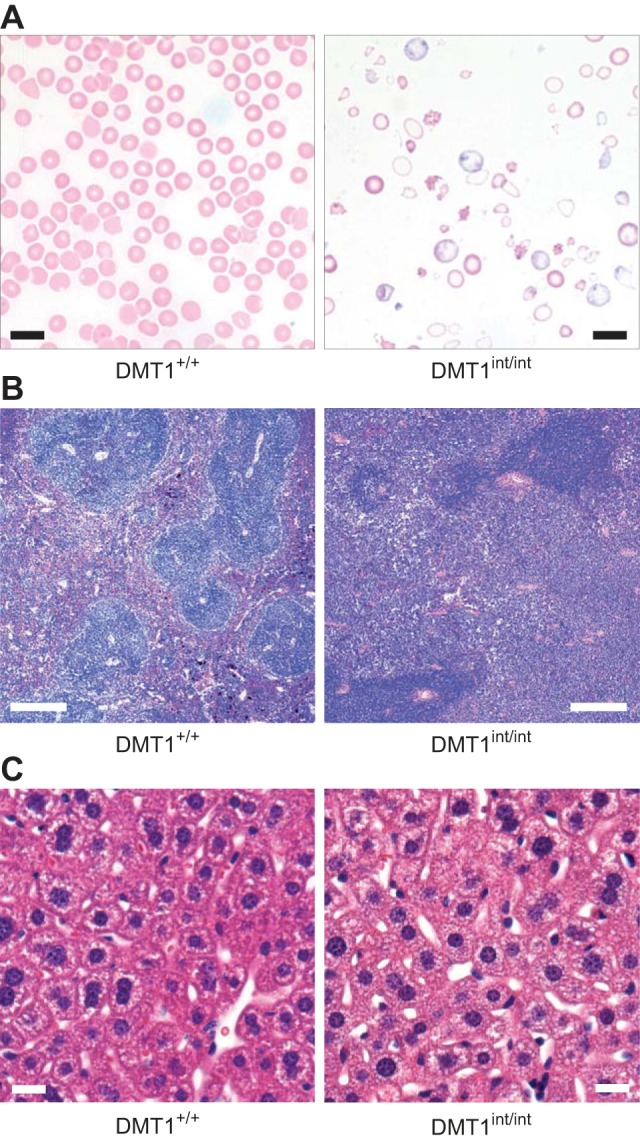
Blood and tissues of control (DMT1+/+) and intestinal DMT1 knockout (DMT1int/int) mice. A: peripheral blood smears prepared by using Wright-Giemsa stain (scale bars, 10 μm). B: histology of spleen prepared by using hematoxylin and eosin stain (scale bars, 100 μm). C: histology of liver prepared by using hematoxylin and eosin stain (scale bars, 40 μm).
Fig. 4.

Tissue weights and tissue nonheme iron content in male control and intestinal DMT1-null mice. Male control (DMT1+/+) and intestine-specific DMT1 knockout (DMT1int/int) mice (∼120 days of age) were untreated (solid bars) or given Fe (ip) at age approximately 28 and 56 days (hatched bars). A–C: spleen, heart, and kidney weight normalized by body weight (n = 4–12 per group): 2-way ANOVA revealed interactions (P ≤ 0.029) for each organ; within Fe (ip)-injected, DMT1int/int did not differ from DMT1+/+ (P ≥ 0.65). D: liver nonheme iron content (n = 4–12 per group): 2-way ANOVA revealed an interaction (P < 0.001); within untreated, DMT1int/int differed from DMT1+/+ (P < 0.001). E: spleen nonheme iron content (n = 3–12 per group): 2-way ANOVA revealed main effects of genotype (P < 0.001) and Fe (ip) injection (P < 0.001) but no interaction (P = 0.12). F–H: tissue nonheme iron content (n = 4–12 per group): 2-way ANOVA revealed no effects of genotype for heart (P = 0.098), kidney (P = 0.57), or skeletal muscle (gastrocnemius) (P = 0.57).
Fig. 6.

Tissue metal content in male control and intestinal DMT1-null mice. Tissue metal content (in units ng per mg of wet tissue) measured by inductively coupled plasma mass spectrometry (ICP-MS) in male control (DMT1+/+, n = 7) and intestine-specific DMT1 knockout (DMT1int/int, n = 8) mice (∼120 days of age). A–E: iron content in liver, heart, spleen, kidney, and muscle, respectively. F–J: copper content in liver, heart, spleen, kidney, and muscle, respectively. K–O: manganese content in liver, heart, spleen, kidney, and muscle, respectively. P–T: zinc content in liver, heart, spleen, kidney, and muscle, respectively. Data were analyzed using Student's t-tests controlled by the FDR procedure for 20 comparisons: aDMT1int/int differed from DMT1+/+ (P ≤ 0.011). No other significant differences: b(Pi = 0.041) and c(Pi = 0.029) were found not significant by the FDR procedure (i.e., DMT1int/int did not differ from DMT+/+); all other comparisons, 0.057 ≤ P ≤ 0. 57.
Fig. 8.

Gene expression in male control and intestinal DMT1-null mice. Gene expression of intestinal ferrireductases and transporters (A–E) and the hepatic iron-regulatory hormone hepcidin (Hamp1) in male control (DMT1+/+) and intestine-specific DMT1 knockout (DMT1int/int) mice (∼120 days of age) that were untreated (n = 6–10, dark gray bars) or given Fe (ip) at age approximately 28 and 56 days (n = 3–4, light gray bars). Data are expressed as geometric mean (and SD) relative fold expression and are unitless. A: intestinal divalent metal-ion transporter-1 (DMT1), primer upstream of the first loxP site. Two-way ANOVA revealed an interaction (P = 0.028); iron-treated groups did not differ by genotype (P = 0.14). B: intestinal Cybrd1 (DcytB), interaction (P = 0.047); iron-treated groups did not differ by genotype (P = 0.25). C: intestinal Steap2, no effects (0.26 ≤ P ≤ 0.99). D: intestinal ferroportin (Fpn), no effects (0.24 ≤ P ≤ 0.28). E: intestinal Ctr1, main effect of iron treatment (P = 0.002) but not of genotype (P = 0.27), and no interaction (P = 0.52). F: liver Hamp1 (hepcidin), interaction (P < 0.001); iron-treated groups did not differ by genotype (P = 0.61).
Fig. 5.

Effects of intestinal ablation of DMT1 in female mice. Body and tissue weights, hematological and blood-iron variables, and tissue nonheme iron were analyzed in female control (DMT1+/+, n = 8–13) and intestine-specific DMT1 knockout (DMT1int/int, n = 3–6). Mean (SD) age of DMT1+/+ mice was 170 (53) days (n = 13) and that of DMT1int/int mice, 103 (18) days (n = 6). A: body weight. B: hematocrit. C: red blood cell count. D: mean corpuscular volume. E: hemoglobin (Hgb) concentration. F: mean corpuscular hemoglobin (MCH). G: serum iron concentration. H: serum transferrin saturation. I and J: spleen and heart weight normalized by body weight. K: liver nonheme iron content. L: spleen nonheme iron content. Student's t-tests revealed that DMT1+/+ and DMT1int/int differed for all variables (P ≤ 0.006) except MCH (P = 0.078).
Fig. 7.
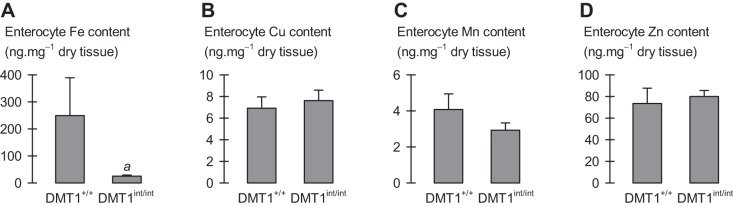
Enterocyte metal content in female control and intestinal DMT1-null mice. Enterocyte metal content measured by inductively coupled plasma mass spectrometry (ICP-MS) in female control (DMT1+/+, n = 4) and intestine-specific DMT1 knockout (DMT1int/int, n = 3) mice (∼120 days of age). A–D: enterocyte total iron, copper, manganese, and zinc content, respectively. Data were analyzed using Student's t-tests: aDMT1int/int differed from DMT1+/+ (P = 0.043); no other effects (0.089 ≤ P ≤ 0.50).
Blood and tissue analyses.
Automated complete blood count (CBC) was performed by Antech Diagnostics (Oak Brook, IL). Serum iron (SI) and unsaturated iron-binding capacity (UIBC) were assayed by using the Iron-SL and UIBC kits (Sekisui Diagnostics, San Diego, CA) according to manufacturer's protocols, and transferrin saturation (%) was computed as SI/(SI + UIBC) × 100.
Nonheme iron content of liver, heart, spleen, kidney, and skeletal muscle (gastrocnemius) was determined by using a standard acid-digestion, chromogen-based colorimetric assay as described (55), and normalized by wet tissue weight. Total metal (iron, copper, manganese, and zinc) content was measured by using high-resolution inductively coupled plasma mass spectrometry (ICP-MS) of nitric acid-digested wet tissues from male DMT1+/+ and DMT1int/int mice. We collected an enterocyte-enriched preparation from female DMT1+/+ and DMT1int/int mice by lightly scraping the luminal surface of the proximal 1–5 cm of small intestine (i.e., duodenum) with the edge of a glass microscope slide. Total metal content was normalized by tissue wet weight, except in the case of enterocytes (dry weight).
qPCR analyses.
Freshly isolated small intestine was flushed with ice-cold saline, and duodenal enterocytes sampled as described above. Enterocytes and liver tissue were collected into TRIzol reagent (Life Technologies, Carlsbad, CA), homogenized, and frozen at −80°C prior to their use in quantitative real-time PCR (qPCR) analysis. We performed reverse transcription by using 50 μg/ml oligo(dT)-20 primer and reverse transcriptase (Qiagen, Valencia, CA) according to the manufacturer's instructions. Sample cDNA concentrations were determined by measuring absorbance at 260 nm by using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). We amplified 200 ng sample cDNA with Fast SYBR Green Real-Time PCR master mix (Life Technologies) in a final volume of 20 μl by using the ABI Step One Machine. Gene expression was determined by using the delta delta CT method (36), with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA in each tissue sampled as reference, by using primers described in Table 1 to determine gene expression. qPCR data were normalized by the mean −ΔΔCT in untreated control (DMT1+/+) mice and expressed as 2−ΔΔCT, i.e., relative fold expression.
Table 1.
Primers used in real-time quantitative PCR assays of isolated mouse tissues
| Gene | NCBI Reference Sequence | Forward Primer | Reverse Primer | Ref. |
|---|---|---|---|---|
| Cybrd1 | AF_354666 | 5′–GCA GCG GGC TCG AGT TTA–3′ | 5′–TTC CAG GTC CAT GGC AGT CT–3′ | 41 |
| Ctr1 | NM_175090 | 5′–TAT GAA CCA CAC GGA CGA CAA–3′ | 5′–GCC ATT TCT CCA GGT GTA TTG A–3′ | N/A |
| DMT1 | NM_008732 | |||
| Upstream of first loxP site | 5′–CGC TCG GTA AGC ATC TCG AA–3′ | 5′–TGT TGC CAC CGC TGG TAT CT–3′ | 41 | |
| Within floxed region | 5′–TCC TCA CCA TCG CAG ACA CTT–3′ | 5′–TCC AAA CGT GAG GGC CAT GAT AGT–3′ | 56 | |
| Fpn | NM_016917 | 5′–CAT TGC TAG AAT CGG TCT T–3′ | 5′–GCA ACT GTG TCA CCG TCA AAT–3′ | 41 |
| Hamp1 | AF_503444 | 5′–AAG CAG GGC AGA CAT TGC GAT–3′ | 5′–CAG GAT GTG GCT CTA GGC TAT GT–3′ | 41 |
| GAPDH | NM_008084 | 5′–CAT GGC CTT CCG TGT TCC TA–3′ | 5′–CCT GCT TCA CCA CCT TCT TGA–3′ | 20 |
| Steap2 | BC150881.1 | 5′–ACG GAA AAC TGA AGG ACA GAA GA–3′ | 5′–GAG ATG GAT CGG GGG TTG TG–3′ | N/A |
N/A, not applicable.
Histology.
Liver and spleen tissue samples were rinsed in phosphate-buffered saline, fixed by using 4% (wt/vol) paraformaldehyde (Thermo Fisher Scientific), and embedded in paraffin. Sections of thickness 4 μm were stained with hematoxylin and eosin. Images were acquired using the Olympus BH2 microscope and Olympus Magnafire digital image-capture system with the aid of AxioVision version 4.8.1 software (Zeiss).
Absorption of iron, copper, and manganese in the DMT1int/int mouse.
We measured radiotracer metal absorption in female control mice and in DMT1int/int mice made iron replete by two intraperitoneal injections (28 and 56 days) of iron dextran (12.5 mg elemental Fe per kg body wt) in phosphate-buffered saline solution (pH 7.4). 59Fe (specific activity 61.6 mCi/mg) and 54Mn (specific activity 196 mCi/mg) were obtained from Perkin-Elmer (Boston, MA), and 64Cu (specific activity 0.081–0.25 mCi/mg) was obtained from Washington University-St. Louis. Conscious mice were administered 59Fe (0.1 μCi per gram body wt as FeSO4 in 1 mM NaCl, 1 mM l-ascorbic acid, 1 mM l-glutamine), 64Cu (1 μCi per gram body wt as CuCl2 in 10 mM NaCl, 1 mM l-ascorbic acid, 1 mM l-histidine), or 54Mn (0.1 μCi per gram body wt as MnCl2 in 1 mM NaCl, 1 mM l-ascorbic acid, 1 mM l-glutamine) via oral−intragastric gavage following an overnight fast.
Blood was collected into heparinized hematocrit tubes by tail-nick incision at 15 min, 45 min, 2 h, and 4 h. Blood samples were centrifuged for measurement of hematocrit and then expelled into 20-ml scintillation vials containing 1 ml Solvable solution (Perkin-Elmer). Mice were euthanized by CO2 asphyxiation at 4 h after which we collected ≈100 mg liver into 1 ml Solvable. Freshly isolated small intestine was flushed with ice-cold wash solution containing 130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM nitrilotriacetic acid (NTA), 5 mM 2-(N-morpholino)ethanesulfonic acid, and buffered to pH 7.4 by using N′,N′-diethylpiperazine, except that 1 mM l-ascorbate and 1 mM l-histidine were used in place of NTA for the 64Cu study. Duodenal enterocytes were collected (as described above) into 1.5-ml microcentrifuge tubes containing 0.99 ml Solvable plus 10 μl protease inhibitor cocktail I (EMD Biochemicals). One-half milliliter of this sample was transferred to a 20-ml scintillation vial with 1 ml Solvable and the remainder was used to quantify protein concentration by using the Pierce BCA assay (Thermo Fisher Scientific). Tissue samples in Solvable were further processed according to the manufacturer's protocol. We added Scintisafe−30% cocktail (Thermo Fisher Scientific) and measured sample radioactivity by using liquid-scintillation counting.
Statistics.
Statistical analyses were performed using SigmaPlot version 13 (Systat Software) with critical significance level α = 0.05. Data are presented as arithmetic mean and standard deviation (SD) for n independent observations, with the following exceptions: 1) qPCR data (relative fold expression) are expressed as geometric mean and SD, and 2) enterocyte and liver radiotracer content (Fig. 9) are presented as box plots (median, interquartile range, and whiskers representing 10th and 90th percentiles). Except where noted, between-group comparisons were made by using 1) two-way analysis of variance (ANOVA) or Kaplan-Meier log-rank test (survival analysis) followed by pairwise multiple comparisons by using the Holm-Šidák test when appropriate, or 2) Student's t-tests controlled by a false-discovery rate (FDR) procedure (11) in which we declare that a significant effect exists only when the individual P value (Pi) is less than the individual critical significance level (di*) computed for each comparison tested. Red blood cell morphology scores were tested by using one-way ANOVA on ranks; blood radiotracer content and hematocrit as a function of time post intragastric dose were analyzed by using repeated-measures two-way ANOVA; and tissue radiotracer content (data for which were not normally distributed) was analyzed by using the Mann-Whitney rank-sum test.
RESULTS
Intestine-specific DMT1 knockout mouse model.
We generated an intestine-specific DMT1 knockout mouse model by crossing the floxed DMT1 (DMT1fl/fl) (22) and villin-Cre transgenic (Vil-Cre) (15) mouse lines. In postnatal Vil-Cre mice, Cre/loxP recombination is observed exclusively and homogeneously in intestinal epithelial cells (15). Introduction of the Cre transgene (Cre+) alone in male 129S6 mice had no effect on body weight, hematological variables, blood-iron variables, spleen or heart weights, or iron stores (Table 2). Gunshin et al. (22) previously found that introduction of the loxP sites into the DMT1 (SLC11A2) gene (i.e., DMT1fl/fl) did not result in any observable phenotype.
In subsequent experiments, we used DMT1fl/fl mice without the Cre transgene as control mice (for simplicity, hereafter described as DMT1+/+) and DMT1fl/fl|Cre+ mice as our intestine-specific DMT1 knockout model (hereafter described as DMT1int/int). We confirmed efficient knockout of intestinal DMT1 by using qPCR analysis of an enriched enterocyte preparation with the use of primers targeting the floxed region of DMT1 (Table 1). Intestinal expression of full-length DMT1 in the untreated DMT1int/int mouse was ∼5% that in DMT1+/+ (Fig. 1A).
Previously, immunoblot analysis indicated that DMT1 was absent from duodenal epithelial cells of DMT1int/int mice, and Southern blot analysis of several tissues isolated from DMT1int/int mice revealed that the deletion of the floxed region was confined to intestinal tissues (22).
Ablation of intestinal DMT1 resulted in early mortality (Fig. 1B). Mean (± SE) survival time in DMT1int/int mice was 186 ± 7 days (n = 125) compared with 565 ± 14 days (n = 215) in DMT1+/+ mice (P < 0.001).
Intestine-specific knockout of DMT1 produces a severe hypochromic-microcytic anemia resulting from iron deficiency.
Intestine-specific knockout of DMT1 in the mouse resulted in a hypochromic-microcytic anemia accompanied by splenomegaly, cardiomegaly, and depleted blood iron and nonheme-iron stores.
Peripheral blood smears of male DMT1int/int mice (aged ∼120 days) revealed marked reticulocytosis (Fig. 2A) and a hypochromic-microcytic anemia, judged from the reduction in erythrocyte size and the increased area of central pallor in erythrocytes compared with DMT1+/+ mice (Fig. 2A). DMT1int/int mice exhibited marked polychromasia (P < 0.001), marked anisocytosis (P < 0.001), and moderate poikilocytosis (P = 0.005) (severity scores 0–4 from visual inspection, data not shown). Automated CBC revealed significant reductions in hematocrit, red blood cell count, mean corpuscular volume (MCV), hemoglobin concentration, and mean corpuscular hemoglobin (MCH) in DMT1int/int mice compared with DMT1+/+ mice (Fig. 3, A–E). We observed no effect on mean corpuscular hemoglobin concentration (MCHC) in DMT1int/int mice compared with DMT1+/+ mice (Fig. 3F), a result that may be explained by the coupled effects of reductions in MCV and MCH.
Fig. 3.

Hematological and blood-iron variables in male control and intestinal DMT1-null mice. Male control (DMT1+/+) and intestine-specific DMT1 knockout (DMT1int/int) mice (approximately 120 days of age) were untreated (solid bars) or given Fe (ip) at age approximately 28 and 56 days (hatched bars). A: hematocrit. B: red blood cell count. C: mean corpuscular volume. D: hemoglobin (Hgb) concentration. E: mean corpuscular hemoglobin. F: mean corpuscular hemoglobin concentration (MCHC). G: serum iron concentration. H: transferrin saturation. Statistical analyses (untreated, n = 5–12 per group; Fe-injected, n = 3–4 per group): with the exception of MCHC (P = 0.063), 2-way ANOVA revealed interactions for all variables (serum iron, P = 0.030; all other comparisons, P < 0.001). In A–E, G, H: within DMT1int/int, Fe (ip) injection produced a significant difference (P ≤ 0.003) and, within Fe (ip)-injected, DMT1int/int did not differ from DMT1+/+ (P ≥ 0.35).
Serum iron and transferrin saturation were decreased in DMT1int/int mice compared with DMT1+/+ mice (Fig. 3, G and H). Parenteral administration of iron dextran (12.5 mg Fe intraperitoneally at age approximately 28 and 56 days) was sufficient to substantially increase blood-iron variables (Fig. 3, G and H), and prevented or reversed the changes in hematological variables and early mortality observed in untreated DMT1int/int mice (Figs. 1B and 3, A–E).
Mean body weight of untreated DMT1int/int mice was 19.3 g (SD 3.1 g) (n = 12), significantly lower than that of their DMT1+/+ littermates, 29.8 (2.7) g (n = 12), whereas body weight did not differ between DMT1int/int and DMT1+/+ mice that had received parenteral Fe (interaction, P < 0.001).
The DMT1int/int mouse exhibited marked splenomegaly and cardiomegaly, and a modest increase in kidney weight compared with DMT1+/+ mice (Fig. 4, A–C). Histological examination of the spleen in the DMT1int/int mouse revealed a relative contraction and darkening of the white pulp, and an expansion of red pulp, in contrast to a greater relative volume of white pulp in the spleen of DMT1+/+ mice (Fig. 2B). Splenomegaly and red-pulp expansion are consistent with an anemia and may indicate increased extramedullary hematopoiesis in the DMT1int/int mouse (6).
Liver nonheme iron was depleted in DMT1int/int mice compared with DMT1+/+ mice (Fig. 4D), an effect not associated with any observed morphological change in the liver (Fig. 2C). Nonheme iron was also depleted in the spleen of DMT1int/int mice compared with DMT1+/+ mice (Fig. 4E). We found no significant changes in nonheme iron content in heart, kidney, and skeletal muscle of untreated mice (Fig. 4, F–H); however, modest reductions in heart and kidney nonheme iron content of untreated DMT1int/int mice may have been masked by the variability associated with large increases in tissue nonheme iron following parenteral iron treatment.
We found similar results in female mice (3–4 mo of age) lacking intestinal DMT1. Female DMT1int/int mice were growth limited (Fig. 5A) and exhibited a hypochromic-microcytic anemia characterized by reductions in hematocrit, RBC count, MCV, and Hgb concentration (Fig. 5, B–E); however, we observed no significant difference in MCH (Fig. 5F). We attribute this hypochromic-microcytic anemia in female DMT1int/int mice to severe iron deficiency since it was associated with substantially lower serum iron and transferrin saturation compared with DMT1+/+ mice, splenomegaly, cardiomegaly, and depleted nonheme iron stores (liver, spleen) (Fig. 5, G–L).
Although our statistical analyses did not directly compare variables between male and female mice, some differences in iron distribution are apparent between male and female control but not DMT1int/int mice (see Fig. 4, D and E, cf. Fig. 5, K and L), possibly an effect arising from the use here of a mixed background. Nevertheless, our study reveals no sexual divergence in the critical functional role of intestinal DMT1 in iron homeostasis.
Effect of DMT1 intestinal knockout on metal homeostasis in mice.
Tissue levels of total iron (i.e., heme iron and nonheme iron) were markedly decreased in the liver, heart, spleen, kidney, and skeletal muscle of male DMT1int/int mice compared with DMT1+/+ mice (Fig. 6, A–E). The effects were more profound in liver, spleen, and kidney than in heart and skeletal muscle, consistent with the depletion of iron stores; nevertheless total iron (which also includes myoglobin iron) was markedly depleted even in the critical muscle mass (cardiac muscle and gastrocnemius).
We found no meaningful changes in the tissue levels of copper, manganese, or zinc (Fig. 6, F–T). A modest decrease in skeletal muscle copper content of male DMT1int/int mice compared with DMT1+/+ mice was accompanied by an increase in copper content of the spleen (Fig. 6, H and J), and kidney manganese content was only modestly decreased in DMT1int/int mice (Fig. 6N). Enterocyte total iron content was decreased in female DMT1int/int mice compared with DMT1+/+ mice but there were no differences in the levels of copper, manganese, or zinc (Fig. 7).
Effect of intestinal DMT1 ablation on the regulation of genes involved in iron absorption and homeostasis.
We examined the effect of intestine-specific ablation of DMT1 on the expression of genes involved in iron and copper homeostasis. We used a DMT1 primer complementary to a region upstream of the first loxP site (and downstream of the transcription start site) such that it would serve as a reporter of the regulatory signal independent of expression of functional DMT1. We found that the DMT1 gene was upregulated (30-fold) and the intestinal ferrireductase Cybrd1 (DcytB) upregulated 48-fold in enterocytes of DMT1int/int mice compared with DMT1+/+ mice (Fig. 8, A and B); however, we found no change in the expression of the Steap2 gene coding a second candidate brush-border ferrireductase (Fig. 8C). mRNA expression of the basolateral iron exporter ferroportin was unchanged in DMT1int/int mice (Fig. 8D), as was the expression of the Ctr1 gene coding the apical copper transporter (Fig. 8E). Ablation of intestinal DMT1 downregulated the expression of the iron-regulatory hormone liver Hamp1 gene (hepcidin) to ≈1% that of DMT1+/+ mice (Fig. 8F). The changes in expression levels of DMT1, Cybrd1, and Hamp1 in DMT1int/int mice were corrected by parenteral Fe injection. Iron treatment increased the expression of Ctr1 in both DMT1int/int and DMT1+/+ mice, i.e., the effect of iron on Ctr1 was independent of DMT1.
Role of intestinal DMT1 in the absorption of iron, copper, and manganese.
To examine directly the role of intestinal DMT1 in the absorption of iron, copper, and manganese, we measured metal absorption in control (DMT1+/+) and DMT1int/int mice fed a radiotracer metal dose via oral-intragastric gavage. Although serial blood collections were small, we measured hematocrit over the 4-h time course of these experiments to ensure against large changes in hematocrit or the onset of hypovolemic shock. We observed in the 59Fe and 64Cu studies only very modest reductions in hematocrit over time and no time-dependent differences between DMT1+/+ and DMT1int/int (Fig. 9, D and H); we observed no changes in hematocrit in the 54Mn study (Fig. 9L).
In DMT1+/+ mice, 59Fe appeared in the blood within 15 min after the oral dose and peaked around 2 h. In contrast, 59Fe appearance in the blood of DMT1int/int mice was substantially blunted, 12% that of DMT1+/+ mice (area under the curve, 0–4 h) (Fig. 9A). We obtained for DMT1+/+ mice at 4 h robust 59Fe signals in enterocytes and liver (the latter tissue being expected to rapidly clear 59Fe from the portal and peripheral circulation); however, 59Fe content of enterocytes and liver of DMT1int/int mice was only a small fraction of that observed for DMT1+/+ mice (Fig. 9, B and C). These data indicate that intestinal DMT1 is required for iron absorption. The lack of 59Fe within enterocytes 4 h after an oral dose provides the first demonstration that the primary lesion in the DMT1int/int mouse is abolished iron uptake at the intestinal brush border (apical membrane). (We assume that we had bypassed the need for luminal reduction of iron by providing the 59Fe as FeSO4 in ascorbic acid.)
Blood 64Cu content in DMT1+/+ mice peaked later than did 59Fe (the 64Cu dose was ten-fold greater than the 59Fe given) and was no different in DMT1int/int mice (Fig. 9E). 64Cu content of enterocytes and liver did not differ between DMT1+/+ and DMT1int/int mice (Fig. 9, F and G). 54Mn appeared in blood within 15 min and did not differ between DMT1int/int and DMT1+/+ mice (Fig. 9I). Tissue 54Mn content in enterocytes and liver of DMT1int/int mice were no different from DMT1+/+ mice (Fig. 9, J and K). These data indicate that copper and manganese absorption do not rely upon intestinal DMT1.
Haplosufficiency of intestinal DMT1 in mice.
To determine the effect of omitting a single copy of the DMT1 gene in the intestine, we examined heterozygous mice (i.e., DMT1+/int). Mean body weight in DMT1+/int mice (≈150 days of age) was 27.8 g (SD 4.6 g), no different from control (DMT1+/+), 31.7 (6.5) g (n = 11–14). Spleen and heart sizes were normal (data not shown). Hematological variables, serum iron, transferrin saturation, and liver nonheme iron in DMT1+/int mice did not differ from those of DMT1+/+ mice (Fig. 10, A–F). Therefore, heterozygosity of intestinal DMT1 produced no observed phenotypic change in healthy mice fed a standard diet.
We examined the expression of genes involved in iron absorption and metabolism to test whether the normal-iron status of the heterozygote was achieved by upregulation of the iron-absorptive machinery. Intestinal mRNA levels of DMT1 (upstream of loxP), Cybrd1, and Fpn in DMT1+/int mice did not differ from those of DMT1+/+ mice (Fig. 10, G–I). Although we observed no regulation at the mRNA level, it is possible that protein levels of DMT1, Cybrd1, or ferroportin are elevated in the intestine of heterozygous DMT1+/int mice, e.g., as a result of increased protein stability. Nevertheless, we found no change in the hepatic expression of Hamp1 (Fig. 10J) coding hepcidin, the primary regulator of ferroportin protein levels (21).
DISCUSSION
Intestinal DMT1 is required for mammalian iron absorption and homeostasis.
We have performed a comprehensive characterization of a mouse model lacking intestinal DMT1. Our key findings were that 1) intestine-specific ablation of DMT1 produces a severe iron-deficiency anemia in the mouse; 2) absorption of 59Fe from an oral dose was virtually abolished in the DMT1int/int mouse whereas absorption of 64Cu and 54Mn did not differ from that in DMT1+/+ mice; and 3) tissue levels of copper, manganese, and zinc in adult DMT1int/int mice were normal whereas serum iron, and tissue levels of nonheme iron and total iron were profoundly depleted.
That the anemia phenotype of the DMT1int/int mouse could be prevented or rescued by intraperitoneal iron injection, thus bypassing the intestine, indicated that the lesion was specific to the intestine. In a similar manner, intraperitoneal copper administration rescued the lethality observed in a Ctr1int/int mouse (43). We found no evidence to support a defect in the ability of the liver to regulate iron metabolism since 1) the morphology of the DMT1int/int mouse liver was normal, and 2) hepcidin expression in the liver was strongly suppressed, as expected when serum iron and hepatic iron stores are extremely low (21).
More precisely, the primary defect in the DMT1int/int mouse was an inability to take up iron at the intestinal brush-border membrane. This conclusion is consistent with the immunolocalization of DMT1 on the apical membrane of enterocytes in mouse, rat, and human intestine (5, 47, 60). No other nonheme-iron transporter has been identified at the mammalian intestinal brush border. Whereas Zrt-like, Irt-like protein-14 (ZIP14) exhibits iron-uptake activity in vitro (35, 48) and is strongly expressed in the intestine (57), its expression is enriched on the basolateral membrane (25). Moreover, ZIP14-mediated iron uptake in vitro is optimal at pH 7.5 (Ref. 48) and therefore a functional role for ZIP14 in intestinal apical iron uptake from an acidic microenvironment (51) is doubtful, at least in the mature mammal.
Our study demonstrates that intestinal DMT1 is essential for iron homeostasis in the adult mouse and its ablation results in early mortality. We observed cardiomegaly in the DMT1int/int mouse by 4 mo of age, and preliminary evidence (J. Rubinstein, A. Shawki, J. N. Lorenz, B. Mackenzie, unpublished data) indicates that intestinal ablation of DMT1 produces left-ventricular dilation by 180 days of age (cf. mean survival of 186 days). We therefore suspect that a dilated cardiomyopathy explains the early mortality in the DMT1int/int mouse.
Whereas the DMT1int/int mouse is born iron replete—it has normal liver nonheme iron stores at birth (22)—we have nevertheless considered what may account for its survival to several months of age and for the residual 59Fe absorption from an intragastric dose. It is possible that DMT1 is not required in the suckling mammal which may obtain sufficient dietary iron by one of the following mechanisms: 1) apical uptake of lactoferrin, 2) apical uptake of free iron via an unknown transporter active in the newborn, or 3) absorption of iron via a paracellular pathway in the immature intestine. Nevertheless, we have preliminary evidence of reduced serum iron in the DMT1int/int mouse by 14 days of age and a mild anemia by 21 days (49). We think it is more likely that the residual iron absorption results from incomplete Cre-mediated excision of the floxed DMT1 gene in the enterocytes of the DMT1int/int mouse, based on the following observations: 1) the expression of full-length DMT1 mRNA in an enriched enterocyte preparation from untreated DMT1int/int mice was ∼5% that of DMT1+/+ mice (Fig. 1A), and 2) intestinal mRNA expression of DMT1 is virtually confined to enterocytes and is not detectable in goblet cells or lamina propria (23). Our data therefore support the notion that DMT1 is the only mechanism by which nonheme iron is taken up at the intestinal brush border in the adult mammal.
Haploinsufficiency of intestinal DMT1 had been suggested previously (22). We tested this possibility more directly in mice heterozygous for intestinal DMT1 and found no evidence of intestinal DMT1 haploinsufficiency. We found no change in hepcidin expression in liver nor of iron-related genes in the intestine; however, we did not test the possibility that transport protein levels were increased independently of hepcidin. Rare mutations in human DMT1 result in a severe hypochromic-microcytic anemia (52). All probands identified so far have been homozygous or compound heterozygous for DMT1 mutations. That parents and siblings of the proband patients appear healthy further supports the view that one wild-type SLC11A2 allele is sufficient for mammalian iron absorption.
Intestinal DMT1 is not required for copper, manganese, or zinc absorption.
In contrast to the deficiency in tissue iron levels in DMT1int/int mice, we found no meaningful change in tissue levels of copper, manganese, or zinc. We concluded that intestinal DMT1 is not required for zinc homeostasis, an anticipated outcome since 1) Zn2+ is a weak DMT1 substrate in vitro, relative to Fe2+ (Ref. 27), and 2) ZIP4, a metal transporter expressed at the intestinal brush border, appears to be sufficient for zinc absorption (10, 28). Homozygous knockout of ZIP4 in the mouse is embryonic lethal and heterozygotes display hypersensitivity to zinc deficiency (13). Hereditary defects in human ZIP4 result in zinc deficiency and acrodermatitis enteropathica (58).
Given that a Ctr1-independent copper-transport activity was previously characterized but not identified in Ctr1−/− mouse embryonic fibroblasts, and that DMT1 was previously suggested to import Cu+ or Cu2+ in intestinal cells or membrane vesicles, we evaluated a potential role for DMT1 in intestinal copper acquisition. The absorption of 64Cu from an intragastric dose did not differ between DMT1int/int and DMT1+/+ mice, confirming that copper does not rely on DMT1 for its absorption. We previously found that Ctr1, a high-affinity copper transporter (32), is localized to both the apical membrane and endosomal compartments of mammalian enterocytes (44). Intestinal ablation of Ctr1 in the mouse produced a severe copper deficiency and early lethality (43), establishing Ctr1 as the primary mechanism serving copper uptake at the intestinal brush border, and these studies together indicate no role for DMT1 in this process.
Moreover, intestinal ablation of DMT1 had no effect on 54Mn absorption or tissue manganese levels except for a modest decrease in renal manganese content. Others have observed decreased manganese transport in two intestinal preparations from the Belgrade rat model, by using closed duodenal loops in situ or intestinal brush-border membrane vesicles (7, 30). That the manganese concentrations used in the studies just cited were supraphysiological and higher than the intestinal luminal concentration expected following oral gavage in the present study may explain in part this discrepancy. Furthermore, DMT1 exhibits higher affinity for Fe2+ than it does for Mn2+ in vitro (27). Our data indicate that DMT1 plays no physiologically relevant role in manganese absorption; however, the possibility remains that DMT1 could contribute to manganese absorption in manganese overexposure or dietary iron deficiency. Alternative intestinal uptake systems for manganese have not been identified but candidates include ZIP14 which is strongly expressed in the intestine and exhibits manganese transport activity in vitro (48, 57). Consistent with the strong preference of DMT1 for iron over any other physiological substrate tested in vitro (27), our present data support the notion that intestinal DMT1 is specific to iron absorption, at least under normal physiological conditions.
Model of severe iron deficiency and iron-deficiency anemia.
We attributed the severe hypochromic-microcytic anemia, splenomegaly, and cardiomegaly of the DMT1int/int mouse to iron deficiency. The effects of intestinal ablation of DMT1 on hematological and blood-iron variables were more profound than those typically observed in mice fed an iron-restricted diet (including the 129S6 strain) (24). Meanwhile, more severe models such as phlebotomy or phenylhydrazine-induced hemolysis may reflect the erythropoietic stress more so than they do iron deficiency per se. The DMT1int/int mouse may therefore serve as a superior model of severe iron deficiency and iron-deficiency anemia. Moreover, the iron deficiency is so severe that we may also anticipate marked defects in biochemical pathways relying on iron-containing enzymes (4), among which are enzymes involved in energetic pathways (21). In addition, our data indicate that the DMT1int/int mouse ought to serve as a robust model in which to test whether 1) forms of dietary iron other than Fe(II) and Fe(III) salts and chelates (e.g., transferrin, heme), and 2) supplemental iron formulations utilize DMT1 for their intestinal absorption.
We observed compensatory regulation in the expression of some iron-related genes in untreated DMT1int/int mice. Among these was the expected downregulation of hepatic hepcidin expression. Both decreased serum iron and decreased liver nonheme iron result in suppression of this iron-regulatory hormone (21). Regulation of systemic iron by hepcidin is mediated primarily by its effect on ferroportin—binding of hepcidin by ferroportin induces its internalization and degradation (42). We found no evidence of regulation of intestinal ferroportin steady-state mRNA levels by iron deficiency in the intestinal DMT1-null model. Although we did not measure protein, we expect that the marked suppression of liver hepcidin expression permits very high ferroportin protein levels in enterocytes of the DMT1int/int mouse. Expression of DMT1 (upstream of the loxP site) was strongly upregulated in enterocytes of DMT1int/int mice. A DMT1 isoform initiated in exon 1A and containing in its mRNA 3′-untranslated region an iron-responsive element [i.e., 1A/IRE(+) isoform] predominates in the intestine, and its expression is exquisitely upregulated in vitro by low-iron status (5, 23, 26, 53).
DMT1 transports ferrous ion (Fe2+) but not ferric ion (Fe3+) (27) and, since most dietary iron is in the form of ferric ion, dietary iron must be reduced by luminal ascorbic acid (37) or by surface ferrireductases prior to its DMT1-mediated uptake into the enterocyte. We found that Cybrd1 (DcytB), a protein that exhibits ferrireductase activity in vitro (39, 59), was upregulated in the DMT1int/int mouse, consistent with other mammalian models of iron deficiency that reveal Cybrd1 as strongly iron-responsive (8, 9, 40). Some redundancy of intestinal ferrireductases had been suggested by the observation that ablation of Cybrd1 in the mouse had little or no observable effect even in animals fed a low-iron diet (24). Another ferrireductase, Steap3, appears to be required for the reduction of endosomal iron taken up via the transferrin-receptor mediated pathway in erythroid precursors (45). A second family member, Steap2, is expressed in intestine and has been implicated as a putative intestinal metalloreductase (31, 46); however, our findings that the iron deficiency of the DMT1int/int mouse did not induce Steap2 expression in enterocytes, nor did parenteral iron injection suppress Steap2 expression, provide no evidence in support of a role for Steap2 in intestinal ferrireduction.
An effect of parenteral iron injection on the expression of Ctr1 was not of direct interest in this study since intestinal ablation of DMT1 had no effect on Ctr1 expression in untreated animals. Nevertheless, our data suggest that excess systemic iron may upregulate intestinal Ctr1, possibly involving iron-copper competition in a system that is capable of driving Ctr1 expression.
Conclusions.
Intestine-specific ablation of DMT1 in the mouse produces a severe iron deficiency and iron-deficiency anemia, confirming a critical role for intestinal DMT1 in systemic iron homeostasis. DMT1 is essential for the uptake of iron at the intestinal brush-border membrane. Intestinal DMT1 is specific for iron absorption under physiological conditions and is not required for the absorption of copper, manganese, or zinc.
GRANTS
This study was supported primarily by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant R01-DK-080047 (to B. Mackenzie). E. J. Niespodzany was supported by the Univ. of Cincinnati Medical Student Summer Research Program, funded in part by NIDDK Short-Term Medical Student Training Grant DK-060444. Use of core services was supported in part by NIDDK Grant P30-DK-078392 (to Digestive Health Center, Cincinnati Children's Hospital and Univ. of Cincinnati). Additionally, this work was supported by Swedish Research Council (SRC) Grant 524–2014-1 (to H. Öhrvik) and NIDDK Grant R01-DK-074192 (to D. J. Thiele). The production of copper-64 at Washington Univ.-St. Louis School of Medicine is supported by the U.S. Department of Energy (DoE), Nuclear Physics Isotope Program. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIDDK, the National Institutes of Health, the DoE, or the SRC.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.S. and B.M. conception and design of research; A.S., S.R.A., Y.N., M.A.E., E.J.N., H.O., and B.M. performed experiments; A.S., S.R.A., Y.N., M.A.E., E.J.N., H.O., and B.M. analyzed data; A.S. and B.M. interpreted results of experiments; A.S. and B.M. prepared figures; A.S. and B.M. drafted manuscript; A.S., S.R.A., Y.N., M.A.E., E.J.N., T.B., H.O., R.T.W., D.J.T., and B.M. edited and revised manuscript; A.S., S.R.A., Y.N., M.A.E., E.J.N., T.B., H.O., R.T.W., D.J.T., and B.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank N. C. Andrews, MD, PhD (Duke Univ.), T. A. Kalfa, MD, PhD (Univ. of Cincinnati and Cincinnati Children's Hospital), and R. Kohli (Univ. of Cincinnati and Cincinnati Children's Hospital) for helpful discussions. The floxed DMT1 mouse line was the kind gift of Dr. Andrews. The villin-Cre transgenic mouse line was supplied by the National Cancer Institute Mouse Repository. We thank A. A. Amratia, R. R. Baik, G. Katsaros, R. S. Kim, P. B. Knight, MD, S. Prakash, T. A. Ruwe, and C. J. Mitchell (Univ. of Cincinnati) for assistance in the laboratory.
Present address of M. A. Engevik: Dept. of Pathology & Immunology, Baylor College of Medicine, Baylor College of Medicine, One Baylor Plaza-BCM 315, Houston, TX 77030.
REFERENCES
- 1.Arredondo M, Mendiburo MJ, Flores S, Singleton ST, Garrick MD. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. Biometals 27: 115–123, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Arredondo M, Muñoz P, Mura CV, Núñez MT. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am J Physiol Cell Physiol 284: C1525–C1530, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Beard JL. Why iron deficiency is important in infant development. J Nutr 138: 2534–2536, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beutler E. Disorders of iron metabolism. In: Williams Hematology, edited by Kaushansky K, Beutler E, Seligsohn U, Lichtman MA, Kipps TJ, Prchal JT. New York: McGraw-Hill Medical, 2010. [Google Scholar]
- 5.Canonne-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 93: 4406–4417, 1999. [PubMed] [Google Scholar]
- 6.Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol 34: 455–465, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Chua ACG, Morgan EH. Manganese metabolism is impaired in the Belgrade laboratory rat. J Comp Physiol [B] 167: 361–369, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Collins JF. Gene chip analyses reveal differential genetic responses to iron deficiency in rat duodenum and jejunum. Biol Res 39: 25–37, 2006. [PMC free article] [PubMed] [Google Scholar]
- 9.Collins JF, Franck CA, Kowdley KV, Ghishan FK. Identification of differentially expressed genes in response to dietary iron deprivation in rat duodenum. Am J Physiol Gastrointest Liver Physiol 288: G964–G971, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Cousins RJ. Gastrointestinal factors influencing zinc absorption and homeostasis. Int J Vitam Nutr Res 80: 243–248, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol 279: R1–R8, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Darshan D, Frazer DM, Anderson GJ. Molecular basis of iron-loading disorders. Expert Rev Mol Med 12: e36, 2010. [DOI] [PubMed] [Google Scholar]
- 13.Dufner-Beattie J, Weaver BP, Geiser J, Bilgen M, Larson M, Xu W, Andrews GK. The mouse acrodermatitis enteropathica gene Slc39a4 (Zip4) is essential for early development and heterozygosity causes hypersensitivity to zinc deficiency. Hum Mol Genet 16: 1391–1399, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Edwards JA, Hoke JE. Defect of intestinal mucosal iron uptake in mice with hereditary microcytic anemia. Proc Soc Exp Biol Med 141: 81–84, 1972. [DOI] [PubMed] [Google Scholar]
- 15.el Marjou F, Janssen KP, Chang BHJ, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39: 186–193, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Espinoza A, Le BS, Olivares M, Pizarro F, Ruz M, Arredondo M. Iron, copper, and zinc transport: Inhibition of divalent metal transporter 1 (DMT1) and human copper transporter 1 (hCTR1) by shRNA. Biol Trace Elem Res 146: 281–286, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 95: 1148–1153, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleming MD, Trenor CC, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16: 383–386, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Fleming RE, Ponka P. Iron overload in human disease. N Eng J Med 366: 348–359, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Franich NR, Fitzsimons HL, Fong DM, Klugmann M, During MJ, Young D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington's disease. Mol Ther 16: 947–956, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganz T. Systemic iron homeostasis. Physiol Rev 93: 1721–1741, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Gunshin H, Fujiwara Y, Custodio ÁO, Direnzo C, Robine S, Andrews NC. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest 115: 1258–1266, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: 482–488, 1997. [DOI] [PubMed] [Google Scholar]
- 24.Gunshin H, Starr CN, Direnzo C, Fleming MD, Jin J, Greer EL, Sellers VM, Galica SM, Andrews NC. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 106: 2879–2883, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guthrie GJ, Aydemir TB, Troche C, Martin AB, Chang SM, Cousins RJ. Influence of ZIP14 (slc39A14) on intestinal zinc processing and barrier function. Am J Physiol Gastrointest Liver Physiol 308: G171–G178, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: Implications for regulation and cellular function. Proc Natl Acad Sci USA 99: 12345–12350, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illing AC, Shawki A, Cunningham CL, Mackenzie B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J Biol Chem 287: 30485–30496, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong J, Eide DJ. The SLC39 family of zinc transporters. Mol Aspects Med 34: 612–619, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang L, Garrick MD, Garrick LM, Zhao L, Collins JF. Divalent metal transporter 1 (Dmt1) mediates copper transport in the duodenum of iron-deficient rats and when overexpressed in iron-deprived HEK-293 cells. J Nutr 143: 1927–1933, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knöpfel M, Zhao L, Garrick MD. Transport of divalent transition-metal ions is lost in small-intestinal tissue of b/b Belgrade rats. Biochemistry 44: 3454–3465, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Knutson MD. Steap proteins: Implications for iron and copper metabolism. Nutr Rev 65: 335–340, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Peña MM, Nose Y, Thiele DJ. Biochemical characterization of the human copper transporter Ctr1. J Biol Chem 277: 4380–4387, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter: Identification of a Ctr1-independent copper transport system. J Biol Chem 277: 40253–40259, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Lin C, Zhang Z, Wang T, Chen C, James Kang Y. Copper uptake by DMT1: A compensatory mechanism for CTR1 deficiency in human umbilical vein endothelial cells. Metallomics 7: 1285–1289, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci USA 103: 13612–13617, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Mackenzie B, Garrick MD. Iron imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol 289: G981–G986, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Mackenzie B, Ujwal ML, Chang MH, Romero MF, Hediger MA. Divalent metal-ion transporter DMT1 mediates both H+-coupled Fe2+ transport and uncoupled fluxes. Pflügers Arch 451: 544–558, 2006. [DOI] [PubMed] [Google Scholar]
- 39.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291: 1755–1759, 2001. [DOI] [PubMed] [Google Scholar]
- 40.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5: 299–309, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 41: 478–481, 2009. [DOI] [PubMed] [Google Scholar]
- 42.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab 4: 235–244, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Nose Y, Wood LK, Kim BE, Prohaska JR, Fry RS, Spears JW, Thiele DJ. Ctr1 is an apical copper transporter in mammalian epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem 285: 32385–32392, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37: 1264–1269, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood 108: 1388–1394, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okazaki Y, Ma Y, Yeh M, Yin H, Li Z, Yeh KY, Glass J. DMT1 (IRE) expression in intestinal and erythroid cells is regulated by peripheral benzodiazepine receptor-associated protein 7. Am J Physiol Gastrointest Liver Physiol 302: G1180–G1190, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinilla-Tenas JJ, Sparkman BK, Shawki A, Illing AC, Mitchell CJ, Zhao N, Liuzzi JP, Cousins RJ, Knutson MD, Mackenzie B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol 301: C862–C871, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prakash S, Shawki A, Niespodzany E, Mackenzie B. Intestinal divalent metal-ion transporter-1 is required for iron homeostasis in the neonatal mouse (Abstract). FASEB J 29: 1011 5, 2015.25466886 [Google Scholar]
- 50.Ramakrishnan U. Prevalence of micronutrient malnutrition worldwide. Nutr Rev 60: S46–S52, 2002. [DOI] [PubMed] [Google Scholar]
- 51.Said HM, Blair JA, Lucas ML, Hilburn ME. Intestinal surface acid microclimate in vitro and in vivo in the rat. J Lab Clin Med 107: 420–424, 1986. [PubMed] [Google Scholar]
- 52.Shawki A, Knight PB, Maliken BD, Niespodzany EJ, Mackenzie B. H+-coupled divalent metal-ion transporter-1: Functional properties, physiological roles and therapeutics. Curr Top Membr 70: 169–214, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabuchi M, Tanaka N, Nishida-Kitayama J, Ohno H, Kishi F. Alternative splicing regulates the subcellular localization of divalent metal transporter 1 isoforms. Mol Biol Cell 13: 4371–4387, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson K, Molina RM, Brain JD, Wessling-Resnick M. Belgrade rats display liver iron loading. J Nutr 136: 3010–3014, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torrance JD, Bothwell TH. Tissue iron stores. In: Iron, edited by Cook JD. New York: Churchill Livingstone, 1980. [Google Scholar]
- 56.Wang CY, Knutson MD. Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology 58: 788–798, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem 287: 34032–34043, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K, Zhou B, Kuo YM, Zemansky J, Gitschier J. A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am J Hum Genet 71: 66–73, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wyman S, Simpson RJ, McKie AT, Sharp PA. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett 582: 1901–1906, 2008. [DOI] [PubMed] [Google Scholar]
- 60.Zoller H, Koch RO, Theurl I, Obrist P, Pietrangelo A, Montosi G, Haile DJ, Vogel W, Weiss G. Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload. Gastroenterology 120: 1412–1419, 2001. [DOI] [PubMed] [Google Scholar]