

Abstract
Dengue is the most prevalent global arboviral disease that affects over 300 million people every year. Brazil has the highest number of dengue cases in the world, with the most severe epidemics in the city of Rio de Janeiro (Rio). The effective control of dengue is critically dependent on the knowledge of population genetic structuring in the primary dengue vector, the mosquito Aedes aegypti. We analyzed mitochondrial and nuclear genomewide single nucleotide polymorphism markers generated via Restriction-site Associated DNA sequencing, as well as traditional microsatellite markers in Ae. aegypti from Rio. We found four divergent mitochondrial lineages and a strong spatial structuring of mitochondrial variation, in contrast to the overall nuclear homogeneity across Rio. Despite a low overall differentiation in the nuclear genome, we detected strong spatial structure for variation in over 20 genes that have a significantly altered expression in response to insecticides, xenobiotics, and pathogens, including the novel biocontrol agent Wolbachia. Our results indicate that high genetic diversity, spatially unconstrained admixing likely mediated by male dispersal, along with locally heterogeneous genetic variation that could affect insecticide resistance and mosquito vectorial capacity, set limits to the effectiveness of measures to control dengue fever in Rio.
Keywords: Aedes aegypti, genetic structure, microsatellites, mito-nuclear discordance, RAD-seq, Rio de Janeiro, vector control
Introduction
Dengue fever, also known as the ‘breakbone fever’, is the most prevalent global arboviral disease that affects over 300 million people every year (Bhatt et al. 2013). Brazil has the highest number of dengue cases reported annually to the World Health Organization (WHO 2012). The spread of dengue in Brazil has been exacerbated by rapid urbanization and underdeveloped water supply infrastructure (Barata et al. 2001). Such conditions are favorable for breeding of the primary dengue vector, the mosquito Aedes aegypti (Linnaeus 1762), that prefers to bite humans in densely populated areas and lay eggs in man-made water containers (Natal 2002). Various insecticide-based control programs are thought to have eradicated Ae. aegypti from Brazil in 1958 (Soper 1965). After a brief period of re-infestation and re-eradication from 1967 to 1973, the mosquito is thought to have returned to Brazil in 1976 (Brathwaite Dick et al. 2012), presumably from the neighboring countries that were never declared free of Ae. aegypti (Monteiro et al. 2014). Dengue appeared soon after the vector's reintroduction and is now endemic in Brazil, representing a major public health threat (Teixeira et al. 2013).
Because an effective vaccine against dengue is still not available, disease control relies almost entirely on suppression of the vector Ae. aegypti (WHO 2012). Sustainable and lasting control of Ae. aegypti in Brazil has proven challenging, despite the extensive efforts of public health organizations and local communities (Claro et al. 2006; Santos et al. 2013). Recent activities that involved intensive mechanical and insecticide applications failed to significantly reduce Ae. aegypti numbers and also led to an increase in insecticide resistance that rapidly spread through the neighboring populations (Maciel-de-Freitas et al. 2014). Such undesirable consequences of traditional control measures have prompted the development and implementation of alternative biocontrol strategies (McGraw and O'Neill 2013), including the replacement of local Ae. aegypti populations with the Wolbachia-infected mosquitoes that have reduced potential for dengue transmission (Hoffmann et al. 2011; Walker et al. 2011).
Efficient implementation of control programs is highly dependent on understanding the dynamics of local mosquito populations, their genetic diversity and spatio-temporal structuring (Endersby et al. 2011; Campos et al. 2012; Rašić et al. 2014a). Genetic structure in Ae. aegypti populations from Brazil has been mostly examined at broad geographic scales using mitochondrial DNA variation and nuclear markers such as RAPDs, allozymes, and microsatellites. Mitochondrial markers (COXI, ND4) revealed co-occurrence of two divergent lineages (Bracco et al. 2007; Scarpassa et al. 2008) and substantial gene flow among Ae. aegypti populations from different regions of Brazil (Gonçalves da Silva et al. 2012). Conversely, nuclear markers have generally demonstrated a high level of genetic structure and isolation of mosquito populations from different regions of the country (Ayres et al. 2004; da Costa-Ribeiro et al. 2007; Monteiro et al. 2014). When genetic structure was analyzed at finer geographic scales (within one region or a city in Brazil), microsatellites and a few single nucleotide polymorphisms (SNPs) revealed genetic homogeneity and high gene flow in the dengue fever mosquito (Campos et al. 2012; Mendonça et al. 2014).
Our objective was to assess the level of genetic structuring in Ae. aegypti from the city of Rio de Janeiro (Rio). Rio is considered a primary entry point for dengue viruses in Brazil from which they spread rapidly throughout the country (Lourenço-de-Oliveira et al. 2004). Furthermore, the largest number of recent dengue epidemics has been concentrated in this city (Roriz-Cruz et al. 2010). High dispersal distances and densities of Ae. aegypti in Rio have been suggested as important contributors to the intense risk of dengue epidemic in the city (Maciel-de-Freitas et al. 2007, 2014). Large populations and weakly restricted dispersal should lead to a low level of genetic structuring (Wright 1943), yet two previous studies found high genetic structure in Ae. aegypti in Rio using a few nuclear markers (isozymes and microsatellites) (da Costa-Ribeiro et al. 2006a,b).
Because ecological data and previously collected genetic data have led to inconsistent views on dispersal, we re-examined population genetic structuring in Ae. aegypti from Rio. We used multiple marker systems: nuclear and mitochondrial genomewide SNPs generated via Restriction-site Associated DNA sequencing (RAD-seq), as well as traditional microsatellite markers. Analyses of multiple marker systems allowed us to compare genetic diversity and structure with different spatio-temporal resolution, because different markers have distinct rates of evolution, levels of variability, and modes of inheritance (Avise 2004). The high density RAD-seq markers also provide an opportunity to consider functionally important variation across populations. We discuss implications of our findings for the implementation of current and future strategies to suppress dengue fever mosquito in Rio.
Methods
Ethics statement
No specific field ethics approval is needed for the collection of wild mosquitoes in the study areas. The Health Municipality of Rio de Janeiro and Niteroi use ovitraps as a routine tool to estimate the frequency of Ae. aegypti and Ae. albopictus throughout the cities as well as to evaluate the efficiency of vector control strategies by measuring post-treatment mosquito infestation. Verbal consent was obtained from residents at each location where collections occurred on private property. The locations were not on protected land, and the collections did not involve endangered or protected species.
Collections of Aedes aegypti
We collected mosquito larvae in 15 locations throughout the city of Rio de Janeiro and the surroundings (Table1, Fig.1). Site location was intended to represent the diversity of urban infrastructure, history of dengue transmission, water storage systems, mosquito, and human density. From each sampling area, larvae were collected using ovitraps. The greatest distance between sample collection points is 29 km, and each location comprised a 500 × 500 m2 area (Fig.1). Mosquitoes were collected within a single 3-week period in November–December 2011 at the beginning of the wet season (wet 2011), followed by the collection in the next wet season during December 2012 (wet 2012) and the dry season during August 2013 (dry 2013). The GPS coordinates of each sampling site and collection date were recorded. No more than five individuals were collected from each ovitrap to reduce the number of sibling individuals obtained, as related individuals tend to co-occur within the same trap (Apostol et al. 1993; Hoffmann et al. 2014; Rašić et al. 2014b). Larvae were then reared in insectary under standard conditions (25 ± 1°C, 80 ± 10% relative humidity and 12 h light/dark cycle) until the third instar stage for species identification. All samples identified as Ae. aegypti were stored in absolute ethanol at 4°C. DNA was extracted from one larva per ovitrap using the Qiagen DNA Blood and Tissue kit with RNAse treatment (Venlo, Limburg, the Netherlands) for SNP typing and the CTAB protocol (Weeks et al. 2002) for microsatellite typing.
Table 1.
Sample sites and sizes
| Location | Code | Lat | Long | Wet 2011 | Wet 2012 | Dry 2013 |
|---|---|---|---|---|---|---|
| Humaitá | HM | −22.955 | −43.198 | 13 | – | 5 |
| Jardim Guanabara | JG | −22.808 | −43.199 | 22 | – | 5 |
| Jurujuba | JU | −22.932 | −43.113 | 14 | 15 | 4 |
| Méier | MI | −22.905 | −43.278 | 31 | – | 9 |
| Olaria | OL | −22.839 | −43.263 | 31 | – | 7 |
| Paquetá | PQ | −22.764 | −43.107 | 19 (17) | 13 | 10 |
| Pavuna | PV | −22.811 | −43.359 | 33 | – | 11 |
| Piratininga | PI | −22.933 | −43.074 | 36 | 9 | 6 |
| Ponta D'Areia | PA | −22.884 | −43.124 | 34 | 10 | – |
| Sao Crístovão | SC | −22.891 | −43.222 | 24 | 7 | 5 |
| Taquara | TQ | −22.924 | −43.381 | 22 | 8 | 8 |
| Tubiacanga | TB | −22.785 | −43.226 | 36 (30) | 15 | 9 |
| Urca | UR | −22.945 | −43.162 | 35 (29) | – | 9 |
| Valqueire | VQ | −22.886 | −43.366 | 31 | 15 | 7 |
| Vaz Lobo | VL | −22.859 | −43.324 | 28 | 11 | 6 |
| Total | 409 | 103 | 101 |
Geographic coordinates (lat/long) and sizes of Aedes aegypti samples collected in Rio de Janeiro, Brazil, during three seasons (wet 2011 and 2012, dry 2013). Sample sizes analyzed at restriction-site associated DNA sequencing (RAD-seq) loci in addition to the microsatellite loci are found in parentheses.
Figure 1.
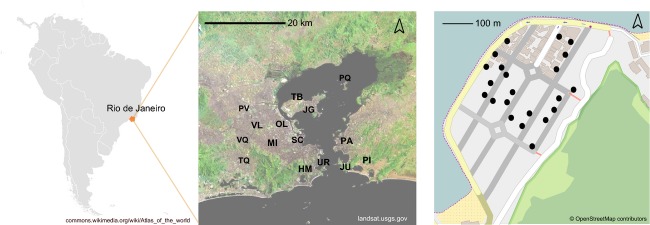
Sampling design. Sites of Rio de Janeiro and trap locations within one such site (500 × 500 m area) where Aedes aegypti were collected. Codes for sampling location are listed in Table1.
RAD-seq data processing and SNP typing
Seventy six individuals collected in Tubiacanga, Urca, and Paquetá island during the wet season in 2011 were screened for nuclear and mitochondrial genomewide SNP variation (Table1). The three locations were chosen as areas with contrasting urbanization levels that are characteristic for different parts of Rio and as sites intended for Wolbachia releases (http://www.eliminatedengue.com/rio-de-janeiro/noticias/article/404/project/rio-de-janeiro). We used a customized double-digest RAD-seq method (Peterson et al. 2012) following the procedure described in Rašić et al. (2014b). Briefly, 100 ng of genomic DNA from each larva was digested with 100 units of restriction enzymes NlaIII and MluCI (New England Biolabs, Beverly MA, USA). One hundred picomolar P1 and 300 pm P2 Illumina adapters with customized barcode sequences were ligated to the genomic fragments using 100 units of T4 ligase at 16°C overnight (New England Biolabs). Following the sample pooling and purification, size selection of fragments 300–450 bp in length was completed with the Pippin Prep protocol (Sage Sciences, Beverly, MA, USA). We used 12 PCR cycles with standard Illumina primers for the final library enrichment: 98°C for 30 s, 12 cycles of 98°C for 10 s, 60°C for 30 s, 72°C for 90 s, and the final elongation at 72°C for 5 min. The libraries were sequenced with HiSeq2000 using the 100-bp paired-end chemistry.
We used FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html by Hannon Lab) to trim all raw FASTQ reads to the same length of 80 bp and retained those with a phred score ≥20 over the entire sequence length. High-quality reads were aligned to the Ae. aegypti reference nuclear genome AaegL1 (Nene et al. 2007) and the mitochondrial genome (Behura et al. 2011) with the short read aligner program Bowtie (Langmead et al. 2009). The alignment parameters included a maximum of three mismatches permitted in the seed, a ‘try-hard’ option to find valid alignments and suppression of reads with more than one optimal alignment. Uniquely, aligned reads were then analyzed with the stacks pipeline v.1.09 (Catchen et al. 2013). Variant and likelihood-based genotype calling was performed using the default parameters on RAD stacks with a minimum depth of reads of five.
Microsatellite genotyping
Eight microsatellite loci previously described (AG5, BbH08, BbA10, AC1, 12ACG1, M201, 69TGA1, BbB19) (Slotman et al. 2006; Chambers et al. 2007; Lovin et al. 2009) were amplified and directly labeled with fluorescent dye as described in Rašić et al. (2014a). The PCR conditions contained: initial incubation step with 94°C for 15 min, 35 amplification cycles of 94°C for 30 s, 60°C for 90 s, 72°C for 60 s, followed by eight labeling cycles with 94°C for 30 s, 53°C for 90 s, 72°C for 60 s, and a final elongation step at 60°C for 30 min. Fragment analysis was carried out with Applied Biosystems (Waltham, MA, USA) 3730 DNA Analyser and 500 LIZ as the size standard. We used GeneMarker v.2.2.0 (Softgenetics, State College, PA, USA) for allele scoring. In total, 613 individuals were genotyped and used for subsequent analyses (Tables1 and S1).
Analyses of genetic diversity
Diversity parameters
The number of mitochondrial haplotypes (mtNh), nucleotide diversity (mtπ), haplotype diversity (mtHd), and theta based on the number of segregating sites (mtθ) was calculated in r package pegas (Paradis 2010). A mitochondrial haplotype network was constructed using the statistical parsimony method implemented in r package TempNet (Prost and Anderson 2011). To assess the level of nuclear genomewide variation, we estimated the observed and expected heterozygosity (HO, HE), nucleotide diversity (nπ), and theta (nθ) using the program stacks v.1.09 (Catchen et al. 2013). For microsatellite loci, we calculated the observed and expected heterozygosity (HO, HE) and allelic richness (NA) using the rarefaction method in r package gstudio (http://cran.rproject.org/web/packages/gstudio/index.html).
Phylogenetic analyses
To assess the evolutionary relationship among Ae. aegypti lineages found in Rio, we performed a maximum likelihood (ML) phylogenetic analysis using (i) the rapid bootstrap heuristic algorithm followed by a thorough ML search implemented in the program RAxML (Stamatakis et al. 2008) and (ii) the model-averaged phylogeny reconstruction in jModelTest 2.1.7 (Darriba et al. 2012). In addition to the samples from Rio, we included a previously published RAD-seq dataset that contains 17 Ae. aegypti individuals from Australia, 15 individuals from Vietnam, and 13 individuals from Indonesia (Rašić et al. 2014b) (NCBI SRA accession numbers: PRJNA241150, PRJNA273913). For each individual, we extracted informative mitochondrial polymorphisms (SNPs detected among individuals) from the RAD tags and concatenated them into the final mitochondrial sequence with 92 variable positions. Informative nuclear polymorphisms were concatenated into the final sequence that contained 5815 variable positions. Intra-individual site polymorphisms (i.e. heterozygous SNP loci) were denoted with ambiguous IUPAC codes (e.g. W for adenine or cytosine, Y for cytosine or thymine, etc.), and the RAxML program considers these ambiguous sites in the phylogenetic reconstruction (Potts et al. 2014). We tested 44 substitution models with the Gamma-distributed rate variation among sites (+G) and calculated the AIC weight as a measure of model support in jModelTest2. These results were used to compute the average phylogenetic reconstruction as the consensus of the ML trees for every model weighted with their AIC weight (Posada 2008). RAxML implements only GTR as the most complex and general model of nucleotide substitution, suitable for datasets with genomewide variation (Stamatakis 2014). We excluded models with the proportion of Invariant sites (+I) in both approaches because invariant sites were absent from our datasets. Separate phylogenetic trees were produced for mitochondrial and nuclear genome data (Files S1 and S2).
Neutrality tests and inferences of demographic change
For the mitochondrial dataset, we calculated the neutrality tests statistics Tajima's D (Tajima 1989) and R2 (Ramos-Onsins and Rozas 2002), determining the statistical significance with 1000 simulations of populations under the drift-mutation equilibrium in pegas (Paradis 2010). Negative Tajima's D value indicates departures from mutation-drift equilibrium due to processes such as population expansions or directional selection, while positive value indicates balancing selection, bottlenecks, and/or cryptic population structure (Tajima 1989). We used R2 test for detecting population growth because it has greater power than a classical mismatch distribution test, particularly for smaller sample sizes (Ramos-Onsins and Rozas 2002).
Due to uncertainty of diploid genotype calls, nuclear data are prone to an artificial inflation of rare polymorphisms (Nielsen et al. 2012). We therefore used the empirical Bayesian approach in calculating the neutrality statistics as implemented in the software angsd (Korneliussen et al. 2013). This approach accommodates the uncertainty of genotype data in statistic calculations and intrinsically deals with missing data and variable depth of loci that are common in next-generation sequencing experiments (Korneliussen et al. 2013). It is particularly beneficial for low- to medium-coverage data (<20×) such as ours. Tajima's D was estimated from the genotype likelihoods obtained using the GATK-based genotype calling method and presence in at least 70% of individuals. We also used this method to calculate the site frequency spectrum (SFS) as folded because information on ancestral polymorphisms is not available for Ae. aegypti. To generate the distribution of population parameters expected under drift-mutation equilibrium, we performed coalescent simulations using the programs msms (Ewing and Hermisson 2010) and angsd. We generated 100 replicates, assuming a recombination rate of 1 cM/Mb (Juneja et al. 2014) and theta that was fitted to the empirical estimate (Table1). We then simulated genotype likelihoods with an 8× read depth (as in the empirical dataset, see Results) and an error rate of 0.5%, from which we then calculated the folded SFS as in Nielsen et al. (2012).
Analyses of genetic structuring
Individual-based inferences
We used two individual-based clustering approaches: (i) discriminant analysis of principal components (DAPC) implemented in the r package Adegenet (Jombart and Ahmed 2011) and (ii) Bayesian clustering implemented in TESS (Chen et al. 2007).
Discriminant analysis of principal components is a nonspatial multivariate method that we applied to the nuclear SNP data and microsatellites. We calculated the a-score as the criterion for determining the optimal number of PCs to be retained in a discriminant analysis. Strength of discrimination was measured using the reassignment of supplemental individuals into the clusters that were predefined with the subset of 10–15 individuals from each sampling location, where higher percentage of correct reassignment indicates stronger discrimination (i.e. higher structure). The optimal number of genetic clusters was calculated using the successive k-means procedure, where different clustering solutions were compared with the Bayesian information criterion (BIC). The lowest BIC value indicates the optimal number of genetic clusters (Jombart et al. 2010).
Spatially explicit Bayesian clustering was applied to microsatellite data only, because the underlying TESS model requires a larger number of spatial sampling points (François and Durand 2010). We used the admixture model with a weighted geographic distance between individuals, 100 000 iterations and 40 000 burn-ins. For consistent visualization of the overall replicate results, outputs from 10 independent replicates for each k were permuted in the program clumpp (Jakobsson and Rosenberg 2007).
Existence of isolation-by-distance between individuals was tested using the Mantel test with 999 permutations on matrices of linearized genetic distance between individuals (Smouse and Peakall 1999) and a corresponding log-geographic distance in GenAlEx 6.5 (Peakall and Smouse 2012).
Group-based inferences
We calculated Nei's GST parameter of genetic structure (Nei 1973) for the nuclear SNPs and microsatellite datasets, assuming sampling sites as genetic groups. Statistical significance of parameter deviation from zero in pairwise comparisons was tested using 1000 permutations in gstudio (http://cran.r-project.org/web/packages/gstudio/index.html). For the mitochondrial dataset, we calculated the genetic structuring parameter PhiST (Excoffier et al. 1992) and used 1000 permutations for significance testing in the r package pegas.
Power analysis for detecting genetic structure
Due to a modest number of microsatellite loci (eight) and low sample sizes in the second and third sampling season that were available for this study (Table1), we assessed the statistical power to detect genetic structure in our microsatellite dataset. We used the program powsim (Ryman and Palm 2006) to simulate the degree of structuring (quantified as Nei's GST) for different effective population sizes and number of generations under isolation, while assuming the empirical number of samples, sample sizes, number of loci and alleles, and allele frequencies. Power to detect a given level of structuring and type I error were estimated using 1000 simulations.
Results
Genetic diversity
Mitochondrial genome diversity
Because Ae. aegypti has abundant mitochondrial pseudogenes in the nuclear genome (Black and Bernhardt 2009; Hlaing et al. 2009), we carefully checked for any mitochondrial RAD loci that were heterozygous or had premature stop codons, indicating paralogs or nonfunctional gene sequences. Validity of our RADseq mitochondrial data was additionally supported by the phylogenetic analyses that produced the same tree topology as the amplicon sequence data from two mitochondrial genes (COXI and ND5) analyzed in mosquitoes from the same locations (H. L. Yeap, unpublished data; File S4). We found 24 mitochondrial RAD tags that were present in ≥80% of individuals and polymorphic among individuals (File S5). RAD tags distributed across 11 mitochondrial genes were concatenated into 39 haplotypes detected in 76 individuals from the three localities in Rio (Table S2). The sequences contained 32 parsimony informative SNPs and 23 singletons. Haplotype diversity in the combined dataset was 0.812, nucleotide diversity was 0.004, and theta was 11.22 (Table2). Each locality, however, had a specific pattern of mtDNA diversity, with Paquetá island having much higher mitochondrial diversity indices than Tubiacanga (Table2).
Table 2.
Genetic diversity parameters for the nuclear and mitochondrial single nucleotide polymorphisms (SNPs) in Aedes aegypti from Rio
| θ | Tajima's D | |||||||
|---|---|---|---|---|---|---|---|---|
| Location | nucHE | nucFIS | nuc | mt | nuc | mt | mtR2 | mtHd |
| Paquetá | 0.249 | 0.034 | 6.11 | 12.72 | 1.06* | −1.82 | 0.07* | 0.948 |
| Tubiacanga | 0.236 | 0.105* | 4.07 | 3.31 | 0.96 | −2.30* | 0.15 | 0.377 |
| Urca | 0.223 | 0.136* | 4.29 | 10.60 | 1.19* | −2.10* | 0.05* | 0.936 |
| All | 0.239 | 0.114* | 5.52 | 11.22 | 1.30* | −2.26* | 0.04* | 0.812 |
Mean expected (HE) heterozygosity and fixation index (FIS) for nuclear loci; mutation-scaled effective population size (θ) and Tajima's D for nuclear (nuc) and mitochondrial (mt) genomewide SNPs; mitochondrial haplotype diversity (mtHd) and R2 parameter for demographic expansion. Parameters are presented for each location separately and for the combined dataset (all).
*Significant parameter estimates (P < 0.05).
Nuclear genome diversity
The final catalog of nuclear SNPs from the Stacks v.1.09 pipeline contained on average 6620 polymorphic RAD tags (SD = 2792) and 9369 SNPs (SD = 3924) per individual (File S3); 5123 RAD tags were present in ≥80% of individuals. After removing biallelic variants with the minor allele frequency lower than 0.05, we retained a subset of 1496 SNPs. For this subset, mean depth of coverage per site per individual was 8.3×. Overall FIS was 0.114 in the pooled dataset and theta (nθ) was 5.52, but these values varied among the three locations (Table2). The sample from Paquetá island had higher estimates of nuclear diversity when compared to Tubiacanga and Urca (Table2).
Microsatellite diversity
All eight analyzed microsatellite loci were polymorphic, with an allelic richness around 4.7 across seasons (Table3). Observed heterozygosity was on average lower than expected, but highly variable across loci (Table3). Three loci (12ACG1, 69TGA1, BbB19) showed significant heterozygote deficiency that was consistent across seasons, indicating the presence of null alleles, while one locus (M201) showed significant excess of heterozygotes (Table3). None of the pairs of loci exhibited significant linkage disequilibrium.
Table 3.
Descriptive statistics for microsatellite data
| F IS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Season | AG5 | BbH08 | BbA10 | AC1 | M201 | 12ACG1 | 69TGA1 | BbB19 | NA (SE) | HO (SE) | HE (SE) |
| Wet 2011 | 0.001 | −0.001 | 0.144 | 0.081 | −0.411* | 0.413* | 0.243* | 0.261* | 4.71 (0.19) | 0.513 (0.069) | 0.551 (0.051) |
| Wet 2012 | 0.050 | −0.111 | 0.071 | 0.021 | −0.258* | 0.443* | 0.224* | 0.426* | 4.53 (0.13) | 0.498 (0.073) | 0.540 (0.047) |
| Dry 2013 | 0.166 | 0.051 | −0.160 | −0.227* | −0.263* | 0.631* | 0.336* | 0.351* | 4.72 (0.14) | 0.549 (0.091) | 0.585 (0.051) |
Overall inbreeding level (FIS) at eight microsatellite loci across Aedes aegypti samples from three seasons. Samples were pooled across localities within a season (15 localities in wet 2011, nine in wet 2012, and 14 in dry 2013). Mean and standard error (SE) for: NA – allelic richness (rarefaction for sample size of 50 individuals), HO – observed heterozygosity, HE – expected heterozygosity, calculated across all individuals and loci within each season.
*Significant departure from Hardy–Weinberg equilibrium.
Phylogenetic analyses
Mitochondrial and nuclear SNP datasets produced contrasting ML phylogenies: samples from Rio had a homogeneous nuclear background divergent from those found in Asia and Australia, but a highly heterogeneous mitochondrial background (Fig.2). These results were obtained with both phylogenetic approaches, the randomized axelerated ML reconstruction in RAxML, and the model-averaged reconstruction in jModelTest (File S6). Samples from Vietnam and Australia also had highly divergent co-occurring mitochondrial genomes, while those from Indonesia fell into one clade (Fig.2). Nuclear data, on the other hand, separated each of the geographic samples into its own highly supported (≥99%) monophyletic clade. Overall level of divergence between the clades was an order of magnitude greater for the mitochondrial than for the nuclear dataset (1.3% vs 0.2%), reflecting the elevated mutation rate for the mitochondrial sequences when compared with the nuclear sequences.
Figure 2.
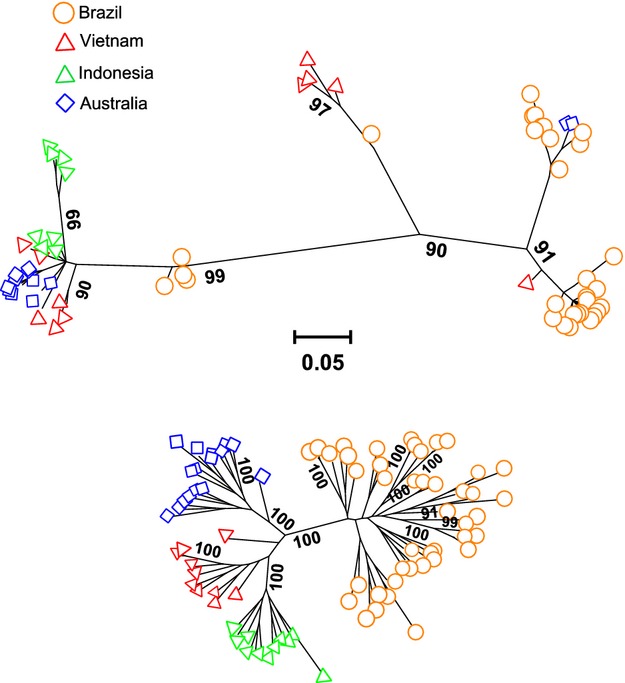
Maximum likelihood (ML) phylogeny. ML phylogenetic reconstructions from RAxML based on mitochondrial genomewide single nucleotide polymorphisms (SNPs) (upper) and nuclear genomewide SNPs (lower) for Aedes aegypti samples from Rio de Janeiro (Brazil), along with samples from Australia, Indonesia, and Vietnam. Only bootstrap support values ≥90% are shown. Scale represents mean number of nucleotide substitutions per site.
Neutrality tests
Neutrality test statistics showed contrasting patterns for the mitochondrial and nuclear datasets. For the mitochondrial genome, significant negative Tajima's D values were present in Paquetá island, Urca, and in the pooled dataset (Table2). The R2 test for population growth was also significant in these samples (Table2). Conversely, Tajima's D was positive for the nuclear genome, and this was recorded for regions within and outside genes (Fig.3). A deficit of rare nuclear alleles was also evident in the folded SFS analysis (Fig.3).
Figure 3.
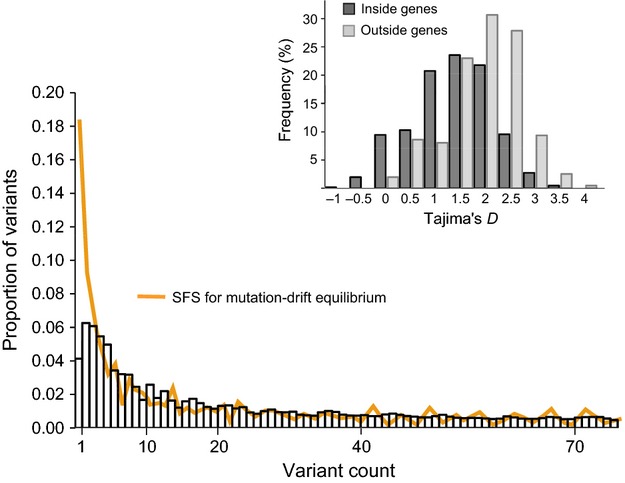
Tajima's D and site frequency spectrum (SFS) for nuclear single nucleotide polymorphisms (SNPs). Tajima's D values for sites in regions inside and outside genes and folded SFS for the genomewide SNPs and in the empirical dataset (Aedes aegypti from Rio de Janeiro) and the dataset simulated under mutation-drift equilibrium.
Genetic structuring
We detected high spatial structure for the mitochondrial variation, with an overall PhiST of 0.163 (P < 0.001). The distribution of different mitochondrial haplotypes within the three localities in Rio is portrayed in the haplotype network analysis (Fig.4).
Figure 4.
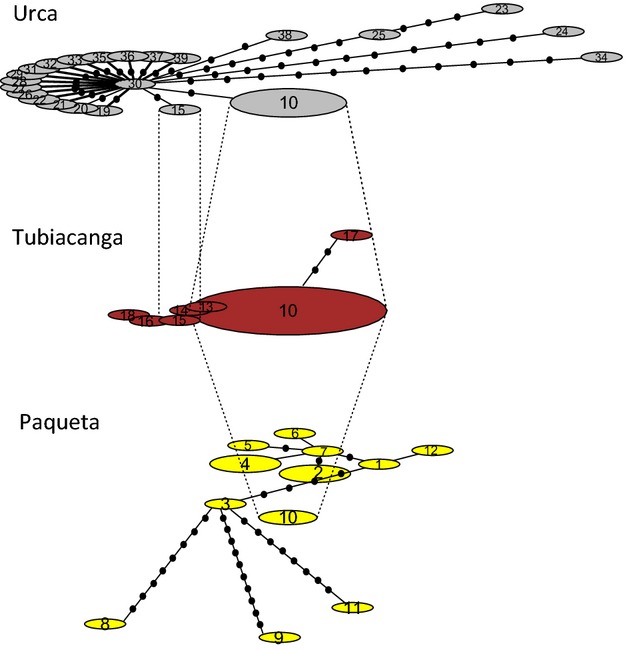
Mitochondrial haplotype network. Haplotype networks in the three localities in Rio. Haplotypes shared among localities are connected by dotted lines.
In contrast, nuclear variation exhibited a low level of spatial structuring and this was observed in both individual- and group-based analyses. In DAPC, the optimal number of genetic clusters was one (k = 1) for both nuclear SNPs and microsatellite datasets, as indicated by the lowest BIC value in the successive k-means procedure (Figure S1). Mosquitoes from Paquetá island did show greater genetic isolation and were more separated from mosquitoes in Tubiacanga and Urca based on their nuclear SNPs but not microsatellites (Fig.5). Overall assignment of individuals to their sampling location was 58% for the nuclear SNP dataset and 46% for the microsatellite dataset (Fig.5). TESS analysis of the microsatellite datasets indicated that mosquitoes sampled across Rio (Table1) had the highest membership probability to one genetic cluster, but several individuals showed different or mixed membership (Figure S2). This was observed in all three sampling seasons regardless of the predefined number of clusters (k) set in the analysis and (Figure S2). We also detected significant but weak isolation-by-distance between individuals across the city (Mantel r = 0.032, P = 0.001). However, after removing individuals from Paquetá island, no isolation-by-distance was evident (Mantel r = 0.019, P = 0.090).
Figure 5.
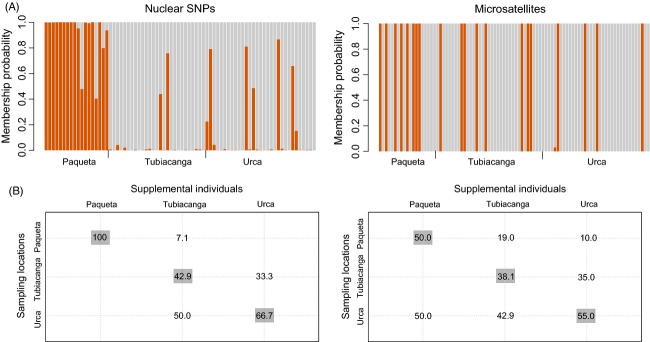
Discriminant analysis of principal components (DAPC). (A) Membership probabilities to two genetic groups for Aedes aegypti individuals, inferred using DAPC of genomewide single nucleotide polymorphism (SNP) and microsatellites; individuals were sampled in Paquetá island, Tubiacanga, and Urca; (B) blind assignment success for supplemental individuals to the three sampling locations; diagonal values in darker boxes represent assignment to the original sampling location (‘correct reassignment’).
Group-based analysis also revealed low overall spatial structuring for the nuclear variation in samples collected across all three seasons (Table S3). In the 2011 dataset, GST for genomewide SNPs was 0.014 (P > 0.05) and GST for microsatellites was 0.008 (P = 0.002). Sample from Paquetá island showed the highest pairwise GST values, but only one of 105 pairwise comparisons in the entire 2011 microsatellite dataset remained statistically significant after Bonferroni correction (Table S3). Power analysis indicated that our microsatellite marker set had an acceptable type I error rate (а = 0.051) and a 100% power to detect GST ≥ 0.008 based on sample sizes from our 2011 collection. Sample sizes of the 2012 and 2013 collections (Table1) decreased the power to detect weak structure to 65% and 30%, respectively.
Despite a very low level of spatial structuring for the entire nuclear marker set, 2.7% of nuclear SNPs (40/1496) had highly significant GST estimates (P ≪ 0.001) that ranged between 0.065 and 0.261 (Table4). Two-thirds of these SNPs (26/40) are located in 26 genes that have a function in nucleic acid, protein and ion binding, transmembrane and neuropeptide signaling, and in protein kinase and oxidoreductase activity (Table4). All but one of these genes (25/26) have a significantly altered expression under experimental conditions such as blood meal and infection with pathogens (dengue viruses, bacteria), including the novel biocontrol agent Wolbachia (wMel and wMelPop), as well as after exposure to insecticides and xenobiotics (Table4).
Table 4.
Within-gene single nucleotide polymorphisms that exhibited significant spatial structuring (GST with P ≪ 0.001) in Aedes aegypti from Rio de Janeiro
| G ST | Gene ID | Molecular function (GOSlim terms) | Experimental factor |
|---|---|---|---|
| 0.261 | AAEL018040 | Transporter activity, ATP binding | Xenobiotic,* insecticides,†‡ bacterial infection,§ blood meal¶ |
| 0.239 | AAEL008354 | Extracellular ligand-gated ion channel activity | Xenobiotic,* insecticides,† bacterial infection,§ blood meal,¶** wMelPop infection†† |
| 0.222 | AAEL008961 | Nucleic acid binding, metal ion binding | Bacterial infection§ |
| 0.218 | AAEL001939 | Nucleic acid binding, ATP binding | Xenobiotic,* bacterial infection,§ blood meal,** wMel/wMelPop infection‡‡ |
| 0.218 | AAEL001996 | Nucleic acid binding, zinc binding | Xenobiotic,* bacterial infection,§ blood meal,** wMel/wMelPop infection‡‡ |
| 0.215 | AAEL001399 | Protein binding | Bacterial infection,§ blood meal,¶** wMelPop infection,†† dengue infection§§ |
| 0.212 | AAEL009666 | DNA binding, ion binding, methyltransferase activity | Insecticides,†‡ blood meal¶** |
| 0.207 | AAEL002055 | Neuropeptide signaling | Xenobiotic,* insecticides,† bacterial infection,§ blood meal¶** |
| 0.201 | AAEL009424 | Protein serine/threonine kinase activity, ATP binding | Xenobiotic,* blood meal** |
| 0.192 | AAEL011309 | Orotidine-5′-phosphate decarboxylase activity | Xenobiotic,* insecticides,† bacterial infection,§ blood meal,¶** wMel/wMelPop infection‡‡¶¶ |
| 0.192 | AAEL009063 | ? | Blood meal,¶ wMelPop infection¶¶ |
| 0.190 | AAEL002852 | ? | Xenobiotic,* insecticides,‡ bacterial infection,§ blood meal¶ |
| 0.170 | AAEL003415 | Structural molecule activity (lamin) | Xenobiotic,* insecticides,‡ bacterial infection,§ blood meal** |
| 0.166 | AAEL002924 | Binding | – |
| 0.164 | AAEL002798 | Ion binding, oxidoreductase activity | Insecticides,† bacterial infection§ |
| 0.160 | AAEL018040 | ATP binding, ATPase activity | Xenobiotic,* insecticides,†‡ bacterial infection,§ blood meal¶ |
| 0.160 | AAEL018133 | ? | Xenobiotic,* insecticides,† bacterial infection,§ blood meal,¶** wMelPop infection†† |
| 0.154 | AAEL005929 | ATPase activity, transmembrane transporter activity | Xenobiotic,* insecticides,†‡ bacterial infection,§ blood meal,¶** wMel/wMelPop infection††‡‡ |
| 0.151 | AAEL013811 | Lysophospholipase activity | Blood meal,** circadian*** |
| 0.132 | AAEL000682 | ? | Xenobiotic,* insecticides,† bacterial infection,§ blood meal¶** |
| 0.131 | AAEL007258 | Protein binding | Blood meal¶** |
| 0.114 | AAEL006301 | Protein binding, metal ion binding | Insecticides,‡ blood meal,¶ wMel/wMelPop infection‡‡ |
| 0.091 | AAEL008318 | Calcium ion binding | Xenobiotic,* wMelPop infection,¶¶ circadian*** |
| 0.088 | AAEL012395 | ATPase activity, transmembrane transporter activity | Xenobiotic,* insecticides,† bacterial infection,§ blood meal,¶ circadian*** |
| 0.087 | AAEL009305 | ATP binding, protein kinase activity | Xenobiotic,* insecticides‡ |
| 0.065 | AAEL004480 | Protein binding (cell division cycle 20) | Bacterial infection,§ blood meal¶ |
?: unknown gene function; –: no experimental data.
Gene ID and ontology terms follow the annotation from VectorBase. Significantly altered expression has been recorded under conditions that are listed as experimental factors.
Discussion
We report novel insights into the complex pattern of genetic diversity and spatial genetic structuring in Ae. aegypti from Rio de Janeiro as revealed by multiple marker systems: RAD-seq generated SNPs in the mitochondrial and nuclear genomes, and microsatellites. We found several divergent mitochondrial lineages and strong spatial structure for mitochondrial variation, in contrast to the overall homogeneity in nuclear markers across Rio. Despite a low overall differentiation for the nuclear markers, we detected strong spatial structuring for variation in over 20 genes that have a significantly altered expression under the exposure to insecticides, xenobiotics, and pathogens, including the novel biocontrol agent Wolbachia.
Low nuclear but high mitochondrial spatial structure
The various analyses we carried out such as spatially explicit Bayesian clustering, DAPC, isolation-by-distance, and GST comparisons all indicated very weak spatial structuring of nuclear variation despite the highly heterogeneous landscape of the Rio de Janeiro city (including mountainous and coastal regions, slums, urban, and rural areas). One exception was the geographically isolated Paquetá Island (Fig.1) that showed significant differentiation from the rest of the city. These results suggest extensive mixing of Ae. aegypti across Rio unlike patterns detected in previous reports (da Costa-Ribeiro et al. 2006a, b), but they are consistent with patterns found in another city and a region in Brazil (Campos et al. 2012; Mendonça et al. 2014). Mitochondrial variation, on the other hand, had a very strong spatial structuring, with only one haplotype shared among locations considered here. Contrasting spatial pattern between mitochondrial and nuclear markers (i.e. mito-nuclear discordance) is not rare in animals, including insect pests (Sun et al. 2015), but is generally found across much broader geographic scales (Toews and Brelsford 2012).
Potential drivers of mito-nuclear discordance in Aedes aegypti from Rio
Endosymbionts like Wolbachia can provide a strong mechanism underlying the discordant spatial patterns between mitochondrial and nuclear variation in many insects (Toews and Brelsford 2012). Because Wolbachia and mitochondria are both maternally inherited, reproductive incompatibilities caused by Wolbachia infections (e.g. cytoplasmic incompatibility) influence the population dynamics of mitochondria (Turelli et al. 1992). Even though Wolbachia are not natively found in Ae. aegypti, we did confirm the Wolbachia-free status of Ae. aegypti in Rio. We tested 683 individuals from our 2011 collection using the quantitative real-time PCR/high-resolution melt assay for Wolbachia detection (Lee et al. 2012) and found no signs of Wolbachia infection in Rio.
Because the mitochondrial genome is haploid and uniparentally inherited in most animals, it has a smaller effective population size than the nuclear genome (Hudson and Turelli 2003). Locally varying population dynamics and stochastic events are reflected in an increased genetic structuring for the mitochondrial markers when compared to nuclear markers, particularly in the early stages of the population divergence (Larsson et al. 2009). Local samples of from Rio had different estimates of effective population size (θ) and parameters of demographic change (Tajima's D, R2) for the mitochondrial genome but not for the nuclear genome (Table2).
FST-like indices should be up to four times higher for mitochondrial than for nuclear markers when populations are diverging under a relatively high gene flow (Larsson et al. 2009). This ratio between the two marker sets (mtDNA FST/nuclear FST) is much greater than four if dispersal is sex-biased such that males show higher dispersal propensity, and much smaller than four if females are the dispersing sex (Karl et al. 2012). In our dataset, the GST ratio for mtDNA and nuclear SNPs was 11.81 (and 9.59 for mtDNA and microsatellites), suggesting that Ae. aegypti males in Rio may have higher dispersal than females.
Male-biased dispersal in Ae. aegypti has not been detected in mark–release–recapture (MRR) studies conducted in Rio or elsewhere (Harrington et al. 2005; Maciel-de-Freitas et al. 2007; Valerio et al. 2012). However, the marking methods and fluorescent dusts in MRR experiments can have a significant impact on differential survival of males and females in Ae. aegypti (Dickens and Brant 2014), biasing the estimates of dispersal in the two sexes. Moreover, genetic parameters such as GST measure effective dispersal that leads to gene flow, unlike the ecological (MRR) parameters. Perhaps Ae. aegypti females that disperse far have a lower reproductive output than females that remain local, while this is not the case in males. Some evidence in support of this hypothesis comes from the MRR experiments that recorded lower daily survival of females in parts of Rio where they showed higher dispersal propensity (Maciel-de-Freitas et al. 2007). These results warrant further investigation where patterns of genetic relatedness in males and females could be compared, which was not possible in our study because we used nonsexed larvae.
Human-assisted introductions are another mechanism contributing to mito-nuclear discordance in animals (Toews and Brelsford 2012). Multiple reintroductions of Ae. aegypti into Brazil from neighboring countries have been suggested in previous studies based on an analyses of mitochondrial genes (COI, ND4) and microsatellite markers. These studies pointed to two divergent mitochondrial clades (Bracco et al. 2007; Scarpassa et al. 2008; Paduan and Ribolla 2009) and two major nuclear genetic groups in larger regions of Brazil (Monteiro et al. 2014). In this study, we found four mitochondrial clades in Rio de Janeiro alone, suggesting that the use of more markers across the mitochondrial genome as in the current study is likely to reveal an additional level of complexity in Ae. aegypti introductions into Brazil. As a comparison, in the city of Yogyakarta, Indonesia, we found only one mitochondrial clade using the same ddRAD approach to screen samples of comparable sizes collected at the same spatial scale (G. Rašić, unpublished data). This suggests that the large ‘gaps’ between clades found in Rio are unlikely to represent missing haplotypes due to insufficient sampling.
We also found an overall deficit of rare SNPs across the nuclear genome of Ae. aegypti from Rio. Introductions from diverse sources coupled with extensive gene flow can lead to these patterns of genetic diversity in invasive populations (Cutter et al. 2012; Lockwood et al. 2013). Positive Tajima's D values can also be interpreted as evidence for population balancing selection, bottlenecks, and/or cryptic population structure (Tajima 1989). A strong influence of balancing selection is unlikely in our dataset, given that Tajima's D remained positive when only regions outside genes were analyzed. Also, the level of nuclear structuring we observed was too low to cause a substantial artifact due to sample pooling (and separate analyses for each of the local samples also gave positive Tajima's D values). In a long-term survey of Ae. aegypti abundance in Rio, there was no major decrease in mosquito numbers (Figure S3), suggesting that bottlenecks are not strong. Furthermore, a dramatic decline in effective population size was not detected in our microsatellite bottleneck tests (Table S4). The pattern of genomewide SNP diversity observed in Ae. aegypti from Rio therefore appears most consistent with complex re-introductions combined with high subsequent gene flow.
Despite high gene flow in Ae. aegypti across Rio, we detected local genetic structure for variation in 25 genes with a putative role in the mosquito immune response and insecticide resistance (Table4). Aedes aegypti in Rio could be responding to locally varying intensities of insecticide application and pathogen load that are strong enough to counteract the effect of high gene flow, but the analyses of selection signatures on a more comprehensive dataset are needed to adequately test this hypothesis.
Implications for the control of Aedes aegypti in Rio
For the past 40 years, the Brazilian National Program for Dengue Control has been using insecticides of the organophosphate and pyrethroid classes as the main strategy to control Ae. aegypti (Lima et al. 2011). Due to intensive insecticide application, Ae. aegypti populations evolved resistance that is now widespread across all regions of Brazil (Montella et al. 2007). New agents, such as Bacillus thuringiensis israelensis toxins (Bti), have been replacing organophosphates in Rio de Janeiro states since 2001 (Lima et al. 2005). Nevertheless, the control of Ae. aegypti populations that intensifies after every new dengue epidemic has generally had a very limited success in suppressing the vector populations or dengue transmission. As an example, recent efforts in the city of Boa Vista not only failed to significantly reduce the mosquito numbers, but led to a rapid increase in pyrethroid resistance even in the adjacent areas that were not directly targeted (Maciel-de-Freitas et al. 2014).
Our study elucidates processes that set limits to the effectiveness of traditional control measures in Rio and possibly other parts of Brazil. Aedes aegypti in Rio harbors high genetic diversity and populations experience spatially unconstrained admixing that is likely mediated by effective male dispersal, but also exhibits locally heterogeneous genetic variation that could affect insecticide resistance. Resistance to organophosphates and pyrethroids in populations from Brazil has been associated with higher fitness cost (Belinato et al. 2012; Brito et al. 2013), limiting the spread of resistance variants into areas that are not under intense insecticide applications. However, the level of gene flow we found in Rio would allow for a rapid spread of resistance if the intensity of insecticide application increases locally. We detected significant spatial structure in the face of high gene flow for genes that have an altered expression in Ae. aegypti that are resistant to or exposed to insecticides such as imidacloprid, permethrin, and Bti (Table4). Even though resistance to Bti has not been reported in Ae. aegypti from Brazil (Araújo et al. 2013), strong spatial structure for several genes that respond to this insecticide could indicate locally selected variants. These findings suggest that it would be worthwhile monitoring and comparing insecticide resistance in Ae. aegypti collected in different parts of Rio.
We also found spatial heterogeneity for variation in genes showing differential expression under the infection with bacteria and dengue viruses, or after a blood meal (Table4), and these genes could affect the vectorial capacity of Ae. aegypti. A recent study found differential vectorial capacity for chikungunya viruses (CHIKV) in Ae. aegypti from different parts of Rio (Vega-Rúa et al. 2014). For example, mosquitoes from Paquetá island and east bay (Jurujuba) showed higher transmission efficacy for CHIKV than mosquitoes from the west bay (Vega-Rúa et al. 2014). Based on that study and our results, a fine scale assessment of Ae. aegypti's vectorial capacity for dengue viruses in Rio may be worthwhile.
Our study also provides useful information for the implementation of novel biocontrol strategies like those that use Wolbachia-infected Ae. aegypti for population replacement or suppression (Hoffmann et al. 2015). If female dispersal is limited, the potential for the spread of Wolbachia infection outside the release zone may be restricted because emigrant individuals are more likely to be males that do not transmit this maternally inherited endosymbiont. However, with male-biased dispersal, the release of Wolbachia-infected males for population suppression may be worth considering (Hancock et al. 2011). In the absence of strong spatial variation in nuclear genes, it may be possible to release mosquitoes in any part of the city with a nuclear background derived from one Rio population rather than being concerned about local backgrounds (Hoffmann et al. 2015). However, with strong spatial structure for genes involved in responses to insecticides and Wolbachia infection (Table4), it would seem prudent to check for phenotypic effects in resistance and endosymbiont parameters that might vary locally as a consequence of population variation.
In conclusion, using multiple marker systems we uncovered a complex genetic structure of Ae. aegypti in Rio de Janeiro, reflecting underlying processes that limit the effectiveness of local measures to control the mosquito populations. These factors need to be considered in future Wolbachia releases, and they contrast with patterns found in other parts of the world. The data we have collected also point to some intriguing patterns of differentiation in genes that may influence the local success of release programs.
Acknowledgments
The authors would like to acknowledge Heng Lin Yeap for providing the COI and ND5 amplicon sequences and Araújo SC from the Laboratório de Biologia Computacional e Sistemas, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro for assisting with the microsatellite genotyping. We are grateful to the Nectar Research Cloud and VIC Node at the University of Melbourne for providing the computing resources for bioinformatics analyses. We would also like to thank Kevin Emerson for valuable comments on the earlier version of the manuscript. This work was funded by a program grant from the National Health and Medical Research Council, a fellowship from the Australian Research Council Australia to AAH, a grant from the Foundation for the National Institutes of Health through the Grand Challenges in Global Health Initiative of the Bill and Melinda Gates Foundation, and a grant from the Rio de Janeiro Science Foundation (FAPERJ) to RS. The funders had no role in study design, data collection, and analysis or preparation of the manuscript.
Data archiving statement
Illumina sequences for the ddRADseq data are archived within the NCBI SRA under accession numbers: PRJNA241150, PRJNA273913. All other data are available as the Supporting Information files in the online version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AAH conceived the study. GR analyzed the data and wrote the manuscript with AAH. GR, RS, RP, NMEH generated the data. IF and GR performed the bioinformatics processing and analysis. RMF, GS, RCM designed the field work and collected the samples. All authors have read and approved the final manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1 DAPC analysis with nuclear genome-wide SNPs in Aedes aegypti from Rio de Janeiro, with the BIC values for the successive number of genetic groups (upper) and the alpha-score optimization process for retaining the optimal number of PCs in the analysis (lower).
Figure S2. TESS results.
Figure S3. Mosquito numbers.
Table S1. Sampling coordinates and microsatellite genotype data.
Table S2. List of Aedes aegypti mitochondrial genes with ddRAD loci.
Table S3.FST. Pairwise FST (bellow diagonal) and the corresponding P-values (from 999 permutations) across eight microsatellite loci among samples of Aedes aegypti collected from 15 location in Rio de Janeiro during wet season 2011.
Table S4.BOTTLENECK analysis.
Files S1 and S2. Relaxed phylip format files used in RAxML. Unique nuclear and mitochondrial SNPs concatenated into the sequences used for the ML phylogenetic analysis.
File S3.VCF for nuclear SNPs.
File S4. Amplicon sequences from mitochondrial genes COXI and ND5 in Ae. aegypti.
File S5.RADseq generated mtDNA sequences.
File S6.jModelTest-2.1.7 results.
Literature cited
- Apostol BL, Black WC, IV, Miller BR, Reiter P. Beaty BJ. Estimation of the number of full sibling families at an oviposition site using RAPD-PCR markers: applications to the mosquito Aedes aegypti. Theoretical and Applied Genetics. 1993;86:991–1000. doi: 10.1007/BF00211052. [DOI] [PubMed] [Google Scholar]
- Araújo AP, Araujo Diniz DF, Helvecio E, de Barros RA, de Oliveira CMF, Ayres CFJ, de Melo-Santos MAV, et al. The susceptibility of Aedes aegypti populations displaying temephos resistance to Bacillus thuringiensis israelensis: a basis for management. Parasites & Vectors. 2013;6:297. doi: 10.1186/1756-3305-6-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC. Molecular Markers, Natural History, and Evolution. 2nd edn. Sunderland, MA: Sinauer Associates, Inc; 2004. [Google Scholar]
- Ayres CEJ, Melo-Santos MAV, Prota JRM, Solé-Cava AM, Regis L. Furtado AE. Genetic structure of natural populations of Aedes aegypti at the micro- and macrogeographic levels in Brazil. Journal of the American Mosquito Control Association. 2004;20:350–356. [PubMed] [Google Scholar]
- Barata EA, Costa AI, Chiaravalloti Neto F, Glasser CM, Barata JM. Natal D. Aedes aegypti (L.) population in an endemic area of dengue in Southeast Brazil. Revista de Saúde Pública. 2001;35:237–242. doi: 10.1590/s0034-89102001000300004. [DOI] [PubMed] [Google Scholar]
- Behura SK, Lobo NF, Haas B, DeBruyn B, Lovin DD, Shumway MF, Puiu D, et al. Complete sequences of mitochondria genomes of Aedes aegypti and Culex quinquefasciatus and comparative analysis of mitochondrial DNA fragments inserted in the nuclear genomes. Insect Biochemistry and Molecular Biology. 2011;41:770–777. doi: 10.1016/j.ibmb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura SK, Gomez-Machorro C, deBruyn B, Lovin DD, Harker BW, Romero-Severson J, Mori A, et al. Influence of mosquito genotype on transcriptional response to dengue virus infection. Functional & Integrative Genomics. 2014;14:581–589. doi: 10.1007/s10142-014-0376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinato TA, Martins AJ. Valle D. Fitness evaluation of two Brazilian Aedes aegypti field populations with distinct levels of resistance to the organophosphate temephos. Memorias do Instituto Oswaldo Cruz. 2012;107:916–922. doi: 10.1590/s0074-02762012000700013. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, et al. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black WC., IV Bernhardt SA. Abundant nuclear copies of mitochondrial origin (NUMTs) in the Aedes aegypti genome. Insect Molecular Biology. 2009;18:705–713. doi: 10.1111/j.1365-2583.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- Bonizzoni M, Dunn WA, Campbell CL, Olson KE, Dimon MT, Marinotti O. James AA. RNA-seq analyses of blood-induced changes in gene expression in the mosquito vector species, Aedes aegypti. BMC Genomics. 2011;12:82. doi: 10.1186/1471-2164-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracco JE, Capurro ML, Lourenço-de-Oliveira R. Sallum MAM. Genetic variability of Aedes aegypti in the Americas using a mitochondrial gene: evidence of multiple introductions. Memorias do Instituto Oswaldo Cruz. 2007;102:573–580. doi: 10.1590/s0074-02762007005000062. [DOI] [PubMed] [Google Scholar]
- Brathwaite Dick O, San Martín JL, Montoya RH, del Diego J, Zambrano B. Dayan GH. The history of dengue outbreaks in the Americas. The American Journal of Tropical Medicine and Hygiene. 2012;87:584–593. doi: 10.4269/ajtmh.2012.11-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito LP, Linss JGB, Lima-Camara TN, Belinato TA, Peixoto AA, Lima JBP, Valle D, et al. Assessing the effects of Aedes aegypti kdr mutations on pyrethroid resistance and its fitness cost. PLoS ONE. 2013;8:e60878. doi: 10.1371/journal.pone.0060878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos M, Spenassatto C, Lourdes da Graça Macoris M, Paduan Kdos S, Pinto J. Ribolla PEM. Seasonal population dynamics and the genetic structure of the mosquito vector Aedes aegypti in São Paulo, Brazil. Ecology and Evolution. 2012;2:2794–2802. doi: 10.1002/ece3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A. Cresko W. Stacks: an analysis tool set for population genomics. Molecular Ecology. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers EW, Meece JK, McGowan JA, Lovin DD, Hemme RR, Chadee DD, McAbee K, et al. Microsatellite isolation and linkage group identification in the yellow fever mosquito Aedes aegypti. The Journal of Heredity. 2007;98:202–210. doi: 10.1093/jhered/esm015. [DOI] [PubMed] [Google Scholar]
- Chen C, Durand E, Forbes F. François O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Molecular Ecology Notes. 2007;7:747–756. [Google Scholar]
- Choi Y-J, Fuchs JF, Mayhew GF, Yu HE. Christensen BM. Tissue-enriched expression profiles in Aedes aegypti identify hemocyte-specific transcriptome responses to infection. Insect Biochemistry and Molecular Biology. 2012;42:729–738. doi: 10.1016/j.ibmb.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claro LBL, Kawa H, Cavalini LT. Rosa MLG. Community participation in dengue control in Brazil. Dengue Bulletin. 2006;30:214–222. [Google Scholar]
- da Costa-Ribeiro MCV, Lourenço-de-Oliveira R. Failloux A-B. Geographic and temporal genetic patterns of Aedes aegypti populations in Rio de Janeiro, Brazil. Tropical Medicine and International Health. 2006a;11:1276–1285. doi: 10.1111/j.1365-3156.2006.01667.x. [DOI] [PubMed] [Google Scholar]
- da Costa-Ribeiro MCV, Lourenço-de-Oliveira R. Failloux A-B. Higher genetic variation estimated by microsatellites compared to isoenzyme markers in Aedes aegypti from Rio de Janeiro. Memorias do Instituto Oswaldo Cruz. 2006b;101:917–921. doi: 10.1590/s0074-02762006000800015. [DOI] [PubMed] [Google Scholar]
- da Costa-Ribeiro MCV, Lourenço-de-Oliveira R. Failloux A-B. Low gene flow of Aedes aegypti between dengue-endemic and dengue-free areas in southeastern and southern Brazil. The American Journal of Tropical Medicine and Hygiene. 2007;77:303–309. [PubMed] [Google Scholar]
- Cutter AD, Wang GX, Ai H. Peng Y. Influence of finite-sites mutation, population subdivision and sampling schemes on patterns of nucleotide polymorphism for species with molecular hyperdiversity. Molecular Ecology. 2012;21:1345–1359. doi: 10.1111/j.1365-294X.2012.05475.x. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R. Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens BL. Brant HL. Effects of marking methods and fluorescent dusts on Aedes aegypti survival. Parasites & Vectors. 2014;7:65. doi: 10.1186/1756-3305-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake SN, Ribeiro JMC, Wang M-H, Dunn WA, Yan G, James AA. Marinotti O. aeGEPUCI: a database of gene expression in the dengue vector mosquito, Aedes aegypti. BMC Research Notes. 2010;3:248. doi: 10.1186/1756-0500-3-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endersby NM, Hoffmann AA, White VL, Ritchie SA, Johnson PH. Weeks AR. Changes in the genetic structure of Aedes aegypti (Diptera: Culicidae) populations in Queensland, Australia, across two seasons: implications for potential mosquito releases. Journal of Medical Entomology. 2011;48:999–1007. doi: 10.1603/me10264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing G. Hermisson J. MSMS: a coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics. 2010;26:2064–2065. doi: 10.1093/bioinformatics/btq322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE. Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François O. Durand E. Spatially explicit Bayesian clustering models in population genetics. Molecular Ecology Resources. 2010;10:773–784. doi: 10.1111/j.1755-0998.2010.02868.x. [DOI] [PubMed] [Google Scholar]
- Gonçalves da Silva A, Cunha ICL, Santos WS, Luz SLB, Ribolla PEM. Abad-Franch F. Gene flow networks among American Aedes aegypti populations. Evolutionary Applications. 2012;5:664–676. doi: 10.1111/j.1752-4571.2012.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock PA, Sinkins SP. Godfray HCJ. Strategies for introducing Wolbachia to reduce transmission of mosquito-borne diseases. PLoS Neglected Tropical Diseases. 2011;5:e1024. doi: 10.1371/journal.pntd.0001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LC, Scott TW, Lerdthusnee K, Coleman RC, Costero A, Clark GG, Jones JJ, et al. Dispersal of the dengue vector Aedes aegypti within and between rural communities. The American Journal of Tropical Medicine and Hygiene. 2005;72:209–220. [PubMed] [Google Scholar]
- Hlaing T, Tun-Lin W, Somboon P, Socheat D, Setha T, Min S, Chang MS, et al. Mitochondrial pseudogenes in the nuclear genome of Aedes aegypti mosquitoes: implications for past and future population genetic studies. BMC Genetics. 2009;10:11. doi: 10.1186/1471-2156-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F, Greenfield M, et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature. 2011;476:454–457. doi: 10.1038/nature10356. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Goundar AA, Long SA, Johnson PH. Ritchie SA. Invasion of Wolbachia at the residential block level is associated with local abundance of Stegomyia aegypti, yellow fever mosquito, populations and property attributes. Medical and Veterinary Entomology. 2014;28:90–97. doi: 10.1111/mve.12077. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Ross PA. Rašić G. Wolbachia strains for disease control: ecological and evolutionary considerations. Evolutionary Applications. 2015;8:751–768. doi: 10.1111/eva.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Turelli M. Stochasticity overrules the “three-times rule”: genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution. 2003;57:182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x. [DOI] [PubMed] [Google Scholar]
- Jakobsson M. Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Jombart T. Ahmed I. adegenet 1. 3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T, Devillard S. Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja P, Osei-poku J, Ho YS, Ariani CV, Palmer WJ, Pain A. Jiggins FM. Assembly of the genome of the disease vector Aedes aegypti onto a genetic linkage map allows mapping of genes affecting disease transmission. PLoS Neglected Tropical Diseases. 2014;8:e2652. doi: 10.1371/journal.pntd.0002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambris Z, Cook PE, Phuc HK. Sinkins SP. Immune activation by life-shortening Wolbachia and reduced filarial competence in mosquitoes. Science. 2009;326:134–136. doi: 10.1126/science.1177531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl SA, Toonen RJ, Grant WS. Bowen BW. Common misconceptions in molecular ecology: echoes of the modern synthesis. Molecular Ecology. 2012;21:4171–4189. doi: 10.1111/j.1365-294X.2012.05576.x. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Moltke I, Albrechtsen A. Nielsen R. Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics. 2013;14:289. doi: 10.1186/1471-2105-14-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M. Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson LC, Charlier J, Laikre L. Ryman N. Statistical power for detecting genetic divergence-organelle versus nuclear markers. Conservation Genetics. 2009;10:1255–1264. [Google Scholar]
- Lee SF, White VL, Weeks AR, Hoffmann AA. Endersby NM. High-throughput PCR assays to monitor Wolbachia infection in the dengue mosquito (Aedes aegypti) and Drosophila simulans. Applied and Environmental Microbiology. 2012;78:4740–4743. doi: 10.1128/AEM.00069-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima JBP, de Melo NV. Valle D. Residual effect of two Bacillus thuringiensis var. israelensis products assayed against Aedes aegypti (Diptera: Culicidae) in laboratory and outdoors at Rio de Janeiro, Brazil. Revista do Instituto de Medicina Tropical de São Paulo. 2005;47:125–130. doi: 10.1590/s0036-46652005000300002. [DOI] [PubMed] [Google Scholar]
- Lima EP, Paiva MHS, de Araújo AP, da Silva EVG, da Silva UM, de Oliveira LN, Santana AEG, et al. Insecticide resistance in Aedes aegypti populations from Ceará, Brazil. Parasites & Vectors. 2011;4:5. doi: 10.1186/1756-3305-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood JL, Hoopes MF. Marchetti MP. Invasion Ecology. 2nd edn. Chichester, UK: Wiley-Blackwell; 2013. [Google Scholar]
- Lourenço-de-Oliveira R, Vazeille M, de Filippis AMB. Failloux AB. Aedes aegypti in Brazil: genetically differentiated populations with high susceptibility to dengue and yellow fever viruses. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2004;98:43–54. doi: 10.1016/s0035-9203(03)00006-3. [DOI] [PubMed] [Google Scholar]
- Lovin DD, Washington KO, deBruyn B, Hemme RR, Mori A, Epstein SR, Harker BW, et al. Genome-based polymorphic microsatellite development and validation in the mosquito Aedes aegypti and application to population genetics in Haiti. BMC Genomics. 2009;10:590. doi: 10.1186/1471-2164-10-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciel-de-Freitas R, Codeço CT. Lourenço-de-Oliveira R. Daily survival rates and dispersal of Aedes aegypti females in Rio de Janeiro, Brazil. The American Journal of Tropical Medicine and Hygiene. 2007;76:659–665. [PubMed] [Google Scholar]
- Maciel-de-Freitas R, Avendanho FC, Santos R, Sylvestre G, Araújo SC, Lima JBP, Martins AJ, et al. Undesirable consequences of insecticide resistance following Aedes aegypti control activities due to a dengue outbreak. PLoS ONE. 2014;9:e92424. doi: 10.1371/journal.pone.0092424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw E. O'Neill SL. Beyond insecticides: new thinking on an ancient problem. Nature Reviews. Microbiology. 2013;11:181–193. doi: 10.1038/nrmicro2968. [DOI] [PubMed] [Google Scholar]
- Mendonça BAA, de Sousa ACB, de Souza AP. Scarpassa VM. Temporal genetic structure of major dengue vector Aedes aegypti from Manaus, Amazonas, Brazil. Acta Tropica. 2014;134:80–88. doi: 10.1016/j.actatropica.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Monteiro FA, Schama R, Martins AJ, Gloria-Soria A, Brown JE. Powell JR. Genetic diversity of Brazilian Aedes aegypti: patterns following an eradication program. PLoS Neglected Tropical Diseases. 2014;8:e3167. doi: 10.1371/journal.pntd.0003167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montella IR, Martins AJ, Viana-Medeiros PF, Lima JBP, Braga IA. Valle D. Insecticide resistance mechanisms of Brazilian Aedes aegypti populations from 2001 to 2004. The American Journal of Tropical Medicine and Hygiene. 2007;77:467–477. [PubMed] [Google Scholar]
- Natal D. Bioecologia do Aedes aegypti. Biológico. 2002;64:205–207. [Google Scholar]
- Nei M. Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:3321–3323. doi: 10.1073/pnas.70.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316:1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Korneliussen T, Albrechtsen A, Li Y. Wang J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS ONE. 2012;7:e37558. doi: 10.1371/journal.pone.0037558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paduan K. Ribolla P. Characterization of eight single nucleotide polymorphism markers in Aedes aegypti. Molecular Ecology Resources. 2009;9:114–116. doi: 10.1111/j.1755-0998.2008.02282.x. [DOI] [PubMed] [Google Scholar]
- Paradis E. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- Peakall R. Smouse PE. GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics. 2012;28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS. Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Molecular Biology and Evolution. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Potts AJ, Hedderson TA. Grimm GW. Constructing phylogenies in the presence of intra-individual site Polymorphisms (2ISPs) with a focus on the nuclear ribosomal cistron. Systematic Biology. 2014;63:1–16. doi: 10.1093/sysbio/syt052. [DOI] [PubMed] [Google Scholar]
- Poupardin R, Riaz MA, Jones CRM, Chandor-Proust A, Reynaud S. David J-P. Do pollutants affect insecticide-driven gene selection in mosquitoes? Experimental evidence from transcriptomics. Aquatic Toxicology. 2012;114–115:49–57. doi: 10.1016/j.aquatox.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Prost S. Anderson CNK. TempNet: a method to display statistical parsimony networks for heterochronous DNA sequence data. Methods in Ecology and Evolution. 2011;2:663–667. [Google Scholar]
- Ptitsyn AA, Reyes-Solis G, Saavedra-Rodriguez K, Betz J, Suchman EL, Carlson JO. Black WC. Rhythms and synchronization patterns in gene expression in the Aedes aegypti mosquito. BMC Genomics. 2011;12:15. doi: 10.1186/1471-2164-12-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Onsins SE. Rozas J. Statistical properties of new neutrality tests against population growth. Molecular Biology and Evolution. 2002;19:2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- Rancès E, Ye YH, Woolfit M, McGraw EA. O'Neill SL. The relative importance of innate immune priming in Wolbachia-mediated dengue interference. PLoS Pathogens. 2012;8:e1002548. doi: 10.1371/journal.ppat.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rašić G, Endersby NM, Williams C. Hoffmann AA. Using Wolbachia-based release for suppression of Aedes mosquitoes: insights from genetic data and population simulations. Ecological Applications. 2014a;24:1226–1234. doi: 10.1890/13-1305.1. [DOI] [PubMed] [Google Scholar]
- Rašić G, Filipović I, Weeks AR. Hoffmann AA. Genome-wide SNPs lead to strong signals of geographic structure and relatedness patterns in the major arbovirus vector, Aedes aegypti. BMC Genomics. 2014b;15:275. doi: 10.1186/1471-2164-15-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz MA, Chandor-Proust A, Dauphin-Villemant C, Poupardin R, Jones CM, Strode C, Régent-Kloeckner M, et al. Molecular mechanisms associated with increased tolerance to the neonicotinoid insecticide imidacloprid in the dengue vector Aedes aegypti. Aquatic Toxicology. 2013;126:326–337. doi: 10.1016/j.aquatox.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Roriz-Cruz M, Sprinz E, Rosset I, Goldani L. Teixeira MG. Dengue and primary care: a tale of two cities. Bulletin of the World Health Organization. 2010;88:244. doi: 10.2471/BLT.10.076935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman N. Palm S. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Molecular Ecology Notes. 2006;6:600–602. doi: 10.1046/j.0962-1083.2001.01345.x. [DOI] [PubMed] [Google Scholar]
- Santos SM, Amorim F, Sousa SS, Ferreira IA, Itria A, Siqueira Junior JB. Toscano CM. Costs of dengue control and prevention program in Brazil. Value in Health. 2013;16:A678. [Google Scholar]
- Scarpassa VM, Cardoza TB. Cardoso Junior RP. Population genetics and phylogeography of Aedes aegypti (Diptera: Culicidae) from Brazil. The American Journal of Tropical Medicine and Hygiene. 2008;78:895–903. [PubMed] [Google Scholar]
- Slotman MA, Kelly NB, Harrington LC, Kitthawee S, Jones JW, Scott TW, Caccone A, et al. Polymorphic microsatellite markers for studies of Aedes aegypti (Diptera: Culicidae), the vector of dengue and yellow fever. Molecular Ecology Notes. 2006;7:168–171. [Google Scholar]
- Smouse PE. Peakall R. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity. 1999;82:561–573. doi: 10.1038/sj.hdy.6885180. [DOI] [PubMed] [Google Scholar]
- Soper FL. The 1964 status of Aedes aegypti eradication and yellow fever in the Americas. The American Journal of Tropical Medicine and Hygiene. 1965;14:887–891. doi: 10.4269/ajtmh.1965.14.887. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P. Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Systematic Biology. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Sun J-T, Wang M-M, Zhang Y-K, Chapuis M-P, Jiang X-Y, Hu G, Yang X-M, et al. Evidence for high dispersal ability and mito-nuclear discordance in the small brown planthopper, Laodelphax striatellus. Scientific Reports. 2015;5:8045. doi: 10.1038/srep08045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123:597–601. doi: 10.1093/genetics/123.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MG, Siqueira JB, Ferreira GLC, Bricks L. Joint G. Epidemiological trends of dengue disease in Brazil (2000–2010): a systematic literature search and analysis. PLoS Neglected Tropical Diseases. 2013;7:e2520. doi: 10.1371/journal.pntd.0002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreau G, Bayyareddy K, Jones CM, Stalinski R, Riaz MA, Paris M, David J-P, et al. Larval midgut modifications associated with Bti resistance in the yellow fever mosquito using proteomic and transcriptomic approaches. BMC Genomics. 2012;13:248. doi: 10.1186/1471-2164-13-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toews DPL. Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology. 2012;21:3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x. [DOI] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA. McKechnie SW. Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics. 1992;132:713–723. doi: 10.1093/genetics/132.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio L, Facchinelli L, Ramsey JM, Bond JG. Scott TW. Dispersal of male Aedes aegypti in a coastal village in southern Mexico. The American Journal of Tropical Medicine and Hygiene. 2012;86:665–676. doi: 10.4269/ajtmh.2012.11-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Rúa A, Zouache K, Girod R, Failloux A-B. Lourenço-de-Oliveira R. High level of vector competence of Aedes aegypti and Aedes albopictus from ten American countries as a crucial factor in the spread of Chikungunya virus. Journal of Virology. 2014;88:6294–6306. doi: 10.1128/JVI.00370-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, Leong YS, et al. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature. 2011;476:450–453. doi: 10.1038/nature10355. [DOI] [PubMed] [Google Scholar]
- Weeks AR, McKechnie SW. Hoffmann AA. Dissecting adaptive clinal variation: markers, inversions and size/stress associations in Drosophila melanogaster from a central field population. Ecology Letters. 2002;5:756–763. [Google Scholar]
- WHO. Report of a World Health Organization Technical Working Group Meeting on Dengue Prevention and Control. Geneva, Switzerland: WHO Headquarters; 2012. pp. 1–6. [Google Scholar]
- Wright S. Isolation by distance. Genetics. 1943;28:114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye YH, Woolfit M, Huttley GA, Rancès E, Caragata EP, Popovici J, O'Neill SL, et al. Infection with a virulent strain of Wolbachia disrupts genome wide-patterns of cytosine methylation in the mosquito Aedes aegypti. PLoS ONE. 2013;8:e66482. doi: 10.1371/journal.pone.0066482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 DAPC analysis with nuclear genome-wide SNPs in Aedes aegypti from Rio de Janeiro, with the BIC values for the successive number of genetic groups (upper) and the alpha-score optimization process for retaining the optimal number of PCs in the analysis (lower).
Figure S2. TESS results.
Figure S3. Mosquito numbers.
Table S1. Sampling coordinates and microsatellite genotype data.
Table S2. List of Aedes aegypti mitochondrial genes with ddRAD loci.
Table S3.FST. Pairwise FST (bellow diagonal) and the corresponding P-values (from 999 permutations) across eight microsatellite loci among samples of Aedes aegypti collected from 15 location in Rio de Janeiro during wet season 2011.
Table S4.BOTTLENECK analysis.
Files S1 and S2. Relaxed phylip format files used in RAxML. Unique nuclear and mitochondrial SNPs concatenated into the sequences used for the ML phylogenetic analysis.
File S3.VCF for nuclear SNPs.
File S4. Amplicon sequences from mitochondrial genes COXI and ND5 in Ae. aegypti.
File S5.RADseq generated mtDNA sequences.
File S6.jModelTest-2.1.7 results.