

Abstract
mRNAs containing premature termination codons (PTCs) are known to be degraded via nonsense-mediated mRNA decay (NMD). Unexpectedly, we found that mRNAs containing any type of PTCs (UAA, UAG, and UGA) are detained in the nucleus, whereas their wild-type counterparts are rapidly exported. This retention is strictly reading-frame dependent. Strikingly, our data indicate that translating ribosomes in the nucleus proofread the frame and detect the PTCs in the nucleus. Moreover, the shuttling NMD protein Upf1 specifically associates with PTC+mRNAs (PTC-containing mRNAs) in the nucleus and is required for nuclear retention of PTC+mRNAs. Together, our data lead to a working model that PTCs are recognized in the nucleus by translating ribosomes, resulting in recruitment of Upf1, which in turn functions in nuclear retention of PTC+mRNA. Nuclear PTC recognition adds a new layer of proofreading for mRNA and may be vital for ensuring the extraordinary fidelity required for protein production.
Keywords: premature termination codons (PTCs), nuclear retention, nuclear frame reading, Upf1
Introduction
Eukaryotic gene expression is a multistep process that begins with transcription and pre-mRNA processing in the nucleus followed by export of the mature mRNA to the cytoplasm for translation. All of these steps in the gene expression pathway are extensively coupled to one another both physically and functionally, and this coupling is thought to maintain the efficiency and fidelity of gene expression [1–4]. For example, mRNA export is coupled to pre-mRNA processing, and failure of any of the processing steps (capping, splicing, and polyadenylation) results in retention of the mRNAs in nuclear speckle domains [5–10].
The fidelity of gene expression is also maintained by numerous RNA surveillance mechanisms. Among these, nonsense-mediated mRNA decay (NMD) is a surveillance mechanism, which detects and rapidly degrades mRNAs containing premature termination codons (PTCs) [11–14]. Eliminating PTC-containing mRNAs (PTC+mRNAs) is vital because these mRNAs encode C-terminally truncated proteins, which may possess dominant-negative or deleterious gain-of-function activity. In current models for NMD, PTCs are recognized relative to a downstream marker on the mRNA [15, 16]. In yeast, the marker is a cis-acting element, whereas in metazoans the marker is the exon–junction complex (EJC), a protein complex recruited to exon–exon junctions during splicing [15, 16]. In metazoans, NMD is thought to be triggered when the translating ribosome stalls at the PTC, and the downstream EJC functions to recruit factors that degrade the mRNA [17–21]. In addition to the downstream markers, PTCs are also thought to be recognized by the aberrant nature of the extended 3′ UTR that results from the presence of the PTC [22]. Recent studies revealed that NMD not only degrades aberrant mRNAs containing PTCs but also functions as an important post-transcriptional regulatory mechanism for specific normal mRNAs that do not contain PTCs [23, 24].
As PTC recognition is tightly coupled to translation, it was believed that NMD is restricted to the cytoplasm. However, recent studies have raised the possibility that NMD can also take place in the nucleus. For example, cell fractionation studies indicated that the levels of PTC+mRNAs are not only reduced in the cytoplasmic, but also in the nuclear fraction [25–28]. A report presenting evidence for nuclear translation has lent additional support to the notion that NMD can take place in the nucleus [29]. Furthermore, multiple observations have linked NMD to nuclear processes. For example, pre-mRNAs containing PTCs accumulate near the site of transcription [30, 31], and PTCs in an alternatively spliced intron induce exclusion of the PTC+intron during splicing [32]. In addition, the presence of a PTC increases the nuclear to cytoplasmic distribution of the PTC+mRNAs [33]. PTCs are also thought to be detected when mRNA translocates through the nuclear pore complex, leading to the view that PTC recognition is nucleus associated [34]. Consistent with this possibility, the ribosome associates with the giant Balbiani ring mRNP when the 5′ end of the mRNA emerges from the nuclear pore complex [35]. These studies notwithstanding, definitive evidence for nuclear PTC recognition is still lacking, and potential mechanisms involved in this process remain to be determined.
In this study, we have obtained compelling evidence that PTC+mRNAs are specifically recognized and detained in the nucleus. Strikingly, we find that this retention occurs with any type of PTCs (UAA, UAG, and UGA) and is strictly reading-frame dependent. Furthermore, we provide strong evidence for our working model that PTCs are detected in the nucleus by translating ribosomes, resulting in recruitment of an EJC-associated factor (Upf1), which functions to detain the PTC+mRNA in the nucleus.
Results
PTC+mRNAs are detained in the nucleus
To investigate the nucleocytoplasmic distribution of mRNA containing a PTC, we microinjected a Smad reporter plasmid containing a PTC at codon 133 (133T Smad) and the corresponding wild-type (WT) Smad plasmid into HeLa cell nuclei and mRNA was detected by fluorescence in situ hybridization (FISH). By 1 h after addition of α-amanitin, most of the WT Smad mRNA was detected in the cytoplasm, and by 2 h, the mRNA was almost completely cytoplasmic (Figure 1b, upper panels, and Figure 1c). In contrast, the 133T Smad mRNA was mainly detected in the nucleus as late as the 2-h time point (Figure 1b, lower panel, and Figure 1c). By the 3 and 4 h time points, 133T mRNA could be detected in the cytoplasm, but the overall FISH signal was low, as expected for an mRNA containing a PTC and undergoing NMD (Figure 1b, lower panels, and Figures 1c and d) [11–14]. To determine whether the mature form of 133T Smad mRNA was retained, we next carried out the same experiment using a probe that specifically detects the spliced mRNA (Supplementary Figure S1). As shown in Figure 1d, significant more 133T Smad mRNA was detected in the nucleus at all time points examined. These results indicate that the spliced 133T mRNA is released to the cytoplasm with a significant delay. However, it was also possible that cytoplasmic NMD of 133T Smad mRNA had occurred, resulting in the seemingly nuclear accumulation phenotype. To investigate this possibility, we co-knocked down the nucleases Dis3L and Xrn1, which function in degradation of PTC+mRNA in the cytoplasm. Co-knockdown of these nucleases significantly inhibited NMD, but did not affect the nucleocytoplasmic distribution of WT and 133T Smad mRNA (Figure 1e and Supplementary Figure S2). These results indicate that nuclear accumulation of the 133T Smad mRNA is not due to cytoplasmic degradation but is a result of slow export kinetics.
Figure 1.

Premature termination codon (PTC)+ Smad mRNA are retained in nuclear speckle domains. (a) Schematic of Smad constructs. The CMV promoter and BGH polyA sites and the location of the fluorescence in situ hybridization (FISH) probe (exon probe, red line; exon–exon probe, green line) are shown. The gray and black bars indicate Myc and HA tag, respectively. The black triangle and hexagon indicate PTC and physiological stop codon, respectively. (b) Equal amount of Smad constructs (50 ng μl−1) were microinjected into nuclei of HeLa cell, and α-amanitin was added to block transcription 15 mins after microinjection. FISH of Smad transcripts were carried out with exon probe at indicated time after addition of α-amanitin. Insets show FITC-conjugated dextran coinjected as an injection marker. Scale bar, 10 μm. (c) N/C ratios were determined for a minimum of 30 cells per construct per time point. The graph shows the average N/C ratios for wild-type (WT) and 133T Smad mRNAs at each time point, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed using the Student’s t-test. *P<0.05, **P<0.01, ***P<0.001. (d) Same as b, except that FISH was carried with with the exon-exon probe. (e) Equal amount of Smad constructs (50 ng μl−1) were microinjected into nuclei of control knockdowned cells or Dis3L and Xrn1 co-knockdowned cells. Fifteen minutes after microinjection, α-amanitin was added to block transcription. FISH was carried out with both exon-exon probe (green) and exon probe (red) by 1 h. (f) Same as b, except that the injection marker was omitted and immunofluorescence using an SC35 antibody was carried out. Confocal microscopy was used to visualize the cells. See also Supplementary Figures S1 and S2.
To investigate whether the prolonged retention of PTC+mRNAs in the nucleus was a general phenomenon, we next microinjected Smad plasmids with nonsense mutations at the 39th or 122nd codons and compared the nucleocytoplasmic distribution of these mRNAs to WT (Supplementary Figure S3). This analysis revealed that nuclear retention was markedly prolonged for both of the PTC+mRNAs relative to WT mRNA. Together, these data indicate that PTC+Smad mRNAs are specifically detained in the nucleus relative to their WT counterparts.
Previous work indicated that WT mRNA initially accumulates in nuclear speckle domains, where it undergoes splicing before release to the cytoplasm [6]. Consistent with this result, confocal microscopic analysis revealed that the nuclear mRNA of both WT and 133T Smad generally colocalized with the splicing factor SC35, which is a standard marker for nuclear speckle domains (Figure 1f and Supplementary Figure S4) [4, 36, 37]. Moreover, kinetic analysis revealed that PTC+mRNAs are retained in nuclear speckle domains longer than WT mRNA (Figure 1f).
To determine whether the prolonged retention of PTC+mRNAs in nuclear speckle domains occurred with a different reporter construct, we examined the nucleocytoplasmic distribution of the most frequently studied NMD substrate, 39T β-globin, comparing it with WT β-globin mRNA (Figure 2a). As expected, WT β-globin mRNA was initially detected in nuclear speckle domains and then accumulated in the cytoplasm over time (Figure 2b, upper panels, Figure 2c) [6]. In striking contrast, the 39T β-globin mRNA was still largely detected in nuclear speckles as late as the 3.5-h time point (Figure 2b, lower panels, and Figure 2c). As observed with Smad PTC+mRNAs, by 5 h after addition of α-amanitin, the mRNA levels of 39T β-globin were noticeably reduced relative to WT mRNA, indicative of NMD of the PTC+mRNAs (Figures 2b and d). Similar results were obtained with a probe that specifically detects β-globin spliced mRNA (Figure 2d). This nuclear accumulation phenotype of 39T β-globin mRNA is unlikely a result of cytoplasmic degradation, as the phenotype was not affected by co-knockdown of Dis3L and Xrn2 (Figure 2e). Together, our data with the β-globin and Smad constructs suggest that prolonged retention of PTC+mRNAs in nuclear speckle domains is general.
Figure 2.

PTC+ β-globin mRNA is detained in the nucleus. (a) Schematic of Smad constructs. The CMV promoter and BGH polyA sites and the location of the fluorescence in situ hybridization (FISH) probe (exon probe, red line; exon-exon probe, green line) are shown. The gray and black bars indicate Myc and HA tag, respectively. The black triangle and hexagon indicate PTC and physiological stop codon respectively. (b) Equal amount of Smad constructs (50 ng μl−1) were microinjected into nuclei of HeLa cell, and α-amanitin was added to block transcription 15 mins after microinjection. FISH of Smad transcripts were carried out with exon probe at indicated time after addition of α-amanitin. Insets show FITC-conjugated dextran coinjected as an injection marker. Scale bar, 10 μm. (c) N/C ratios were determined for a minimum of 30 cells per construct per time point. The graph shows the average N/T and C/T ratios for wild-type (WT) and 133T Smad mRNAs at each time point, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed using the Student’s t-test. *P<0.05, **P<0.01, ***P<0.001. (d) Same as b, except that FISH was carried with the exon-exon probe. (e) Equal amount of Smad constructs (50 ng μl−1) were microinjected into nuclei of control knockdown cells or Dis3L and Xrn1 co-knockdowned cells. Fifteen minutes after microinjection, α-amanitin was added to block transcription. FISH was carried out with both exon–exon probe (green) and exon probe (red) by 1 h.
To investigate whether the prolonged retention of PTC+mRNAs was unique to the microinjection assay, we transfected WT and 39T β-globin constructs into HeLa cells and carried out FISH 12, 24, and 48 h later. As shown in Figure 3a, by 12 h post transfection, most of the WT β-globin mRNA was detected in the cytoplasm, whereas 39T mRNA was mainly detected in the nucleus. At the 24- and 48-h time points, the FISH signal of 39T mRNA was diminished, consistent with degradation via NMD. Of the cells that did contain FISH signal, a nuclear signal was still apparent, although some signal was also detected in the cytoplasm (Figure 3a). These results obtained using transfection were similar to those using microinjection, indicating that the prolonged retention of PTC+mRNAs is not specific to the microinjection assay.
Figure 3.

PTC+mRNAs do not have processing defects. (a) Equal amount of wild-type (WT) and 39T β-globin DNA constructs were transfected into HeLa cells, 12, 24, or 48 h later, fluorescence in situ hybridization (FISH) of β-globin transcripts was carried out. At 12 h post transfection, in 84±5% of cells transfected with WT β-globin plasmid, the mRNA was mainly cytoplasmic, and in 76±7% of cells transfected with 39T β-globin plasmid, the mRNA was mainly nuclear. (b) Schematic of intron-containing and intron-lacking β-globin constructs are shown and the size (in nucleotides (nt)) of the introns and exons are indicated. Primers are indicated by the arrows. The GInExR primer is complementary to the junction of intron2–exon3 and the GExExR primer is complementary to the junction of exon2–exon3. Equal amount of WT and 39T β-globin constructs (intron-containing) were transfected into HeLa cells and 4 h later, α-amanitin was added. Total RNA was extracted at 0, 4, and 8 h after addition of α-amanitin, followed by RT-PCR using the two sets of primers as indicated separately. Equal volume of PCR products were mixed and loaded on the gel. The PCR products amplified from intron-containing and intron-lacking plasmid DNAs were used as size markers of pre-mRNA and mRNA, respectively. The sizes (in kb) of the DNA marker are indicated. (c) Same as b, except that Smad DNA constructs were transfected and PCR was carried out using primers as indicated. See also Supplementary Figure S4.
Prolonged retention of PTC+mRNAs in nuclear speckles is not due to processing defects
Previous studies have reported that PTC+mRNAs are inefficiently spliced, and at least in some cases, this was because the PTC disrupted an exonic splicing enhancer [38, 39]. As splicing defects are known to result in retention of pre-mRNA in nuclear speckle domains [36, 40], we next investigated whether our PTC+mRNAs were defective in splicing over a time course. WT and 39T β-globin constructs were transfected into HeLa cells and α-amanitin was added at 4 h. Total RNA was then extracted at the 0-, 4-, and 8-h time points followed by reverse transcription-PCRs. We used the PCR primer pairs of G1F+GInExR and G1F+GExExR to detect unspliced pre-mRNA and spliced mRNA, respectively. As shown in Figure 3b, the amount of unspliced pre-mRNAs for both WT and 39T mRNAs decreased and the amount of spliced mRNAs increased over time after transfection (Figure 3b). Importantly, there was no difference in the ratios of pre-mRNA to mRNA between WT and 39T mRNA at any time point. Similarly, the splicing kinetics of PTC+Smad mRNAs were identical to the WT counterpart (Figure 3c). These data indicate that defects or altered kinetics of splicing do not account for the prolonged nuclear retention of PTC+mRNAs.
Proper 3′ end processing is also required for efficient mRNA export and mRNA stability [10]. Thus, we next examined the polyA tail length of WT and PTC+mRNAs over a time course using the PAT methods-rapid amplification of cDNA ends-PAT (RACE-PAT) method. This analysis revealed that the polyA tails were always the same for the PTC+β-globin and Smad mRNAs as their corresponding WT mRNAs (Supplementary Figure S5). We conclude that the defects in mRNA processing do not account for the retention of PTC-containing mRNAs in the nucleus.
Retention of PTC+mRNAs in the nucleus is reading-frame dependent
It is possible that the PTC mutations alter some cis-acting mRNA elements or structure that results in the prolonged nuclear retention phenotype that we observed. If this was the case, a missense mutation at the same site would be expected to have similar consequences. To test this possibility, we made a missense mutation at the same codon as the 39T mutation in β-globin mRNA, which changed the codon from UAG to AAG (Figure 4a, 39M). We then compared the nucleocytoplasmic distribution of the missense (39M), 39T, and WT mRNAs (Figure 4a). By 1 h after addition of α-amanitin, both the 39M and WT mRNAs had begun to accumulate in the cytoplasm, whereas the 39T mRNA was detected only in the nucleus (Figure 4a). These data indicate that a PTC is required for the nuclear speckle retention phenotype. To further examine this possibility, we constructed two frame-shift mutants, one that shifted the 39T UAG out of frame by deleting an upstream nucleotide (nt), and the other that shifted a UAA at codon 59 of WT β-globin mRNA into frame by inserting an upstream nucleotide (Figure 4a, 39T-OutF and 59T-InF). In both constructs, the reading frame was restored downstream (see Figure 4a legend). As expected, shifting the PTC out of frame in the 39T-OutF construct eliminated mRNA decay, whereas introducing a new PTC for 59T-InF conferred mRNA decay (Supplementary Figure S6a). Strikingly, the mRNA with the PTC out of the reading frame (39T-OutF) was detected in the cytoplasm as observed for WT and 39M mRNAs (Figure 4a). In contrast, the mRNA (59T-InF) that contained a new in-frame PTC was retained in the nucleus (Figure 4a). These data indicate that retention of PTC+mRNAs in the nucleus is reading-frame dependent.
Figure 4.

Prolonged retention of PTC+mRNAs in the nucleus requires a proper reading frame. (a) Schematic of β-globin constructs. The black boxes indicate the codons that have been changed. For 39T β-globin, the codon at 39 was changed from CAG to TAG, and for missense mutation at codon 39 (39M), CAG was changed to AAG. For 39T-OutF, the frame was shifted up by deleting a T, which is 6 nt (nucleotide) upstream of TAG, and restored by addition of a C, which is 22 nt downstream of the TAG. For 59T-InF, the frame of the WT β-globin was shifted to create a nonsense mutation at 59th codon by adding a C into 19 nt upstream of the TAA, and the frame was corrected by deleting the A into 18 nt downstream of the 59th TAA. (b) PCR products (100 ng μl−1) amplified from β-globin constructs using CMV-F and pCDpAR primers were microinjected into nuclei of HeLa cells, and α-amanitin was added 30 mins after microinjection. fluorescence in situ hybridization (FISH) of β-globin transcripts was carried out at 1 h after addition of α-amanitin. Insets show FITC-conjugated dextran coinjected as an injection marker. The graph shows the average N/C ratios for β-globin mRNAs, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (c) The 39th, 122nd, and 133rd nonsense codon was replaced to create missense mutation, respectively. Smad plasmids (50 ng μl−1) were microinjected into the nuclei of HeLa cells, and α-amanitin was added 15 mins after microinjection. FISH of Smad transcripts was carried out at 1 h after addition of α-amanitin. Insets show FITC-conjugated dextran co-microinjected as an injection marker. The graph shows the average N/C ratios for Smad mRNAs, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c.
We also examined the nucleocytoplasmic distribution of Smad mRNAs containing missense mutations. When the PTCs at the 39th, 122nd, and 133rd positions were replaced with missense mutations, the nucleocytoplasmic distribution of all of the missense mutants was similar to that observed for the WT mRNA (Figure 4b). Together with the β-globin results, our data indicate that the prolonged nuclear retention phenotype is not a consequence of disruption of an RNA element or structure, but rather is caused by recognition of an in-frame PTC.
PTC+mRNAs can be distinguished from WT mRNA in the nucleus
Our data demonstrating that PTCs result in nuclear retention of mRNA raised the possibility that PTC+mRNA can be recognized in the nucleus. However, it was also possible that a small amount of PTC+mRNA expressed early during transcription is exported to the cytoplasm, and this elicits a feedback mechanism to detain the PTC+ mRNAs made subsequently in the nucleus. In the case of the latter possibility, the presence of the PTC+ mRNA would also result in retention of WT mRNA in the same cell, unless there was a nuclear mechanism to distinguish WT versus PTC+mRNA. To investigate this question, we asked whether the PTC+mRNAs have any trans effects on the nucleocytoplasmic distribution of WT mRNA using a mixing experiment. In this experiment, the WT Smad construct was coinjected with the 39T β-globin construct, and as a control the WT Smad and WT β-globin constructs were coinjected (Figure 5a; different fluorophores were used to detect Smad and β-globin mRNAs in the same cell). This analysis revealed that WT Smad mRNA accumulated to the same extent in the cytoplasm in the presence of either WT or 39T β-globin mRNA, indicating that the presence of the PTC+mRNAs had no effect on export of WT mRNA, excluding the possibility that PTC+ mRNAs are recognized in the cytoplasm and feedback to block mRNA export.
Figure 5.

PTC+mRNAs do not affect the nucleocytoplasmic distribution of wild-type (WT) mRNAs. (a) WT Smad DNA was coinjected with either WT or 39T β-globin DNA, and α-amanitin was added 30 mins after microinjection. Fluorescence in situ hybridization (FISH) of β-globin or Smad transcripts was carried out at 1 h after addition of α-amanitin using β-globin (Alexa Flour 488) or Smad (Alexa Flour 647)-specific probes respectively. Scale bar, 10 μm. The graph shows the average N/C ratios for Smad mRNA in the presence of WT or 39T β-globin mRNA, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (b) WT, 133T or the mixture of half WT and half 133T Smad DNA was microinjected into the nuclei of HeLa cells, and α-amanitin was added 15 mins after microinjection. FISH of Smad transcripts was performed at 0.5 or 1 h after addition of α-amanitin. Scale bar, 10 μm. Insets show FITC-conjugated dextran coinjected as an injection marker. The graph shows the average N/C ratios for Smad mRNAs, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (c) Higher magnification of portions of images shown in b for details. Scale bar, 10 μm.
An alternative possibility is that PTC+mRNA is detected in the cytoplasm and feeds back to the nucleus to specifically block the export of itself as well as its corresponding WT mRNA. To test this possibility, we asked whether PTC+mRNA could be distinguished from its WT counterpart in the nucleus. To do this, we microinjected WT Smad, 133T Smad, or a mixture containing half WT and half 133T Smad constructs, and compared the nucleocytoplasmic distribution of the mRNAs. At both 0.5 and 1 h time points after addition of α-amanitin, Smad mRNA was detected in the cytoplasm when cells were microinjected with either the WT Smad construct alone or the mixture of WT and 133T Smad constructs. In contrast, when the 133T Smad construct was microinjected alone, Smad mRNA was only detected in the nucleus (Figure 5b, and see Figure 5c for higher magnification). These results reveal that the PTC+mRNAs do not block export of the corresponding WT mRNA and indicate that PTC+mRNA can be distinguished from the corresponding WT mRNA in the nucleus. Together, our data exclude the possibility that PTCs are recognized in the cytoplasm and feed back to the nucleus to cause nuclear retention of PTC+mRNAs. We conclude that PTC recognition can occur in the nucleus.
The start codon AUG is required for nuclear PTC recognition
Our data demonstrating that PTCs can be recognized in the nucleus indicate that a nuclear frame-reading mechanism exists. To investigate this mechanism, we examined whether the start codon AUG is required for nuclear retention of PTC+mRNAs. WT and 39T β-globin DNA plasmids containing no AUG or only the very first AUG (1AUG) were constructed and transfected into HeLa cells (Figure 6a). As controls, constructs with normal AUGs (all AUG) were also transfected. Twelve hours after transfection, with normal AUGs, WT β-globin mRNA was mainly cytoplasmic, whereas 39T β-globin mRNA was largely retained in the nucleus (Figure 6a, i and ii). Strikingly, however, when all AUGs were removed, both WT and 39T β-globin mRNA efficiently accumulated in the cytoplasm (Figure 6a, iii and iv). In contrast, when only the 1AUG was present, 39T β-globin mRNA was mainly nuclear, whereas the WT mRNA was largely cytoplasmic (Figure 6a, v and vi). These results reveal that the start codon AUG is necessary and sufficient for nuclear retention of PTC+mRNAs.
Figure 6.

Evidence that nuclear premature termination codon (PTC) recognition depends on the ribosome and tRNA. (a) Schematic of β-globin constructs is shown. The small black boxes indicate the ATGs. Equal amount of wild-type (WT) and mutant β-globin constructs were transfected into HeLa cells, and 12 h after transfection fluorescence in situ hybridization (FISH) was carried out. The percentages of cells with similar mRNA nucleocytoplasmic distribution patterns to the images shown were, from left to right, top, 84±4%, 83±3%, 90±3%, and 85±5%; bottom, 80±5%, 83±1%, 82±3%, and 88±3%. We have set the N/T ratio bigger than 1.2 as N-postive and smaller than 0.8 as C-positive. (b) Schematic of β-globin constructs. The gray box indicates the EMCV internal ribosomal entry sites (IRES). Positions of the start codon, the hairpin structure, and the PTC were marked. No hp, β-globin construct contains no hairpin; hp, β-globin construct contains a hairpin in the 5′ UTR; and hp WT/Mut IRES, β-globin construct contains a WT or mutant EMCV IRES downstream of the hairpin. β-Globin constructs were microinjected into the nuclei of HeLa cells, and α-amanitin was added 15 mins after microinjection. One hour after addition of α-amanitin, FISH was carried out to detect the mRNA. Inset pictures show the injection marker. The graph shows the average N/C ratios for β-globin mRNAs, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (c) Schematic of Ser-tRNA constructs whose anticodon either base pair or does not base pair with the PTC. Equal amount of WT and mutant Ser-tRNA constructs were co-transfected with WT or PTC+β-globin construct into HeLa cells, 12 h later, FISH and immunofluoresence with the HA antibody were carried out. The percentages of cells have similar mRNA nucleocytoplasmic distribution pattern to the images shown were, from left to right, 85±3%, 86±4%, 79±4%, 81±9%, 76±3%, and 77±2%. See also Supplementary Figures S5–S7.
Evidence that the ribosome and tRNA function in nuclear PTC recognition
The Kozak sequence functions in recognition of the start codon by the ribosome [41]. To investigate whether this sequence has a role in detaining PTC+mRNAs, we mutated the Kozak sequence in the β-globin reporter constructs, 1AUG WT and 1AUG 39T (Figure 6a, 1AUG WT-Kmut and 1AUG 39T-Kmut). As expected, translation of 1AUG WT-Kmut mRNA and degradation of 1AUG 39T-Kmut mRNA were inhibited (Supplementary Figure S6). Strikingly, analysis of the mRNAs by FISH revealed that 1AUG 39T-Kmut mRNA accumulated in the cytoplasm to the same extent as the WT-Kmut counterpart (Figure 6a, vii and viii). These results indicate that a Kozak sequence is required for nuclear retention of PTC+mRNAs, providing evidence that nuclear PTC detection requires proper recognition of the translation start site by the ribosome.
To investigate whether a scanning mechanism is involved in nuclear recognition of PTCs, we inserted a hairpin structure (hp) into the 5′ UTR of WT and 39T β-globin constructs (Figure 6b). As observed in previous work, translation and mRNA degradation were efficiently blocked by the hp (Supplementary Figure S6) [25, 42, 43]. When FISH analysis was carried out, 39T β-globin mRNA was mainly detected in the nucleus (Figure 6b, ii). Remarkably, however, in the presence of the hp, 39T β-globin mRNA accumulated in the cytoplasm to the same extent as WT (Figure 6b, iii, iv). When the same hp was inserted into the 5′ UTR of Smad constructs, similar results were obtained (Supplementary Figure S7). Together, these data indicate that a scanning mechanism is required for the prolonged nuclear retention of PTC+mRNA. It was also possible that instead of blocking scanning of the mRNA, the hp recruited some factor(s) that functions in releasing the PTC+mRNAs from the nucleus. To test this possibility, we inserted the hp into the 3′ UTR of β-globin constructs and examined its effect on the nucleocytoplasmic distribution of PTC+mRNA (Supplementary Figure S7). Significantly, 3′ hp 39T β-globin mRNA was mainly nuclear, whereas the 5′ hp 39T β-globin mRNA accumulated in the cytoplasm (Supplementary Figure S7). Together, these results indicate that inhibition of a scanning mechanism, rather than insertion of the hp itself, releases the PTC+mRNA from the nucleus.
Internal ribosomal entry sites (IRESs) recruit the ribosome and drive efficient translation initiation when cap-dependent translation is inhibited [43–45]. Our result that the Kozak sequence is required for nuclear PTC recognition suggested that the ribosome is involved in nuclear frame reading. To further investigate this possibility, we examined whether an IRES could rescue nuclear PTC recognition in the presence of the 5′ hp. To this end, the encephalomyocarditis virus (EMCV) IRES was inserted downstream of the hp and, as a control, an IRES harboring three-point mutations that inhibit translation was inserted into the same site [43, 46] (Figure 6b, hp WT/Mut IRES). Significantly, the hp 39T β-globin mRNA containing the WT IRES was retained in the nucleus, whereas the same mRNA containing the mutant IRES accumulated in the cytoplasm (Figure 6b, v–viii). These results indicate that the EMCV IRES can function in nuclear PTC recognition when scanning of the mRNA from the 5′ UTR is inhibited. Together, our data are consistent with the possibility that a ribosome-dependent scanning mechanism is involved in nuclear PTC recognition.
We next asked whether tRNA is involved in the mechanism of nuclear PTC recognition by examining whether a suppressor tRNA affects the nucleocytoplasmic distribution of PTC+mRNA. It is well established that mutation of the anticodon does not affect aminoacylation of Ser-tRNA [47] reviewed in Giege et al. [48]. We constructed a Ser-tRNA mutant (Ser-tRNACUA), in which the anticodon is complementary to the PTC (UAG) of 39T β-globin mRNA (to be clear, indicated as 39 UAG β-globin here) (Figure 6c). As a control, we made a new 39T β-globin construct (39 UGA β-globin), in which the PTC (UGA) is not complementary to the anticodon of Ser-tRNACUA. When this construct, as well as WT or 39 UAG β-globin constructs, was co-transfected with the WT Ser-tRNA (Ser-tRNAAGA), WT β-globin mRNA was mainly cytoplasmic, whereas the PTC+mRNAs were largely nuclear (Figure 6c, i–iii). Strikingly, when co-transfected with the Ser-tRNACUA mutant, 39 UAG β-globin mRNA accumulated in the cytoplasm to the same extent as WT, whereas the 39 UGA β-globin mRNA was still nuclear (Figure 6c, iv–vi). Immunofluoresence (IF) and western analysis with the anti-HA antibody revealed that co-transfection of Ser-tRNACUA resulted in efficient translation of 39 UAG β-globin mRNA, but not 39 UGA β-globin mRNA, and mass spectrometry analysis revealed that the 39 UAG was translated into serine (Figure 6c and Supplementary Table S1). Together, these results indicate that tRNA has a role in nuclear PTC recognition and this role depends on base pairing of its anticodon with the PTC.
We next investigated whether aminoacylation of tRNA is required for nuclear PTC recognition. To do this, we mutated the anticodon of the elongation Met-tRNA, whose aminoacylation strictly depends on the correct anticodon [32, 49, 50]. As shown in Supplementary Figure S8, nucleocytoplasmic distribution of the 39T β-globin mRNAs was not affected by the mutant Met-tRNA regardless of whether the PTC is complementary to the anticodon. Consistent with these results, IF and western analysis revealed that no protein was produced with PTC+β-globin mRNAs. These results indicate that aminoacylation of tRNA is required for nuclear PTC recognition. Taken together, we conclude that the charged tRNA functions in nuclear retention of PTC mRNA.
To further investigate the role of the ribosome and tRNA in nuclear PTC recognition, we treated HeLa cells with cycloheximide (CHX), which inhibits translation by competing with tRNA to bind to the ribosome [51, 52]. In control cells treated with ethanol alone, WT Smad mRNA efficiently accumulated in the cytoplasm, whereas the 133T Smad mRNA was mainly retained in the nucleus (Supplementary Figure S8). In marked contrast, upon CHX treatment, 133T Smad mRNA accumulated in the cytoplasm to the same extent as the WT counterpart, indicating that efficient binding of tRNA to the ribosome is required for the prolonged retention of the PTC+mRNAs (Supplementary Figure S8). In control studies, IF using the HA antibody revealed that translation was efficiently inhibited in cells treated with CHX (Supplementary Figure S8). Although we cannot formally rule out the possibility that inhibition of translation by CHX indirectly affects nuclear retention of PTC+mRNAs, our CHX data are consistent with all of our results, indicating that nuclear recognition of PTCs requires a frame-reading process in which both the ribosome and tRNAs are involved.
Nascent peptide chain and ribosomal proteins colocalize with mRNA in the nucleus
Our data indicating that both the ribosome and tRNA are involved in nuclear frame reading raised the possibility that the mRNA is scanned by the translating ribosome in the nucleus. To test this possibility, we first examined whether a translation product could be detected in the nucleus when mRNA export was blocked, but were unable to detect this product (GFP, luciferase, Smad, and β-globin were tried; data not shown; and see Discussion section). As an alternative approach to detect nuclear translation, we used the ribopuromycylation (RPM) method that visualizes translation in cells via standard microscopy [53]. In this method, puromycin (PMY), added to the cell culture media, enters the ribosome A site and terminates translation by ribosome-catalyzed covalent incorporation into the nascent chain’s C terminus [54]. Emetine, which blocks release of the nascent chain, is also added to immobilize the nascent peptide chain at the translation site [55, 56]. Following fixation and cell permeabilization, a PMY-specific antibody is used to detect the nascent peptide chain and mark the sites of active translation (Supplementary Figure S9). To test whether PMY is incorporated into peptide chains translated from mRNA in the nucleus, cells were microinjected with the WT β-globin construct 30 min before treatment with PMY/emetine, and IF was carried out with the anti-PMY antibody. As shown in Supplementary Figure S8, the PMY IF signal was readily detected in both the nucleus and the cytoplasm of cells treated with PMY. Significantly, PMY colocalizes with the β-globin mRNA in the nucleus (Supplementary Figure S9). In contrast, in control cells microinjected with the same β-globin construct, but not treated with PMY, no PMY IF signal was detected in either the nucleus or the cytoplasm (Supplementary Figure S9). These results suggest that PMY is incorporated into peptide chains in the nucleus.
To further investigate whether the PMY IF signal detected in the nucleus was due to translation of the β-globin mRNA and to test whether RPM also occurs on PTC+mRNA, we compared the PMY IF signal in cells microinjected with 39T β-globin or the untranslatable Kmut β-globin construct to that in cells microinjected with the WT counterpart. As shown in Figure 7a, when the WT and 39T β-globin constructs were microinjected, PMY colocalized with both WT and 39T β-globin mRNAs in the nucleus. In marked contrast, PMY signal was not detected in the nuclei of cells microinjected with the Kmut β-globin construct. This result indicates that translation is required for PMY colocalization with mRNAs and in the nucleus, RPM also occurs on PTC+mRNAs.
Figure 7.
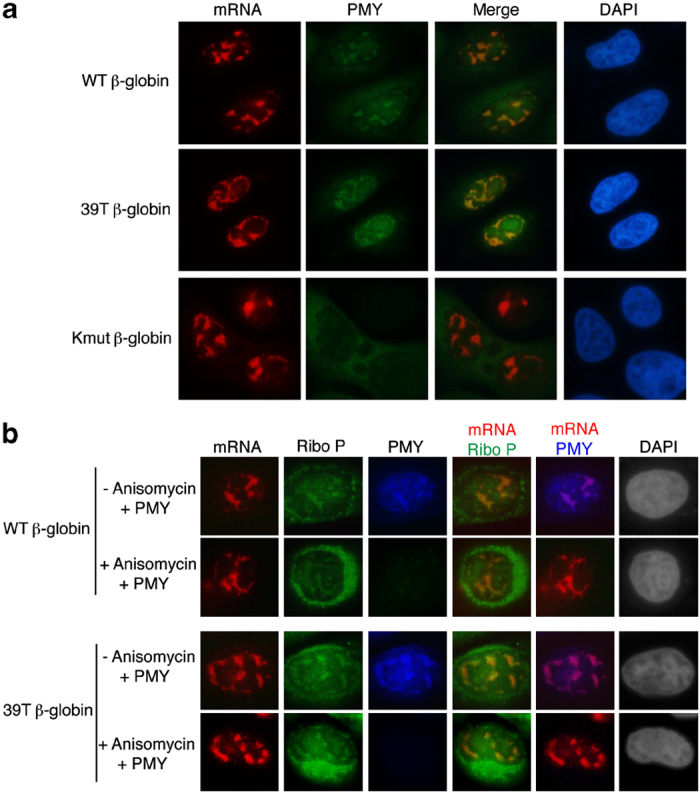
Actively translating ribosomes colocalize with mRNA in the nucleus. (a) HeLa cells were microinjected with the wild-type (WT), 39Τ, or Kmut β-globin construct. 35 mins later, cells were treated with puromycin (PMY) and emetine for 5 mins at 37 °C. Cells were then treated with digitonin for 2 mins followed by fluorescence in situ hybridization (FISH) and immunofluorescence with the PMY antibody. (b) HeLa cells were microinjected with the WT or 39Τ β-globin DNA construct. Twenty minutes later, these cells were or were not treated with anisomycin for 15 mins followed by PMY and emetine treatment for 5 mins at 37 °C. Cells were then treated with digitonin for 2 mins followed by FISH and immunofluorescence with the PMY and Ribo P (ribosomal P proteins) antibodies.
To further determine whether the nuclear PMY IF signal is due to the immobilized puromycylated peptide chain, we tested whether ribosomal proteins colocalized with PMY. To do this, HeLa cells were microinjected with WT or 39T β-globin construct and treated with PMY/emetine, followed by FISH to detect the mRNA. In addition, IF was carried out with the antibody to PMY and antibodies to ribosomal P proteins (Ribo P), which are three highly conserved phosphoproteins located in the 60S ribosomal subunit (Supplementary Figure S9). This analysis revealed that PMY colocalized with both Ribo P and β-globin mRNAs, indicating that the PMY signal is indeed from the puromycylated ribosome. Anisomycin is a competitor of PMY that blocks association of PMY with the A site of the ribosome [57]. To further investigate whether the colocalization of nuclear mRNA and PMY specifically depends on the ribosome, we pretreated the cells with anisomycin before PMY treatment. As shown in Figure 7b, while anisomycin did not affect colocalization of nuclear mRNA with Ribo P proteins, it efficiently blocked the colocalization of PMY, further confirming the specificity of the colocalization of nuclear mRNA and PMY with the ribosome. Together, these results provide strong support for the model in which mRNA is translated by the ribosome in the nucleus, and the translating ribosome proofreads the mRNA to detect PTCs in the nucleus.
A functional downstream intron is required for the prolonged nuclear retention of PTC mRNAs
The EJC, which is recruited during splicing, functions in PTC recognition during NMD in the cytoplasm [15, 17–19, 58]. Thus, we next asked whether splicing is also required for detecting PTCs in the nucleus. To do this, we constructed Smad reporter plasmids lacking the last intron that is located directly downstream from the 122T and 133T PTCs, and microinjected them (Figure 8a). Significantly, both 122T and 133T Smad Δi3 mRNAs accumulated in the cytoplasm to the same extent as WT mRNA (Figure 8a). In contrast, no effect was observed with 39T, which is located upstream from a functional intron. As expected for NMD substrates, deletion of the last intron inhibited degradation of 133T Smad mRNA, but not that of 39T mRNA (Supplementary Figure S10). We conclude that a downstream intron is required for nuclear retention of PTC+mRNAs. Moreover, this intron must be spliced as mutations of the 5′ and 3′ splice sites which inhibit splicing; (Supplementary Figure S10), resulting in cytoplasmic accumulation of 133T (Figure 8b).
Figure 8.

Upf1 is required for nuclear recognition and retention of PTC+mRNAs. (a) Schematic of Smad Δi3 constructs. Wild-type (WT) and PTC+ Smad plasmids (50 ng μl−1) were microinjected into the nuclei of HeLa cells, and α-amanitin was added 15 mins after microinjection. Fluorescence in situ hybridization (FISH) of Smad transcripts was carried out at 1 h after addition of α-amanitin. Insets show the injection marker. The graph shows the average N/C ratios for Smad mRNAs, and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (b) Same as a, except that 5′ and 3′ splicing sites on the last intron of WT, 39T, and 133T Smad plasmids were mutated as indicated. The graph shows the average N/C ratios and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (c) Western blot analysis using whole-cell lysates from control or Upf1 knockdown cells probed with an antibody against Upf1 or an antibody against eIF4AIII as a loading control. (d) WT and 133T Smad DNAs were microinjected into the nuclei of control or Upf1 knockdown HeLa cells, and α-amanitin was added 15 mins after microinjection. FISH of Smad transcripts was performed at 1 h after addition of α-amanitin. Scale bar, 10 μm. The graph shows the average N/C ratios and error bars indicate the standard errors among three independent experiments. Statistical analysis was performed as in Figure 1c. (e) Using an antibody to Upf1 or an unrelated protein, IPs were carried out from nuclear extracts prepared from HeLa cells transfected with WT or 39T β-globin constructs. RNAs were extracted from immunoprecipitates followed by RT-PCR using primers to β-globin or luciferase. The relative mRNA level of β-globin to luciferase was quantified and indicated in the graph. Error bars indicate the standard errors from three independent experiments. Statistical analysis was performed as in Figure 1c. (f) WT and PTC β-globin and Smad constructs were microinjected into the nuclei of HeLa cells, and α-amanitin was added 15 mins after microinjection. One hour after addition of α-amanitin, cells were permeabilized with digitonin (40 mg ml−1) for 2 mins followed by FISH and immunofluorescence with the Upf1 antibody. See also Supplementary Figures S9 and S10.
Detection of PTCs in the nucleus requires the NMD factor Upf1
The observation that nuclear PTC recognition requires splicing of a downstream intron is consistent with the possibility that the EJC and/or associated factors function in nuclear PTC recognition. Upf1 is a nucleocytoplasmic shuttling protein that associates with the EJC and is essential for NMD [59–61]. Moreover, Upf1 is specifically recruited to PTC+mRNA [62]. Thus, we knocked down Upf1 to determine whether it functions in nuclear PTC recognition. The knockdown efficiency was shown in Figure 8c. As expected, this knockdown inhibited degradation of 39T β-globin mRNA (Supplementary Figure S11). Strikingly, when 133T Smad mRNA was microinjected into Upf1 knockdown cells, this mRNA accumulated in the cytoplasm to the same extent as WT Smad mRNA (Figure 8d). In contrast, in the negative control knockdown, 133T mRNA was largely detected in the nucleus, whereas WT mRNA was cytoplasmic (Figure 8d). The same results were obtained with 39T β-globin mRNA (Supplementary Figure S11). We conclude that Upf1 functions in recognition of PTC+mRNAs in the nucleus. We also tested whether knockdown of the core EJC proteins eIF4AIII, Y14, and Magoh affected nuclear retention of PTC+mRNAs. Although knockdown of these proteins inhibited degradation of PTC+mRNA to some extent, the nucleocytoplasmic distribution of PTC+ mRNA was not affected (data not shown). It is possible that the remaining EJC proteins after knockdown are sufficient for nuclear PTC recognition, and further studies are needed to understand the role of the EJC in nuclear retention of PTC+mRNAs.
We next asked whether Upf1 functions to demarcate PTC+mRNAs in the nucleus. To investigate this question, WT or 39T β-globin constructs were co-transfected with luciferase. Cytoplasm-free nuclear extract was then prepared, and immunoprecipitations (IPs) were carried out using a Upf1 antibody. To determine the purity of the nuclear extract, western blot analysis of nuclear and cytoplasmic extracts was carried out using an antibody to tubulin, which is exclusively cytoplasmic. As shown in Supplementary Figure S10, tubulin was only detected in the cytoplasmic extract and not in the nuclear extract. The purity of the nuclear extract was further verified because no tubulin at all could be detected in the purified nuclear extract, whereas tubulin could be detected if we contaminated the nuclear extract so that it contained only 1 or 3% of the cytoplasmic fraction (Supplementary Figure S11). Significantly, when these cytoplasm-free nuclear extracts were used for IPs, 39T β-globin mRNA was efficiently IP'd by the Upf1 antibody, whereas WT β-globin mRNA was not (Figure 8e). In contrast, the control luciferase mRNA was not efficiently IP by the Upf1 antibody (Figure 8e). This preferential association of Upf1 with PTC+mRNA was not due to a difference in the amount of Upf1 IP by the Upf1 antibody (Figure 8e, lower panel). This result indicates that Upf1 specifically and stably associates with PTC+mRNA in the nucleus or nucleus-associated cytoplasm. To distinguish these two possibilities, we next asked whether Upf1 colocalizes with PTC+mRNA in the nucleus. WT and 39T β-globin constructs were microinjected into nuclei, followed by FISH and IF with the Upf1 antibody. As shown in Figure 8f, Upf1 colocalized with the mRNA in the nuclei of cells microinjected with 39T β-globin construct. In contrast, the Upf1 signal was diffuse in the nucleoplasm of cells microinjected with the WT β-globin construct. These results indicate that Upf1 colocalizes with PTC+β-globin mRNA, but not WT mRNA in the nucleus. When Smad constructs were microinjected, similarly, Upf1 colocalizes with 133T mRNA, but not WT mRNA, in the nucleoplasm (Figure 8f). However, it was possible that Upf1 localizes with the PTC+mRNA in the nucleus-associated cytoplasm but not in the nucleoplasm. To investigate this possibility, we used confocal microscope to acquire z-stacks through several cells followed by three-dimensional rendering of the images. As shown in Supplementary Videos S1–S4, three-dimensional movie reveals that Upf1 colocalizes with both 39T β-globin and 133T Smad mRNAs deep inside the nucleoplasm, rather than at the nuclear rim. Together, our data indicate that Upf1 functions both in nuclear recognition and retention of PTC+mRNA.
Discussion
In this study, we report that PTC+mRNAs are specifically detained in the nucleus and this retention is reading-frame dependent. Several lines of evidence as follows support this conclusion: (1) PTC+mRNAs are retained in the nucleus while their WT counterparts are rapidly exported to the cytoplasm. (2) All three nonsense codons (UAG, UAA, and UGA), but not missense codons at the same sites, result in the prolonged retention of mRNAs. (3) If all of the AUGs in the mRNA are mutated, the PTC+mRNA accumulates in the cytoplasm to the same extent as WT mRNA containing no PTC. (4) If the PTC is shifted out of frame, the PTC+ mRNA accumulates in the cytoplasm. (5) If an out-of-frame PTC in WT mRNA is shifted back in-frame, this mRNA is now retained in the nucleus. Together, these observations indicate that the frame of mRNAs is proofread in the nucleus.
Mechanism for nuclear frame reading of mRNA
To investigate the mechanism for the reading-frame-dependent retention of PTC+ mRNA in the nucleus, we first examined potential roles for the ribosome and tRNA in this process. Our data indicate that a Kozak sequence is required for nuclear retention of the PTC+mRNA. Moreover, when a hairpin is inserted into the 5′ UTR, nuclear retention of PTC+mRNAs does not occur. However, when an IRES is inserted downstream of the hairpin, the PTC+ mRNA is now retained again. We also show that nuclear retention of PTC+mRNA does not occur when the cells are treated with CHX. In the case of tRNAs, we found that a charged tRNA that has an anticodon complementary to the PTC blocked nuclear retention of PTC+mRNA, whereas an uncharged tRNA that has the same anticodon did not. Together, these data indicate that both the ribosome and tRNA function in nuclear PTC recognition and suggest that the frame of mRNA might be proofread in the nucleus via translation or a translation-like mechanism. As the gold standard for translation is nascent peptide chain formation, we used the RPM method to visualize active translation in the cells. Strikingly, our RPM data demonstrate that peptide chain synthesis occurs on a translatable mRNA, but not on an untranslatable mRNA, in the nucleus. Together, our data indicate that ribosomes scan the mRNA via at least one round of translation, proofreading the frame and detecting PTCs in the nucleus. We also sought to examine whether a nuclear translation product could be detected, but were unable to detect it (GFP, luciferase, Smad, and β-globin were tried; data not shown). One possible explanation for this negative result is that only a single round of translation may occur in the nucleus and the level of the translation product is too low to be detected. It is also possible that the translated protein is subject to rapid degradation. In either case, the nascent peptide chain would not accumulate to sufficient amount to be detected by traditional IF method using an antibody to the translation product. On top of that, the nascent peptide chain is likely to be released and diffused in the nucleoplasm. Comparing with the traditional IF, the RPM method can be more sensitive in detecting active translation for a couple of reasons. First, it detects both full-length and partially translated peptide chains. Moreover, the nascent peptide chain is now immobilized on mRNAs and thus is more concentrated. Finally, it is also possible that incorporation of PMY at the end of the nascent peptide chain protects it from degradation.
Mechanism for nuclear retention of PTC+mRNA
To investigate the mechanism involved in nuclear PTC+mRNA retention, we examined whether splicing of an intron downstream of PTC is required. We found that this was indeed the case, suggesting a role for the EJC, which is recruited in the nucleus during splicing and functions in NMD [63]. Significantly, we show that the shuttling NMD factor Upf1, which associates with EJC, specifically binds to PTC+mRNA in the nucleus. Furthermore, Upf1 colocalizes with PTC+, but not PTC−, mRNA in the nucleus. Finally, knockdown of Upf1 resulted in accumulation of PTC+mRNA in the cytoplasm. Together, these data indicate that Upf1 functions in both nuclear recognition and retention of PTC+mRNAs.
Collectively, our data lead to a working model that PTCs are detected in the nucleus by the ribosome, resulting in recruitment of the EJC-associated protein Upf1, which functions to detain the PTC+mRNA in the nucleus. Thus, our data indicate that the EJC is not only recruited in the nucleus but also functions in the nucleus. Previous work has tied PTC recognition to nuclear events (see Introduction section). However, a key uncertainty in these studies was whether PTC recognition was actually nuclear or instead occurred on the cytoplasmic face of the nuclear envelope (by the cytoplasmic translation machinery), and this PTC recognition fed back to the nucleus [31]. Importantly, however, our result showing that a PTC+ mRNA can be distinguished from the corresponding WT mRNA in the same nucleus provides compelling evidence that PTC recognition can occur in the nucleus. Furthermore, the mechanism involved in potential nuclear PTC recognition was not known. Our data reveal that this mechanism involves both the ribosome and tRNA. Moreover, our mechanistic studies revealed that Upf1 is a nuclear PTC demarcating factor that also functions in nuclear retention of PTC+mRNAs.
While nuclear recognition of PTCs appears unexpected at first glance, numerous precedents exist for nuclear events preceding functional consequences in the cytoplasm. For example, localized mRNAs recruit nuclear factors that ultimately associate with cytoplasmic machinery for proper mRNA localization (reviewed in Meignin and Davis [64]. In addition, ample precedents exist for multiple recognition events of a specific sequence. For example, each of the pre-mRNA splicing signals is recognized by several factors sequentially throughout the splicing pathway (reviewed in Wahl et al. [65]. Sequential recognition events are most likely critical for ensuring the high fidelity required for most cellular processes. In the current view of translation, PTCs are recognized only once, and this sole recognition occurs via the ribosome reading the frame in the cytoplasm. Our data indicating that PTCs are first recognized in the nucleus before their recognition in the cytoplasm would support the precedents of sequential proofreading steps observed for most cellular machineries.
Materials and Methods
Plasmids and antibodies
The WT β-globin plasmid was described [9] and 39T β-globin and 39M β-globin plasmids were constructed by mutagenesis from the WT β-globin. The WT Smad plasmid was described [9] and PTC-containing Smad plasmids were constructed by mutagenesis from WT Smad. The Smad missense mutants were generated by mutagenesis from the WT Smad plasmid. The UAP56 and GFP antibodies were described previously (Cheng, 2006; Cheng, 2002). For western blot, Upf1 antibody was diluted to 1:500 and antibodies to HA, GFP, CBP80 (ABclonal, China), UAP56 and Tubulin (Sigma, USA) were diluted to 1:1000.
Cell culture and RNA interference
HeLa cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, USA) on 35mm dishes with glass coverslip bottoms (MatTek Corp). The siRNA (Ribobio, China) sequences were shown in Supplementary Table S2. The control nontargeting small interfering RNA was purchased from Dharmacon (USA, catalog no. D0018100420). In the RPM experiment, HeLa cells were cultured in DMEM supplemented with 7.5% FBS and split 24 h before each experiment. Where indicated, translation inhibitors were used at the following concentrations: 208 μM emetine (EMD), 9.4 μM anisomycin (EMD), and 91 μM PMY (Sigma).
The DNA microinjections and FISH
HeLa cells used for microinjection were plated on fibronectin-coated 20-mm coverslip at the bottom of 35-mm dishes. Plasmid DNA (50 ng μl−1) or PCR product (100 ng μl−1) was coinjected with FITC-conjugated 70-kDa dextran. For each experiment, 80–100 cells were microinjected, followed by incubation at 37 °C. After 15 or 30 mins of incubation, transcription was terminated with α-amanitin (1 μg ml−1; Sigma), and incubation was continued for the desired period of time before fixation. The 70-nt FISH probes (sequences are shown in Supplementary Table S3) were labeled at the 5′ end with Alexa Fluor 546 NHS Ester and high-performance liquid chromatography purified. Vector probe was used except in Figure 5a. FISH was carried out as described previously [9]. Images were captured with a DP72-CCD camera (Olympus, Japan) on an inverted microscope using DP-BSW software (Olympus). FISH quantitation was carried out using ImageJ 1.33u software (National Institutes of Health). C/N ratios were calculated as described in Valencia et al. [9]. IF was performed by incubating fixed cells with the primary antibody in blocking buffer (1× phosphate-buffered saline, 0.1% Triton, and 2 mg ml−1 bovine serum albumin) for 30 mins at the room temperature. Cells were then washed three times with phosphate-buffered saline for 5 mins each and incubated with the secondary antibody in blocking buffer for another 30 mins at the room temperature, followed by DAPI staining and three washes in phosphate-buffered saline for 10 mins each. For IF, the SC35 antibody (Sigma) and the HA antibody (Sigma) were diluted to 1:1000 and 1:200, respectively, using the blocking buffer. The Upf1 antibody, which was a kind gift of Jens Lykke-Andersen [59], was diluted to 1:500. The rabbit PMY antibody was diluted to 1:1000. The anti-Ribo P antibody is a human polyclonal autoimmune antisera (ImmunoVision) and diluted to 1:200. The anti-Rabbit Cy5 and anti-Mouse Cy2 antibodies (Jackson Inc.) were diluted to 1:500. The anti-Rabbit Cy2 and anti-Human Cy3 antibodies (Jackson Inc.) were diluted to 1:200 and 1:1000, respectively.
Transfections and RNA isolation
For FISH, HeLa cells were plated on fibronectin-coated 35-mm dishes with a 20-mm coverslip at the bottom and transfected with plasmids DNA using Lipofectamine 2000 (Invitrogen). FISH was carried out at the indicated time. For RNA isolation, HeLa cells were transfected on 12-well plates at 80–90% confluency. Total RNAs were extracted using TRIzol (Invitrogen) at the indicated time.
Reverse transcription and PCR analysis
For RT-PCR, 3 μg of total RNA was treated with 3 units of the RNase-free RQ DNase I (Promega, USA). cDNA was synthesized from 1 μg of the RNA with random primer using M-MLV reverse transcriptase (Promega). To detect the NMD efficiency, 32P-dATP was added in PCR. For assaying the polyadenylation state of mRNAs, total RNA was reverse transcribed with an oligo (dT) primer linked to a G/C-rich anchor sequence [66] using the AMV reverse transcriptase (Promega), followed by PCR with the anchored dT and gene-specific primers. The PCR primers sequences were shown in Supplementary Table S4.
Nuclear and cytoplasmic extracts preparation
Cells were suspended in hypotonic buffer (10 mM HEPES, pH 7.9/1.5 mM MgCl2/10 mM KCl/0.2 mM PMSF/0.5 mM DTT) and incubated for 10 mins on ice. The swollen cells were dounced followed by centrifuge. The supernatant was collected as the cytoplasmic extract and the packed nuclear volume was estimated. The nuclei were resuspended slowly in 1/2 packed nuclear volume of low salt buffer (20 mM HEPES, pH 7.9/1.5 mM MgCl2/20 mM KCl/0.2 mM EDTA/25% glycerol/0.2 mM PMSF/0.5 mM DTT) followed by adding 1/2 packed nuclear volume of high salt buffer (20 mM HEPES, pH 7.9/1.5 mM MgCl2/1.4M KCl/0.2 mM EDTA/25% glycerol/0.2 mM PMSF/0.5 mM DTT) and mixing quickly. The mixture was rotated for 30 mins at 4 oC followed by centrifuge and the supernatant is the nuclear extract.
Immunoprecipitations
For RNA IPs, antibodies were covalently cross-linked to protein A Sepharose beads in dimethylpimelimidate (Sigma) at a 1:1 ratio of beads to antibody. Nuclear extract was rotated with antibody–cross-linked beads overnight at 4 oC. Half of the immunoprecipitates were used for SDS-polyacrylamide gel electrophoresis followed by western blot and half were used for RNAs extraction using phenol/chloroform extraction. For IP and mass spectrometry analysis, HeLa cells co-tranfected with tRNA and β-globin constructs were harvested 24 h after transfection and resuspended in 500 μl NET-2 buffer (50 mM Tris-HCl, pH 7.4/200 mM NaCl/0.1% NP40/1 mM PMSF) followed by sonication. After centrifuge, the supernatants were used for IPs using HA antibody covalently cross-linked to protein A Sepharose beads. The immunoprecipitates were used for liquid chromatography-tandem mass spectrometry analysis on a LTQ mass spectrometry (Thermo Fisher Scientific, USA).
Acknowledgments
We are grateful to Junlin Guan and Haixin Lei for their useful discussions. We thank Matthias Hentze and Joseph Sperling for providing the EMCV IRES β-globin constructs and the WT Met-tRNA plasmids. We thank Jens Lykke-Andersen for providing the Upf1 antibody. This work was funded by the Ministry of Science and Technology (973 project; 2011CB811304, and 2013CB910402), the National Natural Science Foundation of China (31270839), and the Youth Science Fund Project of the National Natural Science Foundation of China (31400676).
References
- Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature 2002; 416: 499–506. [DOI] [PubMed] [Google Scholar]
- Pandit S, Wang D, Fu XD. Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol 2008; 20: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed R. Coupling transcription, splicing and mRNA export. Curr Opin Cell Biol 2003; 15: 326–331. [DOI] [PubMed] [Google Scholar]
- Zhong XY, Wang P, Han J, Rosenfeld MG, Fu XD. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol Cell 2009; 35: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Dufu K, Lee CS, Hsu JL, Dias A, Reed R. Human mRNA export machinery recruited to the 5' end of mRNA. Cell 2006; 127: 1389–1400. [DOI] [PubMed] [Google Scholar]
- Dias AP, Dufu K, Lei H, Reed R. A role for TREX components in the release of spliced mRNA from nuclear speckle domains. Nat Commun 2010; 1: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Dias AP, Reed R. Export and stability of naturally intronless mRNAs require specific coding region sequences and the TREX mRNA export complex. Proc Natl Acad Sci USA 2011; 108: 17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MJ, Reed R. Splicing is required for rapid and efficient mRNA export in metazoans. Proc Natl Acad Sci USA 1999; 96: 14937–14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc Natl Acad Sci USA 2008; 105: 3386–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3' ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 1999; 63: 405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 1999; 8: 1893–1900. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999; 96: 307–310. [DOI] [PubMed] [Google Scholar]
- Li S, Wilkinson MF. Nonsense surveillance in lymphocytes? Immunity 1998; 8: 135–141. [DOI] [PubMed] [Google Scholar]
- Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1995; 1: 453–465. [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 2001; 20: 4987–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltz SW, Brown AH, Jacobson A. mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev 1993; 7: 1737–1754. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo MA, Lee CS, Ler LW et al. A nuclear translation-like factor eIF4AIII is recruited to the mRNA during splicing and functions in nonsense-mediated decay. Proc Natl Acad Sci USA 2004; 101: 4118–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE. Y14 and hUpf3b form an NMD-activating complex. Mol Cell 2003; 11: 939–949. [DOI] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N et al. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 2006; 20: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya T, Tange TO, Stroupe ME, Moore MJ. Mutational analysis of human eIF4AIII identifies regions necessary for exon junction complex formation and nonsense-mediated mRNA decay. RNA 2006; 12: 360–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Izumi N, Kashima I et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev 2009; 23: 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 2004; 432: 112–118. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 2004; 36: 1073–1078. [DOI] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 2008; 133: 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgrader P, Cheng J, Maquat LE. Evidence to implicate translation by ribosomes in the mechanism by which nonsense codons reduce the nuclear level of human triosephosphate isomerase mRNA. Proc Natl Acad Sci USA 1993; 90: 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter MS, Li S, Wilkinson MF. A splicing-dependent regulatory mechanism that detects translation signals. EMBO J 1996; 15: 5965–5975. [PMC free article] [PubMed] [Google Scholar]
- Kessler O, Chasin LA. Effects of nonsense mutations on nuclear and cytoplasmic adenine phosphoribosyltransferase RNA. Mol Cell Biol 1996; 16: 4426–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sun X, Qian Y, LaDuca JP, Maquat LE. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol 1998; 18: 5272–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuret R, Sun M, Lughezzani G et al. A contemporary population-based assessment of the rate of lymph node dissection for penile carcinoma. Ann Surg Oncol 2011; 18: 439–446. [DOI] [PubMed] [Google Scholar]
- de Turris V, Nicholson P, Orozco RZ, Singer RH, Muhlemann O. Cotranscriptional effect of a premature termination codon revealed by live-cell imaging. RNA 2011; 17: 2094–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlemann O, Mock-Casagrande CS, Wang J et al. Precursor RNAs harboring nonsense codons accumulate near the site of transcription. Mol Cell 2001; 8: 33–43. [DOI] [PubMed] [Google Scholar]
- Kamhi E, Raitskin O, Sperling R, Sperling J. A potential role for initiator-tRNA in pre-mRNA splicing regulation. Proc Natl Acad Sci USA 2010; 107: 11319–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla AD, Gudikote JP, Wang J et al. Nonsense codons trigger an RNA partitioning shift. J Biol Chem 2009; 284: 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Li X, Serin G, Maquat LE. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 2001; 106: 607–617. [DOI] [PubMed] [Google Scholar]
- Daneholt B. A look at messenger RNP moving through the nuclear pore. Cell 1997; 88: 585–588. [DOI] [PubMed] [Google Scholar]
- Hall LL, Smith KP, Byron M, Lawrence JB. Molecular anatomy of a speckle. Anat Rec A Discov Mol Cell Evol Biol 2006; 288: 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Bodnar MS, Spector DL. Nuclear neighborhoods and gene expression. Curr Opin Genet Dev 2009; 19: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. Defects in RNA splicing and the consequence of shortened translational reading frames. Am J Hum Genet 1996; 59: 279–286. [PMC free article] [PubMed] [Google Scholar]
- Valentine CR. The association of nonsense codons with exon skipping. Mutat Res 1998; 411: 87–117. [DOI] [PubMed] [Google Scholar]
- Johnson C, Primorac D, McKinstry M, McNeil J, Rowe D, Lawrence JB. Tracking COL1A1 RNA in osteogenesis imperfecta. splice-defective transcripts initiate transport from the gene but are retained within the SC35 domain. J Cell Biol 2000; 150: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986; 44: 283–292. [DOI] [PubMed] [Google Scholar]
- De Gregorio E, Preiss T, Hentze MW. Translation driven by an eIF4G core domain in vivo. EMBO J 1999; 18: 4865–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Gehring NH, Kulozik AE, Hentze MW. Internal ribosome entry sequence-mediated translation initiation triggers nonsense-mediated decay. EMBO Rep 2006; 7: 722–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Davies MV, Kaufman RJ, Wimmer E. Initiation of protein synthesis by internal entry of ribosomes into the 5' nontranslated region of encephalomyocarditis virus RNA in vivo. J Virol 1989; 63: 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5' nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol 1988; 62: 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson ME, Seamons RA, Belsham GJ. A selection system for functional internal ribosome entry site (IRES) elements: analysis of the requirement for a conserved GNRA tetraloop in the encephalomyocarditis virus IRES. RNA 1999; 5: 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achsel T, Gross HJ. Identity determinants of human tRNA(Ser): sequence elements necessary for serylation and maturation of a tRNA with a long extra arm. EMBO J 1993; 12: 3333–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res 1998; 26: 5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabkin HJ, Park HJ, RajBhandary UL. Amber suppression in mammalian cells dependent upon expression of an Escherichia coli aminoacyl-tRNA synthetase gene. Mol Cell Biol 1996; 16: 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal AK, Kohrer C, RajBhandary UL. Twenty-first aminoacyl-tRNA synthetase-suppressor tRNA pairs for possible use in site-specific incorporation of amino acid analogues into proteins in eukaryotes and in eubacteria. Proc Natl Acad Sci USA 2001; 98: 2268–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeehan W, Hardesty B. The mechanism of cycloheximide inhibition of protein synthesis in rabbit reticulocytes. Biochem Biophys Res Commun 1969; 36: 625–630. [DOI] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem 1971; 246: 174–181. [PubMed] [Google Scholar]
- David A, Dolan BP, Hickman HD et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 2012; 197: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S. Inhibitors of ribosome functions. Annu Rev Microbiol 1971; 25: 487–562. [DOI] [PubMed] [Google Scholar]
- Jimenez A, Carrasco L, Vazquez D. Enzymic and nonenzymic translocation by yeast polysomes. Site of action of a number of inhibitors. Biochemistry 1977; 16: 4727–4730. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012; 151: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol 2003; 330: 1061–1075. [DOI] [PubMed] [Google Scholar]
- Shibuya T, Tange TO, Sonenberg N, Moore MJ. eIF4AIII binds spliced mRNA in the exon junction complex and is essential for nonsense-mediated decay. Nat Struct Mol Biol 2004; 11: 346–351. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000; 103: 1121–1131. [DOI] [PubMed] [Google Scholar]
- Mendell JT, ap Rhys CM, Dietz HC. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science 2002; 298: 419–422. [DOI] [PubMed] [Google Scholar]
- Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev 2004; 18: 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Sato H, Tang Y, Matsuda D, Maquat LE. UPF1 association with the cap-binding protein, CBP80, promotes nonsense-mediated mRNA decay at two distinct steps. Mol Cell 2010; 39: 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer J, Iborra JP, Pries P, Clarac JP. [Mid-term behavior of the bone fixation in non-cemented Miller-Galante 1 total knee arthroplasty]. Rev Chir Orthop Reparatrice Appar Mot 2001; 87: 40–49. [PubMed] [Google Scholar]
- Meignin C, Davis I. Transmitting the message: intracellular mRNA localization. Curr Opin Cell Biol 2010; 22: 112–119. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Salles FJ, Richards WG, Strickland S. Assaying the polyadenylation state of mRNAs. Methods 1999; 17: 38–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.