

ABSTRACT
Phenotypic variation is prevalent in the zoonotic pathogen Campylobacter jejuni, the leading agent of enterocolitis in the developed world. Heterogeneity enhances the survival and adaptive malleability of bacterial populations because variable phenotypes may allow some cells to be protected against future stress. Exposure to hyperosmotic stress previously revealed prevalent differences in growth between C. jejuni strain 81-176 colonies due to resistant or sensitive phenotypes, and these isolated colonies continued to produce progeny with differential phenotypes. In this study, whole-genome sequencing of isolated colonies identified allelic variants of two purine biosynthesis genes, purF and apt, encoding phosphoribosyltransferases that utilize a shared substrate. Genetic analyses determined that purF was essential for fitness, while apt was critical. Traditional and high-depth amplicon-sequencing analyses confirmed extensive intrapopulation genetic variation of purF and apt that resulted in viable strains bearing alleles with in-frame insertion duplications, deletions, or missense polymorphisms. Different purF and apt alleles were associated with various stress survival capabilities under several niche-relevant conditions and contributed to differential intracellular survival in an epithelial cell infection model. Amplicon sequencing revealed that intracellular survival selected for stress-fit purF and apt alleles, as did exposure to oxygen and hyperosmotic stress. Putative protein recognition direct repeat sequences were identified in purF and apt, and a DNA-protein affinity screen captured a predicted exonuclease that promoted the global spontaneous mutation rate. This work illustrates the adaptive properties of high-frequency genetic variation in two housekeeping genes, which influences C. jejuni survival under stress and promotes its success as a pathogen.
IMPORTANCE
C. jejuni is an important cause of bacterial diarrheal illness. Bacterial populations have many strategies for stress survival, but phenotypic variation due to genetic diversity has a powerful advantage: no matter how swift the change in environment, a fraction of the population already expresses the survival trait. Nonclonality is thus increasingly viewed as a mechanism of population success. Our previous work identified prominent resistant/sensitive colonial variation in C. jejuni bacteria in response to hyperosmotic stress; in the work presented here, we attribute that to high-frequency genetic variation in two purine biosynthesis genes, purF and apt. We demonstrated selective pressure for nonlethal mutant alleles of both genes, showed that single-cell variants had the capacity to give rise to diverse purF and apt populations, and determined that stress exposure selected for desirable alleles. Thus, a novel C. jejuni adaptive strategy was identified, which was, unusually, reliant on prevalent genetic variation in two housekeeping genes.
INTRODUCTION
The helical Gram-negative microaerophilic epsilonproteobacterium Campylobacter jejuni is notable for extensive intrapopulation genetic variation. As revealed by the first genome sequence of C. jejuni NCTC11168 (1), much of this genetic variation results from phase variation due to lengthening or shortening of simple homopolymeric nucleotide sequence tract repeats, influencing the expression or translation of genes affecting numerous phenotypes. This typically involves genes encoding surface structures, including those required for flagellar motility (2–4), lipooligosaccharide structure (5), and capsular polysaccharide biosynthesis (6), and can also affect genes involved in DNA restriction and modification (RM) systems (1). Variation of these so-called “contingency loci” is accepted as an important survival strategy of C. jejuni and other important pathogens, promoting niche and host adaption, virulence, and antigenic diversity for immune system evasion (7). Thus, genetic variation is thought to enhance the success of C. jejuni as the leading cause of bacterial food-borne diarrheal disease in the developed world and the most frequent antecedent to Guillain-Barré syndrome demyelinating polyneuropathy (8).
Since C. jejuni does not possess homologues of DNA mismatch repair (MMR) systems (mutS, mutL, and mutH) (1, 9), reversible phase variation in homopolymeric tracts is thought to occur via slip-strand mispairing (SSM) during DNA synthesis. The failure to detect and repair SSM is thought to facilitate the high frequency of reversible phase variation, which is experimentally determined to be between 10−3 and 10−5 mutations/cell division (10). However, our previous work identified population-level phenotypic heterogeneity with characteristics that were potentially inconsistent with homopolymeric-tract-based phase variation (11). In the current study, we describe novel high-frequency genetic mutations associated with colony phenotype variation that are unusual in C. jejuni for several reasons: (i) the affected genes were not involved in the biosynthesis of surface-exposed structures or RM systems; (ii) the genetic variation was not mediated by SSM at homopolymeric tracts; and (iii) unlike homopolymeric tract changes, the DNA sequence changes did not result in frameshift truncations but were predicted to produce different alleles of genes with relatively modest amino acid sequence changes. Therefore, the variation did not result in the on-off phenotypic switching behavior characteristic of phase variation but instead enabled a spectrum of phenotypic outcomes among individual bacteria. We demonstrate that this genetic diversity occurred in two purine biosynthesis genes and drove adaptive bet hedging-like phenotypic behavior predicted to enable niche exploitation through multistress resistance/sensitivity and promotion of C. jejuni survival.
RESULTS
Whole-genome-sequencing identification of purF and apt mutations in colonial variants.
We previously identified prevalent phenotypic heterogeneity among single colonies of C. jejuni strain 81-176, observed via their relative fitness for survival following transfer to Mueller-Hinton (MH) agar supplemented with 1.0% NaCl (hyperosmotic stress) (11). In that work, colonies grown on MH agar were isolated and then spotted to NaCl-containing medium and assessed for growth or nongrowth (Fig. 1A). Differences in sensitivity or resistance to hyperosmotic stress were observed among individual colonies. To assess the heritability of these phenotypes, progeny from five sensitive colonies were passaged on MH agar and then tested for sensitivity or resistance to hyperosmotic stress as before. The majority of the progeny colonies retained the sensitive parental phenotype, but frequent phenotypic reversion was evident (Fig. 1B). To quantify the phenotypic differences between the five sensitive isolates and the heterogeneous wild type, optical density (OD)-standardized cultures were plated on MH agar or MH agar with 1.0% NaCl, and colony counts were taken. Approximately 30% of the wild-type heterogeneous population survived hyperosmotic challenge, but only 3 to 7% of the populations derived from the sensitive isolates survived (Fig. 1C). To determine whether these growth defects were due to a genetic cause, whole-genome sequencing of the five sensitive strains and the parental wild type was carried out via 454 pyrosequencing to a depth of ~16×. Variant analysis detected only one nonsynonymous mutation with 100% variation frequency in each of the five sensitive strains, occurring in either of two open reading frames (ORFs), CJJ81176_0227 (encoding the glutamate amidophosphoribosyltransferase PurF) and CJJ81176_0934 (encoding the adenine phosphoribosyltransferase Apt). Of the five mutations, three involved distinct changes in purF (Fig. 1D): variant colony 2 had a G-to-C transversion that resulted in a change of alanine to glycine at position 304 (A304G), colony 3 had a C-to-T transition that resulted in a G292S mutation, and colony 5 had a 3-bp insertion that resulted in the deletion-insertion G95delinsGV. The apt mutation in colonies 1 and 4 was identical, a T-to-C transition resulting in an I61T change in Apt. The other genes in the purF and apt operons encode genes of unrelated or unknown function, and neither these operons nor other purine biosynthesis genes are organized nearby each other on the chromosome (Fig. 1E). PurF and Apt share phosphoribosyltransferase (PRTase) domain homology (Fig. 1F), and both purF and apt are predicted purine biosynthesis pathway genes encoding PRTases that use 5-phospho-α-d-ribose-1-diphosphate (PRPP) in their respective reactions (Fig. 1G). PurF additionally harbors a glutamine amidotransferase domain.
FIG 1 .

Heritable colony stress phenotype variation linked with purF and apt mutations. (A) Schematic illustration depicting 100 single colonies. Blue indicates growth/stress resistance and yellow indicates defective growth/stress sensitivity of replicated colonies grown on Mueller-Hinton (MH) agar with 1.0% NaCl (hyperosmotic stress). Colonies with red outlines on both plates indicate NaCl-sensitive isolates designated for heritability testing. (B) Heritability assessment. Colonies outlined in red in panel A were selected from the MH agar-only plate and correspond to the five NaCl-sensitive isolates also outlined in red on MH agar with 1.0% NaCl. These clones were propagated on MH agar only and then tested for heritability of the NaCl-sensitivity defect. One hundred progeny were tested on MH agar with 1.0% NaCl; shown is a schematic illustration of progeny phenotypes, color coded as shown by the key in panel A. (C) Relative levels of NaCl stress sensitivity of colony isolates compared to that of the parental wild type. Mean results with standard errors of the means (SEM) from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only: **, P ≤ 0.01; ***, P ≤ 0.001. (D) Whole-genome sequencing identification of a single mutation with 100% variant frequency in either purF or apt in each sensitive strain, with anticipated protein effect. (E) Genomic loci and operons of purF and apt. (F) Conserved domains of purF and apt. (G) Purine biosynthesis pathway schematic with purine substrates involved in purF and apt reactions.
Deletion of apt results in growth defects, and deletion of purF is lethal.
To determine whether the mutations detected in purF and apt were equivalent to loss-of-function mutations, deletion of purF and apt was attempted via gene replacement with a nonpolar apramycin antibiotic resistance (aprr) marker (12). Deletion of purF was not possible unless a second copy of the purF gene was provided in trans (not shown). Thus, purF is likely an essential gene, consistent with a previous report (13). Deletion of apt was possible, but the Δapt::aprr strain (Δapt) was severely compromised in its growth, as assessed by plating OD-standardized equivalents from wild-type and Δapt cultures for CFU quantification. In comparison to the wild type, the Δapt strain produced 107-fold fewer CFU (Fig. 2A) and could not be transformed with a plasmid-borne apt complement construct (not shown). Complementation of the mutation was enabled by introducing the apt-containing operon (see below) at a secondary locus before deletion of apt (Δapt/apt). Microscopy of 6-h (log-phase) cultures revealed aberrant Δapt morphologies, which were not seen with apt allele mutants (not shown). Fluorescence microscopy also revealed widespread permeabilization and lysis of Δapt bacteria, indicated via phosphatidylserine-binding fluorescent annexin V staining, and the presence of extracellular DNA, shown by DNA-binding propidium iodide (PI) fluorescence external to bacterial cells (Fig. 2B). Since the Δapt mutant was highly defective for viability, it was not useful for comparative studies versus the wild type. Overall, these data showed that the sequenced apt mutants, which did not display growth defects on MH agar (not shown), were unlike the Δapt deletion strain. Furthermore, at least one functional copy of purF was required for growth; thus, the sequenced mutations resulted in purF and apt alleles that did not confer complete loss of function.
FIG 2 .
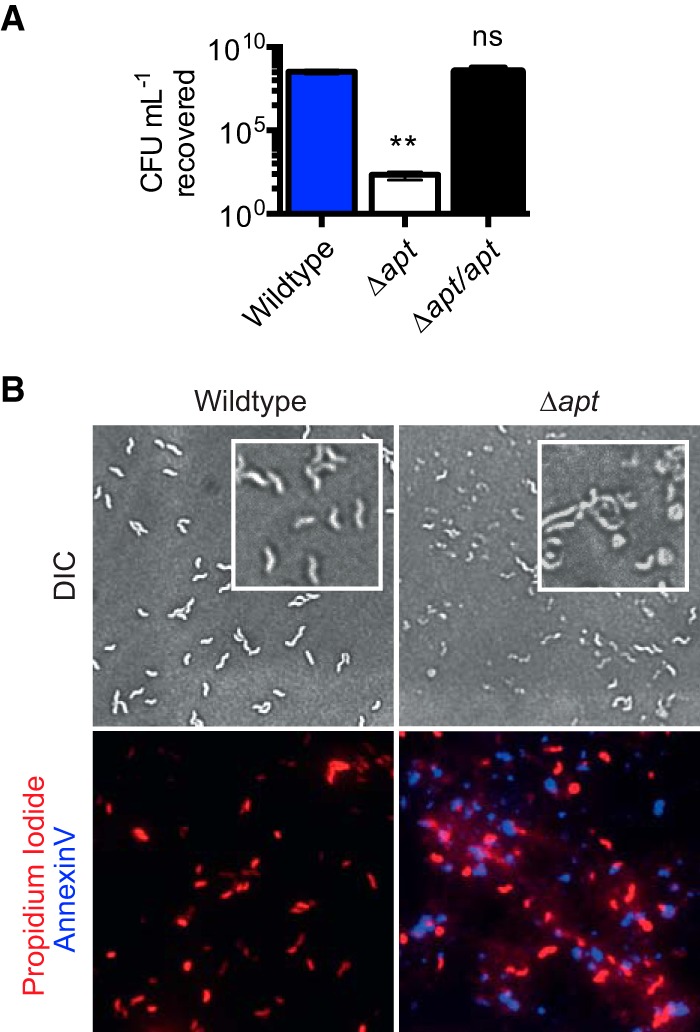
Effect of apt deletion. (A) Relative counts of CFU recovered on MH agar from OD-equivalent cultures of the parental wild-type and Δapt::aprr strains. Mean results with SEM from six independent experiments are shown: **, P ≤ 0.01; ns, not significant. (B) Bright-field (differential interference contrast [DIC]) and fluorescence microscopy examining morphology and integrity of wild-type and Δapt::aprr strains. Fluorescence microscopy samples were costained with propidium iodide for DNA (red) and annexin V conjugate for permeabilization/phosphatidylserine (blue). Representative fields of view from two independent experiments are shown.
Allelic complementation restores resistant phenotypes to sensitive isolates.
To show that purF and apt alleles were responsible for the observed sensitive/resistant phenotypes, allelic complementation was performed. Insufficient expression from the nonnative cat promoter precluded purF or apt single-gene complementation (not shown). As mutations were not detected in any other genes of the purF or apt operons, allelic complementation was carried out via in trans chromosomal integration of purF and apt variant operons (see Fig. S1A in the supplemental material). Integrated PCR-amplified purF or apt operons from wild-type genomic DNA rescued the hyperosmotic defects of purF-A304G and apt-I61T mutant strains (see Fig. S1B). Sequence analysis showed that complementation of apt-I61T was via insertion of the reference apt copy, but complementation of purF-A304G was due to insertion of a purF-T91del allele, not the reference allele. Thus, the purF-T91del allele had been unexpectedly cloned into the allelic complement. Addition of the sensitive purF-A304G or apt-I61T allele into the wild type did not result in hyperosmotic sensitivity, implying that these alleles are not dominant (see Fig. S1C).
Multifarious purF and apt mutations are prevalent in the C. jejuni population.
A comprehensive analysis of allelic variation in the heterogeneous population was carried out to associate purF and apt genotypes with stress-related phenotypes (see below). From the parental heterogeneous population passaged on MH agar, 96 colonies were chosen randomly for growth in MH broth for preservation and genomic extraction. Via traditional Sanger dideoxy methodology, the purF, apt, and prsA genes from each of the 96 single colonies were PCR amplified and sequenced. The prsA sequencing control was chosen because PrsA is the ribose-phosphate pyrophosphokinase encoded by CJJ81176_0925 that acts upstream from purF and apt in purine biosynthesis (Fig. 1G). The 96-colony purF, apt, and prsA concatenated sequence analysis revealed that the reference alleles for purF and apt were not the most prevalent and comprised only 11.5% (11/96) of the population (Fig. 3A). No mutations were identified in the prsA sequencing control. Certain alleles were rare (e.g., purF-S256R [1/96]), and other alleles were abundant (e.g., purF-T91del [14/96]). All of the mutations were missense only, with no apparent transition/transversion bias. Mutations were more common in purF (71/96) than in apt (17/96), and no colonies with mutations in both purF and apt were detected. Multiple mutations of the same gene were uncommon, and all occurred within purF (4/96). The most common mutations in purF were single single-nucleotide polymorphisms (SNPs) (34/71), followed by deletion/insertion of single codons (18/71). The most abundant mutations in apt were large insertions of 48 bp (7/17) or 51 bp (1/17). These insertions were duplications of the sequence flanking the insertion. SNPs were also detected in apt and were typically found in the coding region between the sites of the larger insertions (Fig. 3B). Mutations in purF were concentrated in a hypervariable region in the glutamate binding domain; however, mutations were also found elsewhere in purF (aligned DNA/translated sequences of purF, apt, and prsA from the 96 colonies are shown in Fig. S2 to S4 in the supplemental material).
FIG 3 .
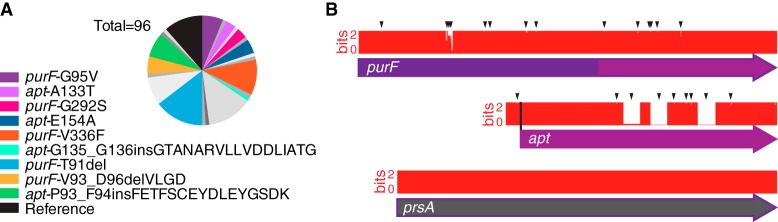
Identification of mutant alleles by sequencing purF and apt from 96 single-colony isolates. (A) Circle graph of relative levels of abundance of representative-allele-bearing strains among 96 colonies, with other alleles shown in shades of gray (see Fig. 4A for the full list). Colors denote strains bearing representative alleles that are used consistently in subsequent figures. “Reference” refers to colonies with both purF and apt identical to the C. jejuni 81-176 genome sequence. (B) Sequence identity plots of 96 aligned sequences of purF, apt, and prsA. Arrows and white areas indicate mutations/loss of identity, and white gaps in apt indicate the presence of 48-bp/51-bp duplication insertions. Not to scale. “Bits” denotes the degree of conservation of aligned sequences, where the maximum sequence conservation per site is log2 4 = 2 bits for DNA.
Differential phenotypes of single-colony isolates are associated with high-frequency variation of purF and apt.
The 96 colonies were tested simultaneously under a variety of stress conditions using OD-standardized equivalent cell numbers. In addition to 48 h of growth on MH agar with or without NaCl, the stresses tested included subjection to atmospheric O2 for 48 h, exposure to 42°C or 45°C for 48 h, and aging for 72 h. O2-stressed and aged bacteria were subsequently grown on MH agar with or without NaCl; heat stress was tested alone or on plates containing NaCl. After single or combination stress exposure, the resulting growth/nongrowth phenotype of each of the 96 strains was assessed, and spot growth was digitized into a heat map and arranged by purF and apt genotype. Results are shown only for conditions with phenotypic differences between isolates (Fig. 4A). Colonies with sensitive phenotypes comprised 20% to 30% of the total colonies tested. Overall, the different stresses were closely related in terms of phenotypic outcome; for instance, a NaCl-sensitive colony was typically sensitive to all stressors tested. Multistress sensitivity was associated with specific allele families (e.g., purF-G95V, purF-G292S, and apt-E154A), as were multiresistant phenotypes (e.g., purF-V336F). However, a number of allele families continued to display phenotypic variation between isolated colonies bearing the same mutation (e.g., purF-V93_D96delVLGD). NaCl-sensitive colonial isolates had improved phenotypic outcomes following aging.
FIG 4 .

Association of purF and apt mutations with stress phenotypes. (A) Heat map of stress phenotypes of 96 colonies (listed on the left), clustered vertically by sequenced purF or apt genotype (listed on the right, with predicted protein effect shown for clarity) and arranged horizontally by stress conditions (top). Each box in the heat map represents the average of the spot growth densitometry measurements from three independent experiments performed with OD-standardized cultures, with each average densitometry value being relative to the average value for all colony isolates grown on MH agar alone (control). Comparatively enhanced growth is shown as blue (resistant) and defective growth is yellow (sensitive), as shown by the key. Colored bars on the right identify allele families from which strains bearing representative mutant alleles are derived and are consistent with the color coding used in Fig. 3A. (B) Quantification of hyperosmotic stress resistance/sensitivity of strains bearing representative mutant alleles. CFU from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl were enumerated. Mean results with SEM from three independent experiments are shown: *, P ≤ 0.05 relative to heterogeneous wild type on MH agar with 1.0% NaCl. (C) ATP determination for OD-standardized strains bearing representative alleles and wild-type strain exposed to MH broth with 1.0% NaCl for 30 s compared to the results for the Δapt strain without NaCl exposure. Luminometric values are relative to average luminometric value of parental wild type. Mean results with SEM from six independent experiments are shown: *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001. Bars in panels B and C are color coded as shown on the right in panel A.
Underproduction of Apt correlates with stress sensitivity.
Colony 15 was found to have a unique mutation in the Shine-Dalgarno ribosome binding site (RBS) of apt, effecting a change (shown in boldface) from CGAGGCA to CGAAGCA (Fig. 4A, red). Since colony 15 was also NaCl sensitive (see Fig. S5A in the supplemental material), we reasoned that modulation of apt translation might have phenotypic outcomes analogous to those of certain primary sequence mutations. To test the translation, a reporter gene, astA, encoding arylsulfatase (14), was fused to the reference or mutant RBS under promoter-equivalent expression control and inserted separately into the strain 81-176ΔastA genetic background (see Fig. S5B). Reporter measurement demonstrated that the mutant RBS reduced translation (see Fig. S5C). Therefore, reduced enzyme production had an effect similar to the effect of mutations in apt that confer NaCl sensitivity.
Mutant alleles contribute to phenotype distribution in the population.
Mutations were associated with multistress resistance, but the initial spot growth/nongrowth phenotypic assay could not quantify enhanced or relative growth defects. To quantify increases/decreases in hyperosmotic stress survival, a more sensitive CFU-based NaCl stress test was carried out on representative strains derived from single-colony isolates harboring representative resistance- or sensitivity-conferring alleles (Fig. 4B). The representative strains had no relative growth defects when grown on MH agar alone. In contrast, the strains with the purF-V336F (12/96) and purF-T91del (14/96) alleles, which conferred resistance to NaCl stress, were recovered at ~15-and ~1.6-fold higher levels of CFU, respectively, than the heterogeneous wild type on NaCl-supplemented MH agar. All other representative-allele-bearing strains tested were sensitive to NaCl, the most sensitive of which (purF-G292S strain) yielded ~105 fewer colonies than the wild type on NaCl. Collectively, the data (Fig. 4A and B) show that the alleles in a heterogeneous population contribute a wide spectrum of specific stress phenotypes to the overall wild-type phenotype.
Given that purF and apt are purine biosynthesis genes, mutations may affect the global purine pool and cell energetics. A luciferase-based ATP determination assay was carried out as a representative assay for the purine pool and to assess respiration and ATP production. Log-phase cultures of the representative-allele-bearing strains were standardized by total cell number, exposed to 1.0% NaCl for 30 s, and lysed for ATP determination. Strains bearing alleles associated with stress sensitivity had less total ATP than the heterogeneous wild-type population or strains bearing the purF-V336F resistance-associated allele (Fig. 4C; see also Fig. S5D in the supplemental material). Without NaCl treatment, no differences in total ATP were observed (not shown). Consistent with all data described thus far, the phenotypic effects of purF and apt mutations were only revealed after exposure to stress.
High-depth amplicon sequencing reveals purF and apt genetic heterogeneity in single-colony isolate progeny.
The high rates of phenotypic reversion in the progeny of single-colony isolates and the prevalence of multiple purF and apt alleles suggested that phenotypic reversion might be due to further mutation of purF and apt or mutation back to the reference sequence. To assess this, genomic extractions from the representative-allele-bearing isolates were used for PCR amplification and high-depth (~5,000-fold) Illumina MiSeq-based amplicon sequencing of purF, apt, and prsA. Sequence variants were determined for each representative isolate and ranked by the frequency of the variation. Hyperosmotic stress testing was also carried out on 96 single-colony progeny from each of the representative-allele-bearing strains to profile progeny phenotypes (Fig. 5A). The NaCl stress phenotypes of the 96 progeny were represented by blue/yellow resistant/sensitive heat maps, which showed that the overall phenotype of the representative strain was related to the abundance of that same phenotype among the colony’s progeny. Phenotypic revertants were not observed for some representative-allele-bearing strains, and amplicon sequencing revealed that those strains either did not mutate further, did so beyond the limit of detection (e.g., apt-E154A, contains 100% variant sequence), or had low-frequency reversion (e.g., purF-G95V, contains 99% variant sequence). Analysis also revealed that certain representative isolate populations (e.g., those bearing purF-V336F and purF-V93_D96delVLGD alleles) contained both reference and other allele sequences at high frequencies. For simplicity, amplicon variants were mapped to the reference 81-176 sequence, and thus, the prevalent large duplication insertions in apt are poorly represented in this analysis. To control for sequencing artifacts, prsA was included in every analysis. No prsA variants differing from the reference sequence were observed (data not shown), thus highlighting the peculiarity of random purF and apt allele frequencies.
FIG 5 .

Identification of genetic heterogeneity within single colonies and other C. jejuni type strains, and effects of stress selection on mutant populations. Each heat map depicts the resistant/sensitive stress phenotypes, color coded as indicated by the key in panel A, of 96 progeny derived from the indicated strains and conditions, with a list of alleles detected by amplicon sequencing and their relative levels of abundance (maximum = 1.0 [100%]; the frequency listed is the ratio of allele to reference sequence at the specific base position) below. Alleles with synonymous mutations are indicated with a red “S”. Each circle in a heat map represents the average of the spot growth densitometry measurements from three independent experiments performed with OD-standardized cultures, with the average densitometry value being relative to the average value for all colony isolates grown on MH agar alone under standard conditions. (A) Stress phenotype variation and heterogeneity of purF and apt alleles within single-colony-derived representative isolates. The original Sanger sequence genotype of the representative variant colony is listed above the phenotypic heat map, with sequence variants detected by Illumina MiSeq-based amplicon sequencing listed below the heat map. (B) Stress phenotype analysis of 96 colonies derived from parental C. jejuni type strains 81116, NCTC11168-GS, NCTC11168-O, and 81-176, and heterogeneity of purF and apt alleles of each strain. Sequence variant analysis was performed using the known reference sequence specific to each type strain. (C) Effects of exposure to atmospheric O2 or repeated treatment with MH broth containing 1.0% NaCl on progeny phenotypes and purF and apt genotypes. For O2 exposure, the parental 81-176 strain was exposed to atmosphere for 48 h and then plated for single colonies on MH agar, from which 96 progeny were derived. The resistant/sensitive phenotype of each of the 96 progeny was retested by exposure to atmospheric O2 for 48 h prior to phenotypic assessment on MH plates. Genomic DNA for amplicon sequencing was obtained from pooled viable colonies after the initial O2 exposure. For successive NaCl exposure, strain 81-176 was grown in MH broth or MH broth with 1.0% NaCl for 24 h, and then NaCl-treated cells were used for repeated treatment. At each treatment endpoint, cultures were plated on MH agar to obtain viable single colonies. Stress phenotypes for 96 random colonies from MH agar were obtained by subsequent testing on MH agar with 1.0% NaCl. Genotypes of purF and apt were assessed by amplicon sequencing of genomic DNA from pooled viable colonies from MH agar.
Purine allele heterogeneity in other C. jejuni type strains.
To determine whether purF and apt variation was also found in other type strains of C. jejuni, phenotypic testing under NaCl stress and amplicon sequencing of purF, apt, and prsA were carried out on strain 81116, the genome-sequenced (GS) and original (O) versions of NCTC11168, and the initial heterogeneous strain 81-176 (Fig. 5B). These analyses demonstrated that 81-176 is the most heterogeneous strain; however, all strains displayed heterogeneity phenotypically and/or in the frequency and type of variation in purF and apt. The two versions of NCTC11168—known for a variety of phenotypic/proteomic differences (15, 16)—also differed in their overall sensitivity to hyperosmotic stress, frequency of phenotypic variation, and purF and apt alleles. For example, apt-C99Y was found in 22% of the NCTC11168-O population and yet was absent in the NCTC11168-GS strain. The phenotypic and sequencing results for strain 81116 revealed that sequence variation in purF did not produce phenotypic variation in this instance, perhaps because purF-S113L was phenotypically silent. Overall, these data confirmed that high rates of variation in purF and apt are generally present in C. jejuni spp.
Stress exposure selects fitter purF and apt alleles.
Assigning phenotypic causation to a single allele is difficult given the potential for high rates of phase variation, reversion, or secondary mutations and other confounding behavior. We hypothesized that if purF and apt mutations affected cellular responses to oxygen or osmotic stress, then oxygen or osmotic stress should select for stress-resistant purF and apt alleles. Cultures of the heterogeneous wild type were thus exposed to atmosphere (O2 stress) or subjected to repeated and increased NaCl treatment. At each endpoint, genomic DNA was harvested for amplicon sequencing, and the NaCl stress phenotypes of 96 colonies grown from the culture were assessed. Exposure to O2 selected for the multistress resistance-conferring purF-T91del and O2-resistant purF-G95V alleles, which increased from undetected to 27% and 19%, respectively, of the post-O2 exposure population (Fig. 5C). For hyperosmotic stress effects, the heterogeneous wild type was exposed to MH broth containing 0.8% NaCl for 24 h, followed by reinoculation into fresh broth and another 24-h exposure to 1.0% NaCl. The allele most differentially represented was purF-V336F, which increased from undetected in the unstressed heterogeneous wild type to 19% and 22% at each successive exposure endpoint. The apt-L92F allele also increased from undetected to 16% and 24% with each successive exposure to NaCl. Phenotypic analyses showed that stress-sensitive progeny decreased in abundance after exposure to either O2 or NaCl. Most of the alleles detected via amplicon sequencing were not previously identified in the screen of 96 colonies.
Intracellular survival in epithelial cells is affected by genetic heterogeneity in purF and apt.
Successful host cell invasion and intracellular survival of C. jejuni are correlated with oxidative stress resistance and virulence (17). Since purF and apt alleles affected O2 and hyperosmotic stress tolerance, the representative-allele-bearing strains were used to infect INT407 host epithelial cells and assessed for intracellular survival by gentamicin protection assay. Invasion and intracellular survival were determined by lysing the epithelial cells and recovering intracellular bacteria for CFU enumeration at relevant time points (Fig. 6A). The counts of CFU recovered from host cells infected with the heterogeneous wild type were normalized to a value of 1.0 at each time point, for comparison to the counts of CFU recovered for representative isolates. The progression from inoculum to 5 h and 8 h intracellular survival is shown (Fig. 6). Representative-allele-bearing strains invaded INT407 host cells equally well (3-h time point; not shown). However, NaCl resistance correlated with intracellular survival, e.g., the strains bearing NaCl stress-resistant alleles purF-V336F and purF-T91del were not significantly different from the heterogeneous wild type. However, at the 8-h time point, strains bearing the purF-V336F or purF-T91del allele yielded 12.1-fold and 6.8-fold more bacteria, respectively, than the strain bearing the NaCl-sensitive allele apt-E154A, which was significantly impaired for intracellular survival compared to the heterogeneous wild type. We next assayed all 96 single-colony isolates in independent host cell infections and compared bacterial recovery from cells to bacterial NaCl resistance. Regardless of the NaCl stress phenotype, the variant strains invaded cells equally well (Fig. 6B, 3-h time point). In contrast, differential intracellular behavior was observed among the 96 single-colony isolates at the 8-h intracellular-survival time point (Fig. 6B, bottom). Furthermore, NaCl sensitivity/resistance phenotypes for allele families or siblings (Fig. 6B, color coded as in Fig. 4A) were generally similar to the intracellular survival trends, e.g., NaCl-sensitive apt-E154A siblings were defective for intracellular survival and NaCl-resistant purF-V336F siblings generally survived better than strains bearing other alleles.
FIG 6 .

Fitness selection and effects of purF and apt mutations in invasion and intracellular survival in infection of INT407 epithelial cells. (A) Invasiveness and intracellular survival of representative-allele-bearing strains in semiconfluent epithelial cells assessed by gentamicin protection assay. The numbers of wild-type CFU recovered following host cell lysis were normalized to 1.0 for each assay time point (0 h, infection inoculum; 5 h and 8 h, intracellular bacteria). Counts of CFU of representative-allele-bearing strains recovered are compared to those of heterogeneous wild type at each relevant time point. Mean results with SEM from three independent experiments are shown, and statistics are shown for 8-h time point: *, P ≤ 0.05; **, P ≤ 0.01; ns, not significant. (B) Adherent/intracellular bacteria (3 h, top), and intracellular survival (8 h, bottom) of 96 purF- and apt-genotyped single-colony isolates in INT407 epithelial cells. Each dot represents an isolate (out of 96); the average CFU count recovered from host cells for an individual isolate relative to the average CFU count recovered for the entire population is shown on the x axis, and growth on NaCl relative to the population average is on the y axis (data are from the experiment whose results are shown in Fig. 4). CFU data are from three independent experiments. CFU recovered at 3 h include bacteria adherent to host cells and intracellular bacteria; CFU recovered at 8 h from gentamicin-treated host cells are from intracellular bacteria only. Dots (B) and bars (A) are color coded to represent the families of representative alleles shown in Fig. 4, and grey dots are used for all other variants (B). (C) CFU recovered from typical INT407 infection with heterogeneous wild-type C. jejuni 81-176 at infection (0 h), adherent and intracellular (3 h), and intracellular survival (5, 8, and 12 h) time points. The infection inoculum is the total number of bacteria used to infect host cells (MOI of 100). (D) Strain selection for allele fitness as shown by intracellular survival and examination of NaCl resistance/sensitivity phenotypes in 96 colonies after liberation from host cells. Amplicon-sequencing analysis of variants was performed on genomic DNA from pooled colonies recovered on MH plates at each intracellular time point. Each pool was the equivalent of three independent experiments with 24 technical replicates (total of 72 infections at each intracellular time point to maximize CFU/allele recovery).
During a typical C. jejuni host cell infection (Fig. 6C), a bottlenecking event occurs in which only a fraction of the total bacterial population adhere to and invade cells and survive intracellularly (in the data shown in Fig. 6A, these data for the wild type were normalized to 1.0 for each time point and do not reflect the actual CFU obtained at each time point). We hypothesized that the cell infection/survival process would select for alleles conferring fitness in epithelial cells. INT407 cells were infected with the heterogeneous wild type, and the purF, apt, and prsA allele sequences were assessed by amplicon sequencing prior to infection and at 5, 8, and 12 h postinfection (Fig. 6D). Sequencing was carried out on pooled colonies obtained by plating harvested epithelial cell lysates. The most selected-for allele in the population was purF-V336F, which increased in abundance from 14% of the preinfection population to 22%, 31%, and 77% at 5, 8, and 12 h postinfection. Since replication of C. jejuni within host cells is not thought to occur (18), this likely represented increased survival rather than growth. The NaCl resistance of 96 colonies recovered at each time point was assessed as described above, and the results showed that passage through INT407 cells selected for variants with NaCl stress-resistant phenotypes. These data show that passage of C. jejuni through epithelial cells selects for stress fit variants.
The putative DNA-binding PolB-like exonuclease Cj1132c promoted spontaneous mutation.
As mutations affecting purine biosynthesis might affect global mutation frequencies (19), the strains bearing representative alleles were tested for differences in the spontaneous mutation rate. Mutation frequency was assessed by enumerating spontaneous resistance to the fluoroquinolone ciprofloxacin, which is conferred by DNA gyrase (gyrA) point mutations (20, 21). No significant differences in the rates of spontaneous ciprofloxacin resistance were detected for the strains bearing representative alleles and the heterogeneous wild-type population (not shown), consistent with prsA-sequencing data. Although the observed mutations in purF were not limited to the hypervariable region, a direct repeat (AATCGCTATAGGGCATAATCGCTAT) was found in purF directly upstream from the hypervariable region, potentially a protein binding site. A similar yet imperfect sequence (AATAGCAACTGG_ins_CGGAACAGCTAT) was found at the boundaries of an apt duplication-insertion. Thus, we reasoned that a specific DNA-binding factor may promote hypervariation. A protein pulldown assay, using as the bait a 5′-biotinylated truncated purF sequence containing the direct repeat and hypervariable region, was performed via stable isotope labeling of amino acids in cell culture (SILAC)-based proteomics (22). Two putative DNA-binding proteins were pulled down in two independent experiments: Cj1132c (encoded by CJJ81176_1149, gene annotated as wlaX), and SSB (single-stranded DNA-binding protein, encoded by CJJ81176_1089) (Fig. 7A). In this assay, pulled-down proteins with nonspecific interactions with the purF DNA probe, such as PorA (Fig. 7B, top), have an equal ratio of [13C6]arginine-labeled and [13C614N4]arginine-labeled peptides, whereas potentially specific interactions are identified by primarily [13C6]arginine-labeled peptides (Fig. 7B, bottom). Since SSB is a ubiquitous protein, Cj1132c was chosen for further assessment; it is predicted to belong to an uncharacterized bacterial group of DEDDy 3′-to-5′ exonucleases, which have homology with the proofreading DNA replication/repair exonuclease domains of family B DNA polymerases (Pfam DNA_pol_B_exo2). To assess roles for Cj1132c in purF variation, cj1132c was deleted (Δcj1132c) in the purF-T91del allele background because T91del is located in the hypervariable region of purF and the differential allele and phenotypic-switching frequencies were known and amenable to testing in this strain. This also avoided the additional complication of single-colony isolation from the heterogeneous wild-type 81-176 strain, which might yield uncharacterized variants during mutant construction. In the purF-T91del background, the Δcj1132c mutation did not seem to affect the rate of NaCl stress phenotypic variation (not shown). However, the Δcj1132c mutation did influence the spontaneous mutation rate, and fewer ciprofloxacin-resistant colonies were produced by the Δcj1132c mutant than by the comparator strain or the mutant with Δcj1132c/cj1132c genetic complementation (Fig. 7C). Thus, Cj1132c did not actively promote hypermutation of purF in these limited analyses but may have a role in DNA maintenance.
FIG 7 .

SILAC-based affinity capture of Cj1132c with purF DNA bait and effect of cj1132c deletion on spontaneous mutation rate. (A) Screen for DNA-binding proteins captured with purF DNA bait. Differentially labeled (light, [13C6]arginine, or heavy, [13C614N4]arginine) C. jejuni total protein lysates were incubated with biotinylated purF DNA or prsA DNA, respectively. A 1:1 light/heavy mixture of DNA/protein complexes was captured on streptavidin beads. Proteins were eluted from beads by digestion with PstI restriction enzyme, and trypsinized proteins were analyzed by gel electrophoresis liquid chromatography-mass spectrometry. Each dot represents an identified peptide. SILAC analysis discriminates the specificity of an interaction; peptides with a low heavy/light ratio indicate higher-affinity capture. Data are representative of two independent experiments statistically assessed by the Benjamini-Hochberg false discovery rate (FDR), and FDR Q significance values are indicated: *, Q = 0.024; **, Q = 0.014. (B) Representative mass spectra for SILAC-labeled captured peptides. Top, equal mass/charge ratio of light and heavy forms of PorA peptide indicating nonspecific interaction; bottom, the light form of Cj1132c is present in a ratio >16.8 times higher than the heavy form, indicating a high probability of interaction with the direct repeat and hypervariable region of purF. (C) Frequency of emergence of spontaneous ciprofloxacin resistance (spontaneous mutation rate) of Δcj1132c and complemented Δcj1132c/cj1132c mutants in purF-T91del genetic background. The numbers of spontaneous ciprofloxacin-resistant mutants relative to total CFU per OD600 equivalent are presented. Mean results with SEM from six independent experiments each with two technical replicates are shown: ****, P ≤ 0.0001.
DISCUSSION
Purines are evolutionarily ancient molecules, and the biosynthesis or uptake of purines is essential to life. Purines are involved in the synthesis of DNA and RNA (adenosine/guanosine), cell energetics (ATP/GTP), and cell signaling alarmones and stress responses [cyclic AMP, cyclic di-GMP, and (p)ppGpp] and are central to metabolism (NADH/coenzyme A) (23). The purine precursor, PRPP, is also important for amino acid biosynthesis (histidine/tryptophan) (24, 25) and for pyrimidine synthesis and salvage (23, 26). PRPP is used by PurF for de novo IMP nucleoside biosynthesis (and thus for AMP/GMP production) and by Apt for the recycling/salvage of the adenine nucleotide to the nucleoside form (AMP), and it provides redundancy in AMP production. Except for apt, the purine salvage pathway is absent in C. jejuni, and the bacterium encodes neither nucleotide/nucleoside transporters nor the array of PRTases, deaminases, or phosphorylases for guanine, thymidine, hypoxanthine, and xanthosine salvage (1). Thus, C. jejuni appears to be incapable of extracellular purine salvage. In agreement with this, we found that ΔpurF mutants could not be generated on medium supplemented with adenine, adenosine, guanine, and/or guanosine, and an exogenous supply of adenosine did not rescue the growth defect of the Δapt mutant (not shown). Thus, our data suggest that de novo purine biosynthesis is essential for C. jejuni and that salvage is absent or dispensable. In contrast, the related Helicobacter pylori does not encode the de novo purine biosynthesis pathway and relies solely on purine salvage, utilizing an outer membrane nuclease not present in C. jejuni to digest extracellular DNA (27). As C. jejuni is a naturally competent organism, whole-DNA uptake is a potential source of nucleobases for Apt salvage, although this remains to be tested.
Mutations in the purine biosynthetic pathway ultimately lead to changes in the availability of purines and intermediates, with complex consequences given the central role of purine metabolites and the integration of stress responses with cellular metabolic pathways. Purine limitation effects are described for many bacterial species. Purine auxotrophs (ΔpurF mutants) of Mycobacterium smegmatis have reduced viability in O2-depleted stationary-phase culture (28). In Bacillus subtilis, purine deprivation promotes sporulation, resulting from a reduction in the synthesis of AMP/GMP (29). Purine auxotrophic mutants of Listeria, Mycobacterium, Francisella, and Brucella species are severely attenuated for virulence/colonization in animal models (30–34), and both Salmonella and Shigella flexneri purine biosynthesis mutants have been evaluated as vaccine strains because of the attenuation of those strains (35, 36).
How might changes in the purine pool affect Campylobacter’s response to osmotic and oxidative stress? In Escherichia coli, hyperosmotic stress (37) or ΔpurF deletion (38) activates the expression of the polyphosphate kinase ppk, which results in the accumulation of polyphosphate, a high-energy phosphate storage polymer (39, 40). In C. jejuni, polyphosphate is required for hyperosmotic stress survival, survival during carbon starvation, and intracellular survival in epithelial cells and is accumulated in the stationary growth phase (41). Furthermore, the guanosine-based alarmones ppGpp and pppGpp are known to enhance polyphosphate accumulation (41), which is also absent in E. coli ΔrelA (p)ppGpp synthetase mutants (37, 42). Additional mechanisms, such as ppGpp-mediated inhibition of the exopolyphosphatase ppx, lead to polyphosphate accumulation (37). Thus, altered polyphosphate reservoirs may account for the phenotypic effects of the mutations in purF and apt, which will be a focus of future work.
In Lactococcus lactis, mutations in guaA (GMP synthetase; de novo purine biosynthesis) and hpt (hypoxanthine guanine PRTase; purine salvage) confer multistress (acid, oxidative, and heat stress) resistance, as do mutations in relA (43). This suggests that flux in the purine biosynthesis pathway and/or ppGpp fluctuations are stress signals that lead to stress resistance. For C. jejuni, we also found that specific purF and apt mutations have the opposite consequence, contributing to multistress sensitivity. Other modified purines, such as the second messengers cyclic AMP and cyclic di-GMP, may also be affected by mutations in purF and apt. In C. jejuni, cyclic di-GMP is implicated in deoxycholate stress resistance (44), and in other bacteria, it has roles in biofilm formation (45). In addition, C. jejuni uses nucleotide-activated sugars in the biosynthesis of glycans, such as the GDP precursors of the O-linked flagellin glycans (46–48). Thus, because purine biosynthesis mutations may influence numerous signaling cascades, metabolic regulation, and specific stress response systems, purF and apt mutations in C. jejuni are likely to have pleotropic consequences for stress resistance and sensitivity mechanisms.
Why are the mutant purF and apt phenotypes only evident under stress? The strains with different purF and apt mutant alleles—except for Δapt strains—all grow equally well under the replete, nonstressed conditions of standard MH medium (not shown). This may explain the prevalence of multiple alleles in the MH-grown population, because there is minimal selective pressure acting on less fit or fitter alleles. In rich MH medium, the growth rate of C. jejuni can be much faster than in many environments where transmission and colonization occur (49–53). High DNA replication rates are known to outpace and saturate postreplicative DNA MMR mechanisms (54, 55). Known MMR mechanisms are absent in C. jejuni (9); thus, accelerated growth rates may contribute to enhanced mutation rates. Since the Δapt null mutant exhibited poor growth in MH medium and the ΔpurF mutant could not grow at all, selective pressure is maintained for alleles conveying overall functionality of both purF and apt in MH medium. The effect of additional selective pressure on the mutant allele population was demonstrated via exposure to NaCl, O2, and the intracellular milieu of INT407 epithelial cells (Fig. 5C and 6D). In each experiment, a stress-fit allele was selected for, and the allele was prominently represented in the poststress population. Since genome-wide association of particular alleles and phenotypes may not conclusively prove a linkage between allele and phenotype, these experiments provide additional support for the specific role of purF and apt mutations in stress fitness.
Highly expressed genes are known to undergo higher mutational frequencies (56), but both purF and apt were expressed at levels similar to that of the nonhypervariable prsA (not shown). Furthermore, transcription of purF and apt was not significantly affected by exposure to osmotic stress (not shown), signifying that the observed phenotypic effects are posttranscriptional or posttranslational. The mutation in the RBS in the representative apt colony 15 demonstrated that reduced translation of Apt had effects synonymous with those of purF and apt mutations that conferred stress sensitivity. Thus, changes in translation efficiency do have a role in this process; however, because the majority of observed mutations altered the primary sequence of purF and apt, this suggests that phenotypes potentially arise from differential enzyme kinetics, insensitivity or hypersensitivity to allosteric feedback, or other, unknown mechanisms (57). As PurF is responsible for purine commitment in the biosynthesis pathway, the enzyme receives feedback from GMP/AMP and from other adenine and guanine nucleotides (58–60). Apt activity is competitively inhibited by various 5′ nucleotides and diphosphate, in cooperation with PRPP, and is subject to end product inhibition by AMP (61–63).
Conserved domain analysis of the purF and apt alleles suggests that the mutations do not occur in conserved active-site residues (not shown). No conclusions can thus far be made about the specific enzymatic effects of altered residues based solely on the different phenotypic outcomes. Although the phenotypes of alleles with low ATP levels correlated with the phenotype of a strain underproducing Apt, ATP levels are a measure of cell energy/respiration and do not necessarily provide insight into the effects of primary sequence mutations in purF and apt on their respective reactions. In the current study, the results of ATP assays generally mirrored major phenotypic differences between single-colony isolates under stress, with more stress-resistant variants showing higher levels of ATP and sensitive variants lower levels of ATP. Addressing the specific effects of mutations on the purF and apt reactions requires in-depth analysis of substrate-to-product ratios. This is an interesting question for future experiments, but the high rates of purF and apt reversion or mutation in most single-colony isolates result in mixed populations that will complicate comparative metabolomic approaches. The caveat of measurements performed on mixed populations is that they reflect the average population behavior (64), which may not be consistent with the specific effect of the original founder allele.
The hypervariability of purF and apt represents a novel phenomenon. A recent study by Mohawk et al. (65) found similar patterns of mutation in the motA flagellar motor protein gene in C. jejuni 81-176, although these mutations abrogated MotA function. This further supports the hypothesis that C. jejuni strain 81-176 is highly variable, exceeding the known phase variation and mutator frequencies of other bacteria (9). Mutator phenotypes in model bacteria typically result from mutator alleles, often mutant MMR genes, with corresponding genome-wide 100- to 1,000-fold increases in transitions, frameshifts, and chromosomal rearrangements (66). Given the absence of MMR genes in Campylobacter, this implies that this is a genus of constitutive mutators. Using ciprofloxacin resistance-based methodologies, the spontaneous mutation rate of our heterogeneous C. jejuni 81-176 parent strain was determined to be ~10−8 (not shown), equivalent to previously established rates (21, 67). Since flaws in purine biosynthesis have been shown to have genome-wide mutational consequences in other organisms (19), purine biosynthesis mutants could be potential mutator alleles. However, despite differences in purF and apt variant frequencies between isolates, the global spontaneous mutation rate was unaffected in each representative isolate (ciprofloxacin resistance; not shown), and other gene mutations were not detected (prsA sequencing). Thus, purF and apt mutations are not classic mutator alleles. Since 85/96 random colonies had various mutations affecting purF and apt and given that contingency locus phase variation occurs at rates of ~10−3 (1, 3, 5, 10), spontaneous mutation rate assessments via ciprofloxacin resistance do not assess the true scope of mutation frequency in the C. jejuni population. Ignoring recombination and horizontal gene transfer, we speculate that the (i) basal mutation frequency, (ii) homopolymeric tract-based phase variation, and (iii) nonhomopolymeric high-frequency variation in purF, apt, and potentially, other genes each contribute to the total variation in the C. jejuni population. The DNA sequences and phenotypes of the purF and apt alleles are examined many generations after the mutation occurred, and during this time, selective pressure and unknown factors have almost certainly acted to confound determination of gene-specific mutation rates. Thus, an important caveat of this study is that it is difficult to measure the mutation rates of specific genes, such as purF and apt.
Why, then, are mutations in purF and apt so prevalent if the same variation is not observed for all genes? Why does the observed selective pressure not ensure sequence fidelity in purF and apt? Current evolutionary opinion is that mutations occur randomly (68), unless a specific mechanism exists to increase the rate of mutation. In the absence of a conventional homopolymeric-tract-based mechanism of phase variation, we assessed whether a specific DNA-binding factor was responsible for purF hypervariation, reasoning that a sequence-specific protein might affect DNA replication fidelity. The putative exonuclease Cj1132c was identified as a candidate purF binding protein. Although cj1132c deletion decreased the emergence of spontaneous ciprofloxacin-resistant mutants, the gene product did not appear to promote purF hypervariability within the limitations of the study performed here. Since the PCR-generated purF probe bait would contain mismatched/single-stranded DNA sequences, the pulldown experiment and subsequent mutation frequency analyses may have identified Cj1132c as a potential repair/DNA maintenance mediator. In C. jejuni, the transcription-repair coupling factor Mfd (mutation frequency decline) promotes spontaneous fluoroquinolone resistance, and Δcj1132c, like Δmfd, decreased the spontaneous mutation rate (20). Despite finding no clear role for Cj1132c in purF and apt hypervariation, our experiments with Cj1132c are included in this study, in part because the SILAC-based affinity screen exemplifies new experimental proteomics approaches available for C. jejuni researchers. These data were also included because they demonstrated challenges posed by heterogeneous populations in reductionist-genetics-based methodologies, discussed below.
Natural populations of bacteria are assumed to be genetically heterogeneous, but laboratory isolates, which are often derived from a single purified colony, are assumed to be reasonably homogenous. Our data are evidence that this is a false assumption for C. jejuni laboratory strains and their progeny. The creation of a deletion mutant necessitates the selection of a single colony, and comparative analyses (often a deletion mutant compared to the original wild type) require otherwise isogenic backgrounds to ensure that phenotypes are not due to secondary mutations. The multiple mutative properties of C. jejuni therefore complicate such analyses, highlighting the importance of phenotype verification from multiple isogenic clones. Homopolymeric-tract-mediated phase variation has been thought to drive the majority of C. jejuni genetic and the resultant phenotypic variation. Phase variation is a stochastic process; thus, we speculate that the effects of phase variation are partly mitigated by the high rates of both on and off switching in homopolymeric tracts, resulting in a predictable on-to-off equilibrium (10). In contrast, since there is no homopolymeric tract or obvious mechanism to revert back to the original sequence, the effects of purF and apt hypervariability may be considerable for a bottlenecked population. Furthermore, phase variation results only in on or off phenotypic states, whereas purF and apt hypervariability has the potential for multiple phenotypic states. Thus, genetic analyses that depend on single-colony procedures that alter population dynamics must be approached with caution by researchers in the field. The potential for high-frequency secondary mutations reinforces the need for (i) phenotypic testing of multiple isogenic clones of deletion mutants and (ii) complementation of mutants.
A population with variable genomes enhances the probability of survival of a bacterial species. In the current study, conclusive evidence was provided for genetic variation in purF and apt and was identified as a mechanism promoting extensive phenotypic heterogeneity in a mixed C. jejuni population. We provided further evidence for the role of this genetic diversity in relevant niche exploitation, and we conclude that purF and apt variability is a novel strategy ensuring C. jejuni success, akin to phenotypic bet hedging (69–71) and yet reliant on the mutation of specific genes. How nonlethal hypervariability of purF and apt occurs remains unexplained and will be the focus of future investigations, as will analyses of the specific cellular effects of various purF and apt alleles. Also of interest is whether any other C. jejuni genes (in addition to purF, apt, and motA) undergo similar hypervariability. Even though this study examined only purF and apt, the genetic heterogeneity in the parental population was so enormous that the prevalence of mutant alleles exceeded that of the wild-type reference allele. This begs the question of what exactly is the wild type? Others have speculated that Campylobacters are a type of quasispecies (1, 72, 73); i.e., a population of genotypes, often organized around more prevalent genotypes of higher fitness (74), with a wild-type sequence that is defined as the average of all present genotypes (75). Our data support this definition and demonstrate the effect of selection on the distribution of mutants. In conclusion, this study identified high-frequency genetic variability in the fitness-requisite housekeeping genes purF and apt, revealing a novel adaptive mechanism important for the life cycle of C. jejuni, with clear consequences for the investigation and containment of this widespread pathogen.
MATERIALS AND METHODS
Bacterial strains, colony isolation, and growth conditions.
Studies were performed with C. jejuni human isolate strain 81-176 (76), unless otherwise stated (see Table S1 in the supplemental material for a full list of bacterial strains and plasmids). C. jejuni was grown on MH agar or broth (Oxoid) supplemented with vancomycin (10 µg·ml−1) and trimethoprim (5 µg·ml−1) in a microaerobic (6% O2) or increased-CO2 (12% CO2) atmosphere in a Sanyo tri-gas incubator (solid medium) or generated via the CampyGen (Oxoid) system (shaken broth cultures). For colony variant identification, strains were plated to give ~150 colonies/plate on MH agar and were grown for 48 h. Individual colonies were selected and inoculated into 200 µl of MH broth in each well of a 96-well plate and grown for 24 h prior to preservation and phenotypic testing.
Deletions, allelic complementation, and fluorescence microscopy.
All PCR was performed with high-fidelity iProof polymerase (Bio-Rad). For replacement deletion, the purF and apt genes were PCR amplified from 81-176 genomic DNA with oligonucleotides 6409/6410 and 6413/6414, respectively, and ligated to pGEM-T vector. Inverse PCR amplification of the resulting plasmids, pGEM-T-purF and pGEM-T-apt, with oligonucleotides 6411 (KpnI) and 6412 (XbaI) and 6415 (KpnI) and 6416 (BamHI), respectively, introduced the restriction sites indicated in parentheses. Similarly digested apramycin resistance cassettes from pAC1A (12) were ligated to the inverse PCR products. The resulting plasmids were introduced into C. jejuni 81-176, and homologous recombinants were selected for on MH agar containing 60 µg·ml−1 apramycin. Transformants were only recovered for the Δapt::aprr mutation. Deletion of cj1132c was carried out by a similar methodology (see Table S2 in the supplemental material for oligonucleotides). Allelic complementations/gene introductions were performed via the introduction of purF and apt variant allele operons or other constructs into the appropriate strain with the genome-insertional gene-delivery plasmid pRRH or pRRA (12). For fluorescence microscopy, 1 µl of bacterial culture was incubated with 30 µM propidium iodide and 1 µl of Alexa Fluor 350-annexin V conjugate (Life Technologies) for 10 min prior to mounting on a 1.0% agarose pad for visualization. Cells were imaged at ×100 magnification with a Nikon TE 2000-U microscope equipped with an argon-ion laser (EXFO X-Cite) and a charge-coupled device camera (Hamamatsu).
Phenotypic stress assessments, ATP determination, and cell infection.
To assess mutant phenotypes, single-colony cultures in 96-well plates were diluted with MH broth to an optical density at 600 nm (OD600) of 0.05, and 5-µl amounts were spotted on test condition plates. Test conditions included growth for a total of 48 h on MH agar with 0.8% NaCl, or with 1.0% NaCl, growth for 48 h at 42°C or 45°C (on plates), or 72 h aging or exposure to atmosphere (O2) for 48 h (in liquid broth) prior to resumption of growth (spotted onto plates) for 48 h under standard conditions, or as indicated in Fig. 4A for combination stresses. Growth/nongrowth of spots was quantified from scanned plates by using ImageJ densitometry tools, and the data converted to heat map visualizations via Matrix2png (77). NaCl sensitivity/resistance phenotypes were analyzed by plating serial dilutions with an OD600 of 0.1 of representative strains on MH agar or MH agar with 1.0% NaCl and enumerating CFU. Luciferase-based ATP determination was carried out on dilutions to an OD600 of 0.2 of C. jejuni exposed to 1.0% NaCl for 30 s that were quantified by using a Varioskan luminometer (Thermo Scientific) according to the manufacturer’s instructions (ATP determination kit; Life Technologies). Cell infection gentamicin protection assays were performed with INT407 intestinal epithelial cells, as previously described (41). Briefly, 1-ml wells containing semiconfluent INT407 epithelial cells were inoculated with logarithmic-phase C. jejuni at a multiplicity of infection (MOI) of 100 (0-h time point; inoculation). Host cell adherence and invasion were assessed prior to the addition of 150 µg·ml−1 gentamicin (Gm) by plating H2O-lysed epithelial cells for bacterial CFU enumeration on MH agar at 3 h postinfection (adherence/invasion time point). Gm treatment was carried out for 2 h to kill extracellular bacteria (5-h time point; intracellular bacteria), and then the medium was replaced with 10 µg·ml−1 of Gm. Intracellular survival was assessed by lysing Gm-treated epithelial cells and plating for bacterial CFU at 8 and 12 h postinfection. Arylsulfatase assays were performed as previously described (78). The viability of the Δapt mutant was assessed by plating for CFU on MH agar using serial dilutions of bacteria standardized to an OD600 of 0.05 after being harvested from bacterial growth on MH plates. All t test statistical analyses were performed in Prism (GraphPad) unless otherwise stated.
Conventional, whole-genome, and amplicon sequencing.
Genomic DNA for all experiments was harvested via Wizard genomic DNA purification (Promega). Conventional dideoxy Sanger sequencing was performed by Genewiz on high-fidelity PCR-amplified DNA obtained using the oligonucleotides listed in Table S2 in the supplemental material and manually assembled and verified. Whole-genome sequencing of five isolates to a depth of ~16× was performed on the 454 GS FLX titanium (Roche 454) sequencing platform. Shotgun library preparation, read mapping, and variant analysis were performed according to established procedures (79), using the sequence deposited in GenBank under accession number CP000538.1 as the reference sequence (The Institute for Genomic Research). Amplicon sequencing of purF, apt, and prsA was performed on the Illumina MiSeq system. To avoid PCR bias, the reaction products of two separate 25-cycle PCRs (52°C annealing temperature, 45-s extension) using optimized semiuniversal oligonucleotides (see Table S2) were pooled for each amplicon. Equimolar amounts of the three amplicons for each sample were pooled for preparation of 24 Nextera XT libraries, according to the manufacturer’s preparation guide (Illumina). Libraries were quantified using KAPA library quantification kit number kk4284 (Kapa) as recommended except with 10-µl volumes and 90-s annealing/extension PCR and then pooled equimolarly and normalized to 4 nM. The libraries were sequenced with a 2 × 300-bp paired-end v3 kit on a MiSeq instrument (Illumina) following the manufacturer’s protocols. Variant discovery was performed using GATK (Broad Institute) software (see variant information in Data Set S1).
SILAC-based DNA-protein interaction screen.
Affinity capture of DNA-binding proteins from SILAC extracts was performed as previously described (22), with modification. Briefly, C. jejuni (ΔargH::cat arginine auxotroph; unpublished) was metabolically labeled with 400 µM either [13C6]arginine (light) or [13C614N4]arginine (heavy) (Cambridge Isotope Laboratories) as the SILAC amino acid in modified Dulbecco's modified Eagle's medium (Caisson) (supplemented with 20 mM glutamine, 10 µM iron ascorbate) and lysed via sonication in 50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Triton X-100. Affinity purifications were performed with biotinylated biotin–TEG (triethyleneglycol, a spacer)–double-stranded DNA purF bait (or prsA control DNA) immobilized on streptavidin beads (Invitrogen). Protein-DNA complexes were pooled, eluted by PstI (NEB) restriction enzyme cleavage, and digested with ArgC, and purified peptides were analyzed by reverse-phase liquid chromatography-mass spectrometry. MaxQuant (80) was used for identification and quantification of data from two independent experiments (see Data Set S2 in the supplemental material); a Benjamini-Hochberg false discovery rate of 0.05 was used for statistical assessment (Perseus). Additional description of methods is in Text S1 in the supplemental material.
SUPPLEMENTAL MATERIAL
Additional methods for isotope labelling of the C. jejuni ΔargH proteome for DNA-protein affinity chromatography, and methods for the identification and statistical analysis of recovered peptides. Download
Amplicon sequencing for variant analysis. Download
SILAC-labeled peptides detected via DNA-protein affinity capture. Download
Effects of allelic complementation with purF and apt sequence variants. (A) Schematic of wild-type gene loci and plasmid-borne constructs for allelic complementation. Nonnative gene expression level of purF or apt alone from the chloramphenicol resistance cassette promoter contained in pRRH (12) was insufficient for allelic complementation (not shown); therefore, allelic complementation was performed with purF and apt variants in native operon/promoter context, inserted into the genome via gene delivery plasmid pRRH into the rRNA locus. (B) Relative NaCl resistance/sensitivity of single-colony isolates after introduction of reference/variant alleles. CFU enumerated from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only: **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significantly different from the wild type. (C) Relative NaCl resistance/sensitivity of the parental wild type after introduction of variant alleles. CFU were enumerated from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only; ns, not significantly different from the wild type. Download
DNA and predicted protein sequence alignments of purF sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
DNA and predicted protein sequence alignments of apt sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
DNA and predicted protein sequence alignments of prsA sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
Effects of differential translation of apt on NaCl stress phenotypes. (A) Relative NaCl resistance/sensitivity of colony 15 of 96. CFU from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl were enumerated. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only: ***, P ≤ 0.001. (B) Schematic of promoter-RBS-reporter fusion constructs. The reference (CGAGGCA) or variant (CGAAGCA) RBS was placed in context to the arylsulfatase reporter gene astA and cloned into pRRA genome-insertional plasmid for delivery into the 81-176ΔastA strain. Expression in both constructs is polycistronic via the promoter of the apramycin antibiotic resistance marker. (C) Spectrophotometric measurement of astA gene product production, via colorimetric arylsulfatase assay. Mean results with SEM from four independent experiments are shown: * P ≤ 0.05. (D) ATP determination of OD-standardized representative apt colony 15 and wild-type strains exposed to MH broth with 1.0% NaCl for 30 s. Luminometric values are relative to average luminometric value of parental wild type. Mean results with SEM from three independent experiments are shown: **, P ≤ 0.01. Download
Bacterial strains and plasmids used in this study.
Oligonucleotides used in this study.
ACKNOWLEDGMENTS
This project was supported by Canadian Institutes of Health Research (CIHR) grants MOP-68981 to E.C.G. and MOP-77688 to L.J.F., by the Burroughs Wellcome Fund (E.C.G.), and USDA Agricultural Research Service CRIS project 5325-42000-047 to C.T.P. N.E.S was supported by a National Health and Medical Research Council of Australia Overseas Biomedical fellowship (no. APP1037373) and a Michael Smith Foundation for Health Research postdoctoral fellowship (no. 5363). A.C. is supported by a CIHR Canada graduate doctoral scholarship.
A.C., E.F., N.E.S., and E.C.G. conceived and designed the experiments. A.C., S.H., and N.E.S. performed the experiments. A.C., S.H., E.F., N.E.S., L.J.F., C.T.P., and E.C.G. analyzed the data. E.C.G., C.T.P., and L.J.F. contributed reagents/materials/analysis tools. A.C., E.F., and E.C.G. wrote the manuscript.
Footnotes
Citation Cameron A, Huynh S, Scott NE, Frirdich E, Apel D, Foster LJ, Parker CT, Gaynor EC. 2015. High-frequency variation of purine biosynthesis genes is a mechanism of success in Campylobacter jejuni. mBio 6(5):e00612-15. doi:10.1128/mBio.00612-15.
REFERENCES
- 1.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. 2000. The genome sequence of the foodborne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 2.Park SF, Purdy D, Leach S. 2000. Localized reversible frameshift mutation in the flhA gene confers phase variability to flagellin gene expression in Campylobacter coli. J Bacteriol 182:207–210. doi: 10.1128/JB.182.1.207-210.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrixson DR. 2006. A phase-variable mechanism controlling the Campylobacter jejuni FlgR response regulator influences commensalism. Mol Microbiol 61:1646–1659. doi: 10.1111/j.1365-2958.2006.05336.x. [DOI] [PubMed] [Google Scholar]
- 4.Hitchen P, Brzostek J, Panico M, Butler JA, Morris HR, Dell A, Linton D. 2010. Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene. Microbiology 156:1953–1962. doi: 10.1099/mic.0.038091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerry P, Szymanski CM, Prendergast MM, Hickey TE, Ewing CP, Pattarini DL, Moran AP. 2002. Phase variation of Campylobacter jejuni 81–176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro. Infect Immun 70:787–793. doi: 10.1128/IAI.70.2.787-793.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacon DJ, Szymanski CM, Burr DH, Silver RP, Alm RA, Guerry P. 2001. A phase-variable capsule is involved in virulence of Campylobacter jejuni 81–176. Mol Microbiol 40:769–777. doi: 10.1046/j.1365-2958.2001.02431.x. [DOI] [PubMed] [Google Scholar]
- 7.Van der Woude MW, Baumler AJ. 2004. Phase and antigenic variation in bacteria. Clin Microbiol Rev 17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaser MJ. 1997. Epidemiologic and clinical features of Campylobacter jejuni infections. J Infect Dis 176(Suppl 2):S103–S105. doi: 10.1086/513780. [DOI] [PubMed] [Google Scholar]
- 9.Ambur OH, Davidsen T, Frye SA, Balasingham SV, Lagesen K, Rognes T, Tønjum T. 2009. Genome dynamics in major bacterial pathogens. FEMS Microbiol Rev 33:453–470. doi: 10.1111/j.1574-6976.2009.00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bayliss CD, Bidmos FA, Anjum A, Manchev VT, Richards RL, Grossier J-, Wooldridge KG, Ketley JM, Barrow PA, Jones MA, Tretyakov MV. 2012. Phase variable genes of Campylobacter jejuni exhibit high mutation rates and specific mutational patterns but mutability is not the major determinant of population structure during host colonization. Nucleic Acids Res 40:5876–5889. doi: 10.1093/nar/gks246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cameron A, Frirdich E, Huynh S, Parker CT, Gaynor EC. 2012. Hyperosmotic stress response of Campylobacter jejuni. J Bacteriol 194:6116–6130. doi: 10.1128/JB.01409-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron A, Gaynor EC. 2014. Hygromycin B and apramycin antibiotic resistance cassettes for use in Campylobacter jejuni. PLoS One 9:e95084. doi: 10.1371/journal.pone.0095084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metris A, Reuter M, Gaskin DJ, Baranyi J, van Vliet AH. 2011. In vivo and in silico determination of essential genes of Campylobacter jejuni. BMC Genomics 12:535. doi: 10.1186/1471-2164-12-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendrixson DR, DiRita VJ. 2003. Transcription of σ54-dependent but not σ28-dependent flagellar genes in Campylobacter jejuni is associated with formation of the flagellar secretory apparatus. Mol Microbiol 50:687–702. doi: 10.1046/j.1365-2958.2003.03731.x. [DOI] [PubMed] [Google Scholar]
- 15.Gaynor EC, Cawthraw S, Manning G, MacKichan JK, Falkow S, Newell DG. 2004. The genome-sequenced variant of Campylobacter jejuni NCTC 11168 and the original clonal clinical isolate differ markedly in colonization, gene expression, and virulence-associated phenotypes. J Bacteriol 186:503–517. doi: 10.1128/JB.186.2.503-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott NE, Marzook NB, Cain JA, Solis N, Thaysen-Andersen M, Djordjevic SP, Packer NH, Larsen MR, Cordwell SJ. 2014. Comparative proteomics and glycoproteomics reveal increased N-linked glycosylation and relaxed sequon specificity in Campylobacter jejuni NCTC11168 O. J Proteome Res 13:5136–5150. doi: 10.1021/pr5005554. [DOI] [PubMed] [Google Scholar]
- 17.Fields JA, Thompson SA. 2008. Campylobacter jejuni CsrA mediates oxidative stress responses, biofilm formation, and host cell invasion. J Bacteriol 190:3411–3416. doi: 10.1128/JB.01928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson RO, Galán JE. 2008. Campylobacter jejuni survives within epithelial cells by avoiding delivery to lysosomes. PLoS Pathog 4:e14. doi: 10.1371/journal.ppat.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang B, McFaline JL, Burgis NE, Dong M, Taghizadeh K, Sullivan MR, Elmquist CE, Cunningham RP, Dedon PC. 2012. Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proc Natl Acad Sci U S A 109:2319–2324. doi: 10.1073/pnas.1118455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, Sahin O, Barton Y, Zhang Q. 2008. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog 4:e1000083. doi: 10.1371/journal.ppat.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanninen ML, Hannula M. 2007. Spontaneous mutation frequency and emergence of ciprofloxacin resistance in Campylobacter jejuni and Campylobacter coli. J Antimicrob Chemother 60:1251–1257. doi: 10.1093/jac/dkm345. [DOI] [PubMed] [Google Scholar]
- 22.Mittler G, Butter F, Mann M. 2009. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome Res 19:284–293. doi: 10.1101/gr.081711.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hove-Jensen B. 1988. Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. J Bacteriol 170:1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ames BN, Martin RG, Garry BJ. 1961. The first step of histidine biosynthesis. J Biol Chem 236:2019–2026. [PubMed] [Google Scholar]
- 25.Galperin MY, Koonin EV. 1997. Sequence analysis of an exceptionally conserved operon suggests enzymes for a new link between histidine and purine biosynthesis. Mol Microbiol 24:443–445. doi: 10.1046/j.1365-2958.1997.3671706.x. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman I, Kornberg A, Simms ES. 1955. Enzymatic synthesis of pyrimidine nucleotides; orotidine-5′-phosphate and uridine-5′-phosphate. J Biol Chem 215:403–451. [PubMed] [Google Scholar]
- 27.Liechti G, Goldberg JB. 2012. Helicobacter pylori relies primarily on the purine salvage pathway for purine nucleotide biosynthesis. J Bacteriol 194:839–854. doi: 10.1128/JB.05757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keer J, Smeulders MJ, Williams HD. 2001. A purF mutant of Mycobacterium smegmatis has impaired survival during oxygen-starved stationary phase. Microbiology 147:473–481. doi: 10.1099/00221287-147-2-473. [DOI] [PubMed] [Google Scholar]
- 29.Freese E, Heinze JE, Galliers EM. 1979. Partial purine deprivation causes sporulation of Bacillus subtilis in the presence of excess ammonia, glucose and phosphate. J Gen Microbiol 115:193–205. doi: 10.1099/00221287-115-1-193. [DOI] [PubMed] [Google Scholar]
- 30.Bacon GA, Burrows TW, Yates M. 1951. The effects of biochemical mutation on the virulence of Bacterium typhosum; the loss of virulence of certain mutants. Br J Exp Pathol 32:85–96. [PMC free article] [PubMed] [Google Scholar]
- 31.Buchmeier NA, Libby SJ. 1997. Dynamics of growth and death within a Salmonella typhimurium population during infection of macrophages. Can J Microbiol 43:29–34. doi: 10.1139/m97-005. [DOI] [PubMed] [Google Scholar]
- 32.Chiang SL, Mekalanos JJ. 1998. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol 27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- 33.Pechous R, Celli J, Penoske R, Hayes SF, Frank DW, Zahrt TC. 2006. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect Immun 74:4452–4461. doi: 10.1128/IAI.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faith NG, Kim JW, Azizoglu R, Kathariou S, Czuprynski C. 2012. Purine biosynthesis mutants (purA and purB) of serotype 4b Listeria monocytogenes are severely attenuated for systemic infection in intragastrically inoculated A/J mice. Foodborne Pathog Dis 9:480–486. doi: 10.1089/fpd.2011.1013. [DOI] [PubMed] [Google Scholar]
- 35.Cersini A, Martino MC, Martini I, Rossi G, Bernardini ML. 2003. Analysis of virulence and inflammatory potential of Shigella flexneri purine biosynthesis mutants. Infect Immun 71:7002–7013. doi: 10.1128/IAI.71.12.7002-7013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Callaghan D, Maskell D, Liew FY, Easmon CS, Dougan G. 1988. Characterization of aromatic- and purine-dependent Salmonella typhimurium: attention, persistence, and ability to induce protective immunity in BALB/c mice. Infect Immun 56:419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ault-Riche D, Fraley CD, Tzeng CM, Kornberg A. 1998. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol 180:1841–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung S, Lee M-S, Kim S-K, Wanner BL. 2012. Effect of purine limitation caused by an amidophosphoribosyl transferase (purF) mutation on polyphosphate kinase 1 (ppk1) gene expression. Genes Genomics 34:27–34. doi: 10.1007/s13258-011-0128-8. [DOI] [Google Scholar]
- 39.Rao NN, Gómez-García MR, Kornberg A. 2009. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- 40.Kornberg A, Rao NN, Ault-Riché D. 1999. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem 68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 41.Candon HL, Allan BJ, Fraley CD, Gaynor EC. 2007. Polyphosphate kinase 1 is a pathogenesis determinant in Campylobacter jejuni. J Bacteriol 189:8099–8108. doi: 10.1128/JB.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuroda A, Murphy H, Cashel M, Kornberg A. 1997. Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem 272:21240–21243. doi: 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- 43.Rallu F, Gruss A, Ehrlich SD, Maguin E. 2000. Acid- and multistress-resistant mutants of Lactococcus lactis: identification of intracellular stress signals. Mol Microbiol 35:517–528. doi: 10.1046/j.1365-2958.2000.01711.x. [DOI] [PubMed] [Google Scholar]
- 44.Raphael BH, Pereira S, Flom GA, Zhang Q, Ketley JM, Konkel ME. 2005. The Campylobacter jejuni response regulator, CbrR, modulates sodium deoxycholate resistance and chicken colonization. J Bacteriol 187:3662–3670. doi: 10.1128/JB.187.11.3662-3670.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garavaglia M, Rossi E, Landini P. 2012. The pyrimidine nucleotide biosynthetic pathway modulates production of biofilm determinants in Escherichia coli. PLoS One 7:e31252. doi: 10.1371/journal.pone.0031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young KT, Davis LM, Dirita VJ. 2007. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol 5:665–679. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- 47.McNally DJ, Hui JP, Aubry AJ, Mui KK, Guerry P, Brisson JR, Logan SM, Soo EC. 2006. Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81–176 using a focused metabolomics approach. J Biol Chem 281:18489–18498. doi: 10.1074/jbc.M603777200. [DOI] [PubMed] [Google Scholar]
- 48.Schoenhofen IC, Vinogradov E, Whitfield DM, Brisson JR, Logan SM. 2009. The CMP-legionaminic acid pathway in Campylobacter: biosynthesis involving novel GDP-linked precursors. Glycobiology 19:715–725. doi: 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- 49.Cools I, Uyttendaele M, Caro C, D’Haese E, Nelis HJ, Debevere J. 2003. Survival of Campylobacter jejuni strains of different origin in drinking water. J Appl Microbiol 94:886–892. doi: 10.1046/j.1365-2672.2003.01916.x. [DOI] [PubMed] [Google Scholar]
- 50.Chynoweth RW, Hudson JA, Thom K. 1998. Aerobic growth and survival of Campylobacter jejuni in food and stream water. Lett Appl Microbiol 27:341–344. doi: 10.1046/j.1472-765X.1998.00453.x. [DOI] [PubMed] [Google Scholar]
- 51.Haddad N, Burns CM, Bolla JM, Prevost H, Federighi M, Drider D, Cappelier JM. 2009. Long-term survival of Campylobacter jejuni at low temperatures is dependent on polynucleotide phosphorylase activity. Appl Environ Microbiol 75:7310–7318. doi: 10.1128/AEM.01366-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hazeleger WC, Wouters JA, Rombouts FM, Abee T. 1998. Physiological activity of Campylobacter jejuni far below the minimal growth temperature. Appl Environ Microbiol 64:3917–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buswell CM, Herlihy YM, Lawrence LM, McGuiggan JT, Marsh PD, Keevil CW, Leach SA. 1998. Extended survival and persistence of Campylobacter spp. in water and aquatic biofilms and their detection by immunofluorescent-antibody and -rRNA staining. Appl Environ Microbiol 64:733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schaaper RM. 1988. Mechanisms of mutagenesis in the Escherichia coli mutator mutD5: role of DNA mismatch repair. Proc Natl Acad Sci U S A 85:8126–8130. doi: 10.1073/pnas.85.21.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horst JP, Wu TH, Marinus MG. 1999. Escherichia coli mutator genes. Trends Microbiol 7:29–36. doi: 10.1016/S0966-842X(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 56.Park C, Qian W, Zhang J. 2012. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep 13:1123–1129. doi: 10.1038/embor.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bera AK, Chen S, Smith JL, Zalkin H. 1999. Interdomain signaling in glutamine phosphoribosylpyrophosphate amidotransferase. J Biol Chem 274:36498–36504. doi: 10.1074/jbc.274.51.36498. [DOI] [PubMed] [Google Scholar]
- 58.Zhou G, Smith JL, Zalkin H. 1994. Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. J Biol Chem 269:6784–6789. [PubMed] [Google Scholar]
- 59.Shimaoka M, Takenaka Y, Kurahashi O, Kawasaki H, Matsui H. 2007. Effect of amplification of desensitized purF and prs on inosine accumulation in Escherichia coli. J Biosci Bioeng 103:255–261. doi: 10.1263/jbb.103.255. [DOI] [PubMed] [Google Scholar]
- 60.Zhou G, Charbonneau H, Colman RF, Zalkin H. 1993. Identification of sites for feedback regulation of glutamine 5-phosphoribosylpyrophosphate amidotransferase by nucleotides and relationship to residues important for catalysis. J Biol Chem 268:10471–10481. [PubMed] [Google Scholar]
- 61.Hochstadt J. 1978. Adenine phosphoribosyltransferase from Escherichia coli. Methods Enzymol 51:558–567. doi: 10.1016/S0076-6879(78)51078-1. [DOI] [PubMed] [Google Scholar]
- 62.Sin IL, Finch LR. 1972. Adenine phosphoribosyltransferase in Mycoplasma mycoides and Escherichia coli. J Bacteriol 112:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hochstadt-Ozer J, Stadtman ER. 1971. The regulation of purine utilization in bacteria. I. Purification of adenine phosphoribosyltransferase from Escherichia coli K12 and control of activity by nucleotides. J Biol Chem 246:5294–5303. [PubMed] [Google Scholar]
- 64.Davidson CJ, Surette MG. 2008. Individuality in bacteria. Annu Rev Genet 42:253–268. doi: 10.1146/annurev.genet.42.110807.091601. [DOI] [PubMed] [Google Scholar]
- 65.Mohawk KL, Poly F, Sahl JW, Rasko DA, Guerry P. 2014. High frequency, spontaneous motA mutations in Campylobacter jejuni strain 81-176. PLoS One 9:e88043. doi: 10.1371/journal.pone.0088043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denamur E, Matic I. 2006. Evolution of mutation rates in bacteria. Mol Microbiol 60:820–827. doi: 10.1111/j.1365-2958.2006.05150.x. [DOI] [PubMed] [Google Scholar]
- 67.Bae J, Jeon B. 2013. Increased emergence of fluoroquinolone-resistant Campylobacter jejuni in biofilm. Antimicrob Agents Chemother 57:5195–5196. doi: 10.1128/AAC.00995-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martincorena I, Seshasayee ASN, Luscombe NM. 2012. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485:95–98. doi: 10.1038/nature10995. [DOI] [PubMed] [Google Scholar]
- 69.de Jong IG, Haccou P, Kuipers OP. 2011. Bet hedging or not? A guide to proper classification of microbial survival strategies. Bioessays 33:215–223. doi: 10.1002/bies.201000127. [DOI] [PubMed] [Google Scholar]
- 70.Dubnau D, Losick R. 2006. Bistability in bacteria. Mol Microbiol 61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- 71.Veening JW, Smits WK, Kuipers OP. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol 62:193–210. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- 72.Thomas DK, Lone AG, Selinger LB, Taboada EN, Uwiera RR, Abbott DW, Inglis GD. 2014. Comparative variation within the genome of Campylobacter jejuni NCTC 11168 in human and murine hosts. PLoS One 9:e88229. doi: 10.1371/journal.pone.0088229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jerome JP, Bell JA, Plovanich-Jones AE, Barrick JE, Brown CT, Mansfield LS. 2011. Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS One 6:e16399. doi: 10.1371/journal.pone.0016399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holmes EC, Moya A. 2002. Is the quasispecies concept relevant to RNA viruses? J Virol 76:460–465. doi: 10.1128/JVI.76.1.460-462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biebricher CK, Eigen M. 2006. What is a quasispecies? Curr Top Microbiol Immunol 299:1–31. [DOI] [PubMed] [Google Scholar]
- 76.Korlath JA, Osterholm MT, Judy LA, Forfang JC, Robinson RA. 1985. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J Infect Dis 152:592–596. doi: 10.1093/infdis/152.3.592. [DOI] [PubMed] [Google Scholar]
- 77.Pavlidis P, Noble WS. 2003. Matrix2png: a utility for visualizing matrix data. Bioinformatics 19:295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 78.Davis L, Young K, DiRita V. 2008. Genetic manipulation of Campylobacter jejuni. Curr Protoc Microbiol Chapter 8:Unit 8A.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kyle JL, Cummings CA, Parker CT, Quiñones B, Vatta P, Newton E, Huynh S, Swimley M, Degoricija L, Barker M, Fontanoz S, Nguyen K, Patel R, Fang R, Tebbs R, Petrauskene O, Furtado M, Mandrell RE. 2012. Escherichia coli serotype o55:H7 diversity supports parallel acquisition of bacteriophage at Shiga toxin phage insertion sites during evolution of the O157:H7 lineage. J Bacteriol 194:1885–1896. doi: 10.1128/JB.00120-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional methods for isotope labelling of the C. jejuni ΔargH proteome for DNA-protein affinity chromatography, and methods for the identification and statistical analysis of recovered peptides. Download
Amplicon sequencing for variant analysis. Download
SILAC-labeled peptides detected via DNA-protein affinity capture. Download
Effects of allelic complementation with purF and apt sequence variants. (A) Schematic of wild-type gene loci and plasmid-borne constructs for allelic complementation. Nonnative gene expression level of purF or apt alone from the chloramphenicol resistance cassette promoter contained in pRRH (12) was insufficient for allelic complementation (not shown); therefore, allelic complementation was performed with purF and apt variants in native operon/promoter context, inserted into the genome via gene delivery plasmid pRRH into the rRNA locus. (B) Relative NaCl resistance/sensitivity of single-colony isolates after introduction of reference/variant alleles. CFU enumerated from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only: **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significantly different from the wild type. (C) Relative NaCl resistance/sensitivity of the parental wild type after introduction of variant alleles. CFU were enumerated from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only; ns, not significantly different from the wild type. Download
DNA and predicted protein sequence alignments of purF sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
DNA and predicted protein sequence alignments of apt sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
DNA and predicted protein sequence alignments of prsA sequenced from 96 single-colony isolates by conventional dideoxy Sanger sequencing. DNA sequence corresponds to protein-coding sequence only. White shading indicates loss of identity. Download
Effects of differential translation of apt on NaCl stress phenotypes. (A) Relative NaCl resistance/sensitivity of colony 15 of 96. CFU from OD-standardized cultures plated on MH agar or MH agar with 0.8% NaCl were enumerated. Mean results with SEM from three independent experiments are shown, presented as the ratio of CFU recovered on MH agar with 1.0% NaCl versus CFU recovered on MH agar only: ***, P ≤ 0.001. (B) Schematic of promoter-RBS-reporter fusion constructs. The reference (CGAGGCA) or variant (CGAAGCA) RBS was placed in context to the arylsulfatase reporter gene astA and cloned into pRRA genome-insertional plasmid for delivery into the 81-176ΔastA strain. Expression in both constructs is polycistronic via the promoter of the apramycin antibiotic resistance marker. (C) Spectrophotometric measurement of astA gene product production, via colorimetric arylsulfatase assay. Mean results with SEM from four independent experiments are shown: * P ≤ 0.05. (D) ATP determination of OD-standardized representative apt colony 15 and wild-type strains exposed to MH broth with 1.0% NaCl for 30 s. Luminometric values are relative to average luminometric value of parental wild type. Mean results with SEM from three independent experiments are shown: **, P ≤ 0.01. Download
Bacterial strains and plasmids used in this study.
Oligonucleotides used in this study.