

ABSTRACT
Stimulation of the antiviral response depends on the sensing of viral pathogen-associated molecular patterns (PAMPs) by specialized cellular proteins. During infection with RNA viruses, 5′-di- or -triphosphates accompanying specific single or double-stranded RNA motifs trigger signaling of intracellular RIG-I-like receptors (RLRs) and initiate the antiviral response. Although these molecular signatures are present during the replication of many viruses, it is unknown whether they are sufficient for strong activation of RLRs during infection. Immunostimulatory defective viral genomes (iDVGs) from Sendai virus (SeV) are among the most potent natural viral triggers of antiviral immunity. Here we describe an RNA motif (DVG70-114) that is essential for the potent immunostimulatory activity of 5′-triphosphate-containing SeV iDVGs. DVG70-114 enhances viral sensing by the host cell independently of the long stretches of complementary RNA flanking the iDVGs, and it retains its stimulatory potential when transferred to otherwise inert viral RNA. In vitro analysis showed that DVG70-114 augments the binding of RIG-I to viral RNA and promotes enhanced RIG-I polymerization, thereby facilitating the onset of the antiviral response. Together, our results define a new natural viral PAMP enhancer motif that promotes viral recognition by RLRs and confers potent immunostimulatory activity to viral RNA.
IMPORTANCE
A discrete group of molecular motifs, including 5′-triphosphates associated with double-stranded RNA, have been identified as essential for the triggering of antiviral immunity. Most RNA viruses expose these motifs during their replication; however, successful viruses normally evade immune recognition and replicate to high levels before detection, indicating that unknown factors drive antiviral immunity. DVGs from SeV are among the most potent natural viral stimuli of the antiviral response known to date. These studies define a new natural viral motif present in DVGs that maximizes viral recognition by the intracellular sensor RIG-I, allowing fast and strong antiviral responses even in the presence of viral-encoded immune antagonists. This motif can be harnessed to increase the immunostimulatory potential of otherwise inert viral RNAs and represents a novel immunostimulatory enhancer that could be used in the development of vaccine adjuvants and antivirals.
INTRODUCTION
Detection of specific viral pathogen-associated molecular patterns (PAMPs) by host pathogen recognition receptors is the first event in the defense against virus infection (1–3). During infection with RNA viruses, the RIG-I-like receptors (RLRs) RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated protein 5) bind viral PAMPs present in the infected cell and initiate a signaling cascade mediated by the mitochondrial antiviral signaling (MAVS) molecule, which culminates in the production of type I interferons (IFNs) and other antiviral proteins (4–6). This primary host response to infection is essential to control virus replication and to initiate protective antiviral immunity (7). Identification of viral structures that effectively trigger this pathway will advance our understanding of the molecular requirements for potent natural RIG-I ligands. Importantly, such knowledge can be used to generate synthetic RLR ligands for strong induction of antiviral responses in the context of vaccination or antiviral therapies.
Studies using synthetic or purified viral RNA, followed by analysis of antiviral activity upon transfection into cells, have revealed that 5′-di- or -triphosphates associated with double-stranded RNA (dsRNA) or with single-stranded RNA (ssRNA), with or without poly(U/UC) ssRNA stretches, trigger RIG-I stimulation (1, 3, 8–10). These structures are predicted in many RNA viruses, and their presence has been validated in influenza A virus (IAV) (8, 11), Sendai virus (SeV) (12), hepatitis C virus (HCV) (13–15), and reovirus (16, 17). MDA5 ligands are much less well characterized and are presumed to be complex secondary RNA structures (18, 19).
Notably, during infection with viruses that are adapted to the host, virus-encoded proteins interfere with RLR activity, allowing the virus to reach high titers prior to the onset of the antiviral response (20–22). The delay in the detection of viral structures that are normally present in the viral genomes during natural infections indicates that additional factors are required for the effective triggering of antiviral responses in vivo. During infection with SeV, a potent stimulus for the initiation of the antiviral response is provided by immunostimulatory defective viral genomes (iDVGs) generated during virus replication at high titers (23–25). SeV iDVGs trigger RLR signaling and initiate strong antiviral immunity both in vitro and during natural infections in vivo (24, 26). SeV iDVGs belong to the copy-back type of RNA DVGs and are produced when the viral polymerase is released from the template strand during replication and copies back the nascent strand (27). iDVGs are unable to replicate in the absence of a helper virus, as they lack the essential replication machinery and they are not transcribed into proteins due to their flanking antigenomic promoter (28–31). A copy-back iDVG of 546 nt (DVG-546) predominant in laboratory stocks of SeV strain Cantell (SeV C) is among the strongest natural triggers of RLR signaling (12, 23, 26, 32). Although this activity largely depends on 5′-triphosphates and the presence of dsRNA structures, it is unclear whether additional RNA motifs optimize the immunostimulatory potential of DVG-546, contributing to its efficient recognition even in the presence of virus-encoded antagonists of the antiviral response (25, 33).
Here we describe an RNA motif, DVG70-114, that is essential for the strong immunostimulatory activity of DVG-546. This motif can be stabilized and engineered into inert viral RNA to confer enhanced immunostimulatory potential. Mechanistically, DVG70-114 facilitates the recognition of viral RNA by RIG-I and promotes the onset of the antiviral response. DVG70-114 represents a novel type of viral PAMP enhancer motif.
RESULTS
Accumulation of positive-sense RNA strands of SeV copy-back DVGs associates with strong stimulation of the antiviral response during infection.
We previously reported that iDVGs naturally generated in vivo trigger potent lung antiviral immunity (24). To examine whether SeV iDVGs induce a broad antiviral state that could protect against unrelated viral infections, mouse cells were infected either with SeV C containing high levels of copy-back iDVGs (SeV HD) or with SeV C depleted of iDVGs (SeV LD) or left untreated (N/T) and reinfected 6 h later with the unrelated IAV. Cells preinfected with SeV HD were significantly more resistant to IAV replication than were cells preinfected with SeV LD (Fig. 1A) and induced higher antiviral gene expression levels (Fig. 1B). These data indicate that SeV iDVGs stimulate a strong antiviral state that can protect against infection with unrelated viruses.
FIG 1 .

The positive-sense DVG strand is associated with the strong antiviral activity induced during infection. (A) MEFs were infected with SeV HD or SeV LD (MOI = 1.5 TCID50/cell) or left untreated (N/T) and challenged 6 h later with IAV (MOI of 1.5). Cells were harvested 12 h after challenge, and the expression of IAV NP mRNA was examined by qRT-PCR. The data are the percentage of IAV NP mRNA relative to that of N/T cells. (B) Expression of antiviral genes examined by RT-qPCR. Results are expressed as the mean ± the standard error of the mean of three independent experiments. (C) Representation of genomic and copy-back DVG RNAs produced during SeV infection. (D) IFNB1 mRNA expression and ratio of positive/negative-sense DVG RNA copy numbers determined by RT-qPCR from A549 cells infected with SeV HD at an MOI of 1.5 TCID50/cell. Experiments were independently repeated at least three times. Each assay was performed in triplicate. A representative graph is shown. (E) Representative staining of positive-sense DVG (green), positive-sense SeV genome or mRNA (red), IRF3 (purple), and nuclei (4′,6-diamidino-2-phenylindole [DAPI], blue) on LLC-MK2 cells at 6 h after SeV HD infection or on mock-treated controls. Magnification, ×100. (F) Quantification of IRF3 nuclear translocation within all of the cells, DVG-containing cells, or DVG-negative cells. Results are expressed as the mean ± the standard error of the mean of three independent experiments. ***, P < 0.001; ns, nonsignificant (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
To better understand the molecular basis of the strong immunostimulatory ability of SeV iDVGs, we looked for molecular motifs responsible for their activity. Because copy-back DVGs maintain the promoter elements necessary for replication, both positive- and negative-sense DVG RNA strands are present in infected cells (Fig. 1C). To narrow the search for molecular motifs that confer strong stimulatory activity on iDVGs, we first assessed whether a specific strand of iDVG preferentially activates the host antiviral response during infection. We quantified the number of positive- and negative-sense RNA strands of a predominant iDVG (DVG-546) present during infection with SeV HD (12, 34) and correlated these amounts with IFNB1 mRNA expression. For the description and validation of this assay, see Fig. S1A and B in the supplemental material. Although copy numbers of negative-sense DVG strands were consistently higher than those of positive-sense DVG strands throughout infection (see Fig. S1C), an increased ratio of positive- to negative-sense DVG strands positively correlated with the induction of IFNB1 expression in infected human A549 cells (Fig. 1D). The correlation between positive-sense DVG RNA and type I IFN expression was recapitulated in LLC-MK2 cells, a cell line highly permissive to SeV replication (see Fig. S1D). Confirming the strong ability of positive-sense DVG RNA to stimulate the antiviral response, RNA fluorescent in situ hybridization (FISH) in combination with immunofluorescent staining showed that IRF3 translocation to the nucleus occurs predominantly in cells showing a strong positive-sense DVG signal (Fig. 1E and F). Thus, accumulation of positive-sense copy-back DVGs is positively associated with the induction of antiviral responses in cells infected with SeV.
Identification of a candidate motif essential for type I IFN induction by positive-sense DVG RNA.
We have reported that alterations in the internal (noncomplementary) region of DVG-546 drastically affect its stimulatory capacity (26). Specifically, we found that a mutant form of DVG (DVG-324) retaining the 5′ end of the internal sequence promoted high levels of expression of type I IFNs upon transfection, while a mutant DVG missing the 5′ end of the internal sequence (DVG-354) showed a significant loss of stimulatory ability. These data suggest that a specific region located at the 5′ end of the internal sequence plays an essential role in maximizing the stimulatory potential of DVG RNA. Additional mutant DVGs further confirmed this prediction (Fig. 2A and B). One mutant DVG that retained a shorter 5′ internal sequence (DVG-268) also showed potent immunostimulatory activity, while mutant DVGs lacking either the complete internal sequence (DVG-200) or both complementary sequences (DVG-IS) demonstrated a reduced ability to stimulate antiviral genes upon transfection into both mouse and human cells (Fig. 2B; see Fig. S2A and B in the supplemental material). This differential activity of mutant DVGs was sustained over a 24 h time course, ruling out the possibility of different kinetics of IFN induction by the different mutant DVGs (see Fig. S2C). For all of these studies, in vitro-transcribed DVG (ivtDVG) RNAs were purified from gels, tested for purity and endotoxin content, and transfected into cells at equal molarity (see Fig. S3A to D).
FIG 2 .

Identification of a candidate motif essential for type I IFN induction by positive-sense DVG RNA. (A) Representation deletion of mutant DVGs. The length of the deletion (dashed line) and the total final length of each molecule are indicated. The mutant DVG with both complementary sequences (gray) deleted is DVG-IS (internal sequence). (B) Expression of IFNB1 and IFIT1 mRNA measured by RT-qPCR from LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG indicated. The data are the mean ± the standard error of the mean of all of the experiments (total n = 5 to 11/group). (C) Folding prediction of highly stimulatory and poorly stimulatory mutant DVGs with the RNAfold ViennaRNA software. A temperature of 37°C was chosen to determine structures based on minimal free energy. The candidate motif (DVG70-114) is circled. (D) Relative position of the candidate stimulatory DVG70-114 motif and its sequence in the genome of DVG-546. The AU content of DVG70-114, compared with that of DVG-546 and the full-length genome, is shown in the inset. (E) Folding predictions for DVG-268 and of the mutants derived from it. The structures of intact DVG-268 and of the DVG70-114 motif deleted (Δ70-114) molecules are shown. The position of DVG70-114 is highlighted. Point mutations within the DVG70-114 motif are indicated in the red square. (F) LLC-MK2 cells were transfected with 4.15 pmol of the ivtDVG indicated, IFNB1 and IFIT1 mRNA expression was analyzed by RT-qPCR at 6 h posttransfection. The data are the mean ± the standard error of the mean of all of the experiments (total n = 3/group). (G) Murine TC-1 cells were transfected with 4.15 pmol of DVG-268 or DVG-268Δ70-114, and IFN-β protein and mRNA levels were measured by ELISA and RT-qPCR, respectively. The data are the mean ± the standard error of the mean of three independent experiments. Each experiment was performed in duplicate. Bars correspond to the standard error of the mean. *, P <0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant (two-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH.
To identify the specific sequence and/or secondary RNA structure responsible for the strong stimulatory activity of iDVGs, we modeled in silico the folding of mutant DVGs with either a strong or a weak ability to induce type I IFN expression. For in silico folding predictions, we excluded the 3′ complementary sequence of the constructs since long complementarities strongly interfere with modeling based on minimal free energy, resulting in predicted structures that significantly deviate from the natural folding of the molecule (Fig. 2C). Under these conditions, we identified a candidate stem-loop structure formed by nt 70 to 114 (DVG70-114) of DVG-546 that was observed only in mutant DVGs showing strong stimulatory ability (Fig. 2D). This region is enriched in A/U nucleotides compared to the positive-sense SeV genome or full-length DVG-546 (Fig. 2D). DVG70-114 spans the 3′ end of the 5′ complementary sequence and the 5′ end of the internal sequence, consistent with a partial requirement for segments of the complementary and internal regions of iDVG for strong induction of IFN expression.
DVG70-114 is necessary for the strong stimulatory activity of SeV DVG RNA.
To determine whether DVG70-114 is necessary for the strong stimulatory ability of SeV DVGs, we removed the DVG70-114 motif from DVG-268 or from the parental DVG-546 and tested these RNAs upon transfection. Removal of DVG70-114 did not significantly affect other secondary structures of these molecules (Fig. 2E; see Fig. S4C in the supplemental material). For the quality controls used, see Fig. S4A and B in the supplemental material. Removal of DVG70-114 nearly abolished the stimulatory activity of DVG-268 at 6 h posttransfection, as cells transfected with DVG-268Δ70-114 expressed significantly lower levels of IFNB1 and IFIT1 mRNA than cells transfected with DVG-268 (Fig. 2F). This difference was recapitulated in murine cells and sustained at the RNA and protein levels over a 24 h time course (Fig. 2G). A reduced ability to induce IFN production was also observed in DVG-546Δ70-114 (see Fig. S4D), demonstrating that DVG70-114 is required for optimal stimulatory activity of DVG-546.
To further confirm the stimulatory role of DVG70-114, we introduced point mutations into this motif. A nucleotide swap within the stem region of DVG70-114 (U106G) disrupted the complementary structure of the DVG70-114 motif (Fig. 2E), resulting in reduced stimulation by DVG-268 (Fig. 2F) and suggesting that the potent activity of SeV iDVGs depends on the integrity of the DVG70-114 motif. To determine if specific noncomplementary structures within DVG70-114 are necessary for stimulation, we performed single-nucleotide swaps in the most distal loops of the DVG70-114 motif (A89U and C97G) while preserving its overall predicted structure (Fig. 2E). Interestingly, the C97G mutation, but not the A89U mutation, decreased the ability of DVG70-114 to trigger IFNB1 mRNA expression compared to that of parental DVG-268 (Fig. 2F), indicating that both the structure and the sequence influence the stimulatory potential of DVG70-114.
DVG70-114 promotes strong antiviral responses during SeV infection, overcoming viral antagonism.
To test whether DVG70-114 is involved in the stimulation of antiviral immunity during infection, we generated SeV stocks including recombinant defective viral particles containing the DVG70-114 motif (rDVG-324) or lacking this motif (rDVG-354) as shown in Fig. 3A (26). Infection with rDVG-324 stimulated significantly higher levels of IFNB1 and IFIT1 mRNA expression in LLC-MK2 cells than did infection with rDVG-354, while the viral protein and DVG RNA expression levels were equivalent (Fig. 3B). These data show that DVG70-114 functions to enhance the antiviral response during infection, demonstrating its biological relevance.
FIG 3 .

Recombinant SeV iDVGs with an intact DVG70-114 motif preserve their stimulatory activity in the context of infection. (A) Schematic of the recombinant SeV DVG generation system. BSR-T7 cells were infected with partially inactivated SeV 52 and transfected with a plasmid encoding either DVG-324 or DVG-354. Cells and supernatants were collected 48 h later and inoculated into 10-day-old embryonated chicken eggs. SeV containing rDVGs was collected from the allantoic fluid. T7 pro, T7 promoter sequence; Riboz., ribozyme; T7 term, T7 polymerase terminator sequence. (B) LL-CMK2 cells were infected with virus rDVG-324 or rDVG-354 at an MOI of 5 TCID50/cell and analyzed at 6 h postinfection by RT-qPCR for the expression of IFNB1 and IFIT1 mRNA, SeV NP mRNA, and DVGs. The relative copy number of DVG RNA was quantified by RT-qPCR with the DVG comp primers (see Table S1 in the supplemental material). The data are the average of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant (one-way ANOVA with Bonferroni’s post hoc test and two-tailed t test in DVG RNA quantification). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. (C and D) LL-CMK2 cells were infected with SeV LD at an MOI of 1.5 TCID50/cell and transfected 24 h later with 4.15 pmol of DVG-268 or DVG-268Δ70-114 RNA or left untreated (NT). Expression of SeV NP and SeV (P/V) (C) and antiviral genes (D) was measured at 6 h posttransfection. The data are the average of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH.
To further evaluate whether DVG70-114 promotes antiviral immunity in the presence of a virus-encoded antagonist, we infected cells with SeV LD 24 h before transfection with either DVG-268 or DVG-268Δ70-114. By the time of DVG RNA transfection, viral protein mRNA, including NP and P/V, had accumulated to high levels in the cells (Fig. 3C; see Fig. S3E in the supplemental material) and no antiviral gene expression was detected (Fig. 3D, NT [nontransfected]). Agreeing with their predicted stimulatory potential, transfection of DVG-268 resulted in the strong expression of antiviral genes while DVG-268Δ70-114 maintained its reduced stimulatory potential under these conditions (Fig. 3D). These data further demonstrate that DVG70-114 has strong immunostimulatory activity even in the presence of potent viral antagonists of detection.
The 3′ complementary end of iDVG RNA is not required for maximal IFN induction.
Contrary to previous implications, our data strongly suggest that complementarity along the 5′ and 3′ ends of copy-back DVGs is not a requirement for their strong stimulatory activity. To directly test if the 3′-complementary sequence is essential, we deleted the complete 3′ complementary sequence (94 nucleotides [nt]) from DVG-324 (Fig. 4A; DVG-324NC). Upon transfection, DVG-324NC induced the same level of expression of antiviral genes as parental DVG-324, and both mutants showed higher potency than DVG-546 (Fig. 4B). As expected, the potent stimulatory activity of DVG-324NC depended on the DVG70-114 motif, as a DVG-324NCΔ70-114 mutant had significantly less stimulatory potential (Fig. 4C and D).
FIG 4 .
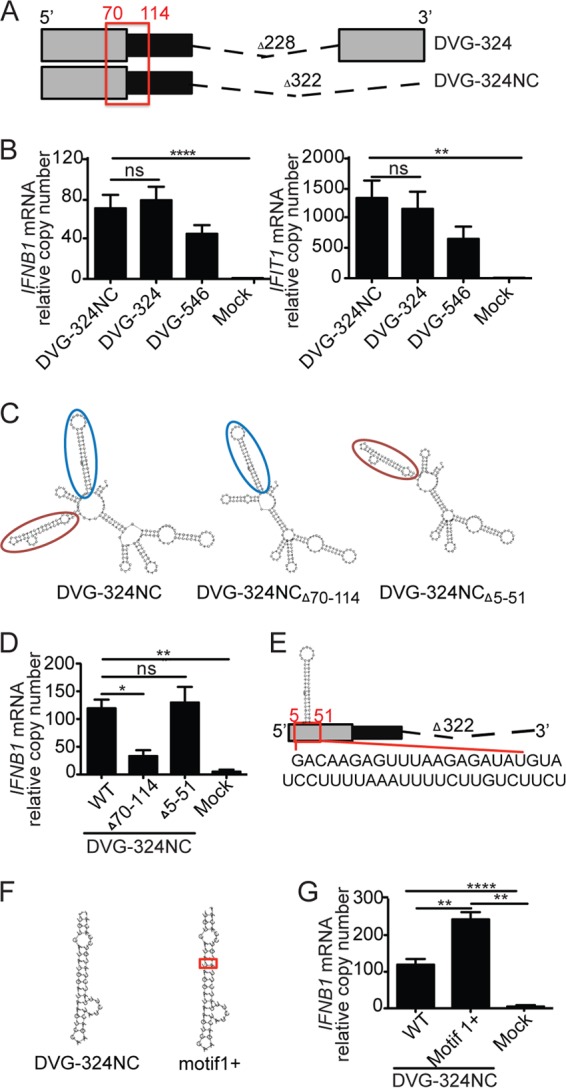
3′-to-5′ complementarity and additional proximal secondary structures are not necessary for IFN induction by DVG-derived RNA. (A) Illustration of DVG-324 and the related 3′ complementary sequence deletion mutant (DVG-324NC). The relative position of the immunostimulatory motif DVG70-114 is highlighted with a red square. The length of the deletion (dashed line) is indicated. (B) Expression of IFNB1 and IFIT1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG indicated. (C) Folding predictions for DVG-324NCΔ70-114, DVG-324NCΔ5-51, and parental DVG-324NC. Motif DVG70-114 is indicated by a red oval. Motif DVG5-51 is indicated by a blue oval. (D) Expression of IFNB1 mRNA by RT-qPCR in LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG indicated. (E) Illustration of the DVG5-51 motif and its relative position in the DVG-324NC construct. The sequence of DVG5-51 is shown below the construct. (F) Folding prediction for DVG-324NC motif1+ compared to that for DVG-324NC. (G) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG illustrated in panel H. All transfection experiments were independently repeated at least three times. The data are the mean ± the standard error of the mean of all of the experiments (total n = 3 to 5/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH.
To control for the role of additional secondary structures in the stimulatory activity of DVG-324NC, we next removed an auxiliary secondary motif (DVG5-51) located more proximal to the 5′-triphosphate end of RNA than DVG70-114 (Fig. 4C, blue circle, and E). This motif is also preserved in all of the mutant DVGs shown in Fig. 2. Deletion of DVG5-51 in DVG-324NC did not affect the integrity of DVG70-114 or the overall structure of the DVG (Fig. 4C) and did not have an impact on the stimulatory activity of DVG-324NC (Fig. 4E). Overall, these data demonstrate that DVG70-114 enhances the immunostimulatory activity of DVG RNA and functions independently of the 3′-end complementary sequence or additional proximal secondary structures.
Stabilization of DVG70-114 enhances its stimulatory potential.
We next evaluated whether stabilization of the DVG70-114 motif could further optimize the stimulatory activity of DVG-derived RNAs. To do this, we introduced an additional A-U base pair into the DVG70-114 long stem region of the DVG-324NC background. This insertion stabilized the stem structure as it increased its minimal free energy from −9.8 to −10.89 kcal/mol (Fig. 4F, DVG-324NC motif1+). Cells transfected with DVG-324NC motif1+ RNA expressed 2-fold higher IFNB1 and IFIT1 mRNA levels than cells transfected with unmodified DVG-324NC (Fig. 4G and data not shown), demonstrating that stabilization of the DVG70-114 motif improves its immunostimulatory potential. The stimulatory enhancement potential of stabilizing DVG70-114 was further validated in DVG-546 (see Fig. S4E and F in the supplemental material).
DVG70-114 motif confers strong immunostimulatory activity to inert RNA molecules.
To assess if the immunostimulatory activity of DVG70-114 could be transferred to inert 5′-triphosphate-containing RNA molecules, we cloned the DVG70-114 motif into the X region of HCV, a well-described nonimmunostimulatory small RNA derived from the virus genome (13). Our cloning strategy preserved the RNA stem-loop structures present in both the DVG70-114 motif and the X region (Fig. 5A). Remarkably, transfection of the ivtDVG RNA X region DVG70-114 (X region-based mutant that includes DVG70-114) led to strong expression of IFNB1 and IFIT1 mRNA, while that of the X region alone did not (Fig. 5B). X region DVG70-114 also demonstrated an ability to stimulate antiviral gene expression equivalent to that of HCV poly(U/UC) (13) (Fig. 5B). To rule out the possibility that the potent activity of X region DVG70-114 resulted from the introduction of an additional stem-loop structure, independently of the sequence of the motif, we also cloned into the X region the auxiliary DVG5-51 motif which is not required for immunostimulatory activity of DVG RNA (Fig. 4G). DVG5-51 has a size similar to that of DVG70-114 and causes equivalent disruption of X region folding (Fig. 5A). Introduction of the DVG5-51 motif did not alter the stimulatory activity of the X region (Fig. 5B), demonstrating that DVG70-114 has exceptional and transferrable immunostimulatory potential.
FIG 5 .
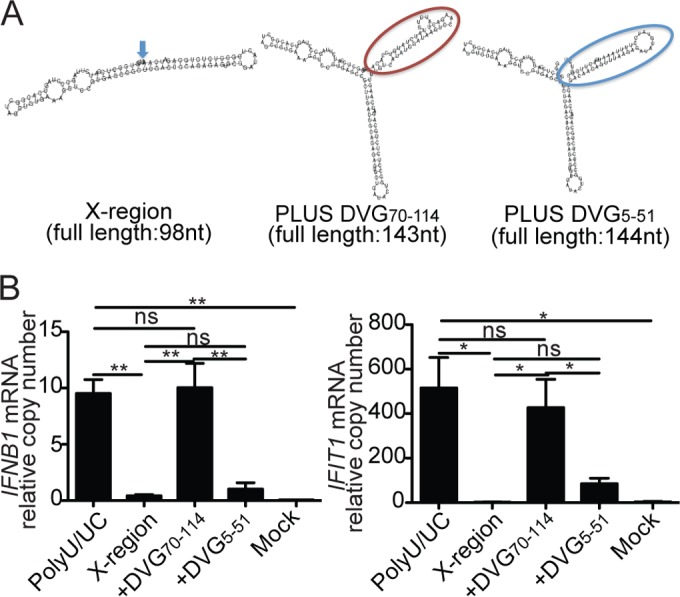
The DVG70-114 motif transfers strong immunostimulatory activity to inert RNA molecules. (A) Folding predictions for HCV X region, X region DVG70-114, and X region DVG5-51. Motif DVG70-114 is in a red balloon, and motif DVG5-51 is in a blue balloon. (B) Expression of IFNB1 and IFIT1 mRNA measured by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of gel-purified poly(U/UC), X region, X region DVG70-114, or X region DVG5-51 ivtDVGs. The data are the mean ± the standard error of the mean of all of the experiments (total n = 3 to 5/group). *, P < 0.05; **, P < 0.01; ns, nonsignificant (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH.
Lastly, we compared the activity of DVG RNA with that of other known RIG-I ligands. As shown in Fig. S5A in the supplemental material, DVG-derived RNA binds to RIG-I with an affinity equivalent to that of poly(U/UC) and RNA containing DVG70-114 stimulates levels of antiviral expression equivalent to that of poly(U/UC) or the viral RNA synthetic analog poly (I:C) (see Fig. S5B and C).
The DVG70-114 motif promotes binding of the viral RNA to RIG-I.
To establish whether the strong stimulatory activity of DVG-268 and DVG-324NC is mediated by RIG-I, we transfected these constructs into mouse embryo fibroblasts (MEFs) lacking the essential RLR MAVS molecule or lacking the RIG-I sensor. Similar to other DVG constructs (26), IFNB1 mRNA expression was dependent on MAVS and RIG-I activity for both mutant DVGs (Fig. 6A), while control poly (I:C) showed only MAVS, but not RIG-I, dependency. In addition, IFNB1 stimulation was largely dependent on the presence of uncapped 5′-triphosphates (Fig. 6B) and on RNA secondary structures (Fig. 6C). To determine if the DVG70-114 motif augmented the binding of DVG RNA to RIG-I, we performed an electrophoretic mobility shift assay (EMSA) with DVG RNA exposed to purified RIG-I proteins that lacked the signaling (CARD) domain (RIG-I deltaCARD). RIG-I deltaCARD was used previously to characterize specific RNA binding signatures and demonstrated RNA binding affinity equivalent to that of the full-length parental protein (10, 35). We tested the binding affinity of RIG-I for iDVGs with or without DVG70-114 in the absence or presence of ATP, which promotes RIG-I polymerization upon association with RNA. DVG RNAs containing the DVG70-114 motif (DVG-268) were more profoundly displaced in the gels than RNAs lacking this motif (DVG-268Δ70-114 and DVG-200) in both the absence and the presence of ATP (Fig. 6D and E; see Fig. S6A in the supplemental material). These data indicate a stronger capacity of RNA containing the DVG70-114 motif to promote RIG-I binding and polymerization to high-molecular-weight complexes, essential events in RLR signaling (36–38). In addition, a slight enhancement of the binding capacity of RNAs containing the DVG70-114 motif to MDA5 deltaCARD was observed (see Fig. S6B), supporting previous reports of a supplementary role for MDA5 in the sensing of DVGs (33). Together, these results demonstrate that DVG70-114 facilitates the binding of viral RNAs to RLRs and is critical for effective triggering of RIG-I signaling in response to SeV iDVGs.
FIG 6 .

The DVG70-114 motif strongly stimulates RLR signaling. (A) Expression of IFNB1 mRNA determined by RT-qPCR from wild-type (WT), Mavs−/−, and Ddx58−/− (RIGI−/−) MEFs transfected for 6 h with 4.15 pmol of ivtDVG or poly (I:C). (B) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with ivtDVGs left untreated or treated with AP. (C) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of DVG-268 not treated (NT) or treated with RNase A, V1, A/V1 or mock transfected. (D and E) EMSA of DVG-268 and DVG-268Δ70-114 RNA with increasing doses of RIG-I deltaCARD in the absence (D) or presence (E) of 1 µM ATP. All of the experiments in panel A to E were independently repeated at least three times. Each RT-qPCR assay was performed in triplicate. The data are the average values of all of the experiments (total n ≥3/group). *, P < 0.05; ***, P < 0.001; ****, P < 0.0001 (two-way [A] or one-way [B and C] ANOVA with Bonferroni’s post hoc test). RT-qPCR data are expressed as the copy number relative to that of the housekeeping gene for GAPDH and/or ACTB (for LL-CMK2 cells) and Rps11 (for MEFs). Representative gel pictures are shown in panels D and E.
DISCUSSION
iDVG-derived RNAs are natural RLR ligands that potentiate host antiviral innate immune responses (12, 26, 39). The strong stimulatory activity of SeV iDVGs has been demonstrated both in vitro and in vivo, but the molecular basis of this activity remains unclear. Because of their outstanding stability, relatively short length, high degree of homogeneity, inability to replicate, and potential to be modified and targeted, iDVG-derived molecules are strong candidates for vaccine adjuvants and immunostimulatory additives for immunotherapy. Here we report the identification of an RNA secondary structure (DVG70-114) that confers exceptional immunostimulatory potential on SeV iDVGs both in the context of infection and when transfected as naked RNA.
The DVG70-114 motif is a stable stem-loop structure present in the positive-sense strand of the SeV DVGs. This motif shows little sequence homology with the standard viral genome, as it forms in the copy-back iDVG “junction region,” where the polymerase starts extending (or copying back) the nascent strand after it has been released from the template strand (12, 40, 41). Similar stem-loop structures are found in other copy-back DVGs generated during infection with SeV C and 52 (data not shown), suggesting that structures similar to DVG70-114 provide enhanced immunostimulatory potential to other SeV iDVGs. Interestingly, folding modeling of the negative-sense strand of the molecule failed to predict the formation similar to DVG70-114 (not shown) corresponding to the observed correlation between the enhanced relative accumulation of positive-sense DVG in infected cells and type I IFN expression. These observations demonstrate that, in addition to the conventional 5′-triphosphate motif, sequences and/or structures present in the positive-sense strand of the molecule maximize its immunostimulatory potential. It can be speculated that the lack of strong immunostimulatory potential on the negative-sense strand of DVG RNA reflects an evolutionary adaptation to avoid sensing of incoming particles before the establishment of infection; however, the biological relevance for a stronger stimulatory potential of one strand over the other requires further investigation.
By using viruses containing recombinant DVGs, we demonstrate that DVG70-114 is functional in the context of infection. In addition, we show that DVGs containing DVG70-114 are better able to overcome SeV immune antagonists, highlighting the crucial role of iDVGs as primary danger signals for the triggering of the antiviral response in infections with viruses that evade detection. Notably, it has been previously reported that viruses bearing a negative-sense RNA genome do not generate dsRNA during infection (42, 43). However, DVG70-114 is 45 nt long and below the detection limit of the dsRNA antibody used in those studies, likely explaining the discrepancy with our data (44).
The immunostimulatory activity of DVG70-114 depends on both its structure and its sequence. Point mutation analyses demonstrated tolerance to nucleotide swaps at the most distal tip of the motif (A89U), while nucleotide stringency was shown in mutations at the bulge (C97G) and some areas of the stem (U106G). In addition, our data demonstrated that the 3′ complementary sequence is not required for iDVG stimulatory activity. DVG-324NC (DVG-324 lacking the 3′ complementary sequence) induced IFN expression equal to that of DVG-324, and this activity depended on intact DVG70-114. This result contradicts a previous study that shows a requirement for both 5′ and 3′ complementary sequences for the maintenance of the immunostimulatory function of SeV DVGs (34). The disparity likely results from an unexpected impact of truncations of the complementary region on the structure of the DVG70-114 motif, leading to a misinterpretation of the need for the complementary region.
Remarkably, the insertion of DVG70-114, but not of DVG5-51, into the inherently inert X region of HCV conferred strong immunostimulatory activity to this molecule, comparable to that of the well-characterized poly(U/UC) region. These data demonstrate that DVG70-114 retains its stimulatory potential even outside the context of iDVGs. Our previous studies demonstrated that 5′-triphosphates are necessary for nonself recognition of SeV iDVG by RIG-I (24). Here we show that 5′-triphosphates are not sufficient and that optimal recognition occurs only in the presence of DVG70-114, as various mutant DVGs, including DVG-IS and DVG-200, maintain their 5′-triphosphate but do not stimulate RLR signaling. We report that DVG70-114 facilitates the formation of high-molecular-weight complexes between iDVG and RIG-I, likely explaining its strong ability to trigger the antiviral response. Further studies using this powerful system may reveal currently unknown requirements for optimal RIG-I activation (45). In addition, we observed a role for DVG70-114 in enhancing the binding of MDA5 to DVG RNA, supporting our previous finding of a role for MDA5 in the early detection of SeV iDVGs (33).
The role of DVGs in promoting strong antiviral responses during infection with a number of viruses has been documented since the 1970s (23), and most recently, we showed that DVGs occurring naturally in the lung during SeV infection drive the antiviral response in vivo (24). Facilitated binding to RLR by DVG70-114, together with the faster accumulation of DVGs over the standard virus genome in infected cells (24), likely explains the ability of iDVGs to overcome the viral antagonism of the immune response. We have identified a natural SeV iDVG molecular motif that facilitates the activation of the antiviral immune response by enhancing the binding of viral RNAs to the intracellular viral sensor protein RIG-I. Importantly, this motif can be harnessed to maximize the immunostimulatory activity of uncapped 5′-triphosphate-containing RNAs, and thus, it represents a novel immunostimulatory enhancer that could be used in the development of vaccine adjuvants or antivirals.
MATERIALS AND METHODS
Cell lines and viruses.
LLC-MK2 rhesus monkey kidney epithelial cells (ATCC catalog no.CCL-7), A549 human type II alveolar cells (ATCC catalog no. CCL-185), and wild-type, Ddx58−/− (RIG-I−/−) and Mavs−/− MEFs (kindly provided by J. Kagan and J. Chen) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM glutamine, and 50 mg/ml gentamicin or penicillin and streptomycin (all from Invitrogen). TC-1 mouse lung epithelial cells (ATCC catalog no.CRL-2785) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM l-glutamine, 2 mM nonessential amino acids, 10 mM HEPES, and 50 mg/ml gentamicin or penicillin and streptomycin (all from Invitrogen). All cell lines were treated with mycoplasma removal agent (MP Biomedicals) before use. SeV Cantell HD and LD (SeV HD, high DVG particle content; SeV LD and 52, low DVG particle content) were prepared in embryonated chicken eggs as described previously (25). Influenza A/PR8/34 virus (IAV) was grown in 10-day-old embryonated chicken eggs (Specific-Pathogen-Free Avian Supplies, Charles River Laboratories) for 40 h at 37°C.
Plasmids and constructs.
A plasmid expressing DVG-546 flanked at the 3′ end by the SpeI-T7 promoter sequence and at the 5′ end by the hepatitis delta virus ribozyme and the T7 polymerase terminator sequence was prepared as described previously (26). To generate mutant DVGs, restriction enzyme sites were introduced into the construct with the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). Specifically, DVG-268 was generated by the introduction of 6 nt to generate a KpnI site at position 163 of the DVG-546 sequence. A second KpnI site at position 448 of the wild-type DVG internal sequence allowed the deletion of a 228-nt-long fragment of DVG. DVG-324 and DVG-354 were generated as previously described (26). DVG-324NC was generated by the introduction of a second KpnI site at position 330 of the DVG-324 sequence, allowing the deletion of a 100-nt-long fragment between positions 230 and 330 of the DVG-324 sequence. Motif DVG70-114 and DVG5-51 deletions, single-nucleotide swaps (T106G, A89U, C97G), and nucleotides additions (motif1+) were generated by site-directed mutagenesis (QuikChange II XL; Agilent Technologies). For nucleotide swaps, primers introduced a T/G mutation at position 89, 97, or 106 of the DVG-268 sequence. Motif1+ includes an A insertion after position 78 and a T insertion after position 104 of the DVG-324NC sequence. For the sequences of all of the mutagenesis primers used, see Table S1 in the supplemental material. Poly(U/UC) and X region plasmids were kindly provided by M. Gale, Jr. (University of Washington).
HCV PAMP preparation and manipulation.
DNA sequences bearing poly(U/UC) or the X region were amplified by PCR (for the sequences of the primers used, see Table S1 in the supplemental material) with a high-fidelity polymerase (Invitrogen). X region mutants bearing DVG motifs were constructed by adding DVG motifs at the 3′ end of the molecule by PCR with the primers shown in Table S1. The resulting PCR products were gel purified (Qiagen) and subjected to in vitro transcription as described below.
ivtDVG preparation.
DVG-expressing plasmids were linearized and in vitro transcribed with the MEGAscript T7 kit (Ambion) in the presence of RNase inhibitor (Fermentas). RNA products were treated with DNase and then precipitated with LiCl. The optical density at 260 nm (OD260)/OD280 ratios of all ivtDVGs were between 2.00 and 2.25, and the OD260/OD230 ratios were between 2.20 and 2.60. Purified PCR products were used for in vitro transcription of poly(U/UC) or X region RNA with the MEGAscript T7 kit (Ambion) in the presence of RNase inhibitor (Fermentas).
RNA treatments and transfection.
To remove 5′-triphosphates, 1 µg of ivtDVG RNA was incubated with 10 U of alkaline phosphatase (AP; Thermo Scientific) for 60 min at 37°C. To cleave ssRNA, 1 µg of RNA was incubated with 1 ng of RNase A (Ambion) for 15 min at room temperature. To cleave dsRNA, it was incubated with 0.1 U of RNase V1 (Ambion) for 15 min at room temperature. After treatment, the RNA was purified with TRIzol or precipitation/inactivation buffer according to the manufacturer’s specifications. AP and RNase dual treatment was accomplished by incubating AP-treated ivtDVG with RNases by using the protocol described above. Prior to transfection, RNAs were heated at 65°C for 5 min, cooled to room temperature for 5 min, and then chilled on ice to promote proper folding of the molecules before transfection into cells. A 4.15-pmol portion of the structured RNA was transfected into 2.5 × 105 cells with Lipofectamine 2000 (Invitrogen). As controls, cells were transfected with an equally molarity of high-molecular-weight poly (I:C) (InvivoGen).
Rescue of recombinant virus.
BSR-T7 cells were infected with partially inactivated SeV 52 at a multiplicity of infection (MOI) of approximately 60 50% tissue culture infective doses (TCID50)/cell for 1 h. Infected cells were transfected with 3 µg of a plasmid encoding DVG with the XtremeGENE transfection reagent (Roche). Cells and supernatant were harvested after 48 h, and 200 µl of the suspension was inoculated into the allantoic cavities of 10-day-old embryonated chicken eggs. After 40 h, allantoic fluid was harvested and egg inoculation was repeated for up to three consecutive passages. The presence of defective viral genome rDVG-324 and rDVG-354 in recombinant virus stock was verified by PCR with a primer specific for each mutant DVG. The relative copy number of DVG RNA presented in each mutant virus upon infection was determined by quantitative PCR (qPCR) with DVG Comp primer (see Table S1 in the supplemental material).
In silico secondary structure prediction.
DVG RNA folding was predicted by the RNAfold server from the ViennaRNA package v.2.1.8 (University of Vienna, Vienna, Austria; http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) (46). For analysis, default parameters were applied for minimum free energy (MFE) and partition functions fold algorithms (Turner 2004 model). The optimal secondary structure with MFE of the thermodynamic ensemble is shown for each DVG RNA prediction.
RT-qPCR.
Reverse transcription (RT)-qPCR was performed as previously described (24). Briefly, 1 µg of RNA was reverse transcribed with the High Capacity RNA-to-cDNA kit (Applied Biosystems). qPCRs were performed with SYBR green PCR master mix (Applied Biosystems) in an Applied Biosystems ViiA 7 Real-time PCR System. For the primers used, see Table S2 in the supplemental material.
RNA FISH and IFA.
Probes complementary to the positive-sense strand of the SeV genome were designed against the full-length genome (from position 1 to position 11630), excluding the 5′ end that encompasses the DVG sequence. A pool of 32 oligonucleotides targeting the positive-sense SeV genome was designed as previously described (47). These probes recognize the positive-sense viral genome, as well as all virus-encoded mRNAs. For probes identifying DVGs, a pool of 15 single-stranded, 20-nt-long oligonucleotides specific for the 3′ end of the positive-sense SeV genome (from position 14965 to position 15416) which covers the DVG sequences was designed by the same methods. These probes bind to DVGs, viral L protein mRNA, and the positive-sense SeV genome. In both pools, oligonucleotides were complementary to a different region of the target RNA, with no fewer than two bases separating any two oligonucleotides. SeV genome probes were labeled with Quasar 570, and DVG probes were labeled with Quasar 670 (Biosearch Technologies). Since L protein mRNA and the positive-sense SeV genome are recognized by both set of probes, pixels containing both colors were considered a SeV genome/protein representation. Pixels stained only with Quasar 670 (pseudocolored green) represent positive-sense DVGs. For combined FISH and immunofluorescence assay (IFA), cells were plated onto plain glass coverslips (Fisher Scientific) at a density of 1 × 106 cells/well and grown overnight at 37°C. RNA FISH was performed according to published protocols, with minor modifications (48). At different times after transfection, the cells were washed with phosphate-buffered saline (PBS) and then fixed with 4% formaldehyde for 10 min at room temperature. Fixed cells were then permeabilized with 70% ethanol for 1 h at room temperature. Permeabilized cells were incubated with anti-human IRF3 antibody (1:100 dilution; Santa Cruz), followed by Alexa Fluor 594-labeled goat anti-rabbit IgG (1:500 dilution; Invitrogen) diluted in 1% bovine serum albumin in the presence of 40 U/ml RNase inhibitor (Invitrogen). Stained cells were incubated in 4% formaldehyde for 10 min prior to FISH, washed with PBS, and then equilibrated in wash buffer containing 10% formamide and 2× saline sodium citrate (SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]). FISH was performed by hybridizing fixed cells with 10 nM probes diluted in hybridization buffer consisting of 10% formamide, 2× SSC, and 10% (wt/vol) dextran sulfate. Hybridization was performed overnight in a humidified chamber at 37°C. Imaging acquisition was performed with a Nikon E600 epifluorescence microscope equipped with a 100×, 1.4 numerical aperture oil immersion objective (Zeiss) and a Zeiss AxioCam MRm camera.
FISH-IFA image quantification.
Imaging quantification was performed with the Volocity Quantitation module (PerkinElmer) (49). Exposure time, gain, and offset were held constant for all images. The fluorescent signal from the nuclei of cells was selected by drawing a region of interest (ROI) around each nucleus. The fluorescent signal of the positive-sense DVG was determined by drawing an ROI around the entire cell and using the procedure described above. The average ROI intensity of IRF3 and positive-sense DVG signals in mock-infected cells was measured and used as a reference to set the threshold for defining nuclear IRF3 and positive-sense DVG-positive cells. Nuclear IRF3-positive cells among positive-sense DVG-positive populations were identified by performing “intersect module,” while the nuclear IRF3-positive cells among positive-sense DVG-negative populations were identified by using the “exclude touching” module. For the analysis, at least 250 cells were quantified in each experimental group through three independent experiments.
EMSA.
The RIG-I deltaCARD and MDA5 deltaCARD proteins (RIG-I or MDA5 without the two N-terminal CARDs) were kindly provided by Sun Hur (Harvard, Boston, MA) and were prepared as described elsewhere (35). EMSA was performed by incubating ivtDVG RNA with 1 µg or the indicated amount of RIG-I deltaCARD or MDA5 deltaCARD protein in buffer A (20 mM HEPES [pH 7.5], 150 mM NaCl, 1.5 mM MgCl2, 2 mM dithiothreitol) for 15 min at 37°C, and the complexes were resolved by 4 to 12% Bis-Tris NativePAGE (Bio-Rad). Gels were stained with SYBR gold (Life Technologies), and fluorescent gel images were recorded and analyzed with the Gel Doc XR+ imaging system (Bio-Rad).
ELISA.
IFN-β in the supernatant of ivtDVG RNA-transfected TC-1 cells was determined by enzyme-linked immunosorbent assay (ELISA) in accordance with the manufacturer’s instructions (PBL Assay Science).
Statistical analysis.
Statistical analysis was performed with GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA). A statistically significant difference was defined as a P value of <0.05 by either one-way analysis of variance (ANOVA) or Student’s t test with or without a post hoc test, as indicated in each figure legend, on the basis of the specific data set.
Additional materials and methods.
For additional materials and methods, see Text S1 in the supplemental material.
SUPPLEMENTAL MATERIAL
Production of positive-sense DVG during DVG replication closely correlates with expression of IFNB1 mRNA. (A) Scheme of PCR amplification strategy utilized to detect and differentiate positive- and negative-sense strands of DVG-546. RT of the positive- and negative-sense DVGs was performed with the primers indicated. RT-qPCR of each sense-specific RT product was performed with a combination of the DVG 1 and DVG J primers to produce identical PCR products at equivalent efficiencies. While the DVG 1 primer will serve to reverse transcribe the positive-sense strand of both DVG-546 and the SeV HD genome, the DVG J primer spans the junction site between the inverse trailer and the internal DVG sequence, which is unique to the DVG-546 sequence and is not present in the SeV HD genome. (B) Positive- and negative-sense DVG qPCR efficiency test of TC-1 mouse lung epithelial cells infected with SeV HD at an MOI of 1.5 TCID50/cell. Total RNA (1 µg) isolated 12 h after infection was reverse transcribed for either positive- or negative-sense DVG. Ten-fold serial dilutions were analyzed for RT-qPCR, and cycle threshold (CT) values for all of the dilutions were graphed relative to the log10 of the number of picograms of initial RNA. The linear region of the standard curve is shown and was used to determine the R2 value and PCR efficiency, which was calculated as 10−1/slope − 1. (C) Copy numbers of positive- and negative-sense DVGs over the course of A549 infection with SeV HD at an MOI of 1.5 TCID50/cell. (D) IFNB1 mRNA expression and ratio of positive/negative-sense DVG RNA determined by RT-qPCR from LLC-MK2 cells infected with SeV HD at an MOI of 1.5 TCID50/cell. All of the experiments shown in panels B to D were independently repeated three times, and each assay was performed in triplicate. A representative data set is shown. Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
ivtDVGs with an intact stimulatory motif stimulate IFN responses in both mouse and human cells, and the responses are sustained for up to 24 h posttransfection. (A) Expression of IFNB1 mRNA determined by RT-qPCR of TC-1 cells transfected for 6 h with 4.15 pmol of the ivtDVGs indicated. (B) Expression of IFNB1 mRNA by RT-qPCR of A549 cells transfected for 6 h with 4.15 pmol of the ivtDVGs indicated. Each RT-qPCR assay was performed in triplicate. The data are the average values of all of the experiments (total n = ≥3/group). *, P <0.05; **, P < 0.001; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). RT-qPCR data are expressed as the copy number relative to that of the housekeeping gene for GAPDH for LL-CMK2 cells and Rps11 for TC-1 cells. (C) Expression of IFNB1 and IFIT1 mRNA at 4, 8, 12, and 24 h posttransfection of LL-CMK2 with 4.15 pmol of the ivtDVG RNA indicated. One representative experiment out of three is shown. Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
Quality control of ivtDVG RNAs. (A) Electrophoretic analysis of ivtDVGs was performed with an Agilent 2100 Bioanalyzer. (B) Endotoxin levels on ivtDVGs used in this study measured by Limulus assay. (C) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of gel-purified ivtDVG. The experiment was independently repeated three times, and each assay was performed in triplicate. A representative qPCR is shown. RT-qPCR data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. (D) The amount of ivtDVGs present upon each transfection was quantified at 6 h posttransfection by RT-qPCR with a common set of primers directed to the 5′-complementary end of the DVGs (DVG comp). Two representative transfections of LL-CMK2 cells are shown (EXPs 1 and 2). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. (E) Immunofluorescence staining of LL-CMK2 cells infected with SeV LD (MOI of 1.5 TCID50/cell) at 24 hpi with SeV NP (red) and nuclei (DAPI, blue). Magnification, ×40. Download
Verification of DVG70-114 motif activity in DVG-546. (A) Electrophoretic analysis for DVG-268 ivtDVGs with an Agilent 2100 Bioanalyzer. (B) Electrophoretic analysis for DVG-546 ivtDVGs with an Agilent 2100 Bioanalyzer. (C) Folding prediction for DVG-546 with the motif deleted (DVG-546Δ70-114) and parental DVG-546. The DVG70-114 motif is indicated by a red oval. (D) Expression of IFNB1 mRNA by RT-qPCR in LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG indicated. (E) Folding prediction for DVG-546 motif1+ and parental DVG-546. The DVG70-114 motif is indicated by a red oval. The mutated DVG70-114 motif in DVG-546 motif1+ is indicated by an orange oval. (F) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of ivtDVG illustrated in panel E. All transfection experiments were independently repeated at least three times. The data are the mean ± the standard error of the mean of all of the experiments (total n = 3 to 5/group). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
DVG-derived ivtDVG RNAs induce levels of antiviral responses equivalent to those induced by known viral RIG-I ligands. (A) EMSA of 6 pmol of HCV poly(U/UC), X region, and DVG-268 in the presence of ATP and increasing doses (0 to 20 pmol) of full-length RIG-I. The product was resolved on a 2% agarose gel. (B and C) Expression of IFNB1 and IFIT1 mRNA measured by RT-qPCR from LLC-MK2 cells transfected for 6 h with 4.15 pmol of ivtDVGs or equivalent amounts of HCV poly(U/UC), X region, or poly(I ⋅ C) (high molecular weight). The data are the mean ± the standard error of the mean of all of the experiments (total n = 3/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
DVG70-114 improves the binding of DVGs to RIG-I and MDA5. (A) EMSA of DVG-268 and DVG-200, which lack immunostimulatory motif DVG70-114, in the presence of ATP and increasing doses of RIG-I deltaCARD. (B) EMSA of DVG-268 and DVG-268Δ70-114 RNA in the presence of increasing doses of MDA5 deltaCARD. Download
Primers used in mutagenesis and mutant DVG verification.
Primers used in qPCR assays.
Additional materials and methods used in this study. Download
ACKNOWLEDGMENTS
We thank Gordon Ruthel of the University of Pennsylvania School of Veterinary Medicine Imaging Core Facility for imaging assistance and the University of Pennsylvania DNA Sequencing Facility for RNA electrophoretic analysis. We also thank Brian Gregory (University of Pennsylvania) for providing invaluable suggestions on RNA modeling and structure analysis. We also thank Wenzhi Song (Bryn Mawr College) and Derek Standlee (University of Pennsylvania) for technical assistance, and Emmanuelle Genoyer for SeV NP immunostaining.
This work was supported by the U.S. National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (R01 AI083284 to C.B.L.), The American Association of Immunologists Careers in Immunology Fellowship Program (EIN 52-2317193 to C.B.L. and J.X.), and NIH National Institute of General Medical Sciences K12GM081295 to J.T.G. The PennVet Imaging Core Facility instrumentation is supported by NIH grant S10 RR027128.
Footnotes
Citation Xu J, Mercado-López X, Grier JT, Kim W, Chun LF, Irvine EB, Del Toro Duany Y, Kell A, Hur S, Gale M, Jr., Raj A, López CB. 2015. Identification of a natural viral RNA motif that optimizes sensing of viral RNA by RIG-I. mBio 6(5):e01265-15. doi:10.1128/mBio.01265-15.
REFERENCES
- 1.Dixit E, Kagan JC. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol 117:99–125. doi: 10.1016/B978-0-12-410524-9.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int Rev Immunol 30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 3.Yoo JS, Kato H, Fujita T. 2014. Sensing viral invasion by RIG-I like receptors. Curr Opin Microbiol 20:131–138. doi: 10.1016/j.mib.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 4.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 6.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. 2015. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol 32:48–53. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Anchisi S, Guerra J, Garcin D. 2015. RIG-I ATPase activity and discrimination of self-RNA versus non-self-RNA. mBio 6:e02349-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC. 2007. 5′-Triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- 12.Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111. doi: 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnell G, Loo YM, Marcotrigiano J, Gale M Jr. 2012. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self recognition by RIG-I. PLoS Pathog 8:e1002839. doi: 10.1371/journal.ppat.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uzri D, Gehrke L. 2009. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol 83:4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witteveldt J, Blundell R, Maarleveld JJ, McFadden N, Evans DJ, Simmonds P. 2014. The influence of viral RNA secondary structure on interactions with innate host cell defences. Nucleic Acids Res 42:3314–3329 doi: 10.1093/nar/gkt1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis e Sousa C. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichlmair A, Schulz O, Tan C-P, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. 2009. Activation of MDA5 Requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 20.Moltedo B, López CB, Pazos M, Becker MI, Hermesh T, Moran TM. 2009. Cutting edge: stealth influenza virus replication precedes the initiation of adaptive immunity. J Immunol 183:3569–3573. doi: 10.4049/jimmunol.0900091. [DOI] [PubMed] [Google Scholar]
- 21.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermesh T, Moltedo B, López CB, Moran TM. 2010. Buying time—the immune system determinants of the incubation period to respiratory viruses. Viruses 2:2541–2558. doi: 10.3390/v2112541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.López CB. 2014. Defective viral genomes: critical danger signals of viral infections. J Virol 88:8720–8723. doi: 10.1128/JVI.00707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tapia K, Kim WK, Sun Y, Mercado-López X, Dunay E, Wise M, Adu M, López CB. 2013. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9:e1003703. doi: 10.1371/journal.ppat.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yount JS, Kraus TA, Horvath CM, Moran TM, López CB. 2006. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J Immunol 177:4503–4513. doi: 10.4049/jimmunol.177.7.4503. [DOI] [PubMed] [Google Scholar]
- 26.Mercado-López X, Cotter CR, Kim WK, Sun Y, Muñoz L, Tapia K, López CB. 2013. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 31:5713–5721. doi: 10.1016/j.vaccine.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Re GG, Gupta KC, Kingsbury DW. 1983. Genomic and copy-back 3′ termini in Sendai virus defective interfering RNA species. J Virol 45:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calain P, Curran J, Kolakofsky D, Roux L. 1992. Molecular cloning of natural paramyxovirus copy-back defective interfering RNAs and their expression from DNA. Virology 191:62–71. doi: 10.1016/0042-6822(92)90166-M. [DOI] [PubMed] [Google Scholar]
- 29.Engelhorn M, Stricker R, Roux L. 1993. Molecular cloning and characterization of a Sendai virus internal deletion defective RNA. J Gen Virol 74:137–141. doi: 10.1099/0022-1317-74-1-137. [DOI] [PubMed] [Google Scholar]
- 30.Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555. doi: 10.1016/0092-8674(76)90223-3. [DOI] [PubMed] [Google Scholar]
- 31.Salinas Y, Roux L. 2005. Replication and packaging properties of short paramyxovirus defective RNAs. Virus Res 109:125–132. doi: 10.1016/j.virusres.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Strähle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. 2007. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J Virol 81:12227–12237. doi: 10.1128/JVI.01300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yount JS, Gitlin L, Moran TM, López CB. 2008. MDA5 Participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai virus defective interfering particles. J Immunol 180:4910–4918. doi: 10.4049/jimmunol.180.7.4910. [DOI] [PubMed] [Google Scholar]
- 34.Patel JR, Jain A, Chou YY, Baum A, Ha T, García-Sastre A. 2013. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 14:780–787. doi: 10.1038/embor.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, Hur S. 2014. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol Cell 55:511–523. doi: 10.1016/j.molcel.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S. 2011. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A 108:21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peisley A, Wu B, Yao H, Walz T, Hur S. 2013. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell 51:573–583. doi: 10.1016/j.molcel.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 38.Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. 2013. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Jain D, Koziol-White CJ, Genoyer E, Gilbert M, Tapia K, Panettieri RA Jr., Hodinka RL, López CB. 2015. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog 11:e1005122. doi: 10.1371/journal.ppat.1005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shioda T, Iwasaki K, Shibuta H. 1986. Determination of the complete nucleotide sequence of the Sendai virus genome RNA and the predicted amino acid sequences of the F, HN and L proteins. Nucleic Acids Res 14:1545–1563. doi: 10.1093/nar/14.4.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plattet P, Strahle L, le Mercier P, Hausmann S, Gracin D, Kolakofsky D. 2007. Sendai virus RNA polymerase scanning for mRNA start sites at gene junctions. Virology 362:411–420. doi: 10.1016/j.virol.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 42.Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 43.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19:2993–3000. doi: 10.1093/nar/19.11.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosai SJ, Foley SW, Wang D, Silverman IM, Selamoglu N, Nelson AD, Beilstein MA, Daldal F, Deal RB, Gregory BD. 2015. Global analysis of the RNA-protein interaction and RNA secondary structure landscapes of the Arabidopsis nucleus. Mol Cell 57:376–388. doi: 10.1016/j.molcel.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deutsch V, Brun G. 1983. Nongenetic complementation in VSV: asymmetric contribution of the L proteins of each parent in the rescue of group I ts mutants. Virology 124:366–379. doi: 10.1016/0042-6822(83)90353-7. [DOI] [PubMed] [Google Scholar]
- 47.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Závada J, Závadová Z, Russ G, Poláková K, Rajcáni J, Stencl J, Loksa J. 1983. Human cell surface proteins selectively assembled into vesicular stomatitis virus virions. Virology 127:345–360. doi: 10.1016/0042-6822(83)90149-6. [DOI] [PubMed] [Google Scholar]
- 49.Peluso RW, Moyer SA. 1983. Initiation and replication of vesicular stomatitis virus genome RNA in a cell-free system. Proc Natl Acad Sci USA 80:3198–3202. doi: 10.1073/pnas.80.11.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Production of positive-sense DVG during DVG replication closely correlates with expression of IFNB1 mRNA. (A) Scheme of PCR amplification strategy utilized to detect and differentiate positive- and negative-sense strands of DVG-546. RT of the positive- and negative-sense DVGs was performed with the primers indicated. RT-qPCR of each sense-specific RT product was performed with a combination of the DVG 1 and DVG J primers to produce identical PCR products at equivalent efficiencies. While the DVG 1 primer will serve to reverse transcribe the positive-sense strand of both DVG-546 and the SeV HD genome, the DVG J primer spans the junction site between the inverse trailer and the internal DVG sequence, which is unique to the DVG-546 sequence and is not present in the SeV HD genome. (B) Positive- and negative-sense DVG qPCR efficiency test of TC-1 mouse lung epithelial cells infected with SeV HD at an MOI of 1.5 TCID50/cell. Total RNA (1 µg) isolated 12 h after infection was reverse transcribed for either positive- or negative-sense DVG. Ten-fold serial dilutions were analyzed for RT-qPCR, and cycle threshold (CT) values for all of the dilutions were graphed relative to the log10 of the number of picograms of initial RNA. The linear region of the standard curve is shown and was used to determine the R2 value and PCR efficiency, which was calculated as 10−1/slope − 1. (C) Copy numbers of positive- and negative-sense DVGs over the course of A549 infection with SeV HD at an MOI of 1.5 TCID50/cell. (D) IFNB1 mRNA expression and ratio of positive/negative-sense DVG RNA determined by RT-qPCR from LLC-MK2 cells infected with SeV HD at an MOI of 1.5 TCID50/cell. All of the experiments shown in panels B to D were independently repeated three times, and each assay was performed in triplicate. A representative data set is shown. Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
ivtDVGs with an intact stimulatory motif stimulate IFN responses in both mouse and human cells, and the responses are sustained for up to 24 h posttransfection. (A) Expression of IFNB1 mRNA determined by RT-qPCR of TC-1 cells transfected for 6 h with 4.15 pmol of the ivtDVGs indicated. (B) Expression of IFNB1 mRNA by RT-qPCR of A549 cells transfected for 6 h with 4.15 pmol of the ivtDVGs indicated. Each RT-qPCR assay was performed in triplicate. The data are the average values of all of the experiments (total n = ≥3/group). *, P <0.05; **, P < 0.001; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). RT-qPCR data are expressed as the copy number relative to that of the housekeeping gene for GAPDH for LL-CMK2 cells and Rps11 for TC-1 cells. (C) Expression of IFNB1 and IFIT1 mRNA at 4, 8, 12, and 24 h posttransfection of LL-CMK2 with 4.15 pmol of the ivtDVG RNA indicated. One representative experiment out of three is shown. Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
Quality control of ivtDVG RNAs. (A) Electrophoretic analysis of ivtDVGs was performed with an Agilent 2100 Bioanalyzer. (B) Endotoxin levels on ivtDVGs used in this study measured by Limulus assay. (C) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of gel-purified ivtDVG. The experiment was independently repeated three times, and each assay was performed in triplicate. A representative qPCR is shown. RT-qPCR data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. (D) The amount of ivtDVGs present upon each transfection was quantified at 6 h posttransfection by RT-qPCR with a common set of primers directed to the 5′-complementary end of the DVGs (DVG comp). Two representative transfections of LL-CMK2 cells are shown (EXPs 1 and 2). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. (E) Immunofluorescence staining of LL-CMK2 cells infected with SeV LD (MOI of 1.5 TCID50/cell) at 24 hpi with SeV NP (red) and nuclei (DAPI, blue). Magnification, ×40. Download
Verification of DVG70-114 motif activity in DVG-546. (A) Electrophoretic analysis for DVG-268 ivtDVGs with an Agilent 2100 Bioanalyzer. (B) Electrophoretic analysis for DVG-546 ivtDVGs with an Agilent 2100 Bioanalyzer. (C) Folding prediction for DVG-546 with the motif deleted (DVG-546Δ70-114) and parental DVG-546. The DVG70-114 motif is indicated by a red oval. (D) Expression of IFNB1 mRNA by RT-qPCR in LLC-MK2 cells transfected for 6 h with 4.15 pmol of the ivtDVG indicated. (E) Folding prediction for DVG-546 motif1+ and parental DVG-546. The DVG70-114 motif is indicated by a red oval. The mutated DVG70-114 motif in DVG-546 motif1+ is indicated by an orange oval. (F) Expression of IFNB1 mRNA by RT-qPCR of LLC-MK2 cells transfected for 6 h with 4.15 pmol of ivtDVG illustrated in panel E. All transfection experiments were independently repeated at least three times. The data are the mean ± the standard error of the mean of all of the experiments (total n = 3 to 5/group). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
DVG-derived ivtDVG RNAs induce levels of antiviral responses equivalent to those induced by known viral RIG-I ligands. (A) EMSA of 6 pmol of HCV poly(U/UC), X region, and DVG-268 in the presence of ATP and increasing doses (0 to 20 pmol) of full-length RIG-I. The product was resolved on a 2% agarose gel. (B and C) Expression of IFNB1 and IFIT1 mRNA measured by RT-qPCR from LLC-MK2 cells transfected for 6 h with 4.15 pmol of ivtDVGs or equivalent amounts of HCV poly(U/UC), X region, or poly(I ⋅ C) (high molecular weight). The data are the mean ± the standard error of the mean of all of the experiments (total n = 3/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (one-way ANOVA with Bonferroni’s post hoc test). Data are expressed as the copy number relative to that of the housekeeping gene for GAPDH. Download
DVG70-114 improves the binding of DVGs to RIG-I and MDA5. (A) EMSA of DVG-268 and DVG-200, which lack immunostimulatory motif DVG70-114, in the presence of ATP and increasing doses of RIG-I deltaCARD. (B) EMSA of DVG-268 and DVG-268Δ70-114 RNA in the presence of increasing doses of MDA5 deltaCARD. Download
Primers used in mutagenesis and mutant DVG verification.
Primers used in qPCR assays.
Additional materials and methods used in this study. Download