

Abstract
Overall survival (OS) has emerged as the definitive regulatory “be-all, end-all” for the demonstration of benefit in cancer clinical trials. The reason and the rationale for why this is so are easily appreciated: literally a “test of time,” OS is a seemingly unambiguous, agenda-free end point, independent of bias-prone variables such as the frequency and methods of assessment, clinical evaluation, and the definition of progression. However, by general consensus, OS is an imperfect end point for several reasons: First, it may often be impractical because of the length, cost, and the size of clinical trials. Second, OS captures the impact of subsequent therapies, both beneficial (i.e., active) and detrimental, on survival but it does not take into account the contribution of subsequent therapies by treatment arm; the postprogression period is treated as an unknown black box (no information about the potential influence of next-line therapies on the outcome) under the implicit assumption that the clinical trial treatment is the only clinical variable that matters: what OS explicitly measures is the destination, that is, the elapsed time between the date of randomization (or intention to treat) and the date of death, not the journey, that is, what transpires in-between.
In long-term maintenance strategies, patients receive treatment in temporally separated but mutually interdependent and causally linked sequences that exert a “field of influence” akin to action-at-a-distance forces like gravity, electricity, and magnetism on both the tumor and each other. Hence, in this setting, a new end point, PFS2, is required to measure this field of influence. This article reviews the definition and use in clinical trials of PFS2 and makes the case for its potential applicability as a preferred end point to measure the mutual influence of individual regimens in long-term maintenance strategies with resensitizing agents in particular.
Introduction
Clinical trial end points serve different purposes. In conventional oncology drug development, early-phase clinical trials evaluate safety and identify evidence of biological drug activity, such as tumor shrinkage. End points for later-phase efficacy studies commonly evaluate whether a drug provides a clinical benefit such as prolongation of survival or an improvement in symptoms. Both the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in their respective guidance point out that confirmatory trials in the evaluation of anticancer medicinal products should demonstrate that the investigational product provides clinical benefit. In the Phase 3 studies conducted, there should thus be sufficient evidence available demonstrating that the chosen primary end point can provide a valid and reliable measure of clinical benefit in the patient population described by the inclusion criteria.
Acceptable primary end points include cure rate, overall survival (OS), and progression- or disease-free survival (PFS/DFS). The choice of primary end point should be guided by the relative toxicity of the experimental therapy, but parameters such as expected survival after progression, available next-line therapies, and the prevalence of the condition must also be taken into account. Irrespective of chosen primary end point, it is the magnitude of the treatment effect on all relevant outcome measures that forms the basis of the benefit–risk assessment.
If PFS/DFS is the selected primary end point, OS should typically be reported as a secondary end point and vice versa. Convincingly demonstrated favorable effects on OS are from both a clinical and methodological perspective the most persuasive outcome of a clinical trial. Although prolonged PFS/DFS is in most cases considered a relevant measure of patients benefit, the magnitude of the treatment effect would need to be sufficiently large to outbalance toxicity and tolerability problems. To capture possible negative effects on the activity of next-line therapies and also treatment-related fatalities, informative data on OS compatible with a trend toward favorable outcome are normally expected at time of submission.
If life is a journey, then the destination is represented by OS, a black box measurement of the time elapsed from a particular point A to point D (death) where what happens between A and D is irrelevant for measurement. For this reason, the end point is susceptible to confounding by subsequent treatments. Nevertheless, by tradition and by default, OS is universally regarded as the most clinically compelling end point in clinical trials, provided quality of life is not adversely affected. Simple to measure and easy to interpret (date of death is ostensibly bias-free), FDA director Richard Pazdur refers to it as “the gold standard” [1].
The FDA’s preference for it notwithstanding, OS, by general consensus, is onerous, expensive, and impractical [2] because it may take years for survival data to mature; this is particularly the case for trials that involve either maintenance therapy strategies or indolent, slow-growing tumors, for example, prostate cancer, where the time on trial itself may be prohibitively long. On the premise that oncology as a whole benefits from faster, shorter, and smaller trials, and “proof of” as soon as possible, consequently increasing treatment options for patients, various end points have been proposed and selected as surrogate end points for OS, including PFS.
The primary advantage of the PFS end point, defined as time from randomization until first evidence of tumor progression or until death from any cause, whichever comes first [3,4], also constitutes a major drawback: because progression, in most cases, is an event that occurs months or years before death, PFS is reached sooner than OS; however, a short-term indication of benefit is not necessarily an indication of long-term impact and/or benefit especially if the experimental treatment induces tumor resistance to subsequent radio- and chemotherapies. Conversely, a short PFS does not necessarily predict a poor OS if exposure to the experimental treatment sensitizes the tumors to the effect of subsequent radio- or chemotherapies. Indeed, PFS ultimately correlates poorly with OS in the majority of cancers [3], likely because it only measures clinical benefit on-therapy, not off-therapy, and because of the possibility that patients receive subsequent anticancer therapies in a nonrandomized and nonstratified manner with potentially unequal or undetermined effects on patient survival, potentially “washing out” a real survival benefit from the experimental therapy.
In addition, PFS, as an imaging-oriented end point, even with independent review and adjudication, may be prone to measurement bias and variation in interpretation, leading to possible treatment discontinuation if progression is not subsequently verified—this is especially evident in the context of pseudoresponse (apparent response without a true cytotoxic effect) and pseudoprogression [5] (apparent radiological progression without clinical deterioration). Pseudoresponse occurs with bevacizumab in gliomas because of vascular normalization, whereas pseudoprogression, where tumor size increases because of treatment-related necrosis and edema, is a common complication with imatinib (Gleevec) in gastrointestinal stromal tumors [6] and with radiotherapy and temozolomide in brain neoplasms. In the case of immunotherapies like the anti–PD-1 and anti–PDL-1 antibodies, as well as the anticytotoxic T-lymphocyte antigen–4 antibody ipilimumab, PFS may also be difficult to interpret as a result of immunologic responses and pseudoprogression [7].
To address these limitations and thereby improve the clarity of the cloudy PFS crystal ball, a recent EMA guidance recommends a substitute end point intermediate to PFS and OS called PFS2, a surrogate for OS when OS cannot be measured (for clinical or financial reasons), which assesses the impact of the experimental therapy on next-line treatment. PFS2, which could be termed PFS deferred, PFS delayed, tandem PFS, or PFS version 2.0, is defined in the EMA guidance as “time from randomisation to objective tumour progression on next-line treatment or death from any cause. In some cases, time on next-line therapy may be used as proxy for PFS.” [8]. For clarity, PFS2 does not refer to the 2-month time point, which distinguishes it from PFS at 6 months (PFS6), PFS at 12 months (PFS12), and PFS at 24 months (PFS24).
This lack of correlation between OS and PFS in most, but not all, tumor types, with ovarian cancer and colorectal cancer as the only exceptions [3] (although biostatisticians [9] and the regulators at the FDA and EMA who depend on their recommendations do not encourage reliance on PFS to assess novel therapies even in these tumor types), may be related to the omnipresent specter of resistance, described by Gatenby [10] and others [11], which invariably develops in response to the selective pressure of treatment. Resistance is an incremental process; each line of therapy not only selects for mutations but also triggers a reparative response [12,13] that reduces treatment susceptibility, resulting in a progressive “dilution” of the PFS effect. The end result is a highly resistant tumor with a legacy of multiple coexisting, resistance-conferring mutations.
In addition to PFS, other time-to-event end points include time to tumor progression (TTP), time to treatment failure (TTF), duration of response (DOR), and duration of disease control (DDC). Similar to PFS, the definition of TTP includes the time from randomization to time of progressive disease; unlike PFS, patients who die without progression are censored [14]. TTF, defined as the time from randomization to treatment discontinuation for any reason, including disease progression, treatment toxicity, patient and/or physician preference, or death, is not an approvable end point [1] on the basis that patient and physician preferences are an invalid criterion for determining efficacy. DOR [15], commonly used in breast cancer and multiple myeloma and defined as the progressive disease–free interval after the date of the first observation of response to the date of progressive disease, only applies to that subset of patients with partial responses. DDC, a close cousin of PFS2, will be discussed at length later in this review (Table 1).
Table 1.
Clinical End Point Key Concepts Defined
| Concept | Abbreviation | Definition |
|---|---|---|
| Progression-free survival | PFS | “The length of time during and after the treatment of a disease, such as cancer, that a patient lives with the disease that may or may not shrink but it’s increase in size does not meet the criteria of progressive disease according to the protocol criteria used” [27] |
| “PFS deferred,” “PFS delayed,” “tandem PFS,” or “PFS version 2.0” | PFS2 | “time from randomisation to objective tumor progression on next-line treatment or death from any cause. In some cases, time on next-line therapy may be used as proxy for PFS” [8] |
| Time to tumor progression | TTP | “The length of time from the date of diagnosis or the start of treatment for a disease until the disease starts to get worse or spread to other parts of the body.” In the context of pivotal clinical studies, this is the time from randomization to objective tumor progression [27]. |
| Time to treatment failure | TTF | “TTF is defined as a composite endpoint measuring time from randomization to discontinuation of treatment for any reason, including disease progression, treatment toxicity, and death.” [28] |
| Duration of response | DOR | “Time from documentation of objective tumor response to objective disease progression” [29] |
| Duration of disease control | DDC | “DDC is defined as the sum of the PFS of each sequence, except when progressive disease is observed at either reintroduction or second-therapy (DDC = PFS1 + PFS2 if treatment 2 achieved stabilization or response).” [30] |
The practice of permanently switching therapies at progression, which defines PFS, implies that each dividing line of therapy, first line, second line, third line, and so on, constitutes an independent event or a fresh start, like a coin toss; in fact, through the ripple effect of drug efflux, overexpression of antiapoptotic proteins, selective loss of cell cycle checkpoints, and epigenetic modifications, tumors over time tend to become increasingly less responsive to subsequent treatments [16] as they acquire a multidrug-resistant phenotype; in other words, the coin has a memory. The flip side of this coin is radio- and chemosensitizing agents, which mitigate or reverse resistance and enhance the activity of subsequent chemotherapy. Examples, which include RRx-001 [11], an experimental pan-epigenetic inhibitor in Phase II clinical trials with radiosensitizing properties [17], or the DNA hypomethylator decitabine [18], may render tumors hypersusceptible, in other words, “prime” them to respond more robustly, to salvage therapies.
In light of the recent EMA guidance, cited earlier, entitled Guideline on the evaluation of anticancer medicinal products in man, PFS2 is anticipated to meet regulatory and reimbursement requirements in Europe and, potentially, the United States as well [19].
This article reviews the clinical use of PFS2 and makes the case for its potential applicability as a preferred end point to measure the extended “field of influence” and the “team effort” of individual agents in a long-term maintenance strategy with resensitizing agents in particular.
End Points for Maintenance Therapies
Metastatic cancer is, by and large, an incurable disease. The goals of treatment are to palliate symptoms, improve quality of life, and potentially prolong survival. The optimal duration of systemic cytotoxic chemotherapy is unclear even in the setting of a complete response, partial response, or stable disease given the potential for continuous treatment to engender resistance, mental fatigue, financial hardships, and cumulative toxicities. One approach is dose-intensive therapy where dose modification or treatment delay only occurs in the context of severe toxicity, whereas another approach is to balance the efficacy and toxicity of intensive chemotherapy with maintenance treatment, where the objective is to “hold serve” against the tumor, that is, not to lose ground and thereby “buy” as much time as possible with a low–side effect regimen, until clinical progression mandates the reintroduction of the prior chemotherapy or initiation of a different one.
The theoretical foundation for maintenance therapy is based on the Goldie-Coldman hypothesis [20], an influential mathematical model from the mid-1980s, which supports the use of alternating cycles of multiple non–cross-resistant treatment modalities—that is, the rotation of one regimen with another—to reduce toxicities and forestall the inevitable development of resistance. In metastatic colorectal cancer, several large randomized clinical trials, OPTIMOX-2, AIO KRK 0207, and CAIRO-3, have investigated the feasibility of two types of maintenance therapy, continuation and switch, as alternatives to distinct lines of continuous treatment. Continuation maintenance therapy refers to the continuation of one or more of the initial treatments, whereas switch involves initiation of an alternate agent.
Clearly, as a discrete “snapshot” tightly framed within the temporal boundaries of randomization and progression, PFS lacks the depth of field necessary for a more panoramic view of the multistage maintenance treatment strategy, making it a nonoptimal end point. The necessary vision is provided by three alternative end points that measure a less circumscribed slice of time from the beginning of the treatment strategy to the end, which were investigated in the OPTIMOX-2, AIO KRK 0207, and CAIRO-3 trials: DDC, time to failure of strategy (TFS), and PFS2, respectively. They will be defined and discussed below.
-
1.
OPTIMOX-2 and DDC
The OPTIMOX-1 study demonstrated the noninferiority of a regimen with oxaliplatin-free intervals versus continuous FOLFOX. In the subsequent OPTIMOX-2 trial, oxaliplatin-free maintenance (5-FU + leucovorin) after FOLFOX chemotherapy was compared with chemotherapy discontinuation until progression, at which point FOLFOX was reintroduced; however, the actual time of chemotherapy reintroduction varied [21]. DCC, the primary end point of both OPTIMOX studies, was nonsignificant and almost identical in both arms (12.9 months vs 11.7 months, in favor of maintenance treatment); the nonsignificant median OS was 19 versus 26 months [22]. DDC is defined as PFSinitial + PFSreintroduction: the PFS during the initial phase of treatment (FOLFOX maintenance or FOLFOX discontinuation) plus the PFS of the reintroduction period (treatment reintroduction, FOLFOX) provided treatment reintroduction achieved stabilization or response (Figure 1).
-
2.
AIO KRK 0207 and TFS
In this Phase III trial, a 24-week induction regimen of fluoropyrimidine/oxaloplatin/bevacizumab was followed with randomization into one of three arms: 1) observation, that is, no treatment; 2) doublet fluoropyrimidine plus bevacizumab; or 3) bevacizumab alone until progression, at which point fluoropyrimidine/oxaloplatin/bevacizumab was reintroduced [23]. At a median follow-up of 27 months, the TFS primary end point was 3.6 months for the no-treatment arm, 6.2 months for the fluoropyrimidine-plus-bevacizumab arm, and 4.6 months for the bevacizumab-alone arm, which suggest that active maintenance is superior to none at all even though TFS did not correlate to OS: no significant difference in OS between the three arms was observed, leaving open to further question which maintenance strategy is best. TFS is defined as time on maintenance to either second progression after reinduction or, in case of no reinduction, after first progression (Figure 2).
-
3)
CAIRO-3 and PFS2
PFS2 is defined by the EMA [8] as “time from randomisation” to progression on a predetermined next-line therapy postmaintenance.
In the CAIRO-3 study [24], six cycles of induction therapy with capecitabine and oxaliplatin (XELOX) plus bevacizumab were followed with randomization to bevacizumab plus capecitabine or observation, that is, no treatment. On disease progression, therapy with XELOX plus bevacizumab was restarted until second disease progression (PFS2), the study’s primary end point (Figure 3). The median PFS2 of 11.7 months with maintenance treatment versus 8.5 months with observation did not translate to a statistically significant OS benefit (median OS of 21.6 months with maintenance therapy vs 18.1 months with observation).
A possible explanation for the weak correlation between these related end points in the OPTIMOX, AIO KRK 0207, COIN, and CAIRO-3 trials, which measure overlapping but not identical concepts, and OS is that the inevitable emergence or (reemergence) of resistance during the resensitization phase of the protocol swamped the small signal of benefit that the DDC, TFS, and PFS2 end points detected for the treatment versus no-treatment arms. Additional trials are either in progress or in the planning stages with these surrogate end points to find a maintenance regimen that confers a clear survival advantage.
Figure 1.
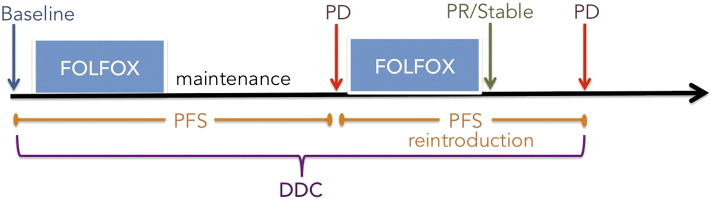
Optimox study and DDC illustrated.
Figure 2.

AIO KRK 0207 study and TFS illustrated.
Figure 3.

CAIRO-3 study and PFS2 illustrated.
Resensitizing Agents
A novel strategy to interrupt or circumvent the vicious cycle of resistance and enhance susceptibility to next-line treatment, thereby potentially conferring a survival advantage, involves the introduction of resensitizing agents. The premise of resensitization is that tumors, which have developed resistance to one or more therapeutic agents as a consequence of repeated exposure to these agents, may regain sensitivity to treatment, that is, become resensitized if the mediator of the resistance is affected by a subsequent therapeutic agent. Data from several recent clinical trials with epigenetic inhibitors demonstrate that despite evidence of intrinsic clinical activity, these agents are particularly well suited for supporting chemosensitization and chemoresensitization roles; pretreatment with epigenetic inhibitors “primes” or preconditions the tumor for response to subsequent therapy due to the partial abrogation of acquired and intrinsic resistance mechanisms [11,25]. Non–small cell lung cancer and colorectal cancer are two of the tumor types where a sensitization/resensitization strategy either has already been explored or is currently under investigation.
-
1.
Entinostat and azacitidine
In a Phase I/II clinical trial of a combination of the DNA demethylator azacitidine and the HDAC inhibitor entinostat in extensively pretreated recurrent metastatic non–small cell lung cancer tumor, preconditioning or priming before subsequent therapies was demonstrated. The median OS of 6.4 months significantly exceeded that of historical controls [26]. This favorable clinical outcome was associated with improved responses to subsequent therapies which included pemetrexed, docetaxel, erlotinib, anti–programmed cell death protein (PD-1) monoclonal antibodies, gemcitabine, irinotecan/bevacizumab, and cisplatin, suggesting that the combination of azacitidine and entinostat reversed the chemoresistant phenotype and enhanced sensitivity to multiple subsequent chemotherapeutic agents.
-
2.
RRx-001
In colorectal cancer, a randomized Phase II study (ROCKET) of the approved multikinase inhibitor regorafenib versus RRx-001, an experimental pan-epigenetic inhibitor and radiosensitizer that resensitizes or “episensitizes” tumors to formerly effective chemotherapies, is designed to investigate sequential cytotoxic rechallenge in irinotecan-refractory third-/fourth-line colorectal cancer patients after a maintenance/priming period with RRx-001. Per protocol, irinotecan-based therapy is restarted on both the regorafenib and the RRx-001 arms, if clinically appropriate (Figure 4). In a 25-patient Phase I trial, RRx-001 demonstrated evidence of single-agent activity with a PFS that would have led to underprediction of OS, which was 8.2 months, based on the prolonged survival of 5/5 patients who were rechallenged with formerly effective chemotherapies (EpicentRx unpublished data).
Figure 4.
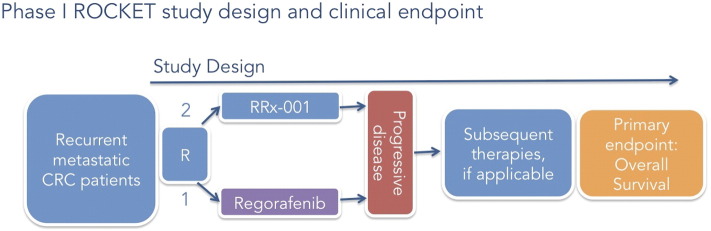
ROCKET study design (clinical end point OS) illustrated.
To date, in the Phase II trial, 7 of 10 evaluable RRx-001 patients have been successfully rechallenged with irinotecan-based therapies after completion of the first priming phase of the study, leading to a decrease in carcinoembryonic antigen (CEA) tumor marker; prolonged stabilization of disease (> 4 months); and, in two cases, a Positron Emission Tomography (PET) partial response. In contrast to the RRx-001–treated arm, the regorafenib patients have been too debilitated to start irinotecan chemotherapy. The primary hypothesis is that the successful reintroduction of previously effective therapies will significantly improve long-term OS rates compared with the regorafenib control arm. In anticipation of a Phase III trial, PFS2 is under consideration as a primary surrogate for OS.
Conclusion
Before Newton discovered gravity, the movement of everyday objects was explainable in terms of local or contact forces that arose through physical interaction like the collision of billiard balls in a game of pool. Gravity, however, was a different matter (no pun intended) altogether: not a contact force but an intangible one exerted between objects in space and invisible to the naked eye except through the prism of advanced mathematics. In the 1850s, Faraday postulated a force field to explain electromagnetism, envisioning lines of force similar to the pattern of iron fillings sprinkled around the poles of a bar magnet, which not only provided an a posteriori mechanism to understand the action-at-a-distance effects of gravity but also indirectly helped to make Einstein’s later E = mc2 epiphany possible.
Just as action-at-a-distance was initially hard to accept in physics, it is similarly unintuitive in oncology that the sequencing of two temporally separated therapies might affect and be affected by each other. The potential for drug–drug interaction makes sense in the context of concomitant exposure. However, it is not a priori obvious that the effects of separate lines of therapy may spill over into subsequent treatment lines, influencing the response of the tumor either positively (in the case of sensitizers) or negatively (in the case of resistance inducers). This mutual and reciprocal influence is analogous to the above-mentioned action-at-a-distance forces in physics like gravity, electricity, and magnetism, which are able to exert a “field of influence” or a “force field.” In oncology, the action-at-a-distance force field impacts the success or failure of long-term maintenance strategies, where the induction phase may have a strong influence on the maintenance phase and vice versa. The problem with the currently used parameter of PFS is that, unlike the DDC, TFS, and PFS2 end points, it does not measure or capture the strength of the “field of influence” on sequential therapies (Figure 5).
Figure 5.

Action-at-a-distance illustrated. Clinical treatments may exert “fields of influence” where sequential treatments are potentially affected by the treatment administered before and/or afterwards. The concept of a clinical “field of influence” is compared with the basic magnetic field pattern.
PFS2 is a preferred end point for the following reasons: First and foremost, it is a validated surrogate end point for OS with the EMA, whereas DDC and TFS are not validated. Second, PFS, an intermediate end point that shares the same name and initials, is a well-known, well-understood surrogate for OS, which may lower the barrier to acceptance of PFS2. Third, according to a 2014 abstract entitled Time to Second Objective Disease Progression (PFS2): An Emerging Clinical Trial Endpoint with Regulatory and Reimbursement Implications [19], several Phase II and III clinical trials in multiple tumor types including breast, prostate, head and neck, colorectal, pancreatic, non–small cell lung cancer, and multiple myeloma are investigating PFS2 as a secondary end point, which may lead to increased reliance on it in the future.
Finally, just as clinical observations of pseudoprogression that were not captured by the World Health Organization (WHO) or Response Evaluation Criteria in Solid Tumors (RECIST) criteria in immunotherapy trials led to the adoption of novel immune-related response criteria, it is anticipated that the unprecedented pattern of chemosensitization and resensitization observed with epigenetic agents will require a similar shift from end points such as PFS to “field of influence” measures like PFS2 that encompass the interactive dynamic between sequential lines of therapy.
In conclusion, Albert Einstein defined insanity as “doing the same thing over and over again and expecting different results.” By that same incontrovertible logic, it is time to rethink the automatic impulse to choose the primary end points of least resistance, PFS and OS, and potentially explore PFS2 as a surrogate for OS especially in the context of maintenance trials with epigenetic agents.
Acknowledgements
The authors acknowledge Harry Lybeck, MD, PhD, and his great granddaughter Brooke Shannon Harper who mutually guide, define, and influence each other in small, unseen ways every day before their first official face-to-face meeting and despite living worlds apart in Scandinavia and the United States, respectively.
References
- 1.Pazdur Richard. Endpoints for assessing drug activity in clinical trials. Oncologist. 2008;13(Suppl 2):19–21. doi: 10.1634/theoncologist.13-S2-19. [DOI] [PubMed] [Google Scholar]
- 2.Kogan Allan Jay, Haren Melissa. Translating cancer trial endpoints into the language of managed care. Biotechnol Health. 2008;5(1):22–35. [PMC free article] [PubMed] [Google Scholar]
- 3.Booth CM, Eisenhauer EA. Progression-free survival: meaningful or simply measurable? J Clin Oncol. 2012;30:1030–1033. doi: 10.1200/JCO.2011.38.7571. [DOI] [PubMed] [Google Scholar]
- 4.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–440. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 5.Hygino da Cruz LC, Jr., Rodriguez I, Domingues RC, Gasparetto EL, Sorensen AG. Pseudoprogression and pseudoresponse: imaging challenges in the assessment of posttreatment glioma. AJNR Am J Neuroradiol. 2011;32(11):1978–1985. doi: 10.3174/ajnr.A2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werewka-Maczuga Agnieszka, Osiński Tomasz, Chrzan Robert, Buczek Magda, Urbanik Andrzej. Characteristics of computed tomography imaging of gastrointestinal stromal tumor (GIST) and related diagnostic problems. Pol J Radiol. 2011;76(3):38–48. [PMC free article] [PubMed] [Google Scholar]
- 7.Chow Laura QM. ASCO Educational Book. 2013. Exploring Novel Immune-Related Toxicities and Endpoints with Immune-Checkpoint Inhibitors in Non–Small Cell Lung Cancer; pp. 280–285. [DOI] [PubMed] [Google Scholar]
- 8.EMA guideline on the evaluation of anticancer medicinal products in man. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/01/WC500137128
- 9.Buyse Marc, Sargent Daniel J, Grothey Axel, Matheson Alastair, de Gramont Aimery. Biomarkers and surrogate end points—the challenge of statistical validation. Nat Rev Clin Oncol. 2010;7(6):309–317. doi: 10.1038/nrclinonc.2010.43. [DOI] [PubMed] [Google Scholar]
- 10.Gillies Robert J, Verduzco Daniel, Gatenby Robert A. Opinion: evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oronsky B, Oronsky N, Scicinski J, Fanger G, Lybeck M, Reid T. Rewriting the epigenetic code for tumor resensitization: a review. Transl Oncol. 2014;7(5):626–631. doi: 10.1016/j.tranon.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122:2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oronsky B, Carter CA, Mackie V, Scicinski J, Oronsky A, Oronsky N, Caroen S, Parker C, Lybeck M, Reid T. The war on cancer: a military perspective. Front Oncol. 2015;26(4):387. doi: 10.3389/fonc.2014.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming Thomas R, Rothmann Mark D, Lu Hong Laura. Issues in using progression-free survival when evaluating oncology products. J Clin Oncol. 2009;27(17):2874–2880. doi: 10.1200/JCO.2008.20.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garnett Sally Anne, Martin Miguel, Jerusalem Guy, Petruzelka Lubos, Torres Roberto, Bondarenko Igor N, Khasanov Rustem, Verhoeven Didier, Pedrini José L, Smirnova Iva. Comparing duration of response and duration of clinical benefit between fulvestrant treatment groups in the CONFIRM trial: application of new methodology. Breast Cancer Res Treat. 2013;138(1):149–155. doi: 10.1007/s10549-012-2395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance—serendipity or a causal relationship? Curr Pharm Des. 2004;10(16):1969–1977. doi: 10.2174/1381612043384411. [DOI] [PubMed] [Google Scholar]
- 17.Ning S, Bednarski M, Oronsky B, Scicinski J, Saul G, Knox SJ. Dinitroazetidines are a novel class of anticancer agents and hypoxia-activated radiation sensitizers developed from highly energetic materials. Cancer Res. 2012;72(10):2600–2608. doi: 10.1158/0008-5472.CAN-11-2303. [DOI] [PubMed] [Google Scholar]
- 18.Scandura Joseph M, Roboz Gail J, Moh Michelle, Morawa Ewelina, Brenet Fabienne, Bose J Robi. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood. 2011;118(6):1472–1480. doi: 10.1182/blood-2010-11-320093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mbanya Zacharie, Chadda Shkun. Time to second objective disease progression (PFS2): an emerging clinical trial endpoint with regulatory and reimbursement implications. Blood. 2014;124(21) [Google Scholar]
- 20.Goldie JH, Coldman AJ, Gudauskas GA. Rationale for the use of alternating non–cross-resistant chemotherapy. Cancer Treat Rep. 1982;66:439–449. [PubMed] [Google Scholar]
- 21.Hochster SH. Stop and go: yes or no? J Clin Oncol. 2009;27:5677–5679. doi: 10.1200/JCO.2009.24.5209. [DOI] [PubMed] [Google Scholar]
- 22.Chibaudel B, Maindrault-Goebel F, Lledo G, Mineur L, André T, Bennamoun M, Mabro M, Artru P, Carola E, Flesch M. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol. 2009;27(34):5727–5733. doi: 10.1200/JCO.2009.23.4344. [DOI] [PubMed] [Google Scholar]
- 23.Tournigand Christophe, André Thierry, de Gramont Aimery, Chibaudel Benoist. Therapeutic strategy in unresectable metastatic colorectal cancer. Ther Adv Med Oncol. 2012;4(2):75–89. doi: 10.1177/1758834011431592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hui EP. Optimising systemic therapy in metastatic colorectal cancer. Hong Kong J Radiol. 2013;16:S17–S22. [Suppl] [Google Scholar]
- 25.Oronsky B, Oronsky N, Knox S, Fanger G, Scicinski J. Episensitization: therapeutic tumor resensitization by epigenetic agents: a review and reassessment. Anticancer Agents Med Chem. 2014;14(8):1121–1127. doi: 10.2174/1871520614666140418144610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B. Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.www.cancer.gov/drugdictionary
- 28.http://www.fda.gov/downloads/Drugs/Guidances/ucm071590.pdf
- 29.http://www.biooncology.com/clinical-trials/clinical-endpoints/advantages-limitations
- 30.Porta C, Tortora G, Linassier C, Papazisis K, Awada A, Berthold D. Maximising the duration of disease control in metastatic renal cell carcinoma with targeted agents: an expert agreement. Med Oncol. 2012;29(3):1896–1907. doi: 10.1007/s12032-011-0016-8. [DOI] [PubMed] [Google Scholar]