

Abstract
Nucleophosmin-anaplastic lymphoma kinase–expressing (NPM-ALK+) T-cell lymphoma is an aggressive form of cancer that commonly affects children and adolescents. The expression of NPM-ALK chimeric oncogene results from the chromosomal translocation t(2;5)(p23;q35) that causes the fusion of the ALK and NPM genes. This translocation generates the NPM-ALK protein tyrosine kinase that forms the constitutively activated NPM-ALK/NPM-ALK homodimers. In addition, NPM-ALK is structurally associated with wild-type NPM to form NPM/NPM-ALK heterodimers, which can translocate to the nucleus. The mechanisms that sustain the stability of NPM-ALK are not fully understood. SUMOylation is a posttranslational modification that is characterized by the reversible conjugation of small ubiquitin-like modifiers (SUMOs) with target proteins. SUMO competes with ubiquitin for substrate binding and therefore, SUMOylation is believed to protect target proteins from proteasomal degradation. Moreover, SUMOylation contributes to the subcellular distribution of target proteins. Herein, we found that the SUMOylation pathway is deregulated in NPM-ALK+ T-cell lymphoma cell lines and primary lymphoma tumors from patients. We also identified Lys24 and Lys32 within the NPM domain as the sites where NPM-ALK conjugates with SUMO-1 and SUMO-3. Importantly, antagonizing SUMOylation by the SENP1 protease decreased the accumulation of NPM-ALK and suppressed lymphoma cell viability, proliferation, and anchorage-independent colony formation. One possible mechanism for the SENP1-mediated decrease in NPM-ALK levels was the increase in NPM-ALK association with ubiquitin, which facilitates its degradation. Our findings propose a model in which aberrancies in SUMOylation contribute to the pathogenesis of NPM-ALK+ T-cell lymphoma. Unraveling such pathogenic mechanisms may lead to devising novel strategies to eliminate this aggressive neoplasm.
Introduction
Nucleophosmin-anaplastic lymphoma kinase–expressing (NPM-ALK+) T-cell lymphoma is an aggressive non-Hodgkin’s lymphoma that is frequently encountered in children and young adults [1]. The expression of NPM-ALK oncogene results from a chromosomal translocation that leads to the fusion of the ALK gene on 2p23 and the NPM gene on 5q35 [2]. The NPM-ALK oncogene encodes the expression of NPM-ALK chimeric tyrosine kinase. NPM-ALK induces lymphomagenic effects through the formation of the constitutively activated NPM-ALK/NPM-ALK homodimers, which reside in the cytoplasm and possess ability to interact with and phosphorylate several survival-promoting proteins including JAK/STAT, PI3K/AKT, MAP kinases, and IGF-IR [3–11]. NPM-ALK is also capable of forming heterodimers that are composed of wild-type NPM and NPM-ALK. Because wild-type NPM contains a nuclear localization signaling domain, the NPM/NPM-ALK heterodimers have ability to translocate to the nucleus [12]. The biological impact of the translocation of NPM-ALK to the nucleus is not completely understood. At least one study suggested that proteins with antiapoptotic potential translocate to the nucleus and interact with NPM-ALK [13]. Notably, the mechanisms that promote the stability and accumulation of NPM-ALK in the lymphoma cells are not completely understood.
SUMOylation is a posttranslational modification that is characterized by the reversible conjugation of small ubiquitin-like modifiers (SUMOs)—SUMO-1, SUMO-2, and SUMO-3—with their target proteins [14–17]. Whereas SUMO-2 and SUMO-3 are 97% identical, they demonstrate only 50% sequence resemblance with SUMO-1. It is unclear whether SUMO-4, another member of the SUMO proteins, is conjugated to target proteins in vivo[18,19]. SUMO-4 is also unique in that its expression is mainly detected in the kidneys, spleen, and lymph nodes, unlike SUMO-1 and SUMO-2/3 that are ubiquitously expressed [18]. The regulation of SUMOylation is ensued via a cascade of SUMO-specific ligases E1, E2, and E3, which warrants that appropriate targets are modified by SUMO [20–24]. SUMOylation is also regulated by the sentrin-specific family of proteases (SENPs) including SENP1-3 and SENP5-7 [25–27]. The role of SENPs encompasses removal of SUMO from target proteins, thus reversing the effects induced by SUMOylation.
Although SUMOylation shares similarities with ubiquitination, it has been shown that SUMO proteins compete with ubiquitin for substrate binding; thus, SUMOylation appears to protect target proteins from proteasomal degradation [28–30]. In addition to enhancing protein stability, SUMO proteins are involved in subcellular localization and distribution of modified proteins as well as inter- and intramolecular interactions of target substrates, which affect processes essential for normal and abnormal cellular homeostasis [31–34].
It has been demonstrated that SUMOylation plays a key role in cancer pathogenesis. For instance, SUMOylation inhibits cancer establishment through stabilization of tumor suppressor genes or promotes cancer development through stabilization of oncogenes [15,35–43]. Because it is also involved in cellular processes that preserve genomic integrity, such as DNA damage repair, it is thought that aberrancies in SUMOylation possess the ability to promote progression and dissemination, and to initiate therapeutic resistance in cancer cells [44–49].
In this study, we hypothesized that SUMOylation aberrancies exist in and contribute to the pathogenesis of NPM-ALK+ T-cell lymphoma. In support of this idea, we found that the SUMOylation pathway is abnormally upregulated in NPM-ALK+ T-cell lymphoma cell lines and ALK+ primary lymphoma tumors from patients. Moreover, we found that SUMO-1 and SUMO-3 are conjugated to two specific lysine residues, namely, Lys24 and Lys32, located in the NPM domain of NPM-ALK. Importantly, antagonism of SUMOylation by the SENP1 protease decreased nuclear and cytoplasmic expression of the NPM-ALK protein. The negative regulatory effects of de-SUMOylation by SENP1 on NPM-ALK led to suppression of lymphoma cell viability, proliferation, and anchorage-independent colony formation.
Materials and Methods
Identifying Potential SUMO Consensus Motifs in NPM-ALK
To identify potential SUMO consensus motifs in NPM-ALK protein, the web-based algorithm SUMOplot (http://www.abgent.com/sumoplot) was used. After entering the amino acid sequence of NPM-ALK (GenBank ID: AAA58698.1) into SUMOplot, potential consensus motifs were identified showing different degrees of probability of conjugation with SUMO. Although the web-based algorithm selects ≥ 0.65 as a high-probability cutoff, we opted for a more stringent probability cut off of ≥ 0.85 to avoid the possibility of false-positive results.
Cell Lines
Five previously characterized NPM-ALK+ T-cell lymphoma cell lines were used in this study including Karpas 299, DEL, SUP-M2, SR-786, and SU-DHL-1 (DSMZ, Germany). The T lymphoblastic leukemia/lymphoma cell line Jurkat (ATCC, Manassas, VA) was used as a positive control (http://www.cellsignal.com/products/primary-antibodies/sumo-1-c9h1-rabbit-mab/4940?hit=productId&ntt=4940), and the renal cell carcinoma cell line 786-O (ATCC) was used as a negative control (unpublished data from our lab) for SUMO protein expression. Jurkat cells were also used as the host cells in the protein degradation experiments. The normal human peripheral blood pan-T lymphocytes were purchased from StemCell Technologies (Vancouver, British Columbia, Canada). Cells were maintained in RPMI 1640 medium (HyClone, South Logan, UT) supplemented with 10% FBS (Sigma, St. Louis, MO), glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in humidified air enriched with 5% CO2.
Antibodies
Antibodies used included SUMO-1 (catalogue number: 4940), SUMO-2/3 (4971), SENP1 (11929), ubiquitin (3933), Myc-tag (2276), and lamin A/C (3032) (Cell Signaling Technology, Danvers, MA); ALK (M7195; DAKO, Carpenteria, CA); Alexafluor 647 anti-mouse secondary antibody (ab150115; Abcam, Cambridge, MA); and β-actin (a5316; Sigma).
Primary Lymphoma Tumors from Patients
Approval of the Institutional Review Board was granted before performing experiments in human tissues. Proteins were extracted from formalin-fixed and paraffin-embedded (FFPE) tissue sections from primary ALK+ T-cell lymphoma tumors by using the Qproteome FFPE Tissue Kit (Qiagen, Germantown, MD). Briefly, 2.0-μm sections were subjected to a sequence of deparaffinization in xylene and concentration gradients of ethanol. Areas of interest, which were previously identified by using hematoxylin and eosin–stained sections and light microscopy, were excised from the slides and transferred to collection tubes. For each extraction procedure, 6.0 μl of β-mercaptoethanol was added to 94 μl of Extraction Buffer EXB Plus to obtain a working solution (final volume: 100 μl). The working solution was added to the tube containing the excised tissue and admixed using vortexing. Tubes were then sealed and incubated on ice for 5 minutes, and then mixed again by vortexing. The samples were incubated on a heating block at 100°C for 20 minutes. Thereafter, samples were incubated in an oven with rotators at 80°C for 2 hours. After incubation, the tubes were held at 4°C for 1 minute and then unsealed. The samples were subjected to centrifugation for 15 minutes at 14,000 × g at 4°C. The supernatant containing the extracted proteins was then transferred to a new 1.5 ml tube. Extracted proteins were used in Western blotting (WB) assays as described below.
Immunoprecipitation and WB
Cells were lysed using lysis buffer containing 25 mM HEPES (pH 7.7), 400 mM NaCl, 1.5 mM MgCl2, 2 mM EDTA, 0.5% Triton X-100, 0.1 mM phenylmethylsulfonyl fluoride, 2 mM dithiothreitol, and 100 × protease and phosphatase inhibitor cocktails (Thermo Scientific). In addition, N-ethylmaleimide (10 mM) (Thermo Scientific) was used to inhibit de-SUMOylation in the immunoprecipitation experiments [50]. For immunoprecipitation, protein A/G agarose beads (Millipore, Billerca, MA) were blocked with 5% bovine serum albumin overnight to reduce nonspecific binding. Lysates were precleared using 2.5 μg normal IgG with rocking for 1 hour at 4°C, followed by centrifugation for 1 minute at 13,000 × g and removal of supernatant. Thereafter, 800 μg of lysate was incubated with 2.5 μg of primary antibody or mouse IgG control antibody along with the blocked protein A/G agarose beads overnight at 4°C. Next day, immunocomplexes were spun and supernatant was removed. The beads were washed three times with cold phosphate-buffered saline solution for 15 minutes each at 13,000 × g and once with lysis buffer, and then resuspended with 2 × sample buffer (Bio-Rad, Hercules, CA). Then, samples were subjected to WB.
For WB, cells were subjected to lysis as described above. Total protein concentrations were measured using the Bio-Rad protein assay. The OD values were obtained using an ELISA plate reader (Bio-Tek Instruments, Winooski, VT). Proteins (50 μg) were subjected to electrophoresis with sodium dodecyl sulfate on 8% polyacrylamide gels (SDS-PAGE). Proteins were transferred to polyvinylidene fluoride membranes and probed with primary antibodies and with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Cardiff, UK), and then detected using a chemiluminescence-based kit (Amersham Life Sciences, Arlington Heights, IL).
Recombinant Proteins
The NPM-ALK recombinant protein was generated using the TnT T7/SP6 Coupled Rabbit Reticulocyte Lysate System (Promega, Fitchburg, WI). The template for the TnT reaction was a previously described and repeatedly used plasmid [9,10]. The following reaction components were assembled in a microcentrifuge tube: TnT Rabbit Reticulocyte Lysate, TnT Reaction Buffer, TnT T7 RNA Polymerase, Amino Acid Mixture Minus Leucine (1.0 mM), Amino Acid Mixture Minus Methionine (1.0 mM), RNasin Ribonuclease Inhibitor (40 U/μl), NPM-ALK plasmid template, Transcend Biotin-Lysyl-tRNA, and nuclease-free H2O. The mixture was incubated at 30°C for 90 minutes. An aliquot of this mixture was analyzed by WB to confirm the correct translation of NPM-ALK.
SUMOylation Assay
In vitro SUMOylation assay was performed using the SUMOlink SUMO-1 and SUMO-2/3 Kits (Active Motif, Carlsbad, CA). Briefly, the following components were assembled in a microcentrifuge tube: protein buffer; SUMOylation buffer (5 ×); NPM-ALK recombinant protein; E1 activating enzyme; E2 conjugating enzyme; SUMO-1, SUMO-2, or SUMO-3 protein; and nuclease-free H2O. In a separate assay, SUMO proteins were substituted with their corresponding forms mutated at a single amino acid (point mutation), which were provided in the kit as controls. The mixtures were incubated at 30°C for 3 hours. The reactions were stopped by adding equal volumes of 2 × SDS-PAGE loading buffer, and proteins were detected by WB.
Site-Directed Mutagenesis and PCR
NPM-ALKK24R, NPM-ALKK32R, and NPM-ALKK24,32R constructs, where lysine was replaced by arginine, were generated by using the QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). Primer sequences are shown in Table 1.
Table 1.
Primers Used to Generate NPM-ALKK24R and NPM-ALKK32R Constructs by Site-Directed Mutagenesis (F: Forward; R: Reverse).
| NPM-ALKK24R (F): 5’-TTTTCGGTTGTGAACTACGGGCCGACAAAGATTATC-3’ |
| NPM-ALKK24R (R): 5’-GATAATCTTTGTCGGCCCGTAGTTCACAACCGAAAAG-3’ |
| NPM-ALKK32R (F): 5’-GCCGACAAAGATTATCACTITCGCGTGGATAATGATGAAAATGAG-3’ |
| NPM-ALKK32R (R): 5’-CTCATTTTCATCATTATCCACGCGAAAGTGATAATCTTTGTCGGC-3’ |
The PCR products were transformed using MaxEfficiency DH5α competent cells (Invitrogen, Carlsbad, CA), and the transformation products were plated on ampicillin-resistant plates. Colonies containing the correct insert were confirmed by direct sequencing and amplified in ampicillin-containing Luria Bertani Broth (LB) overnight at 37°C with shaking. Next day, colonies were processed with the QiaPrep Spin Miniprep Kit (Qiagen) to isolate plasmids. For in vitro SUMOylation assays, mutated plasmids were used as templates for the TnT reactions to create mutated recombinant proteins.
Transfection
Transfection of Jurkat cells using the wild-type NPM-ALK, NPM-ALKK24R, NPM-ALKK32R, or NPM-ALKK24,32R plasmid was performed using electroporation and the Nucleofector System (Solution V, Program X-001; Lonza). Thereafter, cells were incubated for 48 hours. The NPM-ALK+ T-cell lymphoma cell lines were transfected with the SENP1 expression plasmid (Origene, Rockville, MD) using electroporation and the Amaxa 4D nucleofection system (Solution SF, Program CA-150; Lonza, Houston, TX). In some experiments, after transfection of SENP1, cells were treated with MG132 (Sigma) for 24 hours to examine the effects of proteasome inhibition.
Protein Degradation Assay
Cells were transfected, as described above, with wild-type or mutated NPM-ALK plasmids for 48 hours and treated with 100 μg/ml cycloheximide (CHX, Sigma) for 24 and 48 hours. Then, cells were harvested and subjected to lysis and WB.
Subcellular Fractionation
The Nuclear/Cytosol Fractionation kit (BioVision, Milipitas, CA) was used according to the manufacturer’s instructions. Briefly, cells were collected by centrifugation at 600 × g for 5 minutes at 4°C. The CEB-A buffer, containing DTT and protease inhibitors, was added, and samples were spun at 500 × g for 3 minutes at 4°C. The supernatant was removed, and the pellet was resuspended in the CEB-A mix and subjected to vigorous vortexing for 15 seconds to fully resuspend the pellet. Thereafter, samples were incubated on ice for 10 minutes. Ice-cold CEB-B buffer was added to the tube, which was subjected to vortexing and placed on ice for 1 minute, and then spun at maximum speed for 5 minutes. Immediately, the supernatant (containing the cytoplasmic extract) was transferred into a prechilled tube. The residual pellet was washed five times in ice-cold PBS by centrifugation at maximum speed for 1 minute to prevent cytoplasmic extraction contamination. Then, the pellet was resuspended in ice-cold Nuclear Extraction buffer (NEB), subjected to vortexing for 15 seconds, and then returned to ice for 10 minutes. This step was repeated four times. Finally, samples were subjected to centrifugation at maximum speed for 15 minutes, and the supernatant (containing nuclear extract) was transferred into a prechilled tube.
Immunofluorescence Staining
Cells were first transfected with empty vector (EV) or Myc-tagged SENP1 for 24 hours. Thereafter, cytospins were prepared followed by fixation with 4% paraformaldehyde at the following time points: 0 minute, 15 minutes, 30 minutes, 1 hour, 3 hours, 6 hours, 12 hours, 24 hours, and 48 hours. After fixation, cells were subjected to permeabilization with 0.2% Triton-X in PBS for 30 minutes at room temperature. Subsequently, cells were blocked with 0.3% Triton-X/2% BSA in PBS solution for 1 hour at room temperature. Thereafter, Myc-tag primary antibody was added at 1:2000 dilution to detect exogenous SENP1, and slides were incubated at 4°C overnight. The following day, slides were washed three times in PBS, and Alexafluor 647 fluorochrome-conjugated secondary antibody was added at 1:200 dilution for 1.5 hours at room temperature. Thereafter, slides were washed three times in PBS and counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Prolong Gold Antifade Mountant (Invitrogen) was added; slides were coverslipped and sealed with nail polish to prevent dehydration. Images were captured using Deltavision Image Restoration microscope (Olympus IX71; GE Healthcare; total magnification is × 400).
Cell Viability Assay
The CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) kit (Promega) was used. Cells were seeded in 96-well plates at a concentration of 1.0 × 104 cells/well in 100 μl of RPMI supplemented with 10% FBS. The MTS reagent was then added and incubated at 37°C in a humidified chamber containing 5% CO2 in air for approximately 4 hours. OD measurements were obtained by using an ELISA plate reader.
Cell Proliferation Assay
The 5-bromo-2’-deoxyuridine (BrdU) assay kit (ExAlpha, Shirley, MA) was used. Briefly, 2.0 × 105 cells/ml were plated into a 96-well plate. The BrdU label was added at a dilution of 1:500, and the plate was incubated for 24 hours at 37°C. Cells were fixed for 30 minutes at room temperature. The anti-BrdU antibody was added for 1 hour after washing, followed by peroxidase goat anti-mouse IgG conjugate (1:2000 dilution) for 30 minutes. Thereafter, the 3,3’,5,5’-tetramethylbenzidine peroxidase substrate was added, and the plate was incubated for 30 minutes at room temperature in the dark. Finally, the acid Stop Solution was added, and the plate was read at 450 nm by using an ELISA plate reader.
Anchorage-Independent Colony Formation Assay
Transfected cells were resuspended to eliminate clumping and then added in 1:10 (v/v) ratio to 3 ml of methylcellulose (Methocult H4230, StemCell Technologies). Tubes were gently inverted several times. Then, 1.0 ml of the mix was divided into 24-well plates in triplicate. Plates were placed in a humidified incubator at 37°C in 5% CO2 for 7 days, and p-iodonitrotetrazolium violet was added for 24 hours for staining. Colonies were visualized using the AlphaImager system (Alpha Innotech, San Leonardo, CA).
Results
Potential SUMO Consensus Motifs in NPM-ALK Proteins
To determine whether SUMO consensus motifs exist within the NPM-ALK amino acid sequences, we used the web-based algorithm SUMOplot, which identified several lysine residues that could represent potential conjugation between SUMO and NPM-ALK (Supplementary Figure 1). The two sites with the highest probability to conjugate with SUMO proteins were Lys24 (probability = .91) and Lys32 (probability = .85).
Expression of SUMO Proteins and SENP1 Protease in NPM-ALK+ T-Cell Lymphoma
Thereafter, we performed screening WB analysis that showed SUMO-2/3 protein to be universally overexpressed in the NPM-ALK+ T-cell lymphoma cell lines Karpas 299, SR-786, DEL, SUP-M2, and SU-DHL-1 compared with its lower levels in normal human T lymphocytes (Figure 1A). In addition, SUMO-1 is overexpressed in Karpas 299, SR-786, and SU-DHL-1 cells. In contrast, SENP1 protein, the major suppressor of SUMO conjugation, was substantially decreased in all of the lymphoma cell lines (Figure 1A). The Jurkat and 786-O cells were used as positive and negative controls, respectively, for SUMO expression. Densitometry analysis of the expression of each protein normalized to β-actin is also shown (Figure 1B). Similar patterns of expression of SUMO-1, SUMO-2/3, and SENP1 were found in the lymphoma primary tumors from patients when compared with normal human T lymphocytes (Figure 1C). Because the quality of the FFPE tissue sections varied significantly, β-actin showed unequal protein levels. Nevertheless, densitometry analysis of the WB bands for each protein normalized to β-actin supported that SUMO-2/3 is upregulated in all of the patients and SUMO-1 is upregulated in 9 of 15 patients included in our study (Figure 1D). In addition, densitometry showed that SENP1 levels are decreased in the 13 of the 15 patient samples compared with T lymphocytes (Figure 1D). Collectively, these results support that the SUMOylation proteins are deregulated in NPM-ALK+ T-cell lymphoma.
Figure 1.

SUMO proteins are upregulated and SENP1 protease is downregulated in NPM-ALK+ T-cell lymphoma. (A) WB studies show very low levels of expression of SUMO-1 and SUMO-2/3 proteins in normal human T lymphocytes. In contrast, overexpression of SUMO-2/3 protein is present in the five NPM-ALK+ T-cell lymphoma cell lines Karpas 299, SR-786, DEL, SUP-M2, and SU-DHL-1. In addition, increased expression of SUMO-1 protein is seen in majority of the lymphoma cell lines including Karpas 299, SR-786, and SU-DHL-1. Moreover, high levels of expression of the SENP1 protease are detected in the normal human T lymphocytes compared with much lower levels in the five lymphoma cell lines. The Jurkat and 786-O cells were used as positive and negative controls, respectively, for the expression of the SUMO proteins. Notably, Jurkat cells that express high levels of the SUMO proteins demonstrate much lower levels of SENP1 than the 786-O cells that lack the expression of SUMO proteins. β-Actin was used as a loading control. (B) Densitometry studies of SUMO and SENP1 proteins relative to β-actin support the findings of the WB. (C) WB performed on protein extracted from FFPE tissue sections collected from 15 patients’ lymphoma tumors. The patient samples were divided into two groups—1 to 7 (left panel) and 8 to 15 (right panel)—and lysates from normal human T lymphocytes (TL) were analyzed. It is important to notice that the quality of the FFPE tissue sections varied significantly, and therefore, β-actin showed unequal protein levels among these samples. Nonetheless, WB revealed a pattern of expression similar to the cell lines in which SUMO-2/3 was overexpressed in all patients, and SUMO-1 was upregulated in 9 of the 15 patients. In clear contrast, SENP1 was decreased in 13 of the 15 patients. β-Actin was used as a loading control. (D) Densitometry studies of the WB bands of SUMO-1, SUMO-2/3, and SENP1 proteins relative to β-actin bands are shown. Despite the variable levels of β-actin in the lysates from patients’ FFPE tumors, densitometry supported the general increase in SUMO proteins and decrease in SENP1 protease in these samples.
Physical Association and Interaction between SUMO Proteins and NPM-ALK
To test whether SUMO proteins are physically associated with NPM-ALK, immunoprecipitation of endogenous NPM-ALK protein was performed in Karpas 299 and SR-786 cells using an anti-ALK antibody, and WB was then used using an anti–SUMO-1 or SUMO-2/3 antibody. The SUMO proteins were physically associated with NPM-ALK (Figure 2A).
Furthermore, an in vitro SUMOylation assay showed that all SUMO modifiers are capable of SUMOylating NPM-ALK, as indicated by the presence of the higher–molecular weight bands above the baseline NPM-ALK protein bands (Figure 2B). In contrast, SUMO-1 and SUMO-2 mutated at a single amino acid (point mutation) induced much less pronounced SUMOylation of NPM-ALK. In addition, mutated SUMO-3 failed to SUMOylate NPM-ALK (Figure 2B).
Figure 2.

NPM-ALK is conjugated and interacts with the SUMO proteins. (A) NPM-ALK was immunoprecipitated in Karpas 299 and SR-786 cells, and the expression of the SUMO proteins was analyzed using WB. These studies showed that SUMO-1 and SUMO-2/3 are physically associated with NPM-ALK protein as indicated by bands present in lane 2 where NPM-ALK was pulled down using an anti-ALK antibody, but not in lanes 3 where normal mouse IgG was immunoprecipitated or in lane 4 where beads alone were used. In addition, a control was used where NPM-ALK was pulled down using an anti-ALK antibody followed by WB using an anti-ALK antibody. Lane 1 (input) demonstrates the corresponding NPM-ALK bands. (B) In the upper panel, NPM-ALK recombinant protein was generated by using the TnT coupled rabbit reticulocyte kit. Thereafter, WB was performed to confirm the expression of the NPM-ALK recombinant protein. As depicted in lane 1, there was no expression of NPM-ALK in rabbit reticulocyte lysate. In addition, WB demonstrated the presence of recombinant NPM-ALK band in lane 2 and endogenous NPM-ALK band (80 kDa) in the lysate from Karpas 299 cells that were used as control as shown in lane 3. In the lower panel, recombinant NPM-ALK protein was subjected to in vitro SUMOylation assay. Bands with a molecular weight that is ~ 20 kDa higher than baseline NPM-ALK’s 80-kDa molecular weight were present in the lanes where incubations with wild-type SUMO-1 (lane 1), SUMO-2 (lane 3), or SUMO-3 (lane 5) were performed. Importantly, incubation with the SUMO constructs mutated at one amino acid (point mutation) showed much less pronounced higher-molecular weight bands in lanes 2 and 4. This band was lacking in lane 6. did not show higher–molecular weight bands (lanes 2, 4, and 6).
Conjugation and Interactions between SUMO Modifiers and Lys24 and Lys32 Residues of NPM-ALK
The SUMOplot algorithm analysis identified Lys24 and Lys32 of NPM-ALK as having the highest probability to conjugate with the SUMO modifiers (Supplemental Figure 1). To determine whether the SUMO modifiers are indeed capable of conjugating with these sites, wild-type NPM-ALK and NPM-ALK mutated at Lys24 or Lys32 residues were translated in vitro using rabbit reticulocytes, and levels of expression of the three constructs are depicted in Figure 3 (upper panel). Thereafter, an in vitro SUMOylation assay was performed and showed that SUMOylation was abrogated for SUMO-1 and SUMO-3 when NPM-ALKK24R mutant was used and for SUMO-3 only when NPM-ALKK32R was used (Figure 3, lower panel). In contrast, mutations at Lys24 and Lys32 failed to prevent SUMO-2 from conjugation with NPM-ALK, suggesting that SUMO-1 and SUMO-3 are most likely the primary SUMO modifiers for NPM-ALK at these lysine residues.
Figure 3.

NPM-ALK is SUMOylated at Lys24 and Lys32 residues. The mutated NPM-ALK recombinant proteins NPM-ALKK24R and NPM-ALKK32R were generated using the TnT coupled rabbit reticulocyte kit. In the upper panel, WB was performed to confirm the expression of recombinant NPM-ALK proteins. Wild-type NPM-ALK recombinant protein is present in lane 1, NPM-ALKK24R recombinant protein in lane 2, and NPM-ALKK32R recombinant protein in lane 3. In the lower panel, wild-type and mutated recombinant NPM-ALK proteins were subjected to an in vitro SUMOylation assay. SUMOylation was abolished with the K24R mutation for SUMO-1 and SUMO-3 and with the K32R mutation for SUMO-3. The two mutations failed to de-SUMOylate SUMO-2.
SUMOylation Sustains the Stability of NPM-ALK Protein
To this end, we set to analyze the effects of SUMOylation on NPM-ALK protein stability. Jurkat cells, which lack NPM-ALK but express high levels of SUMO-1 and SUMO-2/3 proteins, were transfected with wild-type NPM-ALK, NPM-ALKK24R, NPM-ALKK32R, or NPM-ALKK24,32R plasmid and then treated with the protein synthesis inhibitor CHX for 24 and 48 hours. WB analysis was performed to measure the levels of expression of wild-type NPM-ALK and its mutants (Figure 4). Of note is that wild-type NPM-ALK did not demonstrate any significant degradation even after 48 hours of treating the cells with CHX. At 24 hours, however, NPM-ALKK32R and NPM-ALKK24,32R demonstrated minimal degradation (data not shown). At 48 hours, the NPM-ALKK32R single mutant demonstrated more degradation than the NPM-ALKK24R single mutant. Notably, the degradation was much more pronounced when the NPM-ALKK24,32R double mutant was used (Figure 4). These data strongly suggest that SUMOylation maintains the stability of NPM-ALK oncogenic protein.
Figure 4.
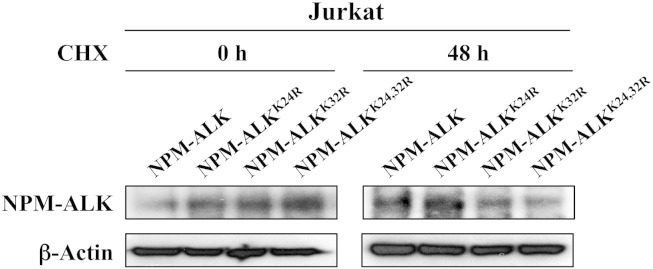
SUMOylation sustains the stability of NPM-ALK protein. Jurkat cells were transfected with wild-type NPM-ALK, NPM-ALKK24R, NPM-ALKK32R, or NPM-ALKK24,32R construct for 0, 24 or 48 hours and then treated with the protein synthesis inhibitor CHX for an additional 48 hours. There was minimal decrease in the levels of NPM-ALK mutants at 24 hours (data not shown). Nonetheless, the decrease in the expressions of NPM-ALKK32R was more pronounced than the decrease in NPM-ALKK24R expression at 48 hours. Importantly, this decrease was much more pronounced when the NPM-ALKK24,32R double mutant was used. In contrast, wild-type NPM-ALK protein expression did not show any evidence of decreased expression even at 48 hours after treatment with CHX. β-Actin shows equal loading of the proteins.
De-SUMOylation by SENP1 Abrogates Nuclear and Cytoplasmic Accumulation of NPM-ALK
To determine whether SUMOylation-mediated stabilization of NPM-ALK occurs in the nucleus and/or the cytoplasm, Karpas 299 and SR-786 cells were transfected with EV or Myc-tagged SENP1 protease. Thereafter, subcellular fractionation and WB assay were performed at 48 hours after transfection. There was more abundant expression of NPM-ALK in the nucleus than the cytoplasm in cells transfected with EV. This observation was more pronounced in the SR-786 cells compared with Karpas 299 cells (Figure 5A). Moreover, the expressions of SUMO-1 and SUMO-2/3 proteins were much more pronounced in the nucleus than the cytoplasm. It is important to note that fractionation and WB studies performed in Karpas 299 and SR-786 cells without transfection of EV demonstrated similar patterns of expression of NPM-ALK and SUMO modifiers (data not shown). These findings indicate that the subcellular distribution of NPM-ALK and SUMO modifiers was most likely not related to the effects of the physical impact resulting from the transfection procedure.
Figure 5.

SENP1-induced de-SUMOylation causes remarkable downregulation of NPM-ALK protein expression. (A) Karpas 299 and SR-786 cells were transfected with EV or Myc-SENP1 expression plasmid. Myc tag was used to distinguish transfected SENP1 from the very low levels of endogenously expressed SENP1 protein. Subcellular fractionation shows that in EV-transfected cells, SUMO-1 and SUMO-2/3 proteins are exclusively localized in the nuclear (N) compartment with lack of expression in the cytoplasm (C). Although the NPM-ALK protein is distributed between the nucleus and cytoplasm, it is slightly more abundant in the nucleus. It is important to mention that similar patterns of expression of the SUMO modifiers as well as NPM-ALK were seen in Karpas 299 and SR-786 cells under the basal conditions without exposing the cells to the transfection stress (data not shown), supporting that these patterns of expression are physiological. Notably, transfection of SENP1 induced remarkable downregulation of SUMO-1 and SUMO-2/3 proteins in the two cell lines. The decrease in SUMO modifiers was associated with marked decrease in the nuclear and cytoplasmic expression of NPM-ALK. Lamin A/C and β-actin were used as nuclear and cytoplasmic loading controls, respectively. (B) To examine whether adequate transfection was achieved, immunofluorescence staining for Myc-SENP1 was performed, and its levels of expression were monitored for up to 48 hours. Transfection of EV shows total lack of SENP1. Similarly, SENP1 was not detected either in Karpas 299 or in SR-786 cells immediately after transfection at baseline. After 15 minutes, low levels of expression of SENP1 were identified in the cytoplasm. At 30/60 minutes, the cytoplasmic expression of SENP1 became much more pronounced. Gradual shuttling of SENP1 into the nucleus occurred, and it became distributed between the nucleus and cytoplasm at 3/6 hours. However, at 24 hours, most of SENP1 appeared to be shuttling back to the cytoplasm, and yet at 48 hours, much higher levels of SENP1 were distributed between the nucleus and cytoplasm.
The transfection of SENP1 was associated with a remarkable decrease in the levels of SUMO-1 and SUMO-2/3 proteins. This decrease was associated with substantial downregulation of NPM-ALK protein expression (Figure 5A). To evaluate the efficiency of SENP1 transfection, an immunofluorescence staining using an anti-Myc antibody was performed at baseline (0 minute) and various time points in Karpas 299 and SR-786 cells. Up to 1 hour, SENP1 was predominantly localized in the cytoplasm, but then at 3 to 6 hours, it became predominantly localized in the nucleus. At 24 hours, most of SENP1 shuttled back to the cytoplasm, whereas at 48 hours, very high levels of SENP1 were found to be distributed between the nucleus and cytoplasm (Figure 5B).
De-SUMOylation by SENP1 Directs NPM-ALK Protein to Ubiquitination
To investigate possible explanations for the decrease in NPM-ALK levels after de-SUMOylation, pull down of the NPM-ALK protein was performed followed by WB using an anti-SENP1, ALK, or ubiquitin antibody. Transfection of EV shows high levels of expression of basal NPM-ALK protein with small fractions associated with SENP1 and ubiquitin (Figure 6). When SENP1 was exogenously expressed, not only was the fraction of NPM-ALK associated with SENP1 remarkably increased, but also the fraction associated with ubiquitin was increased as well. Importantly, basal levels of NPM-ALK decreased significantly after SENP1 transfection (Figure 6). These results suggest that de-SUMOylation redirects the NPM-ALK protein to the ubiquitination system, which leads to its degradation and removal from the cell.
Figure 6.

SENP1-induced de-SUMOylation directs NPM-ALK to ubiquitination. Transfection of EV demonstrates that most of NPM-ALK protein is present in unbound form with much smaller fractions conjugated to SENP1 or ubiquitin. To this end, exogenous transfection of SENP1 expression plasmid induced remarkable increase in NPM-ALK fraction that is associated with SENP1 or ubiquitin, which was associated with substantial downregulation in its unconjugated form. In lane 3, normal mouse IgG was used as a negative control and immunoprecipitated instead of NPM-ALK.
Cellular Effects of De-SUMOylation by SENP1 in NPM-ALK+ T-Cell Lymphoma
To study the cellular impact of antagonizing the SUMOylation pathway in NPM-ALK+ T-cell lymphoma, forced expression of EV or SENP1 was performed in Karpas 299, DEL, SR-786, and SU-DHL-1 cells. The SENP1-mediated downregulation of SUMO-1 and SUMO-2/3 proteins as well as NPM-ALK (as shown in Figure 5A) was associated with decreased NPM-ALK+ T-cell lymphoma cell viability (Figure 7A), proliferation (Figure 7B), and anchorage-independent colony formation (Figure 7C).
Figure 7.

De-SUMOylation of NPM-ALK by SENP1 decreases cell viability, proliferation, and anchorage-independent colony formation of NPM-ALK+ T-cell lymphoma. (A) Transfection of Karpas 299, DEL, SR-786, and SU-DHL-1 cells with the SENP1 protease expression plasmid resulted in a significant decrease in their viability after 48 hours (*P < .001, **P < .0001, ***P < .00001). (B) In addition, transfection of the lymphoma cells with SENP1 decreased their proliferation (*P < .05, **P < .001, ***P < .00001). (C) Transfection of SENP1 also decreased the anchorage-independent colony formation potential of the different lymphoma cells (*P < .05, **P < .01). (D) Representative examples of the colonies from each cell line are shown at 7 days after transfection with EV or SENP1. Results shown in panels A, B, and C represent the means ± SE of three independent experiments.
Discussion
In this paper, we provide novel evidence showing that there is an upregulation of the SUMO modifiers SUMO-1, SUMO-2, and SUMO-3 in NPM-ALK+ T-cell lymphoma cell lines and ALK+ T-cell lymphoma primary patient tumors compared with normal human T lymphocytes. In contrast, the expression of the SENP1 protease, which physiologically induces de-SUMOylation through removal of SUMO modifiers from their target proteins, was substantially decreased in these lymphoma cell lines and primary tumors. To our knowledge, only one screening study reported upregulation of the SUMO-1 gene, among several other genes, in NPM-ALK+ T-cell lymphoma, but functional studies to characterize the role of SUMO-1 were not performed in that study [51]. Herein, we found that NPM-ALK can be SUMOylated through conjugation with the SUMO modifiers, which leads to sustaining the stability of NPM-ALK protein. In support of this idea, de-SUMOylation through exogenous expression of SENP1 downregulated the SUMO modifiers and was associated with substantial decrease in NPM-ALK protein expression, which reduced its accumulation in the nucleus and cytoplasm. Our data suggest that the de-SUMOylating effects of SENP1 and the subsequent decrease in NPM-ALK could be attributed, at least partially, to promoting the association between NPM-ALK and ubiquitin. At the cellular level, downregulation of the SUMO modifiers resulted in negative biological effects, attesting to the idea that the disruption of the SUMOylation pathway conveys tumor-suppressing effects in NPM-ALK+ T-cell lymphoma.
SUMOylation is a dynamic process that causes important modifications of target proteins, which may alter their subcellular localization and functional activity [16,17,31–34]. It has also been proposed that an important outcome of SUMOylation is the maintenance of protein stability by protecting targets from proteasomal degradation [28–30]. Although SUMOylation is important for physiological processes, it has also been shown to be commonly deregulated in cancer cells [44–49]. In line with this idea, our data showed that the SUMO proteins were overexpressed and SENP1 protease was decreased in NPM-ALK+ T-cell lymphoma relative to normal T lymphocytes. Indeed, SUMO-1, SUMO-2, and SUMO-3 modifiers were all capable of conjugation with NPM-ALK. The conjugation with the SUMO modifiers can occur through an acceptor Lys residue within a ΨKX(D/E) SUMO consensus motif present within the target protein, where Ψ is a large hydrophobic amino acid residue [52]. In addition to the SUMO consensus motifs, target proteins can sometimes conjugate noncovalently with the SUMO modifiers through SUMO interacting motifs that are characterized by a short stretch of hydrophobic amino acids flanked by acidic residues [53]. Herein, we were able to identify Lys24 and Lys32 as acceptor residues located within potential SUMO consensus motifs in NPM-ALK. A point mutation of Lys24 or Lys32 to arginine abrogated the conjugation and interactions between NPM-ALK and SUMO-1 and SUMO-3. In addition, this point mutation resulted in degradation of NPM-ALK. The finding that the degradation of the NPM-ALKK32R single mutant was more pronounced than the degradation of the NPM-ALKK24R suggests that, individually, the Lys32 residue may play a more significant role in sustaining the stability of NPM-ALK protein. Notably, the degradation of the NPM-ALK protein became much more pronounced when the two lysine residues were simultaneously mutated to arginine. These observations testify to the stabilizing effects of SUMO-1 and SUMO-3, specifically through Lys24 and Lys32 residues, on NPM-ALK protein. In contrast, mutations at the same residues failed to abrogate the conjugation and interactions between NPM-ALK and SUMO-2, suggesting that the interactions between these two proteins could occur through other SUMO consensus motifs that are present in NPM-ALK. The preferred conjugation and interactions between NPM-ALK and SUMO-3 compared with SUMO-2 may prove to be biologically relevant considering that several recent studies demonstrated that SUMO-2 and SUMO-3 possess distinct biological functions in different types of cells [54–57]. Further studies are required to explore this point in NPM-ALK+ T-cell lymphoma.
SENPs have two primary functions including the conversion of SUMO precursors to mature SUMO via removal of a portion of their C-terminus and the reversal of the SUMOylation process via removal of SUMO modifiers from target proteins. These SENP-controlled processes contribute to the dynamic nature of SUMOylation. We elected to evaluate SENP1 because of its ability to universally de-SUMOylate the three SUMO modifiers, unlike other SENPs that have preferential de-SUMOylation activities [58–61]. In our hands, reestablishment of SENP1 in NPM-ALK+ T-cell lymphoma cells resulted in decreasing the expression of the SUMO proteins, which was associated with downregulation of NPM-ALK levels. Because NPM-ALK plays central roles in driving the survival of this lymphoma [1,2,62], its downregulation through de-SUMOylation by SENP1 decreased lymphoma cell viability, proliferation, and anchorage-independent colony formation.
The role of NPM in the pathogenesis of NPM-ALK+ T-cell lymphoma remains intriguing. Previous studies showed that the nuclear localization signal in NPM is not conserved in the NPM-ALK chimeric protein. Therefore, nuclear translocation of NPM-ALK occurs primarily because of the formation of heterodimers between wild-type NPM and NPM-ALK [12]. Herein, we identified for the first time SUMO consensus motifs that contain the Lys24 and Lys32 that are located within the NPM domain as the sites where SUMO modifiers conjugate with NPM-ALK to maintain its stability. Similar to NPM-ALK, previous studies showed that SENP1 is also capable of shuttling between the nucleus and cytoplasm [58,63]. Indeed, overexpression of SENP1 protein in NPM-ALK+ T-cell lymphoma cells was associated with robust shuttling between the nucleus and cytoplasm. The predominant localization of the SUMO modifiers in the nucleus suggests that the interactions between the SUMO modifiers, NPM-ALK, and SENP1 occur primarily in the nucleus. The tendency of SUMO modifiers to be colocalized with their targets in the nucleus has been previously reported [31,33,52,64]. Our results suggest a model in which the NPM/NPM-ALK heterodimers translocate from the cytoplasm to the nucleus where they conjugate and interact with the SUMO modifiers. SUMOylation provides stabilization of NPM/NPM-ALK, which leads to its nuclear accumulation, followed by its shuttling back to the cytoplasm (Figure 8). In line with our findings in NPM-ALK, previous studies showed that SUMOylation plays an important role in the stability and nuclear accumulation of several survival-regulatory proteins [65–69]. However, it cannot be ruled out that, in addition to SUMOylation, the localization of NPM/NPM-ALK heterodimers in the nucleus may also be required for other yet unidentified biological functions that are directly relevant to the survival of this lymphoma. In addition to our novel data demonstrating direct contribution of NPM to sustaining the stability of NPM-ALK protein through SUMOylation, it is still possible that NPM contributes indirectly to global SUMOylation of survival regulatory proteins in this lymphoma in a fashion similar to what has been recently proposed in other biological systems [70].
Figure 8.
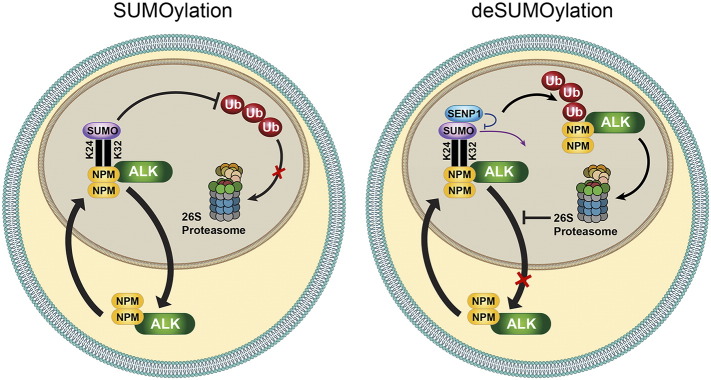
Proposed model for NPM-ALK SUMOylation. NPM-ALK forms heterodimers with wild-type NPM. These NPM/NPM-ALK heterodimers possess ability to shuttle between the nucleus and cytoplasm. The association between NPM/NPM-ALK, through its Lys24 and Lys32 residues, with the SUMO proteins occurs predominantly in the nucleus, which is the cellular compartment where SUMO proteins appear to be predominantly expressed in NPM-ALK+ T-cell lymphoma cells. In turn, conjugation with the SUMO modifiers confers stability on NPM-ALK oncogenic protein by preventing its ubiquitination and degradation. Therefore, NPM/NPM-ALK is capable of shuttling back to the cytoplasm. To this end, SUMOylation-mediated stability leads to accumulation and abundant expression of NPM-ALK in the nucleus and cytoplasm. At the other hand, SENP1-mediated de-SUMOylation decreases significantly the expression and accumulation of NPM/NPM-ALK in the nucleus and cytoplasm through the increase in NPM-ALK breakdown that occurs through the switch from the protective effects of SUMOylation to the ubiquitination-induced degradation.
It has been shown that SUMOylation hinders protein-protein interactions including the interactions with ubiquitin E1–activating enzyme [71]. Furthermore, there have been studies indicating that de-SUMOylation exposes ubiquitin-acceptor lysine residues located within the target protein [72–74]. In the NPM-ALK+ T cell lymphoma cells, de-SUMOylation of NPM-ALK led to an increase in its association with ubiquitin, suggesting that SUMOylation has the ability to prevent NPM-ALK from entering the proteasomal degradation pathway. NPM-ALK has been previously demonstrated to be ubiquitinated. Treating NPM-ALK+ T-cell lymphoma cells with 7-AAG causes NPM-ALK to undergo proteasomal degradation through Hsp70-dependent mechanism, although a specific Lys residue for ubiquitination has not been identified [75]. In our study, transfection with SENP1 resulted in de-SUMOylation, and therefore it is possible that the Lys24 and Lys32 residues on NPM-ALK became available for interaction with ubiquitin and subsequent proteasomal degradation. This idea was further illustrated when the proteasome inhibitor MG132 abrogated the SENP1-mediated downregulation of NPM-ALK (Supplemental Figure 2). To this end, we currently do not have evidence that ubiquitin binds to these specific residues, but entering NPM-ALK amino acid sequence in ubiquitination prediction software (UbPred; www.ubpred.org) demonstrated that it contains a potential ubiquitination site at Lys32. Whether NPM-ALK binds ubiquitin at this residue or other residues that may potentially interact with the SUMO proteins constitutes the subject of future studies.
In summary, we have identified for the first time that aberrant SUMOylation exists and contributes to the pathogenesis of NPM-ALK+ T-cell lymphoma through maintaining the stability of NPM-ALK oncogenic tyrosine kinase, which appears to promote its nuclear localization and cellular accumulation. Forced expression of SENP1 protease caused disruption of SUMOylation and subsequent degradation of NPM-ALK, and thereby reduced the tumorigenic potential of these lymphoma cells. Hence, SUMOylation may represent a potential target for drug development to treat this aggressive cancer.
Acknowledgements
This study was performed as a partial fulfillment for the requirement of a Ph.D. degree by D. V., who was at the time of the study a graduate student at The University of Texas Graduate School of Biomedical Sciences in Houston. This work is supported by an R01 CA151533 grant from the National Cancer Institute (NCI) to H. M. A., a P30 CA125123 grant from the NCI to The Pathology and Histology Core at Baylor College of Medicine, and by Shanghai Scientific and Technological Innovation Project (14520720700). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NCI or the National Institutes of Health.
Competing Interests
The authors declare no competing financial interests.
Authors’ Contributions
D. V. developed the concept of the study, designed experiments, performed research, analyzed data, and wrote the paper; C. V. C., P. S., and S. A., provided essential experimental tools and analyzed data; H. M. A. developed the concept of and supervised the study, provided essential experimental tools, designed experiments, analyzed data, and wrote the paper. The authors read and approved the manuscript.
Footnotes
This work is supported by an R01 CA151533 grant from the National Cancer Institute (NCI) to H. M. A., a P30 CA125123 grant from the NCI to The Pathology and Histology Core at Baylor College of Medicine, and by Shanghai Scientific and Technological Innovation Project (14520720700).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2015.09.005.
Contributor Information
Deeksha Vishwamitra, Email: dtvishwa@mdanderson.org.
Hesham M. Amin, Email: hamin@mdanderson.org.
Appendix A. Supplementary data
Supplemental Figure 1. NPM-ALK amino acid sequence shows its SUMO consensus motifs. The online software SUMOplot revealed several potential SUMO conjugation sites that exist within the NPM-ALK amino acid sequence. We elected to focus on Lys24 and Lys32 because they appeared to have the highest probability of conjugation with the SUMO proteins (≥ 0.85).
Supplemental Figure 2. SENP1-induced decrease in levels of NPM-ALK is reversed by MG132. Treatment of Karpas 299 cells with SENP1 induced de-SUMOylation of NPM-ALK protein (Figure 5), which was associated with a remarkable decrease in the basal levels of NPM-ALK protein most likely through an increase in its association with ubiquitin (Figure 6). In this experiment, we show that SENP1-mediated downregulation of NPM-ALK protein levels was reversed when the proteasome inhibitor MG132 was simultaneously used to treat the cells with SENP1. These data further suggest that de-SUMOylation directs NPM-ALK protein to ubiquitination and proteasomal degradation.
Supplemental Figure 2.

SENP1-induced decrease in levels of NPM-ALK is reversed by MG132. Treatment of Karpas 299 cells with SENP1 induced de-SUMOylation of NPM-ALK protein (Figure 5), which was associated with a remarkable decrease in the basal levels of NPM-ALK protein most likely through an increase in its association with ubiquitin (Figure 6). In this experiment, we show that SENP1-mediated downregulation of NPM-ALK protein levels was reversed when the proteasome inhibitor MG132 was simultaneously used to treat the cells with SENP1. These data further suggest that de-SUMOylation directs NPM-ALK protein to ubiquitination and proteasomal degradation.
References
- 1.Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110:2259–2267. doi: 10.1182/blood-2007-04-060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 3.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, Levy DE, Inghirami G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 4.Amin HM, McDonnell TJ, Ma Y, Lin Q, Fujio Y, Kunisada K, Leventaki V, Das P, Rassidakis GZ, Cutler C. Selective inhibition of STAT3 induces apoptosis and G1 cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene. 2004;23:5426–5434. doi: 10.1038/sj.onc.1207703. [DOI] [PubMed] [Google Scholar]
- 5.Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol-3-kinase/Akt antiapoptotic signaling pathway. Blood. 2000;96:4319–4327. [PubMed] [Google Scholar]
- 6.Rassidakis GZ, Feretzaki M, Atwell C, Grammatikakis I, Lin Q, Lai R, Claret FX, Medeiros LJ, Amin HM. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105:827–829. doi: 10.1182/blood-2004-06-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu L, Lai R, Lin Q, Lau E, Thomazy DM, Calame D, Ford RJ, Kwak LW, Kirken RA, Amin HM. Autocrine release of interleukin-9 promotes Jak3-dependent survival of ALK+ anaplastic large-cell lymphoma. Blood. 2006;108:2407–2415. doi: 10.1182/blood-2006-04-020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leventaki V, Drakos E, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Claret FX, Rassidakis GZ. NPM-ALK oncogenic tyrosine kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood. 2007;110:1621–1630. doi: 10.1182/blood-2006-11-059451. [DOI] [PubMed] [Google Scholar]
- 9.Shi P, Lai R, Lin Q, Iqbal AS, Young LC, Kwak LW, Ford RJ, Amin HM. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood. 2009;114:360–370. doi: 10.1182/blood-2007-11-125658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi B, Vishwamitra D, Granda JG, Whitton T, Shi P, Amin HM. Molecular and functional characterizations of the association and interactions between nucleophosmin-anaplastic lymphoma kinase and type I insulin-like growth factor receptor. Neoplasia. 2013;15:669–683. doi: 10.1593/neo.122012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vishwamitra D, Curry CV, Alkan S, Song YH, Gallick GE, Kaseb AO, Shi P, Amin HM. The transcription factors Ik-1 and MZF1 downregulate IGF-IR expression in NPM-ALK+ T-cell lymphoma. Mol Cancer. 2015;14:53. doi: 10.1186/s12943-015-0324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997;17:2312–2325. doi: 10.1128/mcb.17.4.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quyang T, Bai RY, Bassermann F, von Klitzing C, Klumpen S, Miething C, Morris SW, Peschel C, Duyster J. Identification and characterization of nuclear interacting partner of anaplastic lymphoma kinase (NIPA) J Biol Chem. 2003;278:30028–30036. doi: 10.1074/jbc.M300883200. [DOI] [PubMed] [Google Scholar]
- 14.Shen Z, Pardington-Purtymun PE, Comeaux JC, Moyzis RK, Chen DJ. UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics. 1996;36:271–279. doi: 10.1006/geno.1996.0462. [DOI] [PubMed] [Google Scholar]
- 15.Boddy MN, Howe K, Etkin LD, Solomon E, Freemont PS. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukemia. Oncogene. 1996;13:971–982. [PubMed] [Google Scholar]
- 16.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 17.Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, Zheng W, Purohit S, Podolsky RH, Muir A. A functional variant of SUMO4, a new IκBα modifier, is associated with type 1 diabetes. Nat Genet. 2004;36:837–841. doi: 10.1038/ng1391. [DOI] [PubMed] [Google Scholar]
- 19.Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun. 2005;337:517–520. doi: 10.1016/j.bbrc.2005.09.090. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz SE, Matuschewski K, Liakopoulos D, Scheffner M, Jentsch S. The ubiquitin-like proteins SMT3 and SUMO-1 are conjugated by the UBC9 E2 enzyme. Proc Natl Acad Sci U S A. 1998;95:560–654. doi: 10.1073/pnas.95.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampson DA, Wang M, Matunis MJ. The small ubiquitin-like modifier–1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- 22.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 23.Kotaja N, Karvonen U, Jänne OA, Palvimo JJ. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol. 2002;22:5222–5234. doi: 10.1128/MCB.22.14.5222-5234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A. 2002;99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itahana Y, Yeh ET, Zhang Y. Nucleocytoplasmic shuttling modulates activity and ubiquitination-dependent turnover of SUMO-specific protease 2. Mol Cell Biol. 2006;26:4675–4689. doi: 10.1128/MCB.01830-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaikkonen S, Jääskeläinen T, Karvonen U, Rytinki MM, Makkonen H, Gioeli D, Paschal BM, Palvimo JJ. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol Endocrinol. 2009;23:292–307. doi: 10.1210/me.2008-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CH, Chang CC, Lee TH, Luo M, Huang P, Liao PH, Wei S, Li FA, Chen RH, Zhou XZ. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013;73:3951–3962. doi: 10.1158/0008-5472.CAN-12-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bayer P, Arndt A, Metzger R, Melchior F, Jaenicke R, Becker J. Structure determination of the small ubiquitin-related modifier SUMO-1. J Biol Chem. 1998;280:275–286. doi: 10.1006/jmbi.1998.1839. [DOI] [PubMed] [Google Scholar]
- 29.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 30.Buschmann T, Fuchs SY, Lee CG, Pan ZQ, Ronai Z. SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53. Cell. 2000;101:753–762. doi: 10.1016/s0092-8674(00)80887-9. [DOI] [PubMed] [Google Scholar]
- 31.Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and nuclear pore complex. J Cell Biol. 1996;135:1457–1470. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahajan R, Gerace L, Melchior F. Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. J Cell Biol. 1998;140:259–270. doi: 10.1083/jcb.140.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, Solomon E, de Thé H, Hay RT, Freemont PS. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci. 1999;112:381–393. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]
- 34.Sternsdorf T, Jensen K, Reich B, Will H. The nuclear dot protein Sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J Biol Chem. 1999;274:12555–12566. doi: 10.1074/jbc.274.18.12555. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999;18:6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Endter C, Kzhyshkowska J, Stauber R, Dobner T. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc Natl Acad Sci U S A. 2001;98:11312–11317. doi: 10.1073/pnas.191361798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood LD, Irvin BJ, Nucifora G, Luce KS, Hiebert SW. Small ubiquitin-like modifier conjugation regulates nuclear export of TEL, a putative tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:3257–3262. doi: 10.1073/pnas.0637114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smolen GA, Vassileva MT, Wells J, Matunis MJ, Haber DA. SUMO-1 modification of the Wilms’ tumor suppressor WT1. Cancer Res. 2004;64:7846–7851. doi: 10.1158/0008-5472.CAN-04-1502. [DOI] [PubMed] [Google Scholar]
- 39.Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T. The SUMO modification pathway is involved in BRCA1 response to genotoxic stress. Nature. 2009;462:886–890. doi: 10.1038/nature08593. [DOI] [PubMed] [Google Scholar]
- 40.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911. doi: 10.1038/ncomms1919. [DOI] [PubMed] [Google Scholar]
- 42.Li R, Wei J, Jiang C, Liu D, Deng L, Zhang K, Wang P. Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer Res. 2013;73:5742–5753. doi: 10.1158/0008-5472.CAN-13-0538. [DOI] [PubMed] [Google Scholar]
- 43.Ren YH, Liu KJ, Wang M, Yu YN, Yang K, Chen Q, Yu B, Wang W, Li QW, Wang J. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget. 2014;5:7093–7104. doi: 10.18632/oncotarget.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JH, Choi HJ, Kim B, Kim MH, Lee JM, Kim IS, Lee MH, Choi SJ, Kim KI, Chung CH. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nat Cell Biol. 2006;8:631–639. doi: 10.1038/ncb1415. [DOI] [PubMed] [Google Scholar]
- 45.Ganesan AK, Kho Y, Kim SC, Chen Y, Zhao Y, White MA. Broad spectrum identification of SUMO substrates in melanoma cells. Prtoeomics. 2007;7:2216–2221. doi: 10.1002/pmic.200600971. [DOI] [PubMed] [Google Scholar]
- 46.Driscoll JJ, Pelluru D, Lefkimmiatis K, Fulcniti M, Prabhala RH, Greipp PR, Barlogie B, Tai YT, Anderson KC, Shaughnessy JD., Jr. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115:2827–2834. doi: 10.1182/blood-2009-03-211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, Bille K, Dessen P, d’Hayer B, Mohamdi H, Remenieras A. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal cell carcinoma. Nature. 2011;480:94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 48.Bonacci T, Audebert S, Camoin L, Baudelet E, Bidaut C, Garcia M, Witzel II, Perkins ND, Borg JP, Iovanna JL. Identification of new mechanisms of cellular response to chemotherapy by tracking changes in post-translational modifications by ubiquitin and ubiquitin-like proteins. J Proteome Res. 2014;13:2478–2494. doi: 10.1021/pr401258d. [DOI] [PubMed] [Google Scholar]
- 49.Bogachek MV, Chen Y, Kulak MV, Woodfield GW, Cyr AR, Park JM, Spanheimer PM, Li Y, Li T, Weigel RJ. Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell. 2014;25:748–761. doi: 10.1016/j.ccr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarge KD, Park-Sarge OK. Detection of proteins sumoylated in vivo and in vitro. Methods Mol Biol. 2009;590:265–277. doi: 10.1007/978-1-60327-378-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villalva C, Trempat P, Greenland C, Thomas C, Girard JP, Moebius F, Delsol G, Brousset P. Isolation of differentially expressed genes in NPM-ALK-positive anaplastic large cell lymphoma. Br J Haematol. 2002;118:791–798. doi: 10.1046/j.1365-2141.2002.03671.x. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification and nuclear targeting. J Biol Chem. 2001;276:12654–12659. doi: 10.1074/jbc.M009476200. [DOI] [PubMed] [Google Scholar]
- 53.Song J, Zhang Z, Hu W, Chen Y. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J Biol Chem. 2005;280:40122–40129. doi: 10.1074/jbc.M507059200. [DOI] [PubMed] [Google Scholar]
- 54.Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014;15:878–885. doi: 10.15252/embr.201438534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Won TJ, Lee YJ, Hyung KE, Yang E, Sohn UD, Min HY, Lee DI, Park SY, Hwang KW. SUMO2 overexpression enhances the generation and function of interleukin-17–producing CD8+ T cells in mice. Cell Signal. 2015;27:1246–1252. doi: 10.1016/j.cellsig.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Lee JE, Kim JH. SUMO modification regulates the protein stability of NDRG1. Biochem Biophys Res Commun. 2015;459:161–165. doi: 10.1016/j.bbrc.2015.02.090. [DOI] [PubMed] [Google Scholar]
- 57.Kim EY, Zhang Y, Ye B, Segura AM, Beketaev I, Xi Y, Yu W, Chang J, Li F, Wang J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. Biochim Biophys Acta. 2015;1852:1388–1399. doi: 10.1016/j.bbadis.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 58.Gong L, Millas S, Maul GG, Yeh ET. Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J Biol Chem. 2000;275:3355–3359. doi: 10.1074/jbc.275.5.3355. [DOI] [PubMed] [Google Scholar]
- 59.Xu Z, Au SW. Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochem J. 2005;386(Pt 2):325–330. doi: 10.1042/BJ20041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gong L, Yeh ET. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J Biol Chem. 2006;281:15869–15877. doi: 10.1074/jbc.M511658200. [DOI] [PubMed] [Google Scholar]
- 61.Mikolajczyk J, Drag M, Békés M, Cao JT, Ronai Z, Salvesen GS. Small ubiquitin-related modifier (SUMO)–specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem. 2007;282:26217–26224. doi: 10.1074/jbc.M702444200. [DOI] [PubMed] [Google Scholar]
- 62.George SK, Vishwamitra D, Manshouri R, Shi P, Amin HM. The ALK inhibitor ASP3026 eradicates NPM-ALK+ T-cell anaplastic large-cell lymphoma in vitro and in a systemic xenograft lymphoma model. Oncotarget. 2014;5:5750–5763. doi: 10.18632/oncotarget.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bailey D, O’Hare P. Characterization of the localization and proteolytic activity of the SUMO-specific protease, SENP1. J Biol Chem. 2004;279:692–703. doi: 10.1074/jbc.M306195200. [DOI] [PubMed] [Google Scholar]
- 64.Vertegaal AC, Ogg SC, Jaffray E, Rodriguez MS, Hay RT, Andersen JS, Mann M, Lamond AI. A proteomic study of SUMO-2 target proteins. J Biol Chem. 2004;279:33791–33798. doi: 10.1074/jbc.M404201200. [DOI] [PubMed] [Google Scholar]
- 65.Ross S, Best JL, Zon LI, Gill G. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol Cell. 2002;10:831–842. doi: 10.1016/s1097-2765(02)00682-2. [DOI] [PubMed] [Google Scholar]
- 66.Kishi A, Nakamura T, Nishio Y, Maegawa H, Kahiwagi A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am J Physiol Endocrinol Metab. 2003;284:E830–E840. doi: 10.1152/ajpendo.00390.2002. [DOI] [PubMed] [Google Scholar]
- 67.Lin X, Liang M, Liang YY, Brunicardi FC, Feng XH. SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J Biol Chem. 2003;278:31043–31048. doi: 10.1074/jbc.C300112200. [DOI] [PubMed] [Google Scholar]
- 68.Bensault-Mascard L, Leprince C, Aufferdou MT, Meunier B, Bourgeade MF, Camonis J, Lorenzo HK, Vazquez A. Caspase-8 sumoylation is associated with nuclear localization. Oncogene. 2005;24:3268–3273. doi: 10.1038/sj.onc.1208448. [DOI] [PubMed] [Google Scholar]
- 69.Du JX, Bialkowska AB, McConnell BB, Yang VW. SUMOylation regulates nuclear localization of Krüppel-like factor 5. J Biol Chem. 2008;283:31991–32002. doi: 10.1074/jbc.M803612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yun C, Wang Y, Mukhopadhyay D, Backlund P, Kolli N, Yergey A, Wilkinson KD, Dasso M. Nuclear protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J Cell Biol. 2008;183:589–595. doi: 10.1083/jcb.200807185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pichler A, Knipscheer P, Oberhofer E, van Dijk WJ, Körner R, Olsen JV, Jentsch S, Melchoir F, Sixma TK. SUMO modification of the ubiquitin-conjugating enzyme E2-25 K. Nat Struct Mol Biol. 2005;12:264–269. doi: 10.1038/nsmb903. [DOI] [PubMed] [Google Scholar]
- 72.Klenk C, Humrich J, Quitterer U, Lohse MJ. SUMO-1 controls the protein stability and the biological function of phosducin. J Biol Chem. 2006;281:8357–8364. doi: 10.1074/jbc.M513703200. [DOI] [PubMed] [Google Scholar]
- 73.de Cristofaro T, Mascia A, Pappalardo A, D’Andrea B, Nitsch L, Zannini M. Pax8 protein stability is controlled by sumoylation. J Mol Endocrinol. 2009;42:35–46. doi: 10.1677/JME-08-0100. [DOI] [PubMed] [Google Scholar]
- 74.Mooney SM, Grande JP, Sailsbury JL, Janknecht R. Sumoylation of p68 and p72 RNA helicases affects protein stability and transactivation potential. Biochemistry. 2010;49:1–10. doi: 10.1021/bi901263m. [DOI] [PubMed] [Google Scholar]
- 75.Bonvini P, Dalla Rosa H, Vignes N, Rosolen A. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70–interacting protein. Cancer Res. 2004;64:3256–3264. doi: 10.1158/0008-5472.can-03-3531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. NPM-ALK amino acid sequence shows its SUMO consensus motifs. The online software SUMOplot revealed several potential SUMO conjugation sites that exist within the NPM-ALK amino acid sequence. We elected to focus on Lys24 and Lys32 because they appeared to have the highest probability of conjugation with the SUMO proteins (≥ 0.85).
Supplemental Figure 2. SENP1-induced decrease in levels of NPM-ALK is reversed by MG132. Treatment of Karpas 299 cells with SENP1 induced de-SUMOylation of NPM-ALK protein (Figure 5), which was associated with a remarkable decrease in the basal levels of NPM-ALK protein most likely through an increase in its association with ubiquitin (Figure 6). In this experiment, we show that SENP1-mediated downregulation of NPM-ALK protein levels was reversed when the proteasome inhibitor MG132 was simultaneously used to treat the cells with SENP1. These data further suggest that de-SUMOylation directs NPM-ALK protein to ubiquitination and proteasomal degradation.